 Linjun Yan
Linjun Yan Huanli Wang1†
Huanli Wang1†- 1Jiangsu Key Laboratory for the Research and Utilization of Plant Resources, Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing Botanical Garden, Memorial Sun Yat-Sen, Nanjing, China
- 2State Key Laboratory of Tree Genetics and Breeding, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, China
Tilia is a complex genus in the family Malvaceae that has high ecological and economical values. Owing to the lack of sufficient distinguishable morphological and molecular characteristics, interspecific relationships in this genus are not clear. Chloroplast (cp) genomes are small, meanwhile most angiosperms usually undergo matrilineal inheritance. Consequently, they can be used in molecular systematics and phylogenetic analyses. Here, we sequenced and assembled cp genomes from T. endochrysea, T. tomentosa, T. miqueliana, T. americana and T. cordata, and compared them with those of seven previously reported Tilia species. Similar gene contents, gene orders and GC contents existed among the 12 cp genomes, which ranged from 162,564 to 162,855 bp and encoded 113 unique genes. Abundant simple sequence repeats (119–127) and dispersed repeats (97–135) were detected in Tilia cp genomes. In total, 11 hypervariable regions were identified that could be suitable for species identification and phylogenetic studies. A phylogenetic analysis of Malvaceae based on 5 hypervariable genes (matK + ndhF + rpoB + rpoC2+ycf1) revealed that all eight subfamilies were monophyletic groups. Additionally, the genus Tilia was divided into three groups on the basis of all 521 molecular variation loci. The current study provides valuable insights into the genomic evolution of the genus Tilia.
Introduction
Malvaceae, the most diverse family within Malvales, consists of 244 genera and 4,225 species. It is divided into nine subfamilies by phenotypic traits, which are Brownlowioideae, Bombacoideae, Byttnerioideae, Dombeyoideae, Grewioideae, Helicteroideae, Malvoideae, Tilioideae and Sterculioideae. Tilia L. (linden, basswood or lime-tree) is a genus of the subfamily Tiliodeae of family Malvaceae, which is widely distributed across the Northern Hemisphere. It has a unique trait that is the bracts adnate to the peduncle of inflorescence, which is intermittently distributed in the temperate and subtropical regions of East Asia and North America, as well as from Europe to western Siberia (Pigott, 2012; Bennett and Alarcón, 2015). Tilia are valuable woody species that have long been cultivated around the world, because they are a source of honey, materials for furniture and attractive landscaping trees. Tilia also provides good medicinal effects, with its inflorescences and bracts being used as a medicine to treat coughing, anxiety and insomnia-related disorders (Negri et al., 2013; Delnavazi et al., 2015). In addition, higher polyene fatty acids found in the fruit of Tilia are considered bioactive compounds (Siger et al., 2021).
In traditional taxonomy, they are usually classified using fruit and leaf characteristics, geographical distribution and cytological features (number of chromosomes) (Mabberley, 2017). Some scholars divided the genus Tilia into three groups that is Sect. Trichophilyra, Sect. Lindnera and Sect. Tilia according to a more stable traits, whether the outer pericarp of the fruit is dehiscence or not (Tang and Zhuge, 1996). However, physical traits vary and cross between species, so their relationships and evolutionary histories of Tilia species, cannot be adequately differentiated. Tilia conservation and taxonomy revision are thus challenging. As the cost of high throughput sequencing was still going down, a well-supported phylogenetic framework of Malvaceae was built based on the whole cp genomes and the nine subfamilies were retrieved, which is a valuable genomic resource to further investigate the evolutionary history at lower taxonomic levels. The phylogeny of four Tilia species in China was well resolved based on the complete plastid genome sequences, however, the sampled species are too limited to represent the most species in Tilia (Cai et al., 2015).
In eukaryotes, the chloroplast (cp) represents the core organelle for photosynthesis and carbon fixation. It was lack of recombination and slow evolution compared to the nuclear genome, so it was a good candidate for resolving phylogenetic and taxonomic discrepancies and developing species barcoding. It has been widely used in many genera and family phylogenetic analyses, such as those of Poaceae (Feng and Gao, 2021a; Feng et al., 2021b), Cucumis (Zhai et al., 2021) and Adenophora (Kim and Cheon, 2021). Taxonomical discrepancies of some Malvaceae plants had been resolving by identifying part of suitable mutational hotpots. (Abdullah et al., 2019; Abdullah et al., 2020). Compared to the complexity of using a whole cp genome, the hypervariable regions in chloroplast genome could be used as polymorphic and robust markers.
In this paper, we selected five Tilia species distributed respectively in Asia, Europe and America, which included the main origins of Tilia. Whereafter, we sequenced the cp genomes of the above five Tilia species and compared them to the reported cp genomes of seven other Tilia species. Meanwhile, all the 12 species were belonged to the all three fruit traits based on traditional morphological classification. Our study mainly aimed to: 1) comprehensively analyze the characteristics of Tilia cp genome; 2) determine the phylogenetic relationships among the 12 Tilia species; and 3) screen the suitable hypervariable genes or regions for species barcoding and phylogenetic analysis.
Results
Characteristics of Chloroplast Genomes
In this study, five cp genome of Tilia were sequenced, and 300 M (T. tomentosa), 288 M (T. endochrysea), 37 M (T. americana), 38 M (T. miqueliana) and 52 M (T. cordata) 150-bp paired-end reads were used for cp genome assemblies. The annotated genome sequences of T. endochrysea, T. tomentosa, T. miqueliana, T. americana and T. cordata were deposited in GenBank under the accession numbers OK624380, OM908761, OM914582, OM908762 and OM908760, respectively (Figure 1).
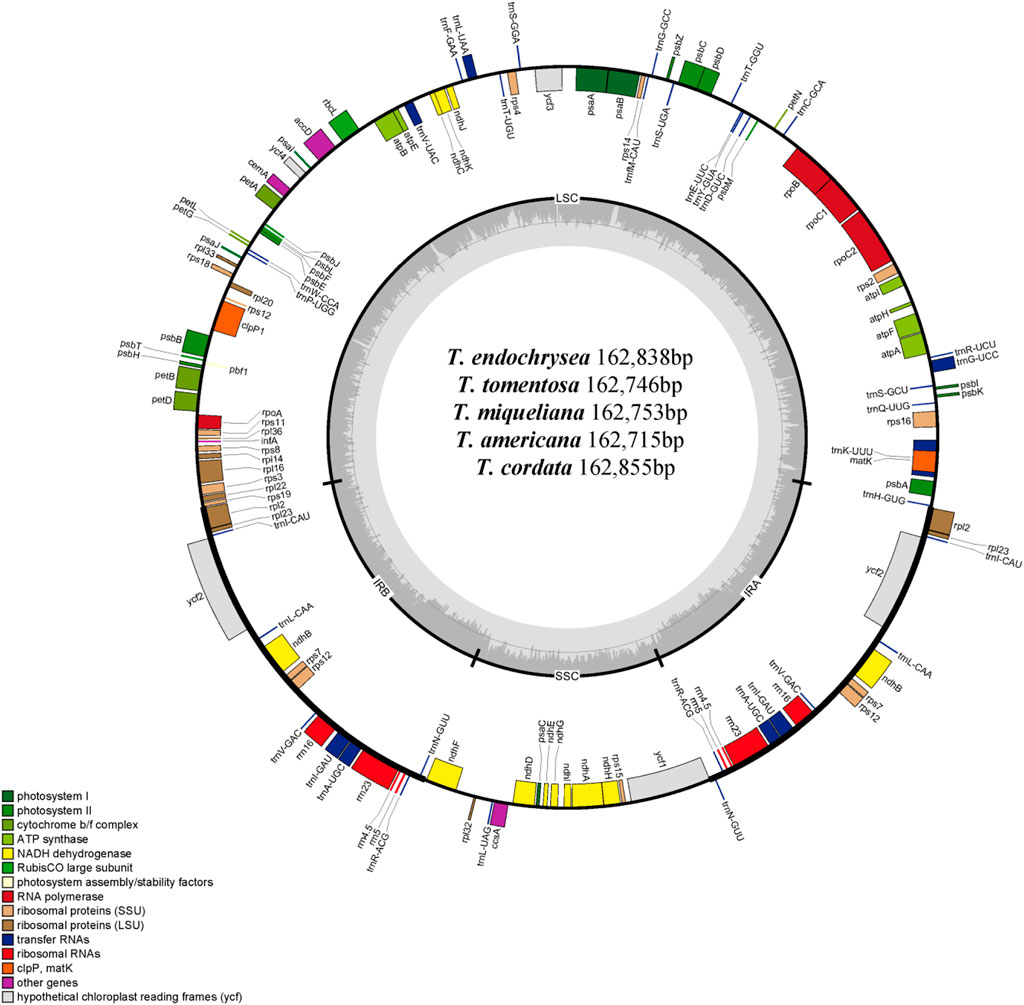
FIGURE 1. Circular gene map of the chloroplast genomes of T. endochrysea, T. tomentosa, T. miqueliana, T. americana and T. cordata. Genes drawn within the circle are transcribed clockwise, whereas those drawn outside are transcribed counterclockwise. Genes are color-coded in accordance with their functional groups. The inner circle represents the GC content.
Complete cp genome lengths of 12 Tilia species ranged from 162,564 to 162,855 bp, and the GC contents were all 36.5%. They exhibited the typical quadripartite structure, consisting of a large single-copy region (LSC, 91,055–91,264 bp, 34.1% GC content), a small single-copy region (SSC, 20,355–20,478 bp, 30.9%–31.0% GC content) and a pair of inverted repeats (IRs, 25,491–25,655 bp, 42.8%–42.9% GC content) (Table 1). All 12 cp genomes contained 130 genes, with 113 being unique, including 79 protein-coding genes, 30 tRNAs and 4 rRNAs. Among them, 17 genes were duplicated in the IR regions, and 18 genes contained a single intron or two introns, including 6 tRNA genes and 12 protein-coding genes (Table 2).
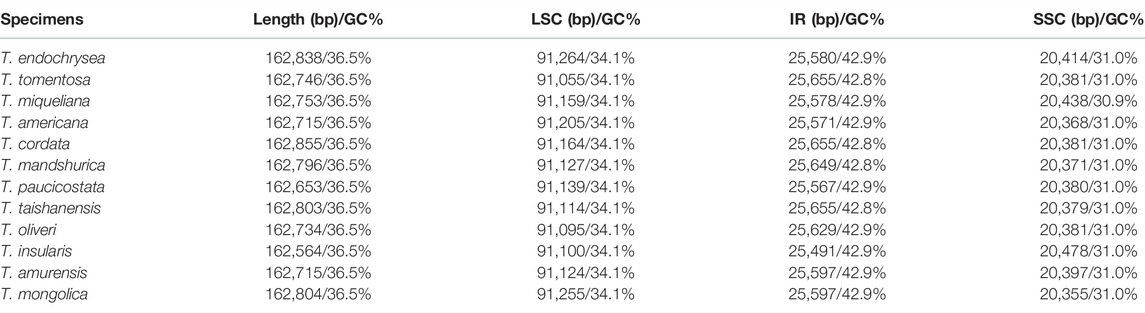
TABLE 1. Characteristics of complete chloroplast genomes of Tilia species.

TABLE 2. Genes encoded in the chloroplast genomes of five Tilia species.
Simple Sequence Repeats and Dispersed Repeats
The number of Simple Sequence Repeats (SSRs) in the 12 cp genomes of Tilia ranged from 119 to 127. The most abundant SSR type was single nucleotide repeats, accounting for 60.00 %–68.55%, followed by dinucleotide repeats (12.10 %–15.20%), pentanucleotide repeats (6.40%–12.10%), and tetranucleotide repeats (7.56%–10.40%). The trinucleotide repeats were the least common (3.20%–5.88%) (Figure 2A). The A/T-type mononucleotides were the most abundant SSRs, and there were no G-type mononucleotides in the species, except T. miqueliana, T. americana and T. mongolica (Figure 2B). Without taking into account mononucleotide repeats, non-coding regions contained 88.2% of the remaining repeats in the genomes, with the repeats being most abundant in the rpl33-rps18 and psbZ-trnG. Meanwhile, there were abundant SSRs in gene ycf1 (Supplementary Table S1).
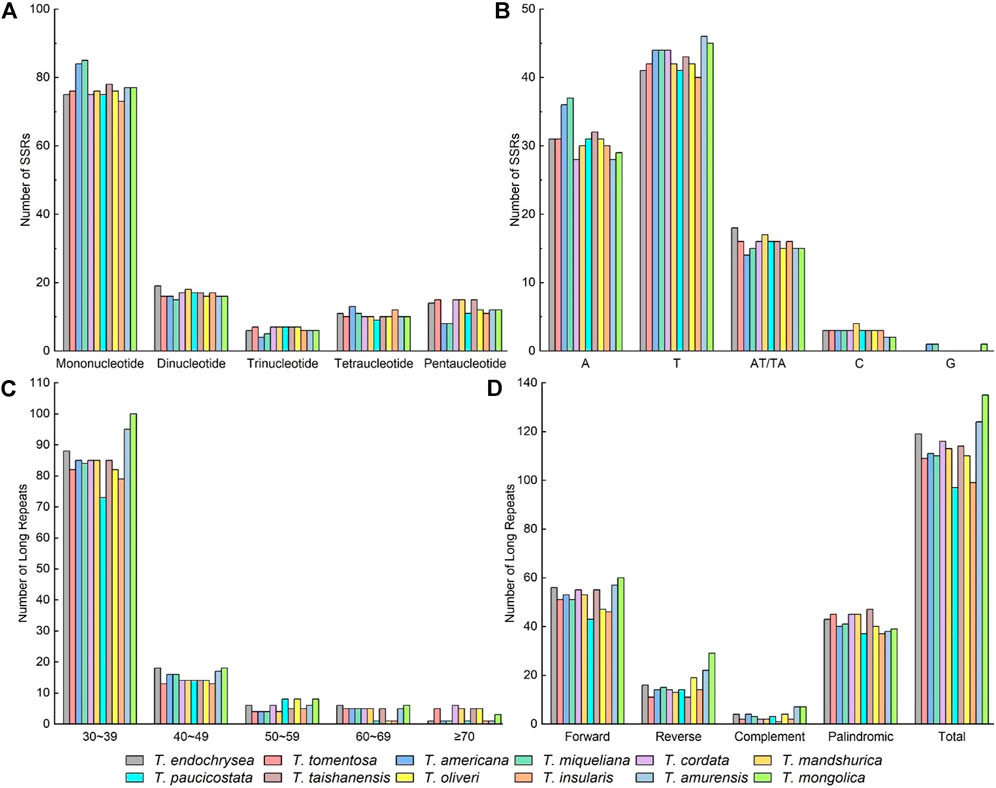
FIGURE 2. Distribution of SSRs and dispersed repeats in the chloroplast genomes of Tilia. (A) Numbers of different SSR types detected in the cp genomes; (B) Numbers of different SSR class types; (C) Numbers of the four dispersed repeat types in Tilia; (D) Numbers of dispersed repeat types having a given length interval (30 to 39, 40 to 49, 50 to 59, 60 to 69 and ≥ 70).
There were 97–135 unique dispersed repeats including forward, complement, reverse and palindromic in the cp genomes of Tilia (Figure 2C). Among all the species, forward and palindromic repeats were the most common types, and complement repeats were the least common type. Tilia amurensis and T. mongolica had the maximum numbers of forward, reverse and complement repeats. Most of the dispersed repeats were less than 40 bp in length, and this was consistent among the 12 species (Figure 2D).
Inverted Repeat Contraction, Expansion, and Interspecific Comparison
We analyzed the junctions of the IRs and the two single-copy regions, along with the placement of adjacent genes in the 12 Tilia cp genomes. The genes located at the junctions included rps19, rpl2, ndhF, ycf1 and trnH. The rps19 and rpl2 genes were detected at the junction of LSC and IRb. The rpl2 gene was entirely located within the IRb region. The rps19 genes of T. mongolica and T. amurensis spanedspaned the LSC and IRb boundary, unlike in the other 10 Tilia cp genomes in which the rps19 gene was located in LSC region with 1 to 19-bp interval before the LSC and IRb boundary. The ndhF genes of 12 Tilia are located in the SSC region with 76 to 146-bp intervals to the IRb and SSC boundary. For all 12 species, the ycf1 genes crossed the SSC and IRa boundary with 36 bp in IRa and the trnH gene was located in the LSC region with a 31 to 56-bp interval to the boundary of Ira and LSC (Figure 3).

FIGURE 3. Comparison of the boundaries of large single-copy (LSC), small single-copy (SSC) an inverted repeat (IR) regions of 12 chloroplast genomes of Tilia.
Sequence Divergence Analysis
Multiple alignments of 12 Tilia plastomes were compared using mVISTA with T. endochrysea as a reference. The results revealed a low divergence level among the 12 sequences. In general, coding regions had higher sequence identities than non-coding regions. The main divergences for the coding regions were in ycf1, matK, ndhF and rpoC2, and for the noncoding regions, the most strongly divergent sequences were psbZ-trnG, trnT-trnL, atpB-rbcL, rpl33-rps18 and ndhF-rpl32 (Supplementary Figure S1).
In total, 521 variation sites and 246 InDels were identified in the whole cp genome sequences of the 12 species. Among them, 140 variation sites were in genes, with 59 synonymous variants in 27 genes, 80 missense variants and 79 intron variant. The coding genes having the most variation sites were ycf1, rpoC2 and rpoB (34, 12 and 7, respectively). The IGS (intergenic region) having most variation sites were trnT-trnL, ndhF-rpl32, psbZ-trnG and trnT-psbD. The coding gene with the most InDels was ycf1 (Figure 4; Supplementary Table S2).
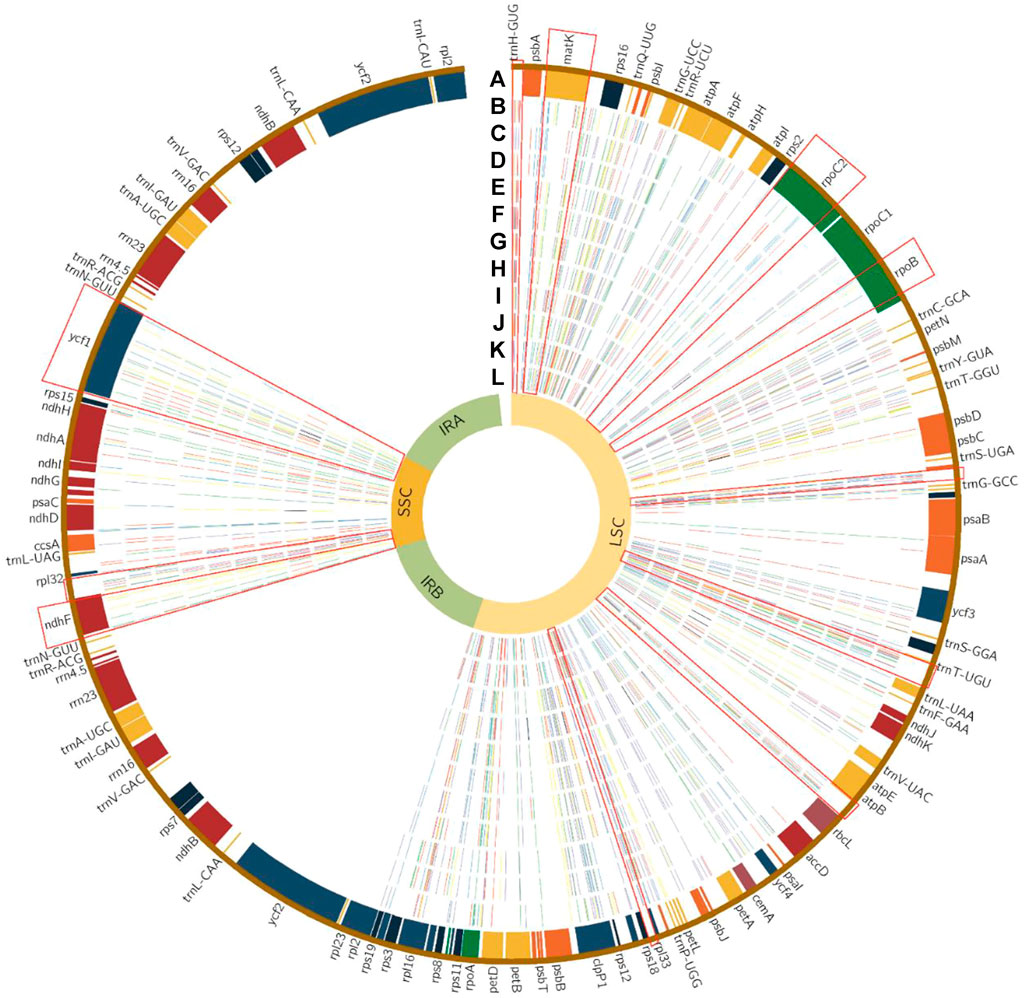
FIGURE 4. Single nucleotide polymorphic sites in 12 chloroplast genomes of Tilia. Track A represents the encoding genes. Tracks B–L represent variant sites in T. tomentosa, T. americana, T. miqueliana, T. cordata, T. mandshurica, T. paucicostata, T. taishanensis, T. oliveri, T. insularis, T. amurensis and T. mongolica compared with T. endorchrysea. The red, blue, yellow and green lines on each track indicate the kinds of variant, A, T, C and G nucleotides, respectively. Purple lines indicate InDel regions. The red rectangles represent the locations of the 11 selected hypervariable regions.
Phylogenetic Analysis
To explore the phylogenetic positions and evolutionary relationships among Tilia species, a maximum-likelihood (ML) tree was constructed using 38 species from 8 subfamilies of Malvaceae. According to the number of mutation loci contained, the maximum five regions were ycf1, matK, nhdF, rpoB, and rpoC2. Five hypervariable region genes were used to construct a phylogenetic tree with two species of Malvales and four species of Brassicales as outgroups, and the variation sites in the 12 Tilia species were used to construct a Tilliodeae tree. The phylogenetic tree based on five hypervariable region genes showed Tilia was sister to Craigia and all eight subfamilies of Malvaceae as monophyletic with strong support. The 12 species of Tilia were divided into 3 clades, with T. americana, T. miqueliana and T. endochrysea forming one clade, T. amurensis and T. mongolica forming one clade, and the remaining 7 species forming the other clade (Figure 5).
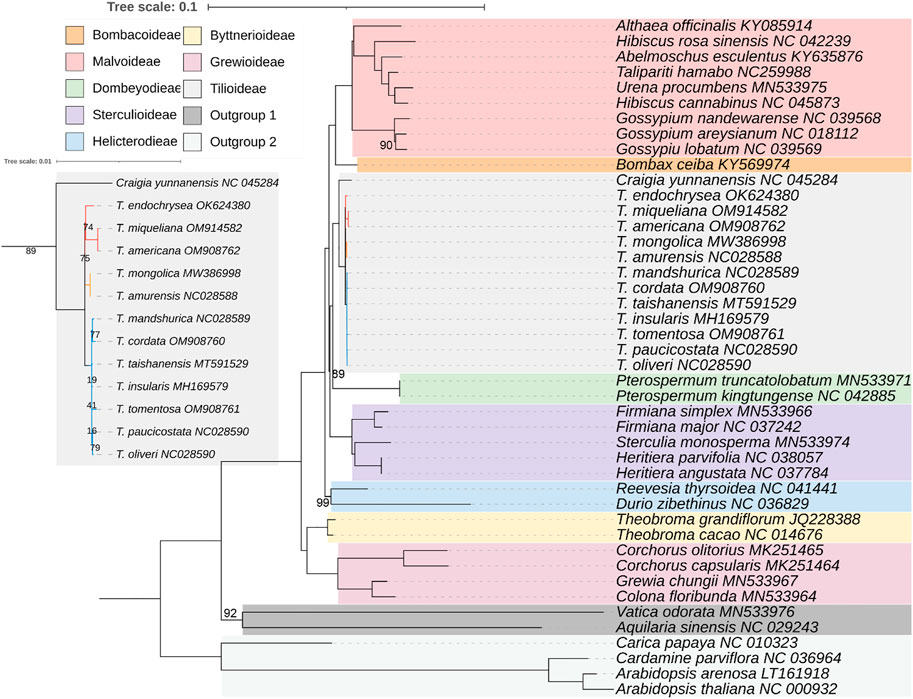
FIGURE 5. Maximum-likelihood phylogenetic inference of 38 Malvaceae species based on matK + ndhF + rpoB + rpoC2 + ycf1 genes. The numbers associated with the nodes are bootstrap support and posterior probability values. All the nodes with bootstrapping equal to 100 was not mentioned. 1000 bootstrap replicates.
In the phylogenetic tree constructed using all variation sites, the 12 Tilia species were divided into 3 clades with a bootstrap value of 1,000 and Craigia yunnanensis as the outgroup. The three clades were as follows: 1) T. americana, T. miqueliana and T. endochrysea; 2) T. amurensis and T. mongolica; 3) T. mandshurica, T. cordata, T. tomentosa, T. insularis, T. taishanensis, T. paucicostata and T. oliveri (Figure 6).
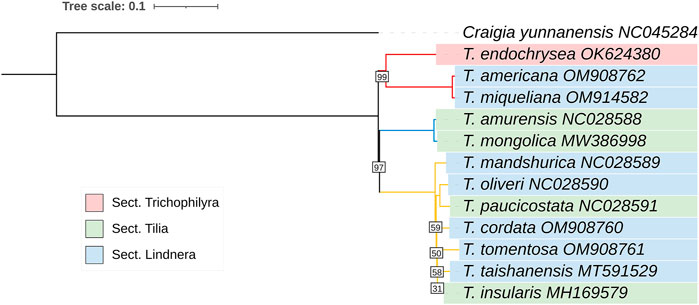
FIGURE 6. Maximum-likelihood treeof 12 Tilia species based on all variation sites. The numbers associated with the nodes are bootstrap support and posterior probability values. All the nodes with bootstrapping equal to 100 was not mentioned. 1000 bootstrap replicates.
Combining the data of the two evolutionary trees, the branch lengths leading to Tilia were short, indicating that Tilia evolved more slowly than other species in Malvaceae.
Discussion
Compared with the seven previously sequenced cp genomes, the five newly sequenced cp genomes had similar lengths and exhibited the typical quadripartite structure. The number of introns and positions of genes were identical in all 12 species, which indicated that the cp genome was highly conserved during evolution. Overall, the lengths of the 12 Tilia cp genomes, ranging from 162,564 to 162,855 bp, were larger than most species of Malvaceae, and the GC contents of the Tilia species (36.5%) was at the low level among the Malvaceae (Wang et al., 2021), being slightly lower than Gossypium (37.2%–37.4%) (Chen et al., 2016), Theobroma (36.9%) (Jansen et al., 2011), Bombax (36.8%) (Gao et al., 2018) and Heritiera (36.8%) (Zhao et al., 2018). In general, the over-all GC content is an important species indicator (Shen et al., 2017). A high GC content is often associated with the earlier divergence of a phylogenetic position (i.e., Nymphaeales and Magnoliids) (Cai et al., 2006). Therefore, we concluded that Tilia differentiated later among the Malvaceae. This conclusion was consistent with divergence time estimation of Malvaceae based on coding gene sequences (Wang et al., 2021).
SSRs often contain 1–6 nucleotides and are primary sources of molecular markers for population genetics and biogeographic studies (Kyalo et al., 2018; Mustafina et al., 2019). In this study it was consistent with previous research results that single nucleotide repeats of A/T were the most common among all the repeat types (Qian et al., 2013; Yi and Kim, 2016; Jiang et al., 2017). A potential reason for the higher frequencies of the A/T repeats is polyadenylation at the end of mRNAs in the cp genes of many species. Additionally, the strand separation for A/Ts is relatively easier than G/Cs during plastome replication, which increases slipped-strand mispairing (Zhao et al., 2020). Meanwhile, SSRs with two to five repeat motifs were found in the 12 cp genomes, with 88.2% of them localized in non-coding areas (88.2% on average). SSRs tend to appear in the cp non-coding regions because of the strong selection against them in coding regions (Menezes et al., 2018). The SSRs identified will be helpful in future population genetic studies and evolutionary analyses. Occasional short sequence repeats and mutations in DNA sequences can be extended into longer tandem repeats through slip-chain mismatch events, and transposon-mediated insertion and copy slip may be responsible for both direct and reverse dispersed repeats (Levinson and Gutman, 1987). In this paper, the forward and palindromic repeats were the most common repeat types and most of the dispersed repeats were less than 40 bp, as the previous studies was reported (Liu et al., 2019; Kirov et al., 2020; Luo et al., 2021).
Although IR regions are highly conserved in most plants, especially within the same genus, structural variations in the IR/SC boundary regions are very common. The contraction and expansion of IR/SC regions are the main reasons for plastid length differences (Zhu et al., 2016; Yu et al., 2019), and they play important roles in evolution (Kim and Lee, 2004; Wang et al., 2008). By comparing boundary regions among the 12 Tilia cp genomes, we found that the numbers and orders of genes were conserved (Li et al., 2018; Ye et al., 2019). Previous studies showed that the rps19 genes in some species of plants (Mu et al., 2018; Xie et al., 2018; Zhang et al., 2020; Li et al., 2021) were pseudogenes, and they were found both in the IRa or the IRa/LSC boundary and in the IRb or the IRb/SSC boundary. However, there is only one copy of rps19 in the Tilia cp genomes. Meanwhile, some differences existed at the boundaries, with the most obvious being for rps19 in T. mongolica and T. amurensis located at the boundary of IRa/LSC. It suggested that T. mongolica and T. amurensis had similar cp gene structures, which were more closely related in evolutionary terms.
As other woody species genera, the 12 whole-cp genome sequences showed low divergence in general. For the Tilia tree, it takes 6 to 40 years for beginning to flower and produce seed. The low mutation rates in these cp genomes may be attributed to their long generation times (Ren et al., 2018). ycf1, ndhF, rpoB, rpoC2, matK, trnH-psbA, psbZ-trnG-, trnT-trnL, atpB-rbcL, rpl33-rps18 and ndhF-rpl32. Our results showed that ycf1, ndhF, rpoB, rpoC2, matK, trnT-psbD, psbZ-trnG, trnT-trnL, atpB-rbcL, rpl33-rps18 and ndhF-rpl32 were the mutational hotspots, which were partly the same as the other Malvaceae species and many other species (Zhao et al., 2015; Mo et al., 2020; Wang et al., 2021). These regions could be good candidate regions for Tilia species identification. In addition, 521 variation sites and 246 InDels were identified in the 12 Tilia species. Most of the variants were in the intron and missense mutations, which might be related to evolution under positive selection (Kim et al., 2017). Through the analysis, we found that the most of mutational hotspots were concentrated in IGS, which was consistent with the occurrence area of SSRs. The variation sites overlapped with the abundant areas of SSRs (ycf1, rpl33-rps18 and psbZ-trnG). Such a phenomenon has been confirmed in Malvaceae (Abdullah et al., 2021a), and be universal in plant cp genomes. This co-occurrence of subsitutions with repeats supported the repeats could be used as identification of mutational hotpots (Li et al., 2020; Abdullah et al., 2021b).
We selected five hypervariable region genes (matK, nhdF, rpoB, rpoC2 and ycf1) for phylogenetic tree construction. All eight subfamilies of Malvaceae were monophyletic with high bootstrap support values, and the genera Tilia was divided into three clades. Thus, some genes were shown to act as a promising plastid genomic barcode, such as ycf1, matK and rbcL (Dong et al., 2015; Amar, 2020). Chloroplast genes were highly conserved sequences in general. Compared with full-length genes, it is more reliable and convenient to construct an evolutionary tree with hypervariable genes. In order to improve the bootstrap support values of the phylogeny tree genus Tilia, we reconstructed a ML tree of Tilia based on all variation sites. It is revealed that the phylogenetic relationship of Tilia based on all variation sites was consistent with tree constructed by the hypervariable five genes. The conflicts between molecular and morphological characteristics existed in genus Tilia. For example, in traditional taxonomy, genus Tilia is divided into three groups based on fruit morphology, Sect. Trichophilyra, Sect. Lindnera and Sect. Tilia. Whereas, Sect. Trichophilyra was a paraphyletic group and Sect. Lindnera and Sect. Tilia are polyphyletic based on molecular evidence. According to the molecular phylogenetic evidence, T. miqueliana and T. americana were more closely related to T. endochrysea. T. insularis is a subspecies of T. amurensis on basis of the vegetative and reproductive morphological evidence (Pigott, 2012; Yang et al., 2018), but the results of this study suggested that T. amurensis and T. insularis are two individual species and T. amurensis is more closely related to T. mongolica. The conflicts between molecular and morphological due to the long reproductive cycles, long history of anthropogenic spread of Tilia and recently rapid differentiation by frequently occurred interspecies hybridization (Pigott, 2012; Cvetković et al., 2021).
In this study, we successfully reconstructed the phylogenetic trees of Malvaceae and Tilia, and they indicated that plastid phylogenomics could be used to determine the interspecific relationships within the genera Tilia. Meanwhile, the hypervariable regions would be selected to build the phylogenetic tree and develop species barcoding.
Materials and Methods
DNA Sequencing, Genome Assembly and Gene Annotation
Fresh leaves were obtained from adult trees of T. endochrysea, T. tomentosa “Sterling”, T. miqueliana, T. americana and T. cordata planted at the Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, China. Total genomic DNA was extracted using a modified cetyltrimethylammonium bromide method and applied to 150-bp paired-end library construction using the NEBNext Ultra DNA Library Prep Kit for Illumina (New England BioLabs, Ltd., United States) sequencing. Sequencing was carried out on the Illumina NovaSeq 6000 platform (BIOZERON Co., Ltd., Shanghai, China). De novo assembly of the cp genome of the closely related species T. oliveri (NC 028590) using NOVOPlasty (Dierckxsens et al., 2017) produced two circular optional contigs of the cp genome. The candidate cp genome was chosen because it had the highest resemblance to T. oliveri cpDNA. BLAST searches against cp genomes of the related species T. oliveri and the NOVOPlasty results were used to retrieve a number of possible cp reads from the pool of Illumina data. Using SPAdes-3.13.0 software (Bankevich et al., 2012), cp Illumina data were collected to perform cp genome de novo assembly. The scaffolds from the SPAdes-3.13.0 (Antipov et al., 2016) result were used to optimize the NOVOPlasty assembly contig, which was then aligned with the original clean Illumina reads using Burrows-Wheeler-Alignment Tool (Li and Durbin, 2009) and base corrected using Pilon v1.22 (Luo et al., 2012). The assembled sequences were then reordered and orientated in accordance with the reference cp genome, resulting in the final assembled cp genomic sequence. The plastid genome annotator tool (Qu et al., 2019) was used to annotate the genomes, and the start and end codon boundaries were manually corrected if necessary.
Identification of Repeat Sequences and Simple Sequence Repeats
REPuter (Kurtz et al., 2001) was used to identify repeat sequences, which had four forms t: forward, reverse, complementary and palindromic, in the cp genomes. The detection parameters were set to a minimum repeat size of 30 bp and a 3-bp edit distance.
The MicroSatellite identification program (Beier et al., 2017) (https://pgrc.ipk-gatersleben.de/misa/) was used to identify SSRs in the cp genome sequences using the following parameter settings: size of a unit (nucleotide) 1_10, 2_5, 3_4, 4_3, 5_3, 6_3 were the minimum repetitions. A minimum spacing of 100 bp was imposed between two SSRs.
Comparison of Genome Structures, and an Inverted Repeat Region Contraction and Expansion Analysis
Using IRscope (https://irscope.shinyapps.io/irapp/), the placements of IR, SSC, and LSC junctions were compared in the 12 cp genomes. mVISTA was used to visualize the variations among the 12 Tilia cp genomes (default parameters and LAGAN mode) (Frazer et al., 2004).
Polymorphism Analysis and Phylogenetic Analysis of Chloroplast Genomes
Each sample was compared with T. endochrysea as a global reference sequence using snippy software (https://github.com/tseemann/snippy).
The cp genomes of 38 species were downloaded from the National Center for Biotechnology Information for the phylogenetic analysis. A phylogenetic tree based on the matK + ndhF + rpoB + rpoC2 + ycf1 matrix of the studied species was constructed using Aquilari sinensis, Vatica odorata, Carica papaya, Arabidopsis arenosa, Arabidopsis thaliana and Cardamine parviflora as outgroups. MAFFT v7.490 (Rozewicki et al., 2019) was used to align the sequences. IQ-tree 1.6.12 (Money and Whelan, 2012), using a ML model with 1,000 bootstrap replicates, constructed phylogenetic trees, and the best-fitting model was TVM + F + R3. The best-fitting model was TVM + F for the phylogenetic tree constructed within genera based on variation positions.
Data Availability Statement
The datasets presented in this study can be deposited in the NCBI repository, accession numbers can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OM908760. https://www.ncbi.nlm.nih.gov/genbank/, OM908761. https://www.ncbi.nlm.nih.gov/genbank/, OM908762. https://www.ncbi.nlm.nih.gov/genbank/, OM914582. https://www.ncbi.nlm.nih.gov/genbank/, OK624380.
Author Contributions
HW conceived and designed the study. LY wrote the manuscript. ZW analyzed the data. XH and YY contributed materials. HW and ST proofed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of Jiangsu Province under Grant Number (BK20170619), National Natural Science Foundation of China under Grant Number (31700477), and Innovation and Extension of Forestry Science and Technology of Jiangsu Province under Grant Number [LYKJ(2019)06].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.925726/full#supplementary-material
Supplementary Figure S1 | A comparison of the cp genomes among the 12 Tilia species. Gray arrows above the alignment indicate gene orientation. Genome regions are color-coded as exons, UTRs and non-coding sequences (CNSs). The vertical scale indicates the percentage of identity, ranging from 50% to 100%.
References
Abdullah Henriquez, C. L., Henriquez, C. L., Croat, T. B., Poczai, P., and Ahmed, I. (2021b). Mutational Dynamics of Aroid Chloroplast Genomes II. Front. Genet. 11, 610838. doi:10.3389/fgene.2020.610838
Abdullah, , Mehmood, F., Shahzadi, I., Ali, Z., Islam, M., Naeem, M., et al. (2021a). Correlations Among Oligonucleotide Repeats, Nucleotide Substitutions, and Insertion-Deletion Mutations in Chloroplast Genomes of Plant Family Malvaceae. J. Syst. Evol. 59 (2), 388–402. doi:10.1111/jse.12585
Abdullah, , Mehmood, F., Shahzadi, I., Waseem, S., Mirza, B., Ahmed, I., et al. (2020). Chloroplast Genome of Hibiscus Rosa-Sinensis (Malvaceae): Comparative Analyses and Identification of Mutational Hotspots. Genomics 112 (1), 581–591. doi:10.1016/j.ygeno.2019.04.010
Abdullah Shahzadi, I., Shahzadi, I., Mehmood, F., Ali, Z., Malik, M. S., Waseem, S., et al. (2019). Comparative Analyses of Chloroplast Genomes Among Three Firmiana Species: Identification of Mutational Hotspots and Phylogenetic Relationship with Other Species of Malvaceae. Plant gene. 19, 100199. doi:10.1016/j.plgene.2019.100199
Amar, M. H. (2020). Ycf1-ndhF Genes, the Most Promising Plastid Genomic Barcode, Sheds Light on Phylogeny at Low Taxonomic Levels in Prunus Persica. J. Genet. Eng. Biotechnol. 18 (1), 42. doi:10.1186/s43141-020-00057-3
Antipov, D., Korobeynikov, A., McLean, J. S., and Pevzner, P. A. (2016). HybridSPAdes: An Algorithm for Hybrid Assembly of Short and Long Reads. Bioinformatics 32 (7), 1009–1015. doi:10.1093/bioinformatics/btv688
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A New Genome Assembly Algorithm and its Applications to Single-Cell Sequencing. J. Comput. Biol. 19 (5), 455–477. doi:10.1089/cmb.2012.0021
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-Web: A Web Server for Microsatellite Prediction. Bioinforma. Oxf. Engl. 33 (16), 2583–2585. doi:10.1093/bioinformatics/btx198
Bennett, B. C., and Alarcón, R. (2015). Hunting and Hallucinogens: The Use Psychoactive and Other Plants to Improve the Hunting Ability of Dogs. J. Ethnopharmacol. 171, 171–183. doi:10.1016/j.jep.2015.05.035
Cai, J., Ma, P. F., Li, H. T., and Li, D. Z. (2015). Complete Plastid Genome Sequencing of Four Tilia Species (Malvaceae): A Comparative Analysis and Phylogenetic Implications. PLoS One 10 (11), e0142705. doi:10.1371/journal.pone.0142705
Cai, Z., Penaflor, C., Kuehl, J. V., Leebens-Mack, J., Carlson, J. E., dePamphilis, C. W., et al. (2006). Complete Plastid Genome Sequences of Drimys, Liriodendron, and Piper: Implications for the Phylogenetic Relationships of Magnoliids. BMC Evol. Biol. 10, 77. doi:10.1186/1471-2148-6-77
Chen, Z., Feng, K., Grover, C. E., Li, P., Liu, F., Wang, Y., et al. (2016). Chloroplast DNA Structural Variation, Phylogeny, and Age of Divergence Among Diploid Cotton Species. PLoS One 11 (6), e0157183. doi:10.1371/journal.pone.0157183
Cvetković, T., Areces-Berazain, F., Hinsinger, D. D., Thomas, D. C., Wieringa, J. J., Ganesan, S. K., et al. (2021). Phylogenomics Resolves Deep Subfamilial Relationships in Malvaceae s.L. G3 (Bethesda) 11 (7), jkab136. Epub ahead of print. doi:10.1093/g3journal/jkab136
Delnavazi, M. R., Shahabi, M., and Yassa, N. (2015). Flavonoids from the Leaves of Iranian Linden; Tilia Rubra Subsp. Caucasica. Res. J. Pharm. 2 (3), 17–22.
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty:de novoassembly of organelle genomes from whole genome data. Nucleic. acids. Res. 45 (4), gkw955. doi:10.1093/nar/gkw955
Dong, W., Xu, C., Li, C., Sun, J., Zuo, Y., Shi, S., et al. (2015). Ycf1, the Most Promising Plastid DNA Barcode of Land Plants. Sci. Rep. 5, 8348. doi:10.1038/srep08348
Feng, L.-Y., and Gao, L.-Z. (2021a). Characterization of the Chloroplast Genome Sequence of Bonia Amplexicaulis (L.C.Chia, H.L.Fung & Y.L.Yang) N.H.Xia (Poaceae). Mitochondrial. DNA. Part B, Res. 6 (10), 2822–2824. doi:10.1080/23802359.2021.1972871
Feng, L.-Y., Shi, C., and Gao, L.-Z. (2021b). The Complete Chloroplast Genome Sequence of Bromus Catharticus Vahl. (Poaceae). Mitochondrial DNA Part B 6 (10), 2825–2827. doi:10.1080/23802359.2021.1970646
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: Computational Tools for Comparative Genomics. Nucleic Acids Res. 32, W273–W279. Web Server issue. doi:10.1093/nar/gkh458
Gao, Y., Wang, H., Liu, C., Chu, H., Yan, Y., and Tang, L. (2018). Complete Chloroplast Genome Sequence of the Red Silk Cotton Tree (Bombax ceiba). Mitochondrial DNA Part B 3 (1), 315–316. doi:10.1080/23802359.2017.1422399
Jansen, R. K., Saski, C., Lee, S.-B., Hansen, A. K., and Daniell, H. (2011). Complete Plastid Genome Sequences of Three Rosids (Castanea, Prunus, Theobroma): Evidence for at Least Two Independent Transfers of Rpl22 to the Nucleus. Mol. Biol. Evol. 28 (1), 835–847. doi:10.1093/molbev/msq261
Jiang, D., Zhao, Z., Zhang, T., Zhong, W., Liu, C., Yuan, Q., et al. (2017). The Chloroplast Genome Sequence of Scutellaria Baicalensis Provides Insight into Intraspecific and Interspecific Chloroplast Genome Diversity in Scutellaria. Genes 8 (9), 227. doi:10.3390/genes8090227
Kim, K.-J., and Lee, H. (2004). Complete Chloroplast Genome Sequences from Korean Ginseng (Panax Schinseng Nees) and Comparative Analysis of Sequence Evolution Among 17 Vascular Plants. DNA Res. 11 (4), 247–261. doi:10.1093/dnares/11.4.247
Kim, K., and Cheon, K. (2021). Complete Chloroplast Genome Sequence of Adenophora Racemosa (Campanulaceae): Comparative Analysis with Congeneric Species. PLoS One 16 (3), e24878. doi:10.1371/journal.pone.0248788
Kim, K., Nguyen, V. B., Dong, J., Wang, Y., Park, J. Y., Lee, S.-C., et al. (2017). Evolution of the Araliaceae Family Inferred from Complete Chloroplast Genomes and 45S nrDNAs of 10 Panax-Related Species. Sci. Rep. 7 (1), 4917. doi:10.1038/s41598-017-05218-y
Kirov, I., Odintsov, S., Omarov, M., Gvaramiya, S., Merkulov, P., Dudnikov, M., et al. (2020). Functional Allium Fistulosum Centromeres Comprise Arrays of a Long Satellite Repeat, Insertions of Retrotransposons and Chloroplast DNA. Front. Plant Sci. 11, 562001. doi:10.3389/fpls.2020.562001
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 29 (22), 4633–4642. doi:10.1093/nar/29.22.4633
Kyalo, C. M., Gichira, A. W., Li, Z. Z., Saina, J. K., Malombe, I., Hu, G. W., et al. (2018). Characterization and Comparative Analysis of the Complete Chloroplast Genome of the Critically Endangered Species Streptocarpus Teitensis (Gesneriaceae). Biomed. Res. Int. 2018, 1507847. doi:10.1155/2018/1507847
Levinson, G., and Gutman, G. A. (1987). Slipped-strand Mispairing: A Major Mechanism for DNA Sequence Evolution. Mol. Biol. Evol. 4 (3), 203–221. doi:10.1093/oxfordjournals.molbev.a040442
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, J., Price, M., Su, D.-M., Zhang, Z., Yu, Y., Xie, D.-F., et al. (2021). Phylogeny and Comparative Analysis for the Plastid Genomes of Five Tulipa (Liliaceae). BioMed Res. Int. 2021, 1–10. doi:10.1155/2021/6648429
Li, L., Jiang, Y., Liu, Y., Niu, Z., Xue, Q., Liu, W., et al. (2020). The Large Single-Copy (LSC) Region Functions as a Highly Effective and Efficient Molecular Marker for Accurate Authentication of Medicinal Dendrobium Species. Acta Pharm. Sin. B 10 (10), 1989–2001. doi:10.1016/j.apsb.2020.01.012
Li, X., Li, Y., Zang, M., Li, M., and Fang, Y. (2018). Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus Acutissima. Ijms 19 (8), 2443. doi:10.3390/ijms19082443
Liu, X.-F., Zhu, G.-F., Li, D.-M., and Wang, X.-J. (2019). Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Spathiphyllum 'Parrish'. PLoS One 14 (10), e0224038. doi:10.1371/journal.pone.0224038
Luo, C., Li, Y., Budhathoki, R., Shi, J., Yer, H., Li, X., et al. (2021). Complete Chloroplast Genomes of Impatiens Cyanantha and Impatiens Monticola: Insights into Genome Structures, Mutational Hotspots, Comparative and Phylogenetic Analysis with its Congeneric Species. Plos One 16 (4), e0248182. doi:10.1371/journal.pone.0248182
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read De Novo Assembler. GigaSci 1 (1), 18. doi:10.1186/2047-217X-1-18
Mabberley, D. J. (2017). Mabberley's Plant-Book: A Portable Dictionary of Plants, Their Classification and Uses. 4th ed. Cambridge: Cambridge University Press.
Menezes, A. P. A., Resende-Moreira, L. C., Buzatti, R. S. O., Nazareno, A. G., Carlsen, M., Lobo, F. P., et al. (2018). Chloroplast Genomes of Byrsonima Species (Malpighiaceae): Comparative Analysis and Screening of High Divergence Sequences. Sci. Rep. 8 (1), 2210. doi:10.1038/s41598-018-20189-4
Mo, Z., Lou, W., Chen, Y., Jia, X., Zhai, M., Guo, Z., et al. (2020). The Chloroplast Genome of Carya Illinoinensis: Genome Structure, Adaptive Evolution, and Phylogenetic Analysis. Forests 11 (2), 207. doi:10.3390/f11020207
Money, D., and Whelan, S. (2012). Characterizing the Phylogenetic Tree-Search Problem. Syst. Biol. 61 (2), 228–239. doi:10.1093/sysbio/syr097
Mu, X., Wang, P., Du, J., Gao, Y. G., and Zhang, J. (2018). The Chloroplast Genome of Cerasus Humilis: Genomic Characterization and Phylogenetic Analysis. PLoS One 13 (4), e0196473. doi:10.1371/journal.pone.0196473
Mustafina, F. U., Yi, D.-K., Choi, K., Shin, C. H., Tojibaev, K. S., and Downie, S. R. (2019). A Comparative Analysis of Complete Plastid Genomes from Prangos Fedtschenkoi and Prangos Lipskyi (Apiaceae). Ecol. Evol. 9 (1), 364–377. doi:10.1002/ece3.4753
Negri, G., Santi, D., and Tabach, R. (2013). Flavonol Glycosides Found in Hydroethanolic Extracts from Tilia Cordata, a Species Utilized as Anxiolytics. Rev. Bras. plantas Med. 15 (15), 217–224. doi:10.1590/S1516-05722013000200008
Pigott, D. (2012). Lime-trees and Basswoods: A Biological Monograph of the Genus Tilia. Cambridge: Cambridge University Press.
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X., et al. (2013). The Complete Chloroplast Genome Sequence of the Medicinal Plant Salvia Miltiorrhiza. PLoS One 8 (2), e57607. doi:10.1371/journal.pone.0057607
Qu, X.-J., Moore, M. J., Li, D.-Z., and Yi, T.-S. (2019). PGA: A Software Package for Rapid, Accurate, and Flexible Batch Annotation of Plastomes. Plant Methods 15, 50. doi:10.1186/s13007-019-0435-7
Ren, T., Yang, Y., Zhou, T., and Liu, Z.-L. (2018). Comparative Plastid Genomes of Primula Species: Sequence Divergence and Phylogenetic Relationships. Ijms 19 (4), 1050. doi:10.3390/ijms19041050
Rozewicki, J., Li, S., Amada, K. M., Standley, D. M., and Katoh, K. (2019). MAFFT-DASH: Integrated Protein Sequence and Structural Alignment. Nucleic Acids Res. 47 (W1), W5–W10. doi:10.1093/nar/gkz342
Shen, X., Wu, M., Liao, B., Liu, Z., Bai, R., Xiao, S., et al. (2017). Complete Chloroplast Genome Sequence and Phylogenetic Analysis of the Medicinal Plant Artemisia Annua. Molecules 22 (8), 1330. doi:10.3390/molecules22081330
Siger, A., Antkowiak, W., Dwiecki, K., Rokosik, E., and Rudzińska, M. (2021). Nutlets of Tilia cordata Mill. and Tilia platyphyllos Scop. - Source of bioactive compounds. Food Chem. 346, 128888. doi:10.1016/j.foodchem.2020.128888
Tang, Y., and Zhuge, R. (1996). Geographical Distribution of Tilia Linn. J. Syst. Evol. 34 (3), 254–264.
Wang, J.-H., Moore, M. J., Wang, H., Zhu, Z.-X., and Wang, H.-F. (2021). Plastome evolution and phylogenetic relationships among Malvaceae subfamilies. Gene 765, 145103. doi:10.1016/j.gene.2020.145103
Wang, R.-J., Cheng, C.-L., Chang, C.-C., Wu, C.-L., Su, T.-M., and Chaw, S.-M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8, 36. doi:10.1186/1471-2148-8-36
Xie, D.-F., Yu, Y., Deng, Y.-Q., Li, J., Liu, H.-Y., Zhou, S.-D., et al. (2018). Comparative analysis of the chloroplast genomes of the chinese endemic genus urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Ijms 19 (7), 1847. doi:10.3390/ijms19071847
Yang, J. Y., Lee, W., Pak, J.-H., and Kim, S.-C. (2018). Complete chloroplast genome of Ulleung Island endemic basswood, Tilia insularis (Malvaceae), in Korea. Mitochondrial DNA. Part B. Resour. 3 (2), 605–606. doi:10.1080/23802359.2018.1473731
Ye, X., Hu, D., Guo, Y., and Sun, R. (2019). Complete chloroplast genome of Castanopsis sclerophylla (Lindl.) Schott: Genome structure and comparative and phylogenetic analysis. PLoS One 14 (7), e0212325. doi:10.1371/journal.pone.0212325
Yi, D.-K., and Kim, K.-J. (2016). The complete chloroplast genome sequences of Pogostemon stellatus and Pogostemon yatabeanus (Lamiaceae). Mitochondrial DNA Part B 1 (1), 571–573. doi:10.1080/23802359.2016.1192509
Yu, X., Zuo, L., Lu, D., Lu, B., Yang, M., and Wang, J. (2019). Comparative analysis of chloroplast genomes of five Robinia species: Genome comparative and evolution analysis. Gene 689, 141–151. doi:10.1016/j.gene.2018.12.023
Zhai, Y., Yu, X., Zhou, J., Li, J., Tian, Z., Wang, P., et al. (2021). Complete chloroplast genome sequencing and comparative analysis reveals changes to the chloroplast genome after allopolyploidization in Cucumis. Genome 64 (6), 627–638. doi:10.1139/gen-2020-0134
Zhang, F., Wang, T., Shu, X., Wang, N., Zhuang, W., and Wang, Z. (2020). Complete Chloroplast Genomes and Comparative Analyses of L. Chinensis, L. Anhuiensis, and L. Aurea (Amaryllidaceae). Ijms 21, 5729. doi:10.3390/ijms21165729
Zhao, F., Li, B., Drew, B. T., Chen, Y.-P., Wang, Q., Yu, W.-B., et al. (2020). Leveraging plastomes for comparative analysis and phylogenomic inference within Scutellarioideae (Lamiaceae). PLoS One 15 (5), e0232602. doi:10.1371/journal.pone.0232602
Zhao, K.-K., Wang, J.-H., Cai, Y.-C., Zhu, Z.-X., López-Pujol, J., and Wang, H.-F. (2018). Complete chloroplast genome sequence of Heritiera angustata (Malvaceae): An endangered plant species. Mitochondrial DNA Part B 3 (1), 141–142. doi:10.1080/23802359.2017.1422398
Zhao, Y., Yin, J., Guo, H., Zhang, Y., Xiao, W., Sun, C., et al. (2015). The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Front. Plant Sci. 5, 696. doi:10.3389/fpls.2014.00696
Keywords: Malvaceae, chloroplast genome, hypervariable regions, comparison analysis, phylogenetic analysis
Citation: Yan L, Wang H, Huang X, Li Y, Yue Y, Wang Z and Tang S (2022) Chloroplast Genomes of Genus Tilia: Comparative Genomics and Molecular Evolution. Front. Genet. 13:925726. doi: 10.3389/fgene.2022.925726
Received: 21 April 2022; Accepted: 17 June 2022;
Published: 08 July 2022.
Edited by:
Ibrar Ahmed, Alpha Genomics Private Limited, PakistanReviewed by:
Abdullah., Quaid-i-Azam University, PakistanJia-Yu Xue, Nanjing Agricultural University, China
Copyright © 2022 Yan, Wang, Huang, Li, Yue, Wang and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shijie Tang, sjtang2021@163.com
†These authors have contributed equally to this work