 Jianli Zhou
Jianli Zhou Qiao Zhang1
Qiao Zhang1 Yuzhen Zhao
Yuzhen Zhao Moxian Chen
Moxian Chen Shaoming Zhou
Shaoming Zhou- 1Department of Gastroenterology, Shenzhen Children’s Hospital, Shenzhen, China
- 2Co-Innovation Center for Sustainable Forestry in Southern China, Key Laboratory of National Forestry and Grassland Administration on Subtropical Forest Biodiversity Conservation, College of Biology and the Environment, Nanjing Forestry University, Nanjing, China
Objective: The aim of the study was to develop the early diagnostic criteria for Wilson’s disease (WD) in young children in southern China by using alanine aminotransferase (ALT) elevation as the first manifestation.
Methods: A cross-sectional retrospective analysis of the clinical data and genetic test results of children with WD in southern China in the past 4 years and the follow-up of their short-term prognosis were performed in this study.
Results: A total of 30 children (5.08 ± 2.06 years old) with elevated ALT as the first manifestation of WD in southern China were enrolled in this study, including 14 females and 16 males. Specifically, in all of the 30 cases (100%), the serum ceruloplasmin (CP) level was decreased, whereas the 24-h urinary copper level was increased. The genetic mutation test of the ATP7B gene was used to confirm the diagnosis. In particular, the two mutation sites, including p.R778L and p.I1148T, had the highest mutation frequencies, approximately 23.0 and 10.7%, respectively. Through follow-up, most of the children had good recovery.
Conclusion: Early diagnosis and treatment of WD would substantially increase the survival rate and have a better prognosis. In addition, in 5-year-old children from southern China, early diagnosis could be performed quickly by referring to the following three parameters: elevated ALT, decreased ceruloplasmin level, and increased 24-h urinary copper level. It lays a foundation for further studies with a larger sample size.
1 Introduction
Hepatolenticular degeneration (HLD), also known as Wilson’s disease (WD; OMIM 277900), is an autosomal recessive disorder of copper metabolism (Huster, 2010; Meranthi et al., 2020). The disease occurs all over the world, and the incidence rate in the human population is about 1:1,500–13,000 in East Asia and 1:7,000 in the United Kingdom (Chen et al., 2019; Xiao et al., 2019; Meranthi et al., 2020). The clinical features of WD include liver function injury, nervous system damage, psychiatric abnormality, corneal Kayser–Fleischer (K–F) ring, and decreased serum ceruloplasmin (Xiao et al., 2019). The onset age of WD ranges from infancy to more than 70 years, with an average age of 15.9 years (Xiao et al., 2019). Late diagnosis and treatment or irregular medication of WD could lead to irreversible brain damage or even death. Therefore, early diagnosis and treatment are crucial to reduce the irreversible sequelae of WD (Xiao et al., 2019).
WD can result from the mutation of the ATP7B (OMIM 606882) gene that encodes the intracellular copper transporter on chromosome 13, leading to an impaired intracellular copper output (Meranthi et al., 2020). ATP7B is a P-type ATPase and is mainly expressed in the liver. It binds copper to its N-terminal domain and is responsible for the transport of copper across the membrane, using ATP as its energy source. Studies have demonstrated that mutations at different sites can affect ATPase activity. Until now, more than 1,000 different mutations of ATP7B have been found in patients with WD in the Human Gene Mutation Database (HGMD v2021.11) (Stenson et al., 2017). The mutation of the ATP7B gene in WD affects the interaction between copper ions and ceruloplasmin and subsequent copper excretion in bile, which is the major way of excreting liver copper. If the copper excretion from bile is reduced, copper is then deposited in places around the liver, causing damage to hepatocytes. In addition, it results in elevated ALT as the primary clinical manifestation. Gradually, copper accumulates in the brain, cornea, and kidneys, causing damage to the corresponding organs and accompanying clinical symptoms (European Association for Study of Liver, 2012). Over time, the liver becomes progressively damaged by copper deposits, and some patients end up with cirrhosis or liver failure, as well as severe nervous and blood system damage (European Association for Study of Liver, 2012). Therefore, early diagnosis with high accuracy is crucial for patients with WD and their prognosis. To this end, the clinical features and genetic characteristics of 30 children diagnosed with WD and treated at Shenzhen Children’s Hospital in the past 4 years were analyzed in this study.
2 Material and Methods
2.1 Clinical Data Collection
This was a single-center cross-sectional retrospective study of 30 patients with WD with elevated ALT as their first manifestation in southern China, from May 2016 to May 2020. Medical history, physical examination, laboratory examination, and imaging findings were all collected as clinical data. Physical examination included jaundice, liver enlargement, K–F ring, and neurological symptoms. Laboratory tests included blood routine, hepatic, renal and immunological function tests, virology tests (hepatitis A, B, C, D, E, cytomegalovirus, and EB virus), ceruloplasmin, and 24-h urinary copper level. Imagological examinations included abdominal (liver) ultrasound, cardiac Doppler ultrasonography, and brain magnetic resonance imaging (MRI).
2.2 Genetic Data Collection
All of the cases were tested with ATP7B targeted gene panel sequencing (TGPS) or whole-exome sequencing (WES). The venous blood (2–5 ml) of the patient was drawn after the results of the serum ceruloplasmin (CP) level and 24-h urinary copper level were available, together with 5 ml of parental venous blood for comparison to verify the source of its pathogenic genes. All test protocols, including DNA extraction, construction of gene library, high-throughput sequencing, data analysis, Sanger sequencing verification, and bioinformatics analysis, were carried out by commercial companies such as BGI (The Beijing Genomics Institute, Shenzhen) and Mykino (Beijing).
2.3 Follow-Up Visit
All of the cases were carried out for follow-up studies using outpatient and telephone recordings, including examination, treatment, and outcome.
3 Results
3.1 Clinical Features (Table 1)
3.3.1 Study Data
The subjects of the present study included 14 female and 16 male patients who were asymptomatic only with an elevated hepatase level. The minimum diagnosed age was 2 years, and the oldest patient was 11 years and 4 months old, with an average age of 5.08 ± 2.06 years. The average duration from the discovery of abnormal liver function to diagnosis was about 4 months, and the longest duration was 4 years and 5 months.
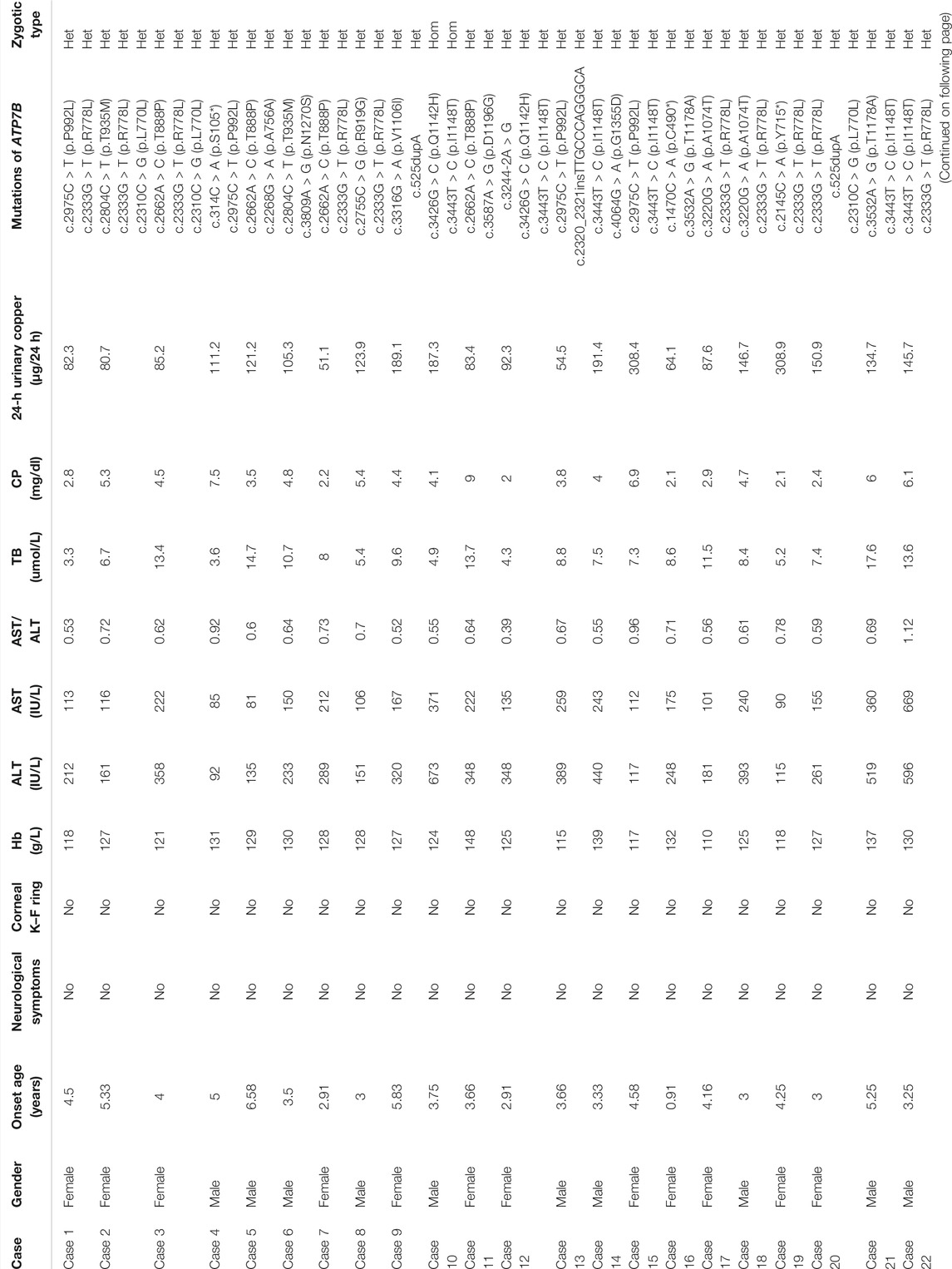
TABLE 1. Detailed clinical information of 30 children with WD.
3.3.2 Blood Biochemical Test
The blood test suggested that ALT was elevated in all of the patients, ranging from 73 to 673 IU/L. In particular, ALT of 11 cases (36.6%) was found to be slightly elevated (increased <5 (upper limit of normal, ULN) times the reference value). In addition, the ALT levels of 14 cases (46.6%) were moderately elevated, that is, 5–10 ULN, whereas 5 cases (16.6%) were found to have severely elevated ALT levels (>10 ULN). Furthermore, the aspartic transaminase (AST) of patients ranged from 75 to 669 IU/L. Specifically, 27 cases (90%) were found to have an AST/ALT ratio of less than 1. In addition, only three cases (10.0%) showed an AST value higher than that of ALT, and the one with an AST/ALT of >2 had jaundice, coagulation dysfunction, liver failure, and eventually died.
3.3.3 Corneal Kayser–Fleischer (K–F) Ring
The 30 children were examined by an ophthalmologist. No corneal K–F ring was found, indicating that there was no eye damage in this group.
3.3.4 Performance of the Nervous System
All of the 30 children had no neurological symptoms. In this group, 12 children underwent brain MRI, and none of them found abnormalities in the basal ganglia, thalamus, and brainstem.
3.3.5 Indicators of Copper Metabolism
The CP level was reduced in all of the 30 cases (100.00%), and the detection value was less than 10 mg/dl. Furthermore, the 24-h urinary copper level was increased in all 30 cases and was more than 40 μg/24 h. In particular, 4 cases (13.3%) reached 40–80 μg/24 h, and the remaining 26 cases (86.6%) were more than 80 μg/24 h.
3.2 Genetic Analysis (Table 2)
The mutation analysis of the ATP7B gene was performed for all of the 30 children, and a total of 65 allelic mutations were detected. This included 51 missense mutations (78.4%), 5 nonsense mutations (7.6%), 4 synonymous mutations (6.1%), 4 frameshift mutations (6.1%), and 1 splicing mutation (1.5%). In our study, there were a total of 28 mutation sites, including 23 reported mutation sites and 5 novel mutation sites. These 28 loci were distributed among different functional regions, including the metal binding units (MBUs), transmembrane domain (TM), actuator domain (A-domain), phosphorylation domain (P-domain), and nucleotide-binding domain (N-domain) (Figure 1). The mutation hot spot was identified as p.R778L, including 15 (23.0%) mutation sites at this spot. Furthermore, the second popular mutation site was p.I1148T, which occurred in 7 (10.7%) patients. The five novel mutations included c.2139C > G (p.Y713*), c.2268G > A (p.A756A), c.2272A > G (p.R758G), c.2320_2321insTTGCCCAGGGCA (p.L776Qfs*695), and c.3220G > A (p.A1074T). Variants p. Y713* and p. L776Qfs*695 can be interpreted as “likely pathogenic” according to the American College of Medical Genetics and Genomics (ACMG) standard (PVS1_strong + PM2+PP3), while the other three mutations can be classified as “variants with uncertain clinical significance” (PM2+PP3) (Richards et al., 2015).
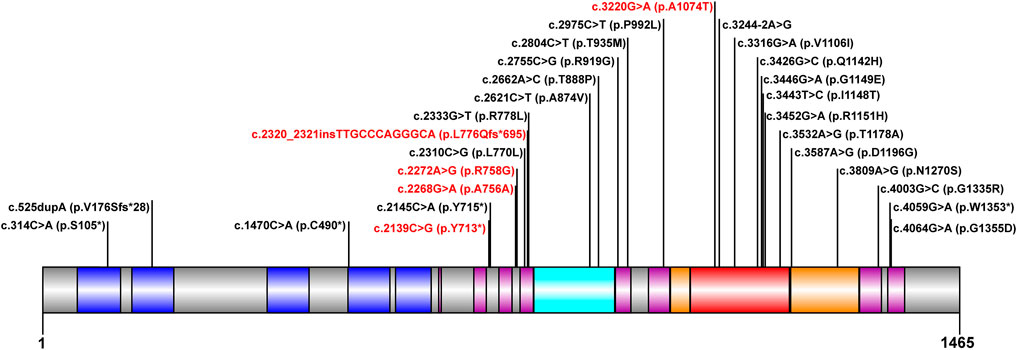
FIGURE 1. Scheme of ATP7B with functional regions and mutations reported in our cohort. The metal binding unit, transmembrane domain, actuator domain, phosphorylation domain, and nucleotide-binding domain are colored in blue, purple, cyan, orange, and red, respectively. The novel mutations detected in this study are in red.
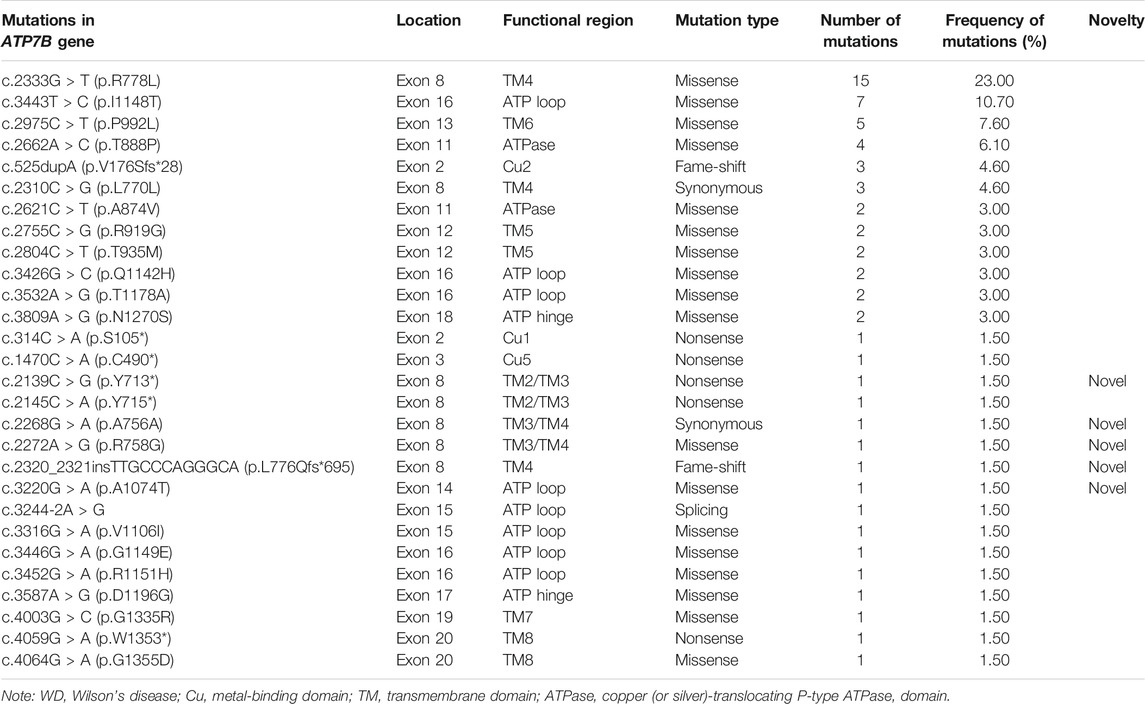
TABLE 2. Information of ATP7B gene mutations in 30 children with WD.
3.3 Criteria of Disease Diagnosis
The diagnosis criteria for WD were according to EASL Clinical Practice Guidelines: Wilson’s disease, from the European Association for Liver Research in 2012 (European Association for Study of Liver, 2012). The parameters used in this evaluation are listed as follows:
(1) Kayser–Fleischer ring (2 points);
(2) neuropsychiatric symptoms suggestive of WD (severe: 2 points and moderate: 1 point);
(3) serum ceruloplasmin content (normal value or >20 mg/dl) normal (0 point), 10–20 mg/dl (1 point), and <10 mg/dl (2 points);
(4) Coombs negative hemolytic anemia with elevated serum copper (1 point);
(5) quantitative determination of liver copper: normal (−1 point), not more than 5 ULN (1 point), and greater than 5 ULN (2 points). Rhodanine staining of hepatocytes is positive (if the quantitative determination of liver copper cannot be obtained) (1 point);
(6) urine copper in the absence of acute hepatitis: normal (0 points), which is 1–2 ULN (1 point), more than 2 ULN (2 points), application of 2 doses of 0.5 g D-penicillamine, and the copper content is more than 5 ULN (2 points);
(7) analysis of gene mutation: pathogenic mutations on both chromosomes (4 points), pathogenic mutations on a single chromosome (1 point), and no pathogenic mutations (0 points).
If the total score is ≥4 points, the possibility of WD is high; if the score is 3 points, it is likely to be WD, but more tests are needed (liver biopsy is required); and if the score is ≤2 points, it is unlikely to be WD. According to the aforementioned criteria, all of the 30 cases were ≥4 points (without checking for liver copper level, the score was from 7 to 9) (Table 3). Since the liver biopsy was invasive, and their parents did not agree to do it, no liver biopsy was performed. Even if the liver biopsy is normal, we subtract one point, and it is still more than four points. Therefore, all of the 30 cases could be diagnosed as WD, and the genetic results confirmed our diagnosis.
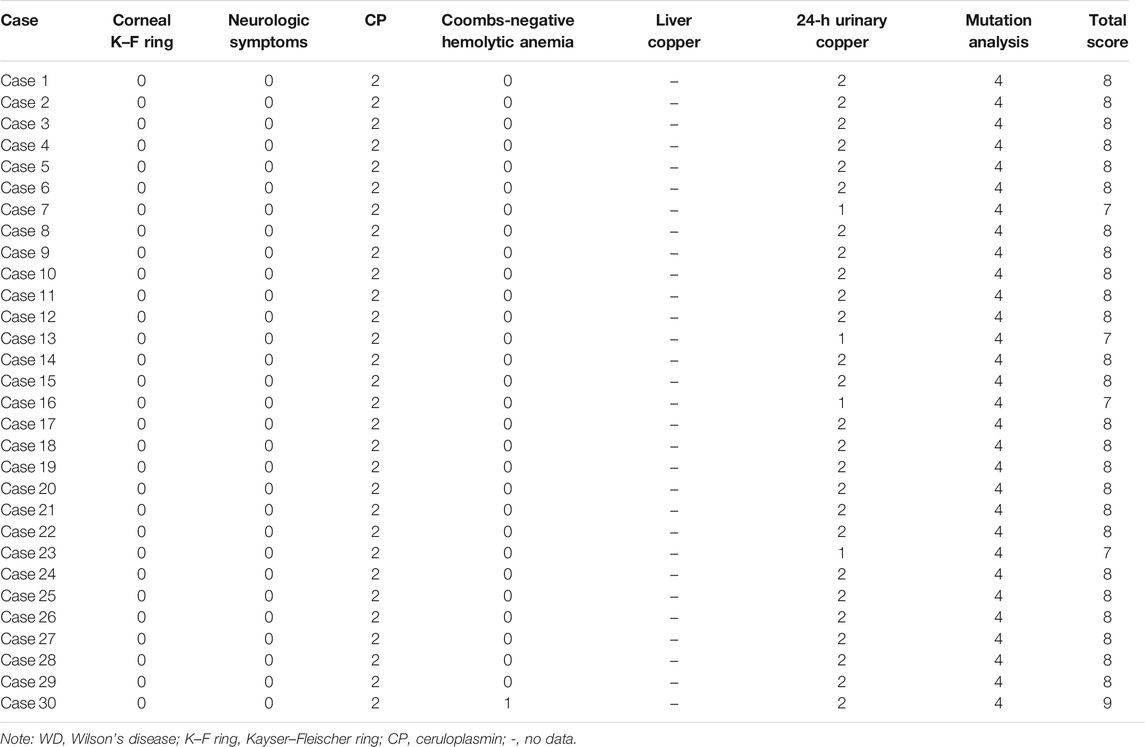
TABLE 3. Diagnosis score of the cases with WD.
3.4 Follow-Up Record and Prognosis Evaluation
The follow-up time was from 1 month to 4 years and 2 months after diagnosis. In detail, 28 of 30 cases (93.3%) were successfully recorded during the follow-up, and the other 2 cases were lost. In particular, 27 cases used basic treatment, including a low-copper diet, oral zinc preparations, and vitamins B, while 25 of them were treated with penicillamine. In total, 27 cases (90%) survived and had good recovery of liver function during the course of treatment, while 20 cases (20/25) were still treated with penicillamine and treatment for 5 cases was stopped. However, 1 child (3.3%) died of acute liver failure (Figure 2).
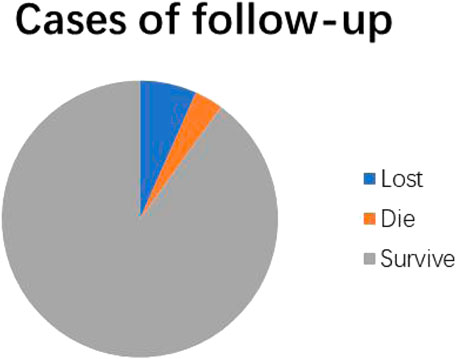
FIGURE 2. Pie chart of statistics in follow-up record and prognosis evaluation.
4 Discussion
The clinical manifestations of children with WD may be diverse due to the starting time of treatment (Lorincz, 2010; Moores et al., 2012). In general, the copper excretion mechanism is not yet fully developed in newborn babies and becomes more effective within the first year after their birth. However, the key pathways of copper excretion in patients with WD fail to develop or have dysfunction, which leads to copper accumulation during the patient’s life, gradually producing various clinical symptoms (Manolaki et al., 2009). Although WD is diagnosed in patients aged 5–35 years (mean, 13 years) (Lin et al., 2014), younger and older patients (>70) are also diagnosed (Stremmel et al., 1991; European Association for Study of Liver, 2012; Lin et al., 2014; Wiernicka et al., 2017). A study of 143 children with WD showed that 21 (15%) of them developed abnormal liver function before the age of 5 years (Wiernicka et al., 2017). At an average age of 9–13 years, the most common initial presentation of children with WD is liver disease (Saito, 1987; Walshe, 1989). Moreover, about 8–10% of children with WD have chronic active hepatitis (Gitlin, 2003). As our cases were from 2 years old to 11 years and 4 months old (5.08 ± 2.06 years), all of them were asymptomatic and just showed up with elevated ALT. Previously, Japanese researchers have suggested that ALT could be the first parameter to screen children with WD between the ages 4 and 8 years (Hayashi et al., 2019), which was similar to our cases. In a previous study involving children and adults, liver presentation was more common in female patients, while neurological presentation was more common in male patients (Ferenci et al., 2019). However, our cases had hepatic presentations only, and the male/female relationship with WD needs to be further investigated in children.
Furthermore, in our group, no corneal K–F ring was detected, making it significantly different from the cases aged 20–30 years old reported in other studies (Bandmann et al., 2015). In addition, older patients (>15 years) are more likely to be diagnosed with neurological manifestations (Oder et al., 1991). The most common age at which WD develops neurological symptoms is 15–21 years (Saito, 1987; Oder et al., 1991; Lorincz, 2010; Žigrai et al., 2020). The discrepancy in the age of WD onset probably reflects variations in gene mutation and penetrance, extragenic factors, and other environmental factors (e.g., diet) (Ala and Schilsky, 2004). However, our cases were younger and were not accompanied by neurological manifestations, and 12 cases of the brain MRI were all negative. We believe that these young children with WD without neurological symptoms do not need to be routinely evaluated by brain MRI.
According to previous reports, the biochemical examination showed that 69.8% of patients with WD have low serum CP, and a serum CP of less than 20 mg/dL has very good accuracy in diagnosing WD (Kim et al., 2015). Furthermore, low CP had a sensitivity of 77–99% and a specificity of 55–88.2% (Ryan et al., 2019). In addition, research reported that 24-h urinary copper levels were increased in all patients (100%), and a level higher than 100 μg/24 h was useful for diagnosing WD (Vieira et al., 2012). In our study, all of the cases had high urinary copper levels (more than 40 μg/24 h) and low serum CP (less than 10 mg/dl). Meanwhile, a high urinary copper level and low serum CP had good diagnostic accuracy for WD (Aksu et al., 2018). Therefore, our results also support this argument.
WD is caused by homozygous or compound heterozygous mutations within ATP7B. At present, the human gene mutation database has more than 1,000 mutations of the ATP7B gene reported, including missense/nonsense mutations, splice site mutations, small deletion/insertion mutations, and frameshift mutations. Mutations can occur anywhere in the gene, including exons, introns, and even promoter regions (Coffey et al., 2013). Furthermore, the mutations of the ATP7B gene have genetic heterogeneity in different races and regions. For example, the most common type of mutation in the European population is p.H1069Q, which is more common in Italy, Sweden, and Romania, with an allele frequency ranging from 30 to 70% (Folhoffer et al., 2007). Contrastingly, the most common type of mutation in the Asian population is the missense mutation p.R778L, which is also the most common mutation in China, South Korea, and Japan, with an allele frequency ranging from 17.3 to 60% (Okada et al., 2000; Liu et al., 2004). Besides p.R778L, other high-frequency mutations include p.P992L and p.Q1399R. Similarly, our study also found that the most abundant mutation type was the missense mutation p.R778L, accounting for 23.0% of the total cases. In addition, the second abundant mutation type in our study was the missense mutation p.I1148T (10.7%). Whether this mutation type is representative of children in southern China remains to be investigated. However, the third abundant mutation type was the missense mutation p.P992L (7.6%), showing consistency with known Asian mutation frequencies. In our study, different gene mutations (affecting different functional domains) of the cases had almost identical clinical phenotypes, which were similar to the previous study (Ferenci et al., 2019). Only one patient died, whose ATP7B had three heterozygous mutations, including c.2333G > T (p.R778L), c.4003G > C (p.G1335R), and c.525dupA, and they included two missense mutations and one frameshift mutation, which affected the functional regions of MBU2, TM4, and TM7. Therefore, the more functional domains are affected, the worse the prognosis may be.
At present, the diagnosis of WD mainly relies on typical clinical manifestations, laboratory tests, and genetic testing (Huster, 2010). Early diagnosis and intervention are essential to delay the progression of the disease and prevent irreversible sequelae. In our study, 30 cases were diagnosed with an elevated ALT level as the first symptom, together with a decreased CP level, an increased 24-h urinary copper level, and ATP7B mutations, suggesting that these three parameters (namely, elevated ALT, decreased CP level, and increased 24-h urinary copper level) are closely related to the early diagnosis of WD in about 5-year-old children in southern China. Thus, we propose that the combined detection of elevated ALT, decreased ceruloplasmin level, and increased 24-h urinary copper level can be useful for an early diagnosis of WD in about 5-year-old asymptomatic children in southern China. In recent years, some researchers thought that genetic screening following serum CP testing reduced costs and facilitated prioritization of non-invasive methods for definitive diagnosis, as well as in asymptomatic or family history cases (Barada et al., 2017; García-Villarreal et al., 2021). Furthermore, other researchers believed that the serum CP level, 24-h urinary copper excretion, and K–F rings could be used to identify patients with WD (Dong et al., 2021). Patients with serum CP levels below 12 mg/dl and children with urinary copper excretion above 40 µg/24 h should undergo genetic testing for WD. As WD needs to identify the diseases, namely, Menkes disease, occipital horn syndrome (OHS), Indian childhood cirrhosis (ICC), and some other diseases and in specific subgroups defined by age, ethnicity, or clinical subgroups, our three parameters (elevated ALT, decreased CP level, and increased 24-h urinary copper level) may not be suitable (Lorincz, 2018; Ryan et al., 2019). However, they can be useful for the early diagnosis of WD in about 5-year-old asymptomatic children in southern China.
Until now, WD was one of the few genetic diseases that could be controlled. The treatment principles are early diagnosis and treatment, lifetime care, and personalized protocol. Current treatment measures include drug therapy, surgical treatment, gene and cell therapy, and rehabilitation (Wiggelinkhuizen et al., 2009). Currently, penicillamine is one of the classic drugs for the treatment of hepatolenticular degeneration due to its effectiveness and cheap price. Studies have shown that certain molecular chaperone drugs (such as 4-phenylbutyric acid) and p38 and JNK inhibitors can correct the mislocalization of the mutant protein and restore the transport function of this protein (Mulligan and Bronstein, 2020). Furthermore, the small-molecule DPM-1001 can effectively reduce the copper deposition in the liver and the brain in the hepatolenticular degeneration mouse model (Krishnan et al., 2018). In particular, personalized cell and (or) gene therapy is the current research hot spot. Its fundamental purpose is to restore the function of ATP7B-mediated hepatic and bile duct excretion of copper (Murillo et al., 2016), and it may be the most promising treatment in the future. The incidence of acute liver failure in WD has previously been reported to be 15–47% (Das et al., 2021; Devarbhavi et al., 2014; Rukunuzzaman, 2015). Among the successful follow-up cases in this group, except for one case (3.3%) with liver failure, the liver function recovered well after the application of penicillamine, oral zinc preparations, B vitamins, and low-copper diet, etc. It showed that as long as early diagnosis and early treatment had been applied, there would be good clinical results in prognosis for children carrying genetic mutations of the ATP7B gene. Our low incidence of acute liver failure may be related to sample size and duration of follow-up.
5 Conclusion
WD is an autosomal recessive genetic disease with diverse clinical manifestations. The group of patients reported in this study came from cities in southern China. Early diagnosis and treatment of WD would substantially increase the survival rate and have a better prognosis. All of these cases had elevated ALT, decreased ceruloplasmin content, and an elevated 24-h urinary copper level, indicating solid first manifestation and potential large-scale screening methods to diagnose WD at an early stage in 5-year-old asymptomatic children in southern China. Although this initial diagnosis can be further confirmed by using genetic testing of the ATP7B gene, it should be confirmed by further research with larger sample sizes.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Shenzhen Children’s Hospital. Written informed consent was obtained from the individual(s) and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YC and SZ designed the experiments. JZ, QZ, YZ, and MC performed the experiments and analyzed data. JZ, YC, and SZ wrote the manuscript. YC critically commented and revised it.
Funding
This work was supported by Shenzhen Fund for Guangdong Provincial High level Clinical Key Specialties (No. SZGSP012) and the Science Technology and Innovation Committee of Shenzhen (2021N062-JCYJ20210324115408023).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We gratefully thank Dr. Di Cui (Running Gene Inc.) for her assistance in genetic analysis.
Abbreviations
ALT, alanine aminotransferase; ALP, alkaline phosphatase; AST, aspartic transaminase; BGI, Beijing Genomics Institute; CP, ceruloplasmin; EEG, electroencephalogram; EASL, European Association for the Study of the Liver; FIB, fibrinogen; GGT, gamma-glutamyl transpeptidase; HLD, hepatolenticular degeneration; INR, international normalized ratio; K–F, Kayser–Fleischer; MRI, magnetic resonance imaging; PT, prothrombin time; ULN, upper limit of normal; WES, whole-exome sequencing; WD, Wilson’s disease.
References
Aksu, A. Ü., Sarı, S., Gürkan, Ö. E., and Dalgıç, B. (2018). Urinary 24-hour Copper Excretion at the Time of Diagnosis in Children with Wilson's Disease. Acta Gastroenterol. Belg. 81, 410–414.
Ala, A., and Schilsky, M. L. (2004). Wilson Disease: Pathophysiology, Diagnosis, Treatment, and Screening. Clin. Liver Dis. 8, 787–805. doi:10.1016/j.cld.2004.06.005
Bandmann, O., Weiss, K. H., and Kaler, S. G. (2015). Wilson's Disease and Other Neurological Copper Disorders. Lancet Neurol. 14, 103–113. doi:10.1016/s1474-4422(14)70190-5
Barada, K., El Haddad, A., Katerji, M., Jomaa, M., and Usta, J. (2017). Wilson's Disease in Lebanon and Regional Countries: Homozygosity and Hepatic Phenotype Predominance. World J. Gastroenterol. 23, 6715–6725. doi:10.3748/wjg.v23.i36.6715
Chen, Y.-C., Yu, H., Wang, R.-M., Xie, J.-J., Ni, W., Zhang, Y., et al. (2019). Contribution of Intragenic Deletions to Mutation Spectrum in Chinese Patients with Wilson's Disease and Possible Mechanism Underlying ATP7B Gross Deletions. Parkinsonism Relat. Disord. 62, 128–133. doi:10.1016/j.parkreldis.2019.01.001
Coffey, A. J., Durkie, M., Hague, S., McLay, K., Emmerson, J., Lo, C., et al. (2013). A Genetic Study of Wilson's Disease in the United Kingdom. Brain 136, 1476–1487. doi:10.1093/brain/awt035
Das, M. C., Sen Sarma, M., Srivastava, A., Yachha, S. K., and Poddar, U. (2021). Effect of Chelation Therapy in Pediatric Wilson's Disease: Liver and Endoscopic Outcome. J. Hepatobiliary Pancreat. Sci. 28, 336–345. doi:10.1002/jhbp.812
Devarbhavi, H., Singh, R., Adarsh, C. K., Sheth, K., Kiran, R., and Patil, M. (2014). Factors that Predict Mortality in Children with Wilson Disease Associated Acute Liver Failure and Comparison of Wilson Disease Specific Prognostic Indices. J. Gastroenterol. Hepatol. 29, 380–386. doi:10.1111/jgh.12356
Dong, Y., Wang, R.-M., Yang, G.-M., Yu, H., Xu, W.-Q., Xie, J.-J., et al. (2021). Role for Biochemical Assays and Kayser-Fleischer Rings in Diagnosis of Wilson's Disease. Clin. Gastroenterol. Hepatol. 19, 590–596. doi:10.1016/j.cgh.2020.05.044
European Association for Study of Liver (2012). EASL Clinical Practice Guidelines: Wilson's Disease. J. Hepatol. 56, 671–685. doi:10.1016/j.jhep.2011.11.007
Ferenci, P., Stremmel, W., Członkowska, A., Szalay, F., Viveiros, A., Stättermayer, A. F., et al. (2019). Age and Sex but Not ATP7B Genotype Effectively Influence the Clinical Phenotype of Wilson Disease. Hepatology 69, 1464–1476. doi:10.1002/hep.30280
Fernando, M., van Mourik, I., Wassmer, E., and Kelly, D. (2020). Wilson disease in children and adolescents. Front. Med. (Lausanne) 105, 499-–505. doi:10.1136/archdischild-2018-315705
Folhoffer, A., Ferenci, P., Csak, T., Horvath, A., Hegedus, D., Firneisz, G., et al. (2007). Novel Mutations of the ATP7B Gene Among 109 Hungarian Patients with Wilson's Disease. Eur. J. Gastroenterol. Hepatol. 19, 105–111. doi:10.1097/01.meg.0000223904.70492.0b
García-Villarreal, L., Hernández-Ortega, A., Sánchez-Monteagudo, A., Peña-Quintana, L., Ramírez-Lorenzo T, T., and Riaño, M. (2021). Wilson Disease: Revision of Diagnostic Criteria in a Clinical Series with Great Genetic Homogeneity. J. Gastroenterol. 56, 78–89. doi:10.1007/s00535-020-01745-0
Gitlin, J. D. (2003). Wilson Disease. Gastroenterology 125, 1868–1877. doi:10.1053/j.gastro.2003.05.010
Hayashi, H., Watanabe, K., Inui, A., Kato, A., Tatsumi, Y., Okumura, A., et al. (2019). Alanine Aminotransferase as the First Test Parameter for Wilson's Disease. J. Clin. Transl Hepatol. 7, 293–296. doi:10.14218/JCTH.2019.00042
Huster, D. (2010). Wilson Disease. Best Pract. Res. Clin. Gastroenterol. 24, 531–539. doi:10.1016/j.bpg.2010.07.014
Kim, J. A., Kim, H. J., Cho, J. M., Oh, S. H., Lee, B. H., Kim, G.-H., et al. (2015). Diagnostic Value of Ceruloplasmin in the Diagnosis of Pediatric Wilson's Disease. Pediatr. Gastroenterol. Hepatol. Nutr. 18, 187–192. doi:10.5223/pghn.2015.18.3.187
Krishnan, N., Felice, C., Rivera, K., Pappin, D. J., and Tonks, N. K. (2018). DPM-1001 Decreased Copper Levels and Ameliorated Deficits in a Mouse Model of Wilson's Disease. Genes Dev. 32, 944–952. doi:10.1101/gad.314658.118
Lin, L.-J., Wang, D.-X., Ding, N.-N., Lin, Y., Jin, Y., and Zheng, C.-Q. (2014). Comprehensive Analysis on Clinical Features of Wilson's Disease: an Experience over 28 Years with 133 Cases. Neurol. Res. 36, 157–163. doi:10.1179/1743132813y.0000000262
Liu, X.-Q., Zhang, Y. F., Liu, T. T., Hsiao, K. J., Zhang, J. M., Gu, X. F., et al. (2004). Correlation of ATP7B Genotype with Phenotype in Chinese Patients with Wilson Disease. World J. Gastroenterol. 10, 590–593. doi:10.3748/wjg.v10.i4.590
Lorincz, M. T. (2010). Neurologic Wilson's Disease. Ann. N. Y Acad. Sci. 1184, 173–187. doi:10.1111/j.1749-6632.2009.05109.x
Lorincz, M. T. (2018). Wilson Disease and Related Copper Disorders. Handb Clin. Neurol. 147, 279–292. doi:10.1016/b978-0-444-63233-3.00018-x
Manolaki, N., Nikolopoulou, G., Daikos, G. L., Panagiotakaki, E., Tzetis, M., Roma, E., et al. (2009). Wilson Disease in Children: Analysis of 57 Cases. J. Pediatr. Gastroenterol. Nutr. 48, 72–77. doi:10.1097/mpg.0b013e31817d80b8
Meranthi, F., Indra, V. M., Evangeline, W., et al. (2020). Wilson Disease in Children and Adolescents. Arch. Dis. Child. 105, 499–505.
Moores, A., Fox, S., Lang, A., and Hirschfield, G. (2012). Wilson Disease: Canadian Perspectives on Presentation and Outcomes from an Adult Ambulatory Setting. Can. J. Gastroenterol. 26, 333–339. doi:10.1155/2012/123431
Mulligan, C., and Bronstein, J. M. (2020). Wilson Disease. Neurol. Clin. 38, 417–432. doi:10.1016/j.ncl.2020.01.005
Murillo, O., Luqui, D. M., Gazquez, C., Martinez-Espartosa, D., Navarro-Blasco, I., Monreal, J. I., et al. (2016). Long-term Metabolic Correction of Wilson's Disease in a Murine Model by Gene Therapy. J. Hepatol. 64, 419–426. doi:10.1016/j.jhep.2015.09.014
Oder, W., Grimm, G., Kollegger, H., Ferenci, P., Schneider, B., and Deecke, L. (1991). Neurological and Neuropsychiatric Spectrum of Wilson's Disease: a Prospective Study of 45 Cases. J. Neurol. 238, 281–287. doi:10.1007/BF00319740
Okada, T., Shiono, Y., Hayashi, H., Satoh, H., Sawada, T., Suzuki, A., et al. (2000). Mutational Analysis ofATP7Band Genotype-Phenotype Correlation in Japanese with Wilson's Disease. Hum. Mutat. 15, 454–462. doi:10.1002/(sici)1098-1004(200005)15:5<454:aid-humu7>3.0.co;2-j
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Rukunuzzaman, M. (2015). Wilson's Disease in Bangladeshi Children: Analysis of 100 Cases. Pediatr. Gastroenterol. Hepatol. Nutr. 18, 121–127. doi:10.5223/pghn.2015.18.2.121
Ryan, A., Nevitt, S. J., Tuohy, O., and Cook, P. (2019). Biomarkers for Diagnosis of Wilson's Disease. Cochrane Database Syst. Rev. 2019, CD012267. doi:10.1002/14651858.cd012267.pub2
Saito, T. (1987). Presenting Symptoms and Natural History of Wilson Disease. Eur. J. Pediatr. 146, 261–265. doi:10.1007/bf00716470
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The Human Gene Mutation Database: towards a Comprehensive Repository of Inherited Mutation Data for Medical Research, Genetic Diagnosis and Next-Generation Sequencing Studies. Hum. Genet. 136, 665–677. doi:10.1007/s00439-017-1779-6
Stremmel, W., Meyerrose, K. W., Niederau, C., Hefter, H., Kreuzpaintner, G., Strohmeyer, G., et al. (1991). Wilson Disease: Clinical Presentation, Treatment, and Survival. Ann. Intern. Med. 115, 720–726. doi:10.7326/0003-4819-115-9-720
Vieira, J., Oliveira, P. V., Juliano, Y., Warde, K. R. J., Deguti, M. M., Barbosa, E. R., et al. (2012). Urinary Copper Excretion before and after Oral Intake of D-Penicillamine in Parents of Patients with Wilson's Disease. Dig. Liver Dis. 44, 323–327. doi:10.1016/j.dld.2011.11.001
Walshe, J. M. (1989). Wilson's Disease Presenting with Features of Hepatic Dysfunction: a Clinical Analysis of Eighty-Seven Patients. Q. J. Med. 70, 253–263.
Wiernicka, A., Dądalski, M., Jańczyk, W., Kamińska, D., Naorniakowska, M., Hüsing-Kabar, A., et al. (2017). Early Onset of Wilson Disease: Diagnostic Challenges. J. Pediatr. Gastroenterol. Nutr. 65, 555–560. doi:10.1097/mpg.0000000000001700
Wiggelinkhuizen, M., Tilanus, M. E. C., Bollen, C. W., and Houwen, R. H. J. (2009). Systematic Review: Clinical Efficacy of Chelator Agents and Zinc in the Initial Treatment of Wilson Disease. Aliment. Pharmacol. Ther. 29, 947–958. doi:10.1111/j.1365-2036.2009.03959.x
Xiao, H., Deng, S., Deng, X., Gu, S., Yang, Z., Yin, H., et al. (2019). Mutation Analysis of the ATP7B Gene in Seven Chinese Families with Wilson's Disease. Digestion 99, 319–326. doi:10.1159/000493314
Keywords: children, southern China, hepatolenticular degeneration, clinical features, genetic mutation
Citation: Zhou J, Zhang Q, Zhao Y, Chen M, Zhou S and Cheng Y (2022) Early Diagnosis of Wilson’s Disease in Children in Southern China by Using Common Parameters. Front. Genet. 13:788658. doi: 10.3389/fgene.2022.788658
Received: 03 October 2021; Accepted: 14 January 2022;
Published: 10 February 2022.
Edited by:
Louis Charles Penning, Utrecht University, NetherlandsReviewed by:
Julnar A. R. Usta, American University of Beirut, LebanonJinchen Li, Central South University, China
Copyright © 2022 Zhou, Zhang, Zhao, Chen, Zhou and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaoming Zhou, zhousm15d@aliyun.com; Yongwei Cheng, 29060628@qq.com