Phylogeography and demographic history of the cyprinid fish Barbodes semifasciolatus: implications for the history of landform changes in south mainland China, Hainan and Taiwan
 Junjie Wang
Junjie Wang JinXian Wu
JinXian Wu Jinquan Yang
Jinquan Yang Jiabo Chen1
Jiabo Chen1  Chao Li
Chao Li Hung-Du Lin
Hung-Du Lin Jun Zhao
Jun Zhao- 1Guangzhou Key Laboratory of Subtropical Biodiversity and Biomonitoring, School of Life Science, South China Normal University, Guangzhou, China
- 2Shanghai Universities Key Laboratory of Marine Animal Taxonomy and Evolution, Shanghai Ocean University, Shanghai, China
- 3The Affiliated School of National Tainan First Senior High School, Tainan, Taiwan
Hainan Island and Taiwan Island are adjacent to the southern margin of mainland China and Vietnam. During glacial periods, global sea levels dropped, allowing that the land bridges connected the continental island and mainland, connecting rivers and providing dispersal opportunities that shaped the origin and diversification of freshwater fishes. Barbodes semifasciolatus is distributed in various water systems of Vietnam, Hainan, Taiwan, and southern mainland China and is restricted to the southern region of the Min River. Our study aimed to evaluate the genetic diversity and phylogeography of B. semifasciolatus using the mtDNA cyt b gene (1,141 bp). A total of 107 haplotypes were identified from 395 specimens in 23 populations, and high haplotype diversity (1.000) and low nucleotide diversity (0.0134) were detected. Mitochondrial phylogenetic analysis and haplotype network analyses revealed three major lineages according to geographical distribution. Lineage A was mainly distributed in Hainan Island, Vietnam and the southern region of the Pearl River in mainland China. Lineage B was distributed only in southeastern Hainan Island. Lineage C was distributed in the coastal rivers of mainland China and Taiwan. We suggest that the river in the Guangdong region is a colonization route in South Taiwan and that the populations distributed in the Pearl River region moved southward to Hainan Island and Vietnam based on the network and Bayesian binary MCMC (BBM) analysis. Our demographic history results indicated that the populations of B. semifasciolatus experienced a bottleneck event following a recent population expansion (DECINC model) supported by ABC analysis. We suggest that sea-level changes exerted pronounced effects on the demography of B. semifasciolatus on the continental island and in the mainland during the late Pleistocene glacial cycles.
1 Introduction
Freshwater fish on continental islands offer excellent opportunities for studying phylogeographic patterns, as their biogeographic relationships reflect historical drainage connections rather than present-day ones (Wang et al., 2021). During glacial periods, particularly the last glacial maximum (LGM) at approximately 20 ka, global sea levels dropped, allowing land bridges to connect islands and mainlands, connecting rivers and providing dispersal opportunities for freshwater fishes (Voris, 2000). Continental islands usually share primary freshwater fishes with the mainland. The pattern of the freshwater fish fauna on islands is essentially an effect of the faunal evolution of the Plio/Pleistocene Palearctic mainland, where zoogeographic fauna lived, mainly triggered by dispersal route events (Bernatchez and Wilson, 1998). The evolutionary diversification of freshwater fishes is strongly affected by possible colonization and landscape evolution (Albert et al., 2018). Studies on the phylogeographic structure of freshwater fish in southern mainland China revealed that the Shiwandashan, Nanling and Wuyi Mountains played a key role in the geographical isolation between the Pearl River, Yangtze River and coastal rivers (e.g., Moyang River, Han River, Jiulong River and Min River). During the glacial period, the continental shelf of the South China Sea was largely above sea level, and the mouths of coastal rivers may have become confluent with each other. The close genetic relationship in the Pearl River, the Jiulong River and Min River revealed that there were temporary connections in palo-drainages, as observed for the genus Acrossocheilus (Hou et al., 2020), Squalidus argentatus (Yang et al., 2013), and Hemibagrus guttatus (Yang and He, 2008). Similarly, the fishes in the Pearl River are closely related to those in the coastal rivers of South China around the Gulf of Tonkin and South China Sea, e.g., the Moyang River and Jian River. These fishes include Garra orientalis (Yang et al., 2016), and Aphyocypris normalis (Huang et al., 2019).
The Pearl River is the second largest river in China and consists of three main tributaries, the Xi River, the Bei River and the Dong River. Previous studies suggested that the Dong River and the Xi/Bei Rivers could be regarded as two distinct rivers owing to their distinct ichthyofaunas (Chen et al., 1986) and phylogeographical structures (e.g., Siniperca scherzeri, Lin et al., 2022; H. guttatus, Yang and He, 2008). However, Chen et al. (2007) found that populations of Glyptothorax fokiensis in the Dong and Bei Rivers were more closely related to those in the Xi River. Some previous studies revealed no population differentiation of some species in the three main tributaries of the Pearl River, such as Opsariichthys bidens (Lin et al., 2016) and Megalobrama terminalis (Chen et al., 2020). The effects of the historical development of the Pearl River drainage on this genetic structure are unclear.
Previous biological studies have also indicated that freshwater species migrate from the southeastern coast of the mainland to islands by different colonization events, including different continental populations, routes, centers, and times (e.g., Wang et al., 2004; Chiang et al., 2010; Chiang et al., 2013; Chang et al., 2016; Lin et al., 2016). The islands of Taiwan and Hainan, the two major continental islands along the mainland Chinese coast, were isolated from the eastern Asian continent by rising sea levels during interglacial periods. Geological evidence indicates that the exposed continental shelf connected both Hainan and Taiwan Islands to the surrounding mainland, initially in the Pliocene and possibly two to three times in the Pleistocene (Gascoyne et al., 1979; Huang et al., 1995; Yu, 1995). Thus, it is an ideal system for studying the phylogeographical origin of freshwater fish on associated continents and islands. According to the essential geohistorical events and previous phylogeographic studies, there are four major phylogeographic regions in Taiwan that originated from different continental populations by various colonization routes in distinct ice ages. For example, Yang et al. (2012) found that the Qiantang River is a source population for colonization of S. argentatus, as found in the Tamsui River in North Taiwan. The freshwater fish distributed only in rivers of western Taiwan indicate that the Miaoli Plateau was the last region isolated from the Min River in mainland China [e.g., Varicorhinus barbatulus (Wang et al., 2004); Cobitis sinensis (Chiang et al., 2010); Acrossocheilus paradoxus (Ju et al., 2018)]. Moreover, the fauna of Hainan Island might have originated from the north (China) and south (Vietnam) through the Qiongzhou Strait and Beibu Gulf during the ice age, respectively. For example, the Northern Hainan region is phylogenetically closer to the mainland China region because the whole region of the Gulf of Tonkin and the Qiongzhou Strait were once part of the coastal plain of the Asian continent during the Pleistocene glaciations [e.g., A. normalis (Huang et al., 2019); G. orientalis (Yang et al., 2016); O. hainanensis (Zhang et al., 2020)]. However, Wang et al. (2021) found that C. gachua inhabited South Hainan (Changhua River) and the Red River, indicating a close relationship of populations between Hainan Island and mainland China. However, few studies have explored the colonization routes of freshwater fish inhabiting southern Taiwan and eastern Hainan. Although colonizations from the eastern Asian continent to Taiwan and Hainan Island have been reported, previous studies have focused on the relationship between a single island and the mainland.
The cyprinid genus Barbodes Bleeker, 1859 (Cyprinidae: Smiliogastrinae) comprises 51 small- to medium-sized valid species inhabiting the lower and middle reaches. Barbodes semifasciolatus is distributed in various water systems of Vietnam, the Red River basin, Hainan, Taiwan and southern mainland China and is restricted to the southern region of the Min River. Within the genus, B. semifasciolatus is the most northerly distributed species (Ren et al., 2020). The broad-scale distribution of B. semifasciolatus is ideal for studying biogeographical questions in eastern Asia, especially providing key insights for understanding the poorly studied colonization routes in southern Taiwan and eastern Hainan. Previous studies on the origin of freshwater fish species in Taiwan and Hainan Island have been limited to the relationship between a single island and the mainland, and there have been no research examples of the same species simultaneously distributed on both islands. As far as we know, this is the first study of freshwater fish species spanning across Taiwan, Hainan Island, and mainland China. In our study, we used the maternally inherited mitochondrial DNA (mtDNA) cytochrome b gene (cyt b) to explore the phylogeography of B. semifasciolatus. Here, we fill this knowledge gap by exploring the patterns of and factors underlying the phylogeographic structure and multiple colonization pathways of freshwater fishes in Vietnam, Hainan, Taiwan and southern mainland China, where the largest land extension occurred during low sea-level periods caused by glacial cycles (Qiu et al., 2011).
The aim of the present study was to assess the phylogeographic structure of the species using a mtDNA marker and address three major questions: (1) What is the genetic diversity and genetic structure of B. semifasciolatus in mainland and island areas. (2) How did B. semifasciolatus colonize the rivers of different geographical districts on the mainland, in Taiwan and on Hainan Island? (3) What were the impacts of Pleistocene sea-level changes on the demographic history of B. semifasciolatus populations?
2 Materials and methods
2.1 Sampling, DNA extraction and sequencing
A total of 395 individuals of B. semifasciolatus were collected from 23 populations of 21 rivers in Vietnam, the southern regions of mainland China, Hainan and Taiwan during 2001–2019 (Table 1; Figure 1). All specimens were collected from field sites with seines, anesthetized by immersion in MS-222 (Sigma, St. Louis, MO), preserved in 95% ethanol, and stored in the laboratory of Jun Zhao, Guangzhou Key Laboratory of Subtropical Biodiversity and Biomonitoring. All procedures were carried out in accordance with the guidelines and approval of the Animal Research and Ethics Committee of the School of Life Science, South China Normal University (permissions, CAMC-2018F). Total genomic DNA was extracted from fin tissue using a genomic DNA purification kit (Gentra Systems, Valencia, CA). The cytochrome b gene (1,141 bp) of mitochondrial DNA (mtDNA) was amplified for all samples using primers L14724 (5’-GACTTGAAAAACCACCGTTG-3’) and H15915 (5’-CTCCGATCTCCGGATTACAAGAC-3’) (Xiao et al., 2001). The PCR program was run on a thermal cycler (Eppendorf Master cycler) as described by Wang et al. (2021). PCR products were cycle sequenced using the BigDye Terminator Kit, purified and read on an ABI PRISM 3730XL sequencer (Applied Biosystems, Foster City, CA, U.S.A.) with the BigDye Terminator Kit (Applied Biosystems). Chromatograms were checked using CHROMAS software (Technelysium Pty Ltd, Australia), and the sequences were aligned manually in BioEdit v.7.2.5 (Hall, 2004) and submitted to GenBank under accession numbers (OP556138-OP556315).
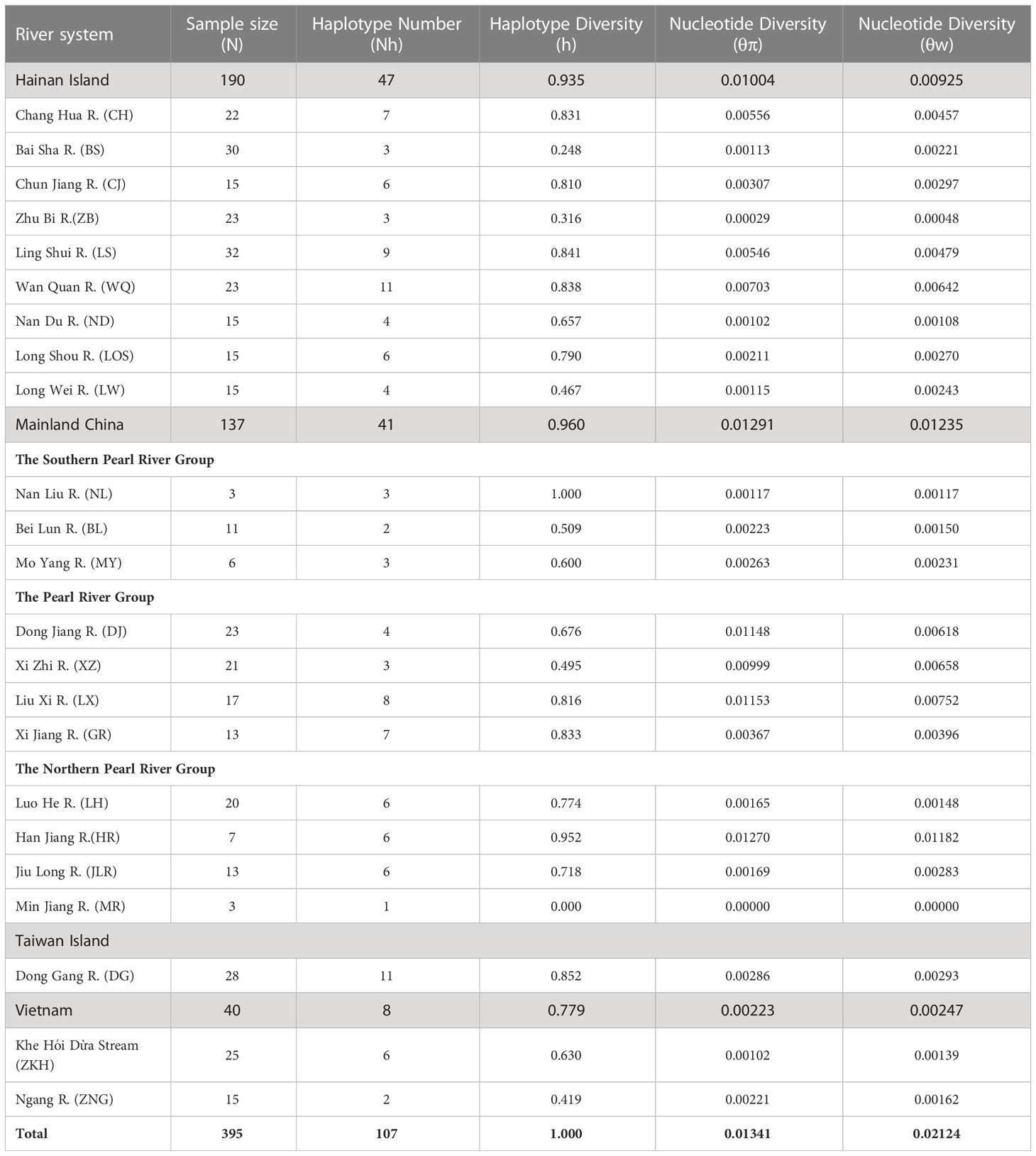
Table 1 Sampling localities, abbreviations and genetic diversity indexes for Barbodes semifasciolatus based on mitochondrial cyt b gene. N, sample size; Nh, haplotype numbers; h, haplotype diversity; θπ, θw, nucleotide diversity.
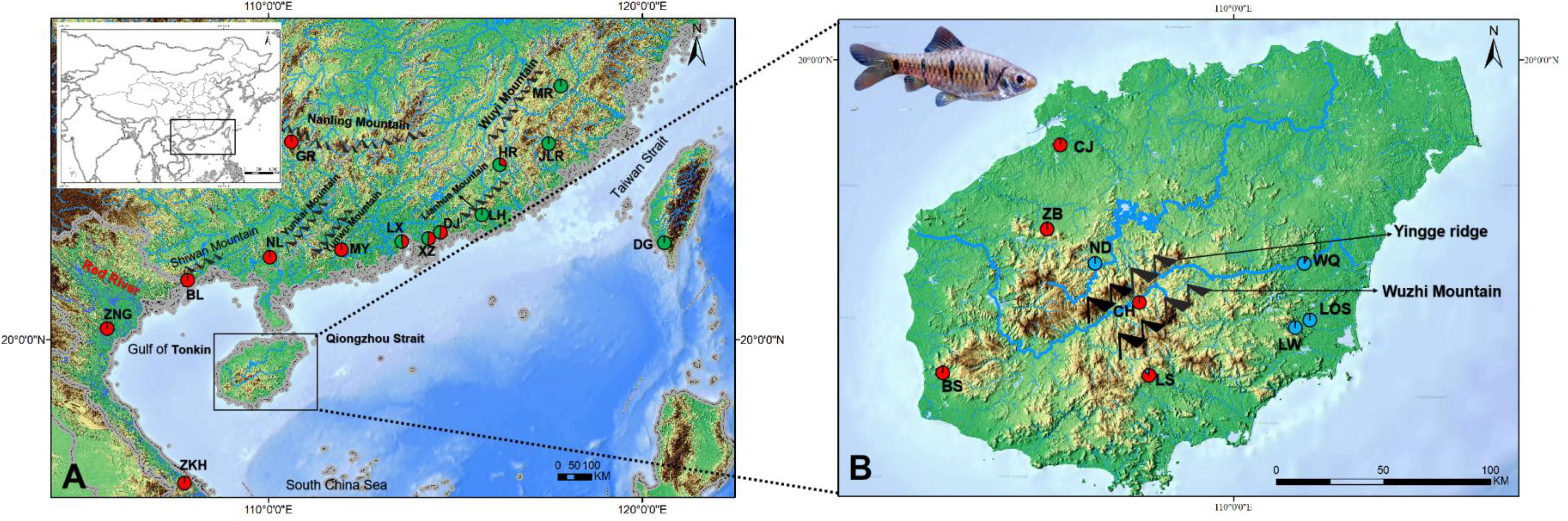
Figure 1 Maps of the study region in mainland China, Taiwan Island, Hainan Island and Vietnam (A) and Hainan Island (B) showing sites where 23 sampling localities of Barbodes semifasciolatus were located (circles). The frequencies of the lineages (Figure 2) in each population are displayed on the map. Red: Lineage A; Blue: Lineage B; Green: Lineage C.
2.2 Sequence variations, diversity and phylogenetic analyses
The evolutionary substitution model for the mitochondrial gene (cyt b) was the TN93 +R model selected using the corrected Akaike information criterion (AICc) in SMS (Smart Model Selection in PhyML) (Lefort et al., 2017). Neighbor-joining (NJ) and maximal likelihood (ML) approaches were used to build phylogenetic trees in MEGA 11 (Tamura et al., 2021) and PhyML 3.0 (Guindon et al., 2010), respectively. We conducted bootstrap analyses with 1000 pseudoreplicates for the maximum likelihood (ML) tree and 10,000 pseudoreplicates for the neighbor-joining (NJ) tree. Puntius titteya and P. snyderi were used as outgroups in phylogenetic analysis. The phylogenetic tree and divergence dates were inferred using a strict clock model in the program BEAST 1.10 (Suchard et al., 2018) with 107 MCMC steps and taking the first 10% as burn-in. The mutation rates for the mitochondrial cyt b gene were based on a divergence rate of 1.05% per million years (Dowling et al., 2002). We checked for convergence of the runs using TRACER v1.6, including checking the effective sample size (ESS) for each parameter greater than 200, and the maximum clade credibility tree was generated in TreeAnnotator v.2.2.1 (Rambaut and Drummond, 2015) in the BEAST package to summarize trees after removing 20% of trees as burn-in, the results of which were displayed in FigTree 1.4.3 (Rambaut, 2016). Haplotype networks among haplotypes for cyt b were computed using the minimum‐spanning network method (MINSPNET algorithm in Arlequin 3.5) (Excoffier and Lischer, 2010).
The indices of molecular diversity of each population, such as the number of haplotypes (Nh), haplotype diversity (h) (Nei and Tajima, 1983) and nucleotide diversity (θπ and θω) (Watterson, 1975; Lynch and Crease, 1990), were determined using DnaSP v5.0 software (Librado and Rozas, 2009). The software DnaSP v5.0 was used to calculate the genetic differentiation coefficients (GST and NST) to determine the existence of phylogeographic structure following the method of Pons and Petit (1996). To examine the spatial partitioning of genetic variation among populations, pairwise FST values and analysis of molecular variance (AMOVA) were obtained and performed in Arlequin version 3.5 (Excoffier and Lischer, 2010), followed by statistical significance testing with 10,000 permutation steps for each comparison. For AMOVA hierarchical analysis, populations defined according to the different geographic barriers were grouped together under six scenarios: (I) two geographical groups primarily divided by the strait: Taiwan + Hainan Island and mainland China + Vietnam; (II) three geographical groups primarily divided by the Taiwan and Qiongzhou Strait: Taiwan, Hainan Island and mainland China + Vietnam; (III) four geographical groups according to the four geographic regions: Vietnam, Hainan Island, Taiwan Island and mainland China; (IV) six geographical groups primarily divided by Shiwandashan, Nanling and Wuyi mountains and the Taiwan and Qiongzhou Strait: the Hainan group (CH, BS, CJ, ZB, LS, WQ, ND, LOS, LW), the Vietnamese group (ZKH, ZNG), the southern Pearl River group (NL, BL, MY), the Pearl River group (DJ, XZ, LX, GR), the Northern Pearl River group (LH, HR, JLR, MR), and the Taiwanese group (DG); (V) seven geographical groups primarily divided by the WY Range, Shiwandashan, Nanling and Wuyi mountains and the Taiwan and Qiongzhou Strait: the North Hainan group (CJ, ZB, WQ, ND, LOS, LW), the South Hainan group (CH, BS, LS), the Vietnamese group (ZKH, ZNG), the southern Pearl River group (NL, BL, MY), the Pearl River group (DJ, XZ, LX, GR), the Northern Pearl River group (LH, HR, JLR, MR), and the Taiwan group (DG); (VI) three geographical groups primarily divided by Shiwandashan, Nanling, and Wuyi Mountain: the Hainan, southern Pearl River and Vietnamese group (CJ, ZB, WQ, ND, LOS, LW, CH, BS, LS, ZKH, ZNG, NL, BL, MY), the Pearl River group (DJ, XZ, LX, GR), the northern Pearl River and Taiwanese group (LH, HR, JLR, MR, DG).
The historical demographic expansions were detected by statistics of neutrality tests [Tajima’s D test (Tajima, 1989) and Fu’s Fs test (Fu, 1997)] and mismatch distributions using DnaSP v5.0 (Librado and Rozas, 2009). We also constructed Bayesian skyline plots (BSPs) in BEAST v1.8.2 (Drummond et al., 2013) for each lineage to determine the effective population size changes over time. A 1.05%/MY divergence rate was calibrated, and plots for each analysis were drawn using Tracer v1.6 (Rambaut et al., 2014) after the first 10% of samples for each chain were discarded as burn-in. For reconstruction of the ancestral state in the area, we performed Bayesian binary MCMC (BBM) analysis implemented in RASP 3.2 (Yu et al., 2015). According to the sampling and distribution range of B. semifasciolatus, seven areas were defined for the biogeographic analyses: (1) the North Hainan group (A; CJ, ZB, WQ, ND, LOS, LW); (2) the South Hainan group (B; CH, BS, LS); (3) the southern Pearl River group (C; NL, BL, MY); (4) the Pearl River group (D; DJ, XZ, LX, GR); (5) (E; LH, HR, JLR, MR); (6) the Vietnamese group (F; ZKH, ZNG); and (7) the Taiwanese group (G; DG).
2.3 Historical population demography and DIY-ABC
To compare concurrent scenarios of demographic history, we used an approximated Bayesian computation (ABC) approach as implemented in the software DIYABC v.2.0 (Cornuet et al., 2014) with mtDNA cyt b data. We simulated five different demographic scenarios to determine the possible historical demography of B. semifasciolatus following the recommendations proposed by Cabrera and Palsbøll (2017) (Figure 2). Such scenarios are described in more detail in the following. In scenario A (CON model), populations of B. semifasciolatus remained constant in size over time. In scenario B (DEC model), populations of B. semifasciolatus experienced a bottleneck event. In scenario C (INC model), populations of B. semifasciolatus expanded recently. In scenario D (INCDEC model), populations of B. semifasciolatus experienced an ancient expansion and then underwent a single instantaneous decrease in population size. In scenario E (DECINC model), populations of B. semifasciolatus experienced an old bottleneck followed by population expansion. For mtDNA cyt b data, we assumed an HKY mutation model, and default settings were used for all other parameters. A total of 3,000,000 simulated datasets in this study were included in the reference table for each scenario using all the summary statistics included in DIYABC software. To determine the best-supported scenario, all scenarios were compared using the posterior distribution probability with logistic regression with the highest posterior probability only. The posterior probability of each model based on 1% of the simulated datasets for each scenario was assessed using logistic approaches, as implemented in DIYABC.
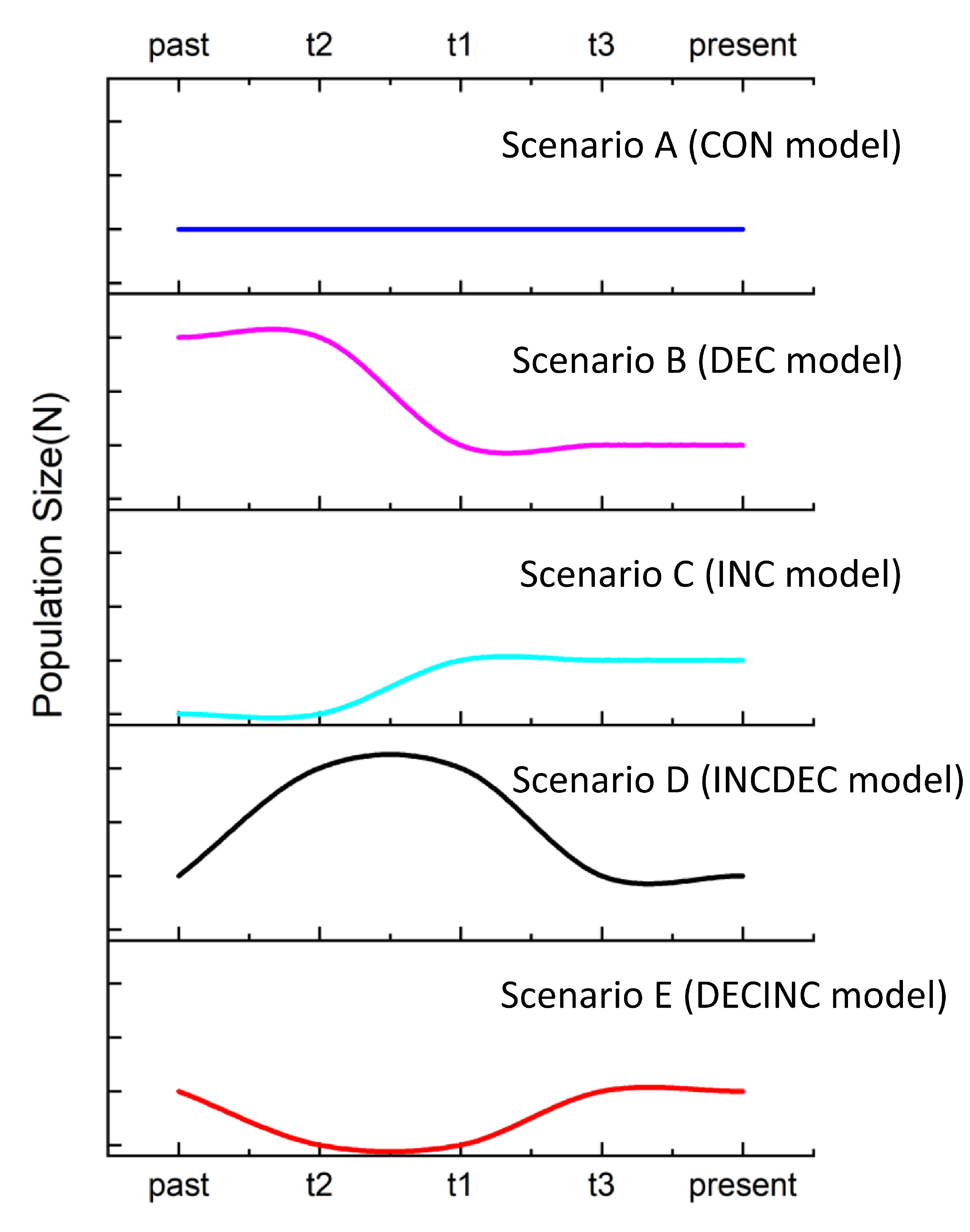
Figure 2 Schematic representation of five demographic scenarios for Barbodes semifasciolatus tested by approximate Bayesian computation (ABC). Time and effective population size are not to scale. In scenario (A): populations remained constant in size over time. In scenario (B): populations experienced a bottleneck event. In scenario (C): populations expanded recently. In scenario (D): populations experienced an ancient expansion followed by bottleneck. In scenario (E): populations experienced an old bottleneck followed by population expansion.
3 Results
3.1 Genetic diversity of Barbodes semifasciolatus
The mitochondrial cyt b gene from B. semifasciolatus comprised 1,141 base pairs (bp), which consisted of 13.8% guanine, 29.5% adenine, 28.4% thymine, and 28.4% cytosine (42.2% GC content). A total of 130 variable sites were observed, of which 111 were parsimony informative. A total of 107 haplotypes were identified from 395 specimens in the 23 populations, with 10 shared haplotypes in the total population. Haplotypes H16 and H21 were shared by 3 (CJ, MY, ZB) and 5 populations (DJ, LX, MY, NL XZ), respectively. The other shared haplotypes were shared by only two populations (Table S1). The average haplotype diversity was high (1.000), ranging from 0.248 (BS) to 1.000 (NL), and the average nucleotide diversity (θπ) within B. semifasciolatus was low (0.0134), ranging from 0.0000 (MR) to 0.0115 (LX) (Table 1). Regarding average haplotype diversity, the mainland China region exhibited the highest (0.960) value among all regions, followed by the Hainan Island region (0.935), the Taiwan region (0.852) and the Vietnam region (0.779) (Table 1). Regarding average nucleotide diversity, the mainland China region exhibited the highest (0.01291) value among all regions, followed by the Hainan Island region (0.01004), the Taiwan region (0.00286) and the Vietnam region (0.00223) (Table 1). Estimates of the current (θπ) and historical (θω) genetic diversity (a higher θω than θπ) for all populations revealed that the overall population of B. semifasciolatus shrank while local populations expanded (Templeton, 1993) (Table 1).
3.2 Phylogenetic reconstruction and genetic structure
The formation of three major lineages (A, B, and C) was inferred in the phylogenetic tree constructed using the mtDNA cyt b gene, based on the distribution patterns observed among different populations (Figure 3). Lineage A was mainly distributed in six populations from Hainan Island (CJ, ZB, CH, BS, LS and WQ), three populations from the southern Pearl River group (NL, BL, MY), four populations from the Pearl River group (DJ, XZ, LX, GR) and two populations from the Vietnam group (ZKH, ZNG). Lineage B was mainly distributed in five populations from Hainan Island (WQ, ND, LOS, LW, LS). Lineage C was composed of individuals from the DJ, XZ and LX populations in the Pearl River group, the LH, HR, JLR and MR populations in the Northern Pearl River group, and the DG population in Taiwan. The network also showed that all mtDNA haplotypes fell into three major lineages (A, B and C), with lineage B being located in the interior and the others being located at the tips (Figure 4). The time to coalescence, estimated in the BEAST analyses, was approximately 1.465 Ma (95% CI = 0.748–3.392 Ma) in B. semifasciolatus. The times for lineages A and B+C were 0.529 Mya (95% CI = 0.348–0.723 Mya) and 0.548 Mya (95% CI = 0.366–0.753 Mya), respectively. The times for lineages B and C were 0.438 Mya (95% CI = 0.247–0.645 Mya) and 0.417 Mya (95% CI = 0.258–0.587 Mya), respectively. Employing ancestral state reconstruction analyses, the results of the BBM analysis indicated that the common ancestor of B. semifasciolatus was distributed on Hainan Island with an occurrence frequency of 62.95%.
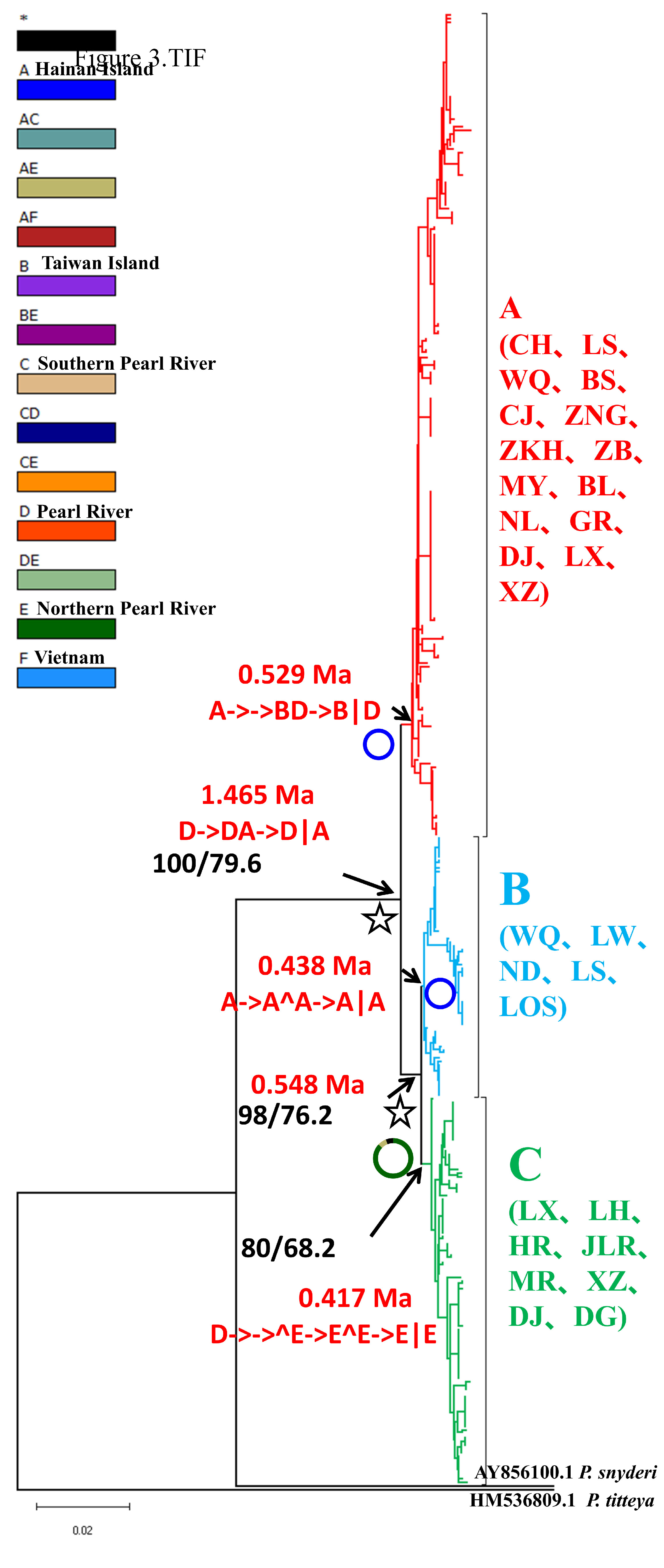
Figure 3 NJ tree of genetic relationships based on the mitochondrial cyt b gene among 23 populations of Barbodes semifasciolatus represented by 395 individuals. The values above the branches are the posterior probabilities of bootstrap values for the NJ, ML and Bayesian analyses. The results are also presented based on Bayesian binary MCMC analysis (BBM) implemented in RASP. ☆: indicates vicariance events; ↘: indicates dispersal events.
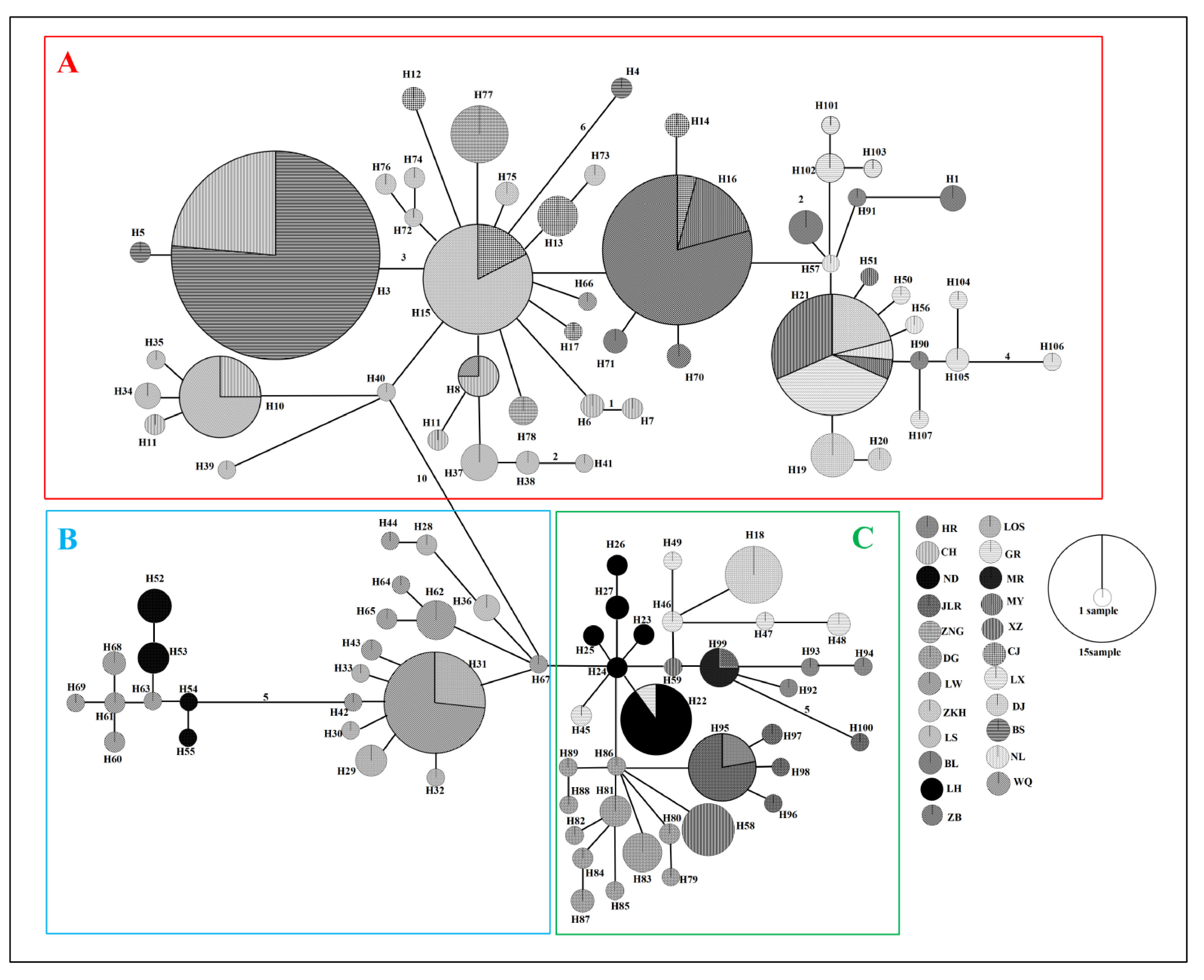
Figure 4 Minimum spanning network (MSN) of 107 haplotypes based on mutations between haplotypes observed in populations of Barbodes semifasciolatus. Haplotype designations are indicated next to each circle (Table S1). Each circle represents a haplotype, and the circle size is proportional to the number of individuals with that haplotype. Black numbers in the bars across the branches are the number of mutational steps between the haplotypes. The results of three major lineages (A–C) in the network analysis are consistent with the phylogenetic analysis.
Our analysis of ancestral area reconstruction (BBM) showed that the common ancestor of B. semifasciolatus was distributed in the Pearl River group (D), dispersed to the North Hainan group (A) and then split. With regard to lineage A, almost all specimens might have originated from ancestors that dispersed to the North Hainan group (A) or dispersed to the South Hainan group (B) and the Pearl River group (D). The populations in lineage C might have originated from the Pearl River group (D), members of which might have also split (Figure 3).
The value of NST was larger than that of GST (0.6836 and 0.3116, respectively), indicating strong phylogeographic structure in B. semifasciolatus. The pairwise FST values ranged from −0.015 (between DJ and XZ) to 0.969 (between ND and ZB), with a mean value of 0.682. The pairwise FST values indicated significant genetic differentiation in all pairwise comparisons, except DJ and XZ (p = 0.25) (Table S2). Moreover, the mean pairwise FST value (0.7259) on Hainan Island was higher than that (0.5087) in mainland China. The results of the hierarchical analyses of molecular variance (AMOVAs) showed significant genetic structure at all hierarchical levels examined (Table 2). The AMOVA results indicated that most of the genetic variation was among populations within groups, i.e., two groups (Scenario I, 65.27%), three groups (Scenario II, 56.75%), four groups (Scenario III, 51.79%) and six groups (Scenario IV, 42.93%) (Table 2). When the populations were divided into two groups (Scenario I), three groups (Scenario II), four groups (Scenario III) and six groups (Scenario IV), 1.11%, 10.96%, 16.25%, and 25.44% of the total variation was found among groups, respectively (Table 2). Moreover, after dividing the populations into seven (Scenario V) and three (Scenario VI) groups, it was found that 29.84% and 27.98%, respectively, of the total variation existed among the groups, as shown in Table 2.
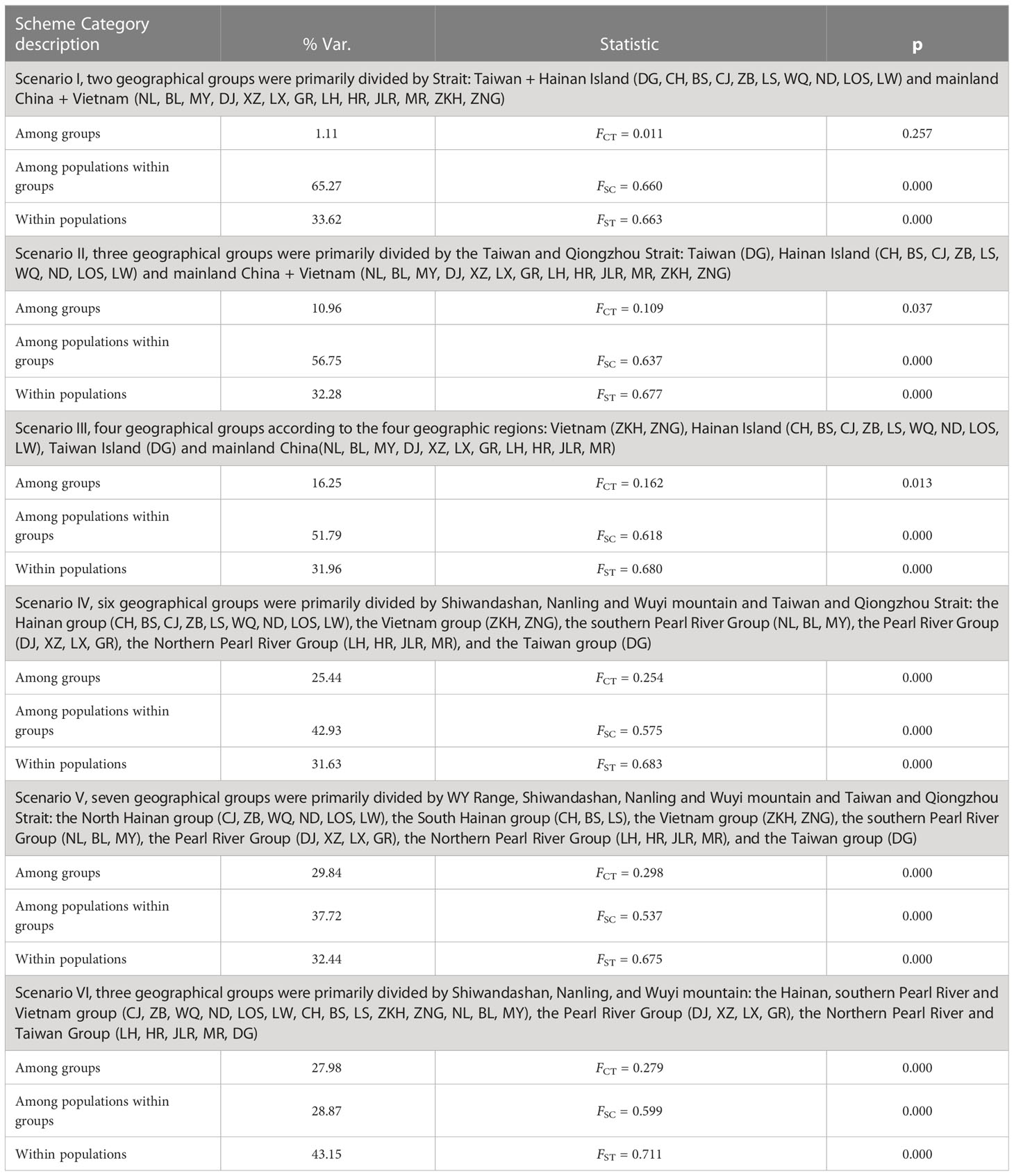
Table 2 Analysis of molecular variance (AMOVA) for Barbodes semifasciolatus based on mitochondrial cyt b gene.
3.3 Historical population demography
The mismatch distribution analysis revealed a multimodal distribution, suggesting that B. semifasciolatus populations are in decline or stable demographic equilibrium. A lower θπ (0.0134) than θω (0.0212) indicated that the population of B. semifasciolatus is in decline. The neutrality tests (Tajima’s D and Fu’s Fs tests) revealed a negative but nonsignificant Tajima’s D value and a significant negative Fu’s Fs value for the total population of B. semifasciolatus (Tajima’s D, −0.873, P > 0.10; Fu’s Fs, −49.448, P <0.01). However, Fu’s Fs test is more sensitive than Tajima’s D test in detecting population expansion (Ramos-Onsins and Rozas, 2002). Bayesian skyline plots for the total population revealed that it remained stable over a long period and that demographic expansion began approximately 300,000 years ago (Figure 5). These results reveal contrasting patterns of admixture and complex demographic histories in B. semifasciolatus. We performed approximate Bayesian computation (ABC) analysis to test explicit hypotheses about the demographic history of current populations based on an mtDNA gene sequence. Our ABC analyses indicated that the “DECINC model” (posterior probability = 0.7156 [0.5941,0.8372]) was highly favored over the “CON model” (posterior probability = 0.1132 [0.0000,0.4712]), the “DEC model” (posterior probability = 0.1712 [0.0000,0.4680]), the “INC model” (posterior probability = 0.0000 [0.0000,0.3290]), and the “INCDEC model” (posterior probability = 0.0000 [0.0000,0.3290]) in B. semifasciolatus. The DIY-ABC results showed that B. semifasciolatus experienced a bottleneck event followed by recent population expansion.
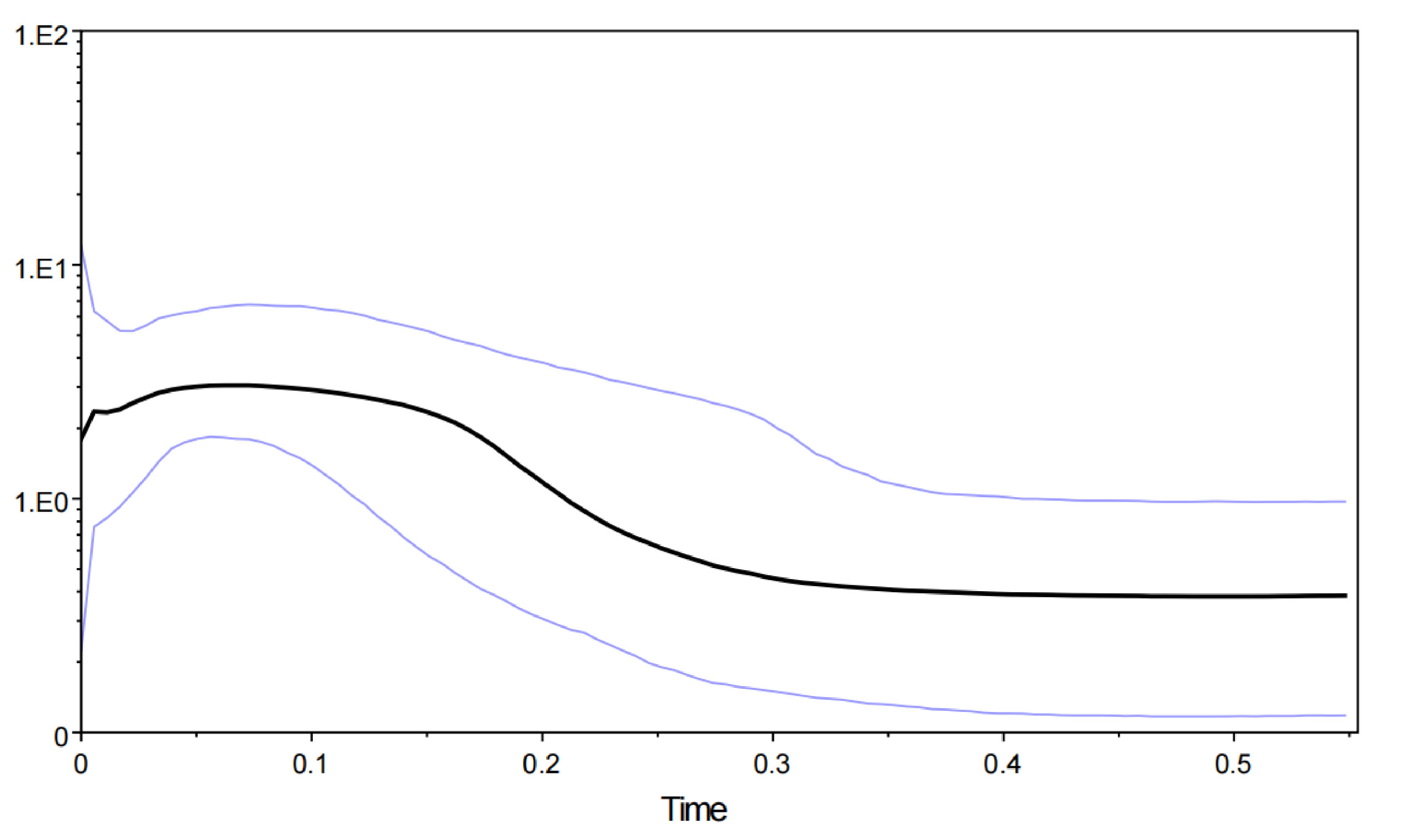
Figure 5 Bayesian skyline plot of effective population sizes over time for Barbodes semifasciolatus based on the mtDNA cyt b gene. The units of the X-axis are millions of years, and the units of the Y-axis are the estimated effective population size in units of Nes (the log-transformed product of effective population size and generation length in years). Median estimated effective population sizes (bold lines) are enclosed within 95% highest posterior density intervals (light gray lines).
4 Discussion
4.1 Genetic diversity of Barbodes semifasciolatus
Due to freshwater habitats being strongly fragmented and isolated owing to the lack of interconnecting surface water, the genetic diversity of freshwater fishes may be strongly influenced by environmental variables, such as habitat degradation, pollution, and introduction of nonnative species or climate change (Kang et al., 2014). Comprehending the extent of genetic diversity is imperative to formulate effective conservation plans, ensuring the persistent viability and adaptability of species or populations (Lande, 1988). Over 1,323 freshwater fish species occur in China, accounting for 9.6% of global fish species, and many freshwater fishes are endangered or critically endangered by human activities, leading to a severe aquatic biodiversity crisis in mainland China (Kang et al., 2014). High haplotype diversity (0.977) and low nucleotide diversity (0.0123) were detected in B. semifasciolatus. In general, high haplotype diversity is a common characteristic of freshwater fishes in south mainland China and on Hainan Island due to their r-selection life history strategies, as observed in Channa gachua (Wang et al., 2021); O. hainanensis (Wang et al., 2022b); A. normalis (Huang et al., 2019); and Onychostoma lepturum (Zhou et al., 2017). Compared with that of other freshwater fishes with similar distributions and ecological habits, the nucleotide diversity of B. semifasciolatus was more similar to that of other cyprinid species (1.076% for O. hainanensis, see Wang et al., 2022b; 1.49% for A. normalis, see Huang et al., 2019). The findings indicate that species with analogous geographic ranges and molecular diversity patterns (i.e., haplotype and nucleotide) have experienced historical events, such as geological shifts and sea level fluctuations, that have influenced their genetic makeup through demographic changes.
4.2 Population structure and demographic history
In general, high levels of genetic differentiation among populations of obligate freshwater fish were expected due to the isolating nature of freshwater systems (Ward, 1994). A high level of genetic differentiation (FST = 0.682) was detected in B. semifasciolatus based on mtDNA, which has been similarly observed in other sympatric freshwater fish with similar life histories and adult habitat preferences, such as A. normalis (FST = 0.530; Huang et al., 2019) and O. hainanensis (FST = 0.682; Wang et al., 2022b). However, the value was lower than that for other freshwater fishes, regardless of their status as upstream species, such as O. lepturum (FST = 0.750; Zhou et al., 2017) and C. gachua (FST = 0.876, Wang et al., 2021). Traditionally, species or populations of freshwater fish that occupy high-elevation, dendritic tributaries are expected to be more physically isolated than those in downstream reaches. The results of this study confirm previous findings in O. hainanensis regarding large pairwise differences between the LW and LOS populations on Hainan Island and all the other populations (Wang et al., 2022b). The effects of isolation and small population size were important factors reflecting the same historical events known to have affected freshwater fish in the same distribution. Additionally, B. semifasciolatus populations in mainland China (FST = 0.5087) exhibit lower genetic differentiation than those on Hainan Island (FST = 0.7259). The results of AMOVA revealed that 1.11%, 10.96%, and 16.25% of the total genetic variation occurred within our geographical groups (Scenarios I, II, and III), respectively. These results revealed that Taiwan and the Qiongzhou Strait were not vicariance barriers for B. semifasciolatus populations. However, 29.84% and 28.43% of the total genetic variation occurred in Scenarios V and VI, primarily divided by the Shiwandashan, Nanling, and Wuyi mountains, respectively. For B. semifasciolatus, mountains serve as a major geographic barrier, not straits. Our results are consistent with those of previous studies on C. gachua and O. hainanensis showing that sea-level change is an important parameter enabling the mixing of populations across this ocean barrier during glacial periods (Wang et al., 2021; Wang et al., 2022b).
Overall, high haplotype diversity (1.000) but low nucleotide diversity (0.013) and star-like networks indicated recent population growth after populations experienced bottleneck effects in B. semifasciolatus (Bowen et al., 2001). In addition, the significant negative values of Fu’s Fs tests and Bayesian skyline plot (BSP) analyses of all populations revealed that B. semifasciolatus might have experienced population expansion, with an effective population size that rapidly increased approximately 30 Kya. However, a multimodal distribution in mismatch distribution analysis and a higher θω than θπ did not support population expansion in B. semifasciolatus. Conflicting results of different population expansion analyses indicated that B. semifasciolatus experienced a complex demographic history. We evaluated five different demographic scenarios in recent timeframes based on the ABC analysis, and the results of the ABC analysis supported that B. semifasciolatus experienced an old shrinkage event followed by a recent population expansion event. Previous demographic studies of freshwater fish in southern mainland China and on Hainan Island revealed a similar scenario, such as that for O. hainanensis (Wang et al., 2022b). However, some previous studies revealed that populations experienced sharp demographic contraction in this region, such as S. scherzeri (Lin et al., 2022) and C. gachua (Wang et al., 2021). During the Late Pleistocene glacial cycles, sea-level changes exerted pronounced effects on the demographic history of freshwater fish. We suggest that the effects of population expansion are more significant for downstream fishes than for upstream fishes due to the more restricted habitat of the latter in the headwaters of the drainage system in south mainland China and on Hainan Island.
4.3 Phylogeography of Barbodes semifasciolatus
In our study, we focused on inferring the historical phylogeography of B. semifasciolatus in mainland China, on Hainan Island, in Taiwan, and in Vietnam. The evolutionary history of freshwater fishes reflects geological reconstructions of past landscapes because the dispersal of freshwater fishes depends on direct connections between river basins. Due to complex geohistorical events, we discuss the phylogeographical structure based on three lineages. Previous phylogeographic studies of freshwater fishes suggested that the Yunkai Mountains are major geographic barriers in southern mainland China, indicating a close evolutionary relationship among populations in the southern region of the Yunkai Mountains, on Hainan Island, and in northern Vietnam (e.g., Glyptothorax Chen et al., 2007; G. orientalis, Yang et al., 2016; O. hainanensis, Lin et al., 2016; Zhang et al., 2020). In our studies, lineage A was distributed in most populations from Hainan Island (except LOS and LW), two populations (ZNG, ZKH) from Vietnam, and six populations in mainland China. These results are consistent with previous phylogeographical findings that populations of B. semifasciolatus migrated from mainland China and Vietnam via the Qiongzhou Strait and the Gulf of Tonkin during Pleistocene glaciations (C. gachua, Wang et al., 2021; O. hainanensis, Wang et al., 2022b). However, populations located on both sides of Yunkai Mountain were also found in lineage A, indicating that the Yunkai Mountains were not phylogeographical breaks for B. semifasciolatus. In general, the fish species were small, weakly swimming, located upstream and recognized as poor dispersers and local residents; thus, they were also most likely to present strong phylogeographic structure (Chen et al., 2020; Wang et al., 2021). On Hainan Island, Wuzhishan Mountain and the Yinggeling Mountain Range (WY Range) act as important barriers limiting gene exchange between populations (e.g., G. orientalis, Yang et al., 2016; O. lepturum, Zhou et al., 2017; C. gachua, Wang et al., 2021; O. hainanensis, Wang et al., 2022b).
The Pearl River, which is the third largest river in the drainage basin area in mainland China, is composed of three main tributaries, namely, the Bei River, Dong River, and Xi River. Previous studies have shown that some species do not have obvious geographic population structure in the Pearl River drainage, such as Anguilla japonica (Zhong et al., 2022), Coilia grayii (Wang et al., 2022a), M. terminalis (Chen et al., 2020), and Hemibagrus guttatus (Yang and He, 2008). However, others had patterns concordant with predicted phylogeographic structure: the Dong and Xi/Bei Rivers contained two phylogeographic groups of S. scherzeri (Lin et al., 2022), and the Xi and Dong/Bei Rivers harbored two phylogeographic groups of G. fokiensis fokiensis (Chen et al., 2007). In our studies, populations from the Pearl River drainage in B. semifasciolatus had haplotypes distributed in lineage A and lineage C. Due to their variable life histories, such as migratory capacity and dispersal ability, freshwater fishes are likely to exhibit dramatic phylogeographic structure because of connectivity for some species and not others. These results revealed that B. semifasciolatus with relatively high dispersal potential is less susceptible to genetic divergence in the Pearl River drainage. Lineage B was located in the southeastern part of Hainan Island, including populations WQ, ND, LS, LW and LOS. Populations LW and LOS were distributed only in lineage B and were located in the small drainage basin, which flows eastward to the South China Sea. These phylogeographic patterns were consistent with those described in previous studies, and the LOS and LW populations may have been colonized by founders from adjacent populations (Wang et al., 2022b). There is potential concordance between phylogeographic patterns in ecologically similar freshwater fishes. Lineage C was mainly distributed in four populations from the coastal rivers of Southeast China, three populations in the Pearl River drainage and one population in Taiwan. The Min River is the northernmost limit of the distribution of B. semifasciolatus. According to previous studies, the Nanling and Wuyi Mountains played a key role as geographical barriers to dispersal between the Yangtze-Pearl River drainage and the coastal rivers of Southeast China (Yang et al., 2012). During glacial phases, freshwater fishes dispersed northward from the paleo-Pearl River drainage to the coastal populations (e.g., Min River and Jiulong River) by the confluence of these rivers when the sea level was low (Yang and He, 2008). Based on the network and BBM analyses, the populations in the coastal rivers of Southeast China may have colonized from population WQ on Hainan Island.
According to previous studies, four major phylogeographical regions have been identified in Taiwan: (1) the Lanyang-Danshuei group, (2) the Touchien-Houlung group, (3) the central group, and (4) the southeastern group (Wang et al., 2004; Lin et al., 2008). During marine regression, freshwater fishes of the mainland dispersed to Taiwan via multiple colonization routes at different time stages (Yang et al., 2012). The majority of this work has focused on northern Taiwan, and there are two major migration routes of freshwater fish from the mainland to northern Taiwan. One major route ran from the Zhejiang region in mainland China to the Tamsui River in the Lanyang-Danshuei group (e.g., Sinibrama macrops, Hsu et al., 2005; Hemibarbus labeo, Lin et al., 2008; S. argentatus, Yang et al., 2012). The other route was from the Min River in mainland China to the Touchien River in the Touchien-Houlung group (e.g., A. paradoxus, Ju et al., 2018; V. barbatulus, Wang et al., 2004; Formosania lacustre, Wang et al., 2007). However, few studies have addressed the possible colonization routes between mainland China and southern Taiwan. B. semifasciolatus is distributed only in the Donggang River in South Taiwan and provides an opportunity to understand the origin of freshwater fish in this region. In our study, the haplotypes in population DG were derived from population LH based on the network, indicating that the coastal river in the Guangdong region is an immigration source in South Taiwan.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
All animal experiments were carried out in accordance with the guidelines and approval of the Animal Research and Ethics Committee of School of Life Science, South China Normal University (permissions, CAMC-2018F).
Author contributions
JW and J-XW performed the data statistical analysis, chart making and drafting of the manuscript. CL and JC performed the sampling and methodology. JMY participated in data collection and DNA sequencing. JQY contributed to the conceptualization and writing manuscript. H-DL contributed to the conceptualization, writing manuscript and writing – review and editing. JZ performed the conceptualization, funding acquisition (lead), project administration (lead) and Resources. All authors contributed to the article and approved the submitted version.
Funding
National Natural Science Foundation of China, Grant/Award Number: 31772430; the China-ASEAN Maritime Cooperation Fund, Grant/Award Number: CAMC-2018F; the Key Project of Science-Technology Basic Condition Platform from the Ministry of Science and Technology of the People’s Republic of China, Grant/Award Number: 2005DKA21402. National Natural Science Foundation of China, Grant/Award Number: 31872207;
Acknowledgments
We thank the Wuzhishan National Nature Reserve, Yinggeling National Nature Reserve, Jianfengling National Nature Reserve, Bawangling National Nature Reserve, Diaoluoshan National Nature Reserve, and Jianling Provincial Nature Reserve for their assistance in the sampling. We thank Dr. Hoang Anh Tuan in Vietnam National Museum of Nature, Vietnam Academy of Science and Technology, for sample collection. We thank Xiaoyi Lin, Xinjing Li and Meiqi Feng in School of Life Science, South China Normal University, for DNA extraction and sequencing. This work was supported financially by the National Natural Science Foundation of China [31772430], the China-ASEAN Maritime Cooperation Fund [CAMC-2018F], the Key Project of Science-Technology Basic Condition Platform from the Ministry of Science and Technology of the People’s Republic of China [2005DKA21402], and National Natural Science Foundation of China, Grant/Award Number [31872207].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1193619/full#supplementary-material
References
Albert J. S., Val P., Hoorn C. (2018). The changing course of the Amazon river in the neogene: center stage for Neotropical diversification. Neotropical Ichthyology 16, e180033. doi: 10.1590/1982-0224-20180033
Bernatchez L., Wilson C.-C. (1998). Comparative phylogeography of nearctic and palearctic fishes. Mol. Ecol. 7, 431–452. doi: 10.1046/j.1365-294x.1998.00319.x
Bowen B. W., Bass A., Rocha L., Grant W., Robertson D. R. (2001). Phylogeography of the trumpetfishes (Aulostomus): ring species complex on a global scale. Evolution 55, 1029–1039. doi: 10.1111/j.0014-3820.2001.tb00619.x
Cabrera A. A., Palsbøll P. J. (2017). Inferring past demographic changes from contemporary genetic data: a simulation-based evaluation of the ABC methods implemented in DIYABC. Mol. Ecol. Resour. 17, e94–e110. doi: 10.1111/j.0014-3820.2001.tb00619.x
Chang H.-Y., Wang W.-K., Chen K.-N., Su J.-K., Hsin C.-Y., Li J., et al. (2016). Phylogeography and genetic structure of the endemic cyprinid fish microphysogobio brevirostris in northern Taiwan. Biochem. Systematics Ecol. 65, 176–184. doi: 10.1016/j.bse.2016.02.020
Chen Y. Y., Cao W. X., Zheng C. Y. (1986). The ichthyofauna of pearl river and its zoogeographic discuss. Acta Hydrobiologia Sin. 10, 228–236. doi: 10.1111/j.1095-8649.2007.01370.x
Chen X.-L., Chiang T.-Y., Lin H.-D., Zheng H.-S., Shao K.-T., Zhang Q., et al. (2007). Mitochondrial DNA phylogeography of glyptothorax fokiensis and glyptothorax hainanensis in Asia. J. Fish Biol. 70, 75–93. doi: 10.1111/j.1095-8649.2007.01370.x
Chen W., Li C., Chen F., Li Y., Yang J., Li J., et al. (2020). Phylogeographic analyses of a migratory freshwater fish (Megalobrama terminalis) reveal a shallow genetic structure and pronounced effects of sea-level changes. Gene 737, 144478. doi: 10.1016/j.gene.2020.144478
Chiang T.-Y., Lin H.-D., Shao K.-T., Hsu K.-C. (2010). Multiple factors have shaped the phylogeography of Chinese spiny loach cobitis sinensis in Taiwan as inferred from mitochondrial DNA variation. J. Fish Biol. 76, 1173–1189. doi: 10.1111/j.1095-8649.2010.02589.x
Chiang T.-Y., Lin H.-D., Zhao J., Kuo P.-H., Lee T.-W., Hsu K.-C. (2013). Diverse processes shape deep phylogeographical divergence in c obitis sinensis (T eleostei: c obitidae) in e ast a sia. J. Zoological Systematics Evolutionary Res. 51, 316–326. doi: 10.1111/jzs.12030
Cornuet J.-M., Pudlo P., Veyssier J., Dehne-Garcia A., Gautier M., Leblois R., et al. (2014). DIYABC v2. 0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30, 1187–1189. doi: 10.1093/bioinformatics/btt763
Drummond A. J., Rambaut A., Suchard M. (2013). BEAST 1.8.0. 2013. Available at: http://beast.bio.ed.ac.uk.
Dowling T. E., Tibbets C. A., Minckley W., Smith G. R. (2002). Evolutionary relationships of the plagopterins (Teleostei: cyprinidae) from cytochrome b sequences. Copeia 2002, 665–678. doi: 10.1643/0045-8511(2002)002[0665:EROTPT]2.0.CO;2
Excoffier L., Lischer H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Fu Y.-X. (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925. doi: 10.1093/genetics/147.2.915
Gascoyne M., Benjamin G., Schwarcz H. P., Ford D. C. (1979). Sea-Level lowering during the illinoian glaciation: evidence from a bahama” blue hole”. Science 205, 806–808. doi: 10.1126/science.205.4408.806
Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hall T. (2004). BioEdit. version 6.0. 7 (Raleigh, North Carolina: Department of Microbiology, North Carolina State University).
Hou X.-J., Lin H.-D., Tang W.-Q., Liu D., Han C.-C., Yang J.-Q. (2020). Complete mitochondrial genome of the freshwater fish acrossocheilus longipinnis (Teleostei: cyprinidae): genome characterization and phylogenetic analysis. Biologia 75, 1871–1880. doi: 10.2478/s11756-020-00440-y
Hsu K., Tsai K., Shao K., Wang J., Lin H. (2005). Phylogeography and population genetic structure of sinibrama macrops based on mtDNA. BioFormosa 40, 58–67.
Huang X.-X., Hsu K.-C., Kang B., Kuo P.-H., Tsai W.-H., Liang C.-M., et al. (2019). Population structure of aphyocypris normalis: phylogeography and systematics. ZooKeys 872, 77. doi: 10.3897/zookeys.872.33105
Huang C.-Y., Yuan P. B., Song S.-R., Lin C.-W., Wang C., Chen M.-T., et al. (1995). Tectonics of short-lived intra-arc basins in the arc-continent collision terrane of the coastal range, eastern Taiwan. Tectonics 14, 19–38. doi: 10.1029/94TC02452
Ju Y.-M., Hsu K.-C., Yang J.-Q., Wu J.-H., Li S., Wang W.-K., et al. (2018). Mitochondrial diversity and phylogeography of acrossocheilus paradoxus (Teleostei: cyprinidae). Mitochondrial DNA Part A 29, 1194–1202. doi: 10.1080/24701394.2018.1431227
Kang B., Deng J., Wu Y., Chen L., Zhang J., Qiu H., et al. (2014). Mapping c hina’s freshwater fishes: diversity and biogeography. Fish Fisheries 15, 209–230. doi: 10.1111/faf.12011
Lande R. (1988). Genetics and demography in biological conservation. Science 241, 1455–1460. doi: 10.1126/science.3420403
Lefort V., Longueville J.-E., Gascuel O. (2017). SMS: Smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Lin H. D., Hsu K. C., Shao K. T., Chang Y. C., Wang J. P., Lin C. J., et al. (2008). Population structure and phylogeography of Aphyocypris kikuchii based on mitochondrial DNA variation. J. Fish Biol. 72, 2011–2025. doi: 10.1111/j.1095-8649.2008.01836.x
Librado P., Rozas J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lin H.-D., Kuo P.-H., Wang W.-K., Chiu Y.-W., Ju Y.-M., Lin F.-J., et al. (2016). Speciation and differentiation of the genus opsariichthys (Teleostei: cyprinidae) in East Asia. Biochem. Systematics Ecol. 68, 92–100. doi: 10.1016/j.bse.2016.07.001
Lin M., Liang X., Gao J., Dou Y., Kuang Y., Zhang Q. (2022). Phylogeographic structure and population demography of the leopard mandarin fish (Siniperca scherzeri) in the pearl river drainage. Environ. Biol. Fishes 105, 477–486. doi: 10.1007/s10641-022-01247-3
Lynch M., Crease T. J. (1990). The analysis of population survey data on DNA sequence variation. Mol. Biol. Evol. 7, 377–394. doi: 10.1093/oxfordjournals.molbev.a040607
Nei M., Tajima F. (1983). Maximum likelihood estimation of the number of nucleotide substitutions from restriction sites data. Genetics 105, 207–217. doi: 10.1093/genetics/105.1.207
Pons O., Petit R. (1996). Measwring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144, 1237–1245. doi: 10.1093/genetics/144.3.1237
Qiu Y.-X., Fu C.-X., Comes H. P. (2011). Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 59, 225–244. doi: 10.1016/j.ympev.2011.01.012
Rambaut A. (2016) FigTree 1.4.3. Available at: http://tree.bio.ed.ac.uk/software/figtree.
Rambaut A., Drummond A. J. (2015) TreeAnnotator. version 2.2.1. Available at: http://beast2.org.
Rambaut A., Suchard M., Xie D., Drummond A. (2014) Tracer v1. 6. computer program and documentation distributed by the author. ac. uk/Tracer. Available at: http://beast.bio.ed.ac.uk/tracer.
Ramos-Onsins S. E., Rozas J. (2002). Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 19, 2092–2100. doi: 10.1093/oxfordjournals.molbev.a004034
Ren Q., Yang L., Chang C.-H., Mayden R. L. (2020). Molecular phylogeny and divergence of major clades in the puntius complex (Teleostei: cypriniformes). Zoologica Scripta 49, 697–709. doi: 10.1111/zsc.12442
Suchard M. A., Lemey P., Baele G., Ayres D. L., Drummond A. J., Rambaut A. (2018). Bayesian Phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016. doi: 10.1093/ve/vey016
Tajima F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595. doi: 10.1093/genetics/123.3.585
Tamura K., Stecher G., Kumar S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Templeton A. R. (1993). The” eve” hypotheses: a genetic critique and reanalysis. Am. Anthropologist 95, 51–72. doi: 10.1046/j.1365-2699.2000.00489.x
Voris H. K. (2000). Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J. Biogeogr. 27, 1153–1167. doi: 10.1046/j.1365-2699.2000.00489.x
Wang J., Li C., Chen J., Wang J., Jin J., Jiang S., et al. (2021). Phylogeographic structure of the dwarf snakehead (Channa gachua) around gulf of tonkin: historical biogeography and pronounced effects of sea-level changes. Ecol. Evol. 11, 12583–12595. doi: 10.1002/ece3.8003
Wang J.-P., Lin H.-D., Huang S., Pan C.-H., Chen X.-L., Chiang T.-Y. (2004). Phylogeography of varicorhinus barbatulus (Cyprinidae) in Taiwan based on nucleotide variation of mtDNA and allozymes. Mol. Phylogenet. Evol. 31, 1143–1156. doi: 10.1016/j.ympev.2003.10.001
Wang G., Tang Q., Chen Z., Guo D., Zhou L., Lai H., et al. (2022a). Otolith microchemistry and demographic history provide new insight into the migratory behavior and heterogeneous genetic divergence of coilia grayii in the pearl river. Fishes 7, 23. doi: 10.3390/fishes7010023
Wang T.-Y., Tzeng C.-S., Teng H.-Y., Chang T. (2007). Phylogeography and identification of a 187-bp-long duplication within the mitochondrial control region of formosania lacustre (Teleostei: balitoridae). ZOOLOGICAL STUDIES-TAIPEI 46, 569.
Wang J., Zhang W., Wu J., Li C., Ju Y.-M., Lin H.-D., et al. (2022b). Multilocus phylogeography and population genetic analyses of opsariichthys hainanensis reveal pleistocene isolation followed by high gene flow around the gulf of tonkin. Genes 13, 1908. doi: 10.3390/genes13101908
Ward J. (1994). Ecology of alpine streams. Freshw. Biol. 32, 277–294. doi: 10.1111/j.1365-2427.1994.tb01126.x
Watterson G. A. (1975). On the number of segregating sites in genetical models without recombination. Theor. Population Biol. 7 (2), 256–276. doi: 10.1016/0040-5809(75)90020-9
Xiao W., Zhang Y., Liu H. (2001). Molecular systematics of xenocyprinae (Teleostei: cyprinidae): taxonomy, biogeography, and coevolution of a special group restricted in East Asia. Mol. Phylogenet. Evol. 18, 163–173. doi: 10.1006/mpev.2000.0879
Yang L., He S. (2008). Phylogeography of the freshwater catfish hemibagrus guttatus (Siluriformes, bagridae): implications for south China biogeography and influence of sea-level changes. Mol. Phylogenet. Evol. 49, 393–398. doi: 10.1016/j.ympev.2008.05.032
Yang J.-Q., Hsu K.-C., Liu Z.-Z., Su L.-W., Kuo P.-H., Tang W.-Q., et al. (2016). The population history of garra orientalis (Teleostei: cyprinidae) using mitochondrial DNA and microsatellite data with approximate Bayesian computation. BMC Evolutionary Biol. 16, 1–15. doi: 10.1186/s12862-016-0645-9
Yang J.-Q., Tang W.-Q., Liao T.-Y., Sun Y., Zhou Z.-C., Han C.-C., et al. (2012). Phylogeographical analysis on squalidus argentatus recapitulates historical landscapes and drainage evolution on the island of Taiwan and mainland China. Int. J. Mol. Sci. 13, 1405–1425. doi: 10.3390/ijms13021405
Yang J.-Q., Tang W.-Q., Sun Y., Tsai K.-C., Zhou Z.-C., Liu Z.-Z., et al. (2013). Microsatellite diversity and population genetic structure of squalidus argentatus (Cyprinidae) on the island of hainan and mainland China. Biochem. Systematics Ecol. 50, 7–15. doi: 10.1016/j.bse.2013.03.023
Yu H.-T. (1995). Patterns of diversification and genetic population structure of small mammals in Taiwan. Biol. J. Linn. Soc. 55, 69–89. doi: 10.1111/j.1095-8312.1995.tb01050.x
Yu Y., Harris A. J., Blair C., He X. (2015). RASP (Reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49. doi: 10.1016/j.ympev.2015.03.008
Zhang W.-J., Wang J.-J., Li C., Chen J.-Q., Li W., Jiang S.-Y., et al. (2020). Spatial genetic structure of opsariichthys hainanensis in south China. Mitochondrial DNA Part A 31, 98–107. doi: 10.1080/24701394.2020.1741564
Zhong Z., Zhu H., Fan J., Ma D. (2022). Mitochondrial DNA and microsatellite analyses showed panmixia between temporal samples in endangered Anguilla japonica in the pearl river basin (China). Animals 12, 3380. doi: 10.3390/ani12233380
Keywords: Barbodes semifasciolatus, demography, DIY-ABC, mitochondria, phylogeography
Citation: Wang J, Wu J, Yang J, Chen J, Yang J, Li C, Lin H-D and Zhao J (2023) Phylogeography and demographic history of the cyprinid fish Barbodes semifasciolatus: implications for the history of landform changes in south mainland China, Hainan and Taiwan. Front. Ecol. Evol. 11:1193619. doi: 10.3389/fevo.2023.1193619
Received: 25 March 2023; Accepted: 26 June 2023;
Published: 11 July 2023.
Edited by:
Wagner Franco Molina, Federal University of Rio Grande do Norte, BrazilReviewed by:
Erik Garcia-Machado, Laval University, CanadaCuizhang Fu, Fudan University, China
Rodrigo Maggioni, Federal University of Ceara, Brazil
Copyright © 2023 Wang, Wu, Yang, Chen, Yang, Li, Lin and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Zhao, zhaojun@scnu.edu.cn; Hung-Du Lin, varicorhinus@hotmail.com
†These authors have contributed equally to this work