 Lin Mao†
Lin Mao† Longyan Yang
Longyan Yang Dong Zhao
Dong Zhao- Beijing Key Laboratory of Diabetes Prevention and Research, Center for Endocrine Metabolic and Immune Diseases, Beijing Luhe Hospital, Capital Medical University, Beijing, China
Atherosclerosis (AS) is a chronic inflammatory disease and leading cause of cardiovascular diseases. The progression of AS is a multi-step process leading to high morbidity and mortality. Hyperglycemia, dyslipidemia, advanced glycation end products (AGEs), inflammation and insulin resistance which strictly involved in diabetes are closely related to the pathogenesis of AS. A growing number of studies have linked AGEs to AS. As one of the risk factors of cardiac metabolic diseases, dysfunction of VSMCs plays an important role in AS pathogenesis. AGEs are increased in diabetes, participate in the occurrence and progression of AS through multiple molecular mechanisms of vascular cell injury. As the main functional cells of vascular, vascular smooth muscle cells (VSMCs) play different roles in each stage of atherosclerotic lesions. The interaction between AGEs and receptor for AGEs (RAGE) accelerates AS by affecting the proliferation and migration of VSMCs. In addition, increasing researches have reported that AGEs promote osteogenic transformation and macrophage-like transformation of VSMCs, and affect the progression of AS through other aspects such as autophagy and cell cycle. In this review, we summarize the effect of AGEs on VSMCs in atherosclerotic plaque development and progression. We also discuss the AGEs that link AS and diabetes mellitus, including oxidative stress, inflammation, RAGE ligands, small noncoding RNAs.
Introduction
With the development of global economy and production technology, there were significant changes in human dietary structure and lifestyle, which have aggravated the global burden of cardiovascular disease (1, 2). Atherosclerosis (AS) is a chronic inflammatory disease, which is a major cause of cardiovascular events (3). The risk factors causing AS are divided into physiological and pathological risk factors, the former includes unhealthy dietary intake, lack of physical activity, genetics, specific population (age, race, nationality and gender) and smoking, the latter includes hyperlipidemia, pre-diabetes/diabetes, hypertension, obesity and renal dysfunction (4). AS is the most common type of disabling and fatal disease among the complications of diabetic macrovascular disease (5). Dysfunction of lipid metabolism accompanying systemic metabolic diseases in AS is an important risk factor and characteristic (6). Hyperglycemia, dyslipidemia, advanced glycation end products (AGEs), inflammation and insulin resistance, which strictly involved in diabetes, are closely related to the pathogenesis of AS (7, 8). Compared with non-diabetic patients, diabetic patients have a 2-5 fold increased risk of developing AS, the degree of plaque lesions is severe which often involves multivessel disease and is characterized by extensity, acceleration, vulnerability (9, 10). Sustained high-glucose environment provides sufficient reaction substrates and increases the oxidation of the microenvironment of vascular wall, aggravating vascular wall damage and atherosclerotic plaque vulnerability (11). In addition to lipid content, atherosclerotic plaques include different cell types, including vascular smooth muscle cells (VSMCs) and inflammatory cells (such as macrophages, dendritic cells, and lymphocytes), extracellular matrix (ECM) proteins, and cell fragments (12).
VSMCs are important participant in early and late-stage AS (13). VSMCs are exquisitely sensitive to stimulation, and responsible for contraction and relaxation in physiological state, thus regulating intravascular blood flow (14). After endothelial cell dysfunction, a variety of bioactive mediators released from the injured site induce phenotype change of VSMCs from the quiescent “contractile” phenotype state to the active “synthetic” state, which is the key segment affecting the progression of AS (15–18). Diabetes, a disease characterized by persistent high glucose status, continuously induces the production of reactive oxygen species (ROS), breaks the redox balance, induces inflammatory response and phenotypic transformation, leads to vascular remodeling (19, 20). Many studies have reported that long term glycemic control is beneficial to the development and progression of diabetic cardiovascular complications (21–24).
Recent studies suggest that AS is related to the “metabolic memory” theory marked by AGEs, which means that even blood glucose concentration in the normal range, the vascular events risk is amplified (25, 26). AGEs are a class of complexes produced by non-enzymatic reactions of glucose and derivatives, which exacerbate AS in a direct or indirect manner, direct manner means the capture and cross-linking of proteins, and indirect manner by binding to specific receptors on the cell surface (27). Binding of specific receptor for advanced glycation end-product (RAGE) and AGEs carries a big weight in the function of atherosclerotic VSMCs. RAGE expressed on VSMCs surface and mediates multiple damage mechanisms, thereby affecting the progression of AS (28–30). AGEs also have adverse effects on erectile dysfunction (ED), a risk factor for cardiovascular disease in diabetes, and diabetes retinopathy (31, 32). Decreased NO and increased prethrombotic factors result in endothelial dysfunction and thrombosis, while an increase in nuclear factor kappaB (NF-κB) with inflammatory reaction (33, 34). AGEs induce apoptosis of retinal pericytes, differentiation of osteoblasts and calcification, and exert toxic effects on retinal capillaries (35). In this review, we aimed to explore the effects of AGEs on VSMCs function in AS and discuss the AGEs that link AS and diabetes mellitus, including oxidative stress, inflammation, RAGE ligands, small noncoding RNAs and other aspects.
VSMCs in different stages of AS
As the main functional cells of vascular, VSMCs play different roles in each stage of atherosclerotic lesions. Abnormal proliferation of VSMCs is a common and important mechanism involved in the pathogenesis of many vascular diseases, including AS and aortic aneurysm formation (36).
AS can be divided into pre-atherosclerotic and atherosclerotic stage, in which the atherosclerotic stage can be subdivided into early AS, late AS and clinical sequelae. The prominent features of pre-atherosclerotic stage are diffuse intimal thickenings (DITs) and intimal lipid streaks, in which DITs considered to be the precursor of AS (37, 38). Different from the normal VSMCs in the media, VSMCs in the thickened intima showed a synthetic phenotype with low contractile protein expression and high extracellular matrix (ECM) expression (39, 40). At this stage, VSMCs slightly decreased the content of cholesterol esterase and ATP binding cassette transporter (ABCA1) (41, 42), which led to the production of VSMCs-derived foam cells (43), and established the pathological basis for the subsequent AS progress. In the early stage of AS, pathological intimal thickenings (PITs) occur, which are manifested intimal deep lipid pools formation on the basis of abundant VSMCs and ECM (12, 38). Complex pathological processes such as lipid accumulation, inflammatory stimulation and phenotype transformation of VSMCs promote DITs to PITs. Studies have confirmed that VSMCs are the main source of ECM that plays a central role in initiation of AS (44–46). The development from DITs to PITs is accompanied by loss of VSMCs marker α-smooth muscle actin (α-SMA), which may be the result of phenotypic transformation (47, 48) or death (49, 50). Macrophages can be absent in early PITs, and are essential to the progression of late PITs to fibroatheromas (12). The co-expression of α-SMA and CD68 cells in human atherosclerotic plaques suggesting that VSMCs also play an important role in the cells expressing macrophage markers (51, 52). In addition, there is an interaction between macrophages and VSMCs (53). VSMCs secrete diversified active factors to recruit macrophages to form PITs (54–56), and macrophages have an effect on VSMCs proliferation, migration and phenotypic transformation (57, 58). The next stage of PITs is fibroatheromas, in which fibrous caps as well as necrotic cores appear, which originate from the deep intima lipid pools as well as VSMCs and macrophages with insufficient cell clearance (59, 60). Macrophages phagocytize deposited lipids to become foam cells (61), after exceeding the scavenging capacity of macrophages, they resulted in sustained inflammatory response, caused the death of macrophages- and VSMCs-derived foam cells, then secondary necrosis (62, 63). Fibrous cap is a healing reaction accompanied by injury, proliferation and migration of medial VSMCs stimulated by a variety of materials (47, 64, 65). In addition, VSMCs has long been considered to contribute to calcification. In early fibroatheromas, the causes of calcification include calcified microvesicles from macrophages and VSMCs (66, 67), release of apoptotic bodies and activity of osteochondrogenic cells (68, 69). In the final phases, The main characteristics are increased cholesterol and calcification in necrotic core and MMPs related fiber cap decomposition and remodeling (70). Both VSMCs death in the fibrous cap and necrotic core enlargement will lead to fibrous cap thinner, which makes the plaque easier to rupture (71, 72). Plaque rupture is conversely correlated with VSMCs, and the proliferation, migration and death of VSMCs are important segments (73). The role of VSMCs in different stages of AS was summarized in Table 1.

Table 1 VSMCs in different stages of AS.
Mechanisms of VSMCs in AS
As is a chronic inflammatory disease, which is closely related to VSMCs function. VSMCs mainly affect the progression of AS through proliferation, migration and phenotypic transformation. In VSMCs, TNF-α induce nuclear translocation of p65 and STAT3, which may be associated with TNF-α-regulated target promoters, such as monocyte chemoattractant protein-1 (MCP-1) and intercellular cell adhesion molecule (ICAM-1) (74). It is reported that 17β-estradiol (E2) downregulate of tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL) expression via suppression of NF-κB, thereby inhibiting rat VSMCs proliferation and migration (75). Systemic knockout of IL-1β or IL-1 receptor type 1 (IL-1R1) reduced plaque formation, whereas knockout of the IL-1 receptor 1 antagonist gene (IL-1Ra) increased plaque development. Furthermore, treatment with IL-1β neutralizing antibody western diet feeding to apoE -/- mice reduces overall plaque burden (76–79). However, there are currently no preclinical studies showing benefits of inhibiting IL-1β in advanced atherosclerotic stage.
Recently the effect of noncoding RNAs on regulating AS has drawn attention. Continued improvements in molecular sequencing technologies have led to a greater understanding of AS at the single-cell, chromatin, and epigenetic levels. Circular RNAs (circRNAs) are endogenous regulatory RNAs, which are covalently closed loops after reverse splicing due to the lack of 3’-poly-a tail and 5’- cap (80). Studies have demonstrated that most circRNAs regulate gene expression post-transcriptionally through sponging microRNAs (miRNAs), for example circACTA2, circ-SATB2, circDiaph3, circ_0020397, circTET3, circCCDC66, and miR-541, miR-195, miR-146a, miR-133, miR-214, miR-34a (81, 82). It is suggested that circRNAs and miRNAs may be a new target spot and a new hot point in preventing dysfunctional VSMCs in AS. Connective tissue growth factor (CTGF) plays a crucial role in the VSMCs proliferation and migration to response stimulation of by hyperglycemia (83, 84), AGEs (85), and hypoxia (86). Silence of CTGF increases VSMCs proliferation time by prolonging cell G0/G1 phase, blocking VSMCs into S phase (85). VSMCs proliferation was associated with increased expression of RAGE and its ligands. High mobility group box-1 (HMGB1) plays a pivotal role after vascular injuries. Human VSMCs were treated with HMGB1 (100 ng/ml), markedly increased osteoprotegerin production which mediated by activator protein 1(AP-1), as well as also affected the migration ability of VSMCs (87, 88). Knockdown of S100B by shRNA inhibits PDGF-BB-induced VSMCs proliferation and migration in vitro (89). The phenotypic transformation of VSMCs is also a key mechanism in AS progress. Recent studies have reported that Nidogen-2 and MEF2B play an important role for maintenance of VSMCs identity. Nidogen-2 is a basement membrane glycoprotein that enhances the interaction between Jagged1 and Notch3 and subsequent Notch3 activation. Compared with wild-type mice, Jagged1 overexpression attenuated the inhibition of neointima formation in nidogen-2-/- mice (90). Cyclic stretch enhances Nox1 mediated ROS production through MEF2B activation signal and causes VSMCs to switch to a synthetic phenotype (91). Atherosclerotic plaque and vascular calcification constitute two features of AS. MFN2 and LncRNA H19 have been shown to promote calcification of VSMCs (92, 93). The stability of later period of atherosclerotic plaque was dominated by FAM172a, RAGE, galectin-3, autophagy and apoptosis (94–97). Trans-differentiation of VSMCs into a macrophage-like state to phagocytize lipids and form more foam cells are both an interfering factor affecting plaque stability (30, 97). The above mechanisms of VSMCs in AS were summarized in Table 2.

Table 2 Mechanisms of VSMCs in AS.
Diabetes and VSMCs
The rapid increase of diabetes and its underlying patients led to a substantial increase in the number of diabetic cardiovascular complications (98). In fact, the effect of diabetes on atherosclerotic VSMCs is not simply regulated by a certain mechanism, but the result of the interaction of multiple factors (99). The simple mechanism of diabetes on atherosclerotic VSMCs was summarized in Figure 1. With the development of genetic engineering technology, it is possible to trace the lineage of VSMCs, study its fate map, and further study its developmental origin, plasticity, clonality and function in plaque, which provides reliable evidence for the complex role of VSMCs and VSMCs-derived cells in AS. Numerous studies demonstrate that regulating the phenotypic transformation of VSMCs and affecting their function alleviate AS severity (100–102).
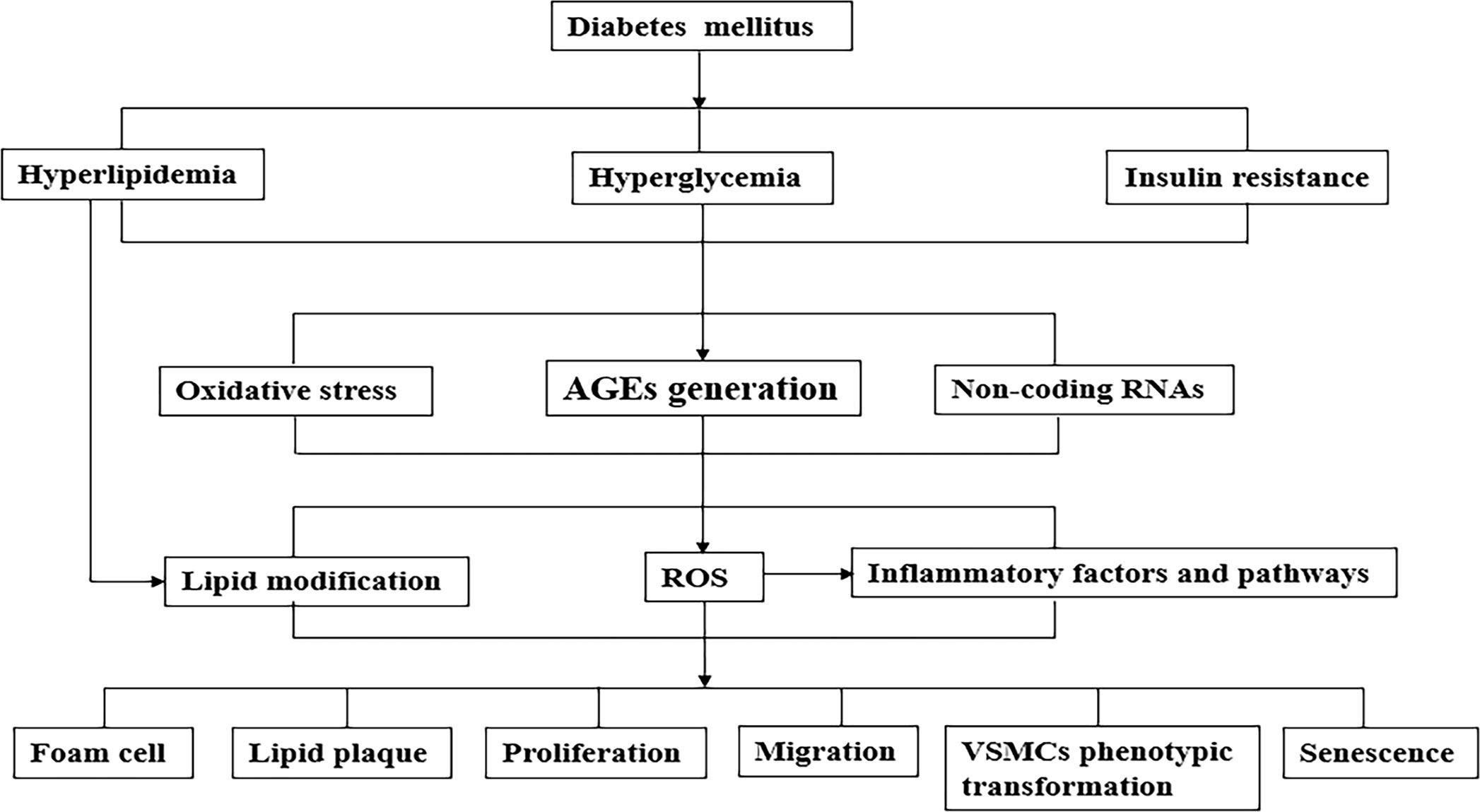
Figure 1 A simplified mechanism of the pathophysiological connection of the effect of diabetes on atherosclerotic VSMCs. Metabolic abnormalities (such as hyperglycemia, hyperlipidemia) and insulin resistance caused by diabetes cause a variety of pathological changes in blood vessels, including atherogenic lipoproteins, the formation of AGEs, oxidative stress and various inflammatory factors/pathways. As the above-mentioned synergistic effects of mechanisms cause phenotypic transformation of VSMCs which are functional cells of blood vessels, lead to aggravate AS development. AGEs, advanced glycation end products; VSMCs, vascular smooth muscle cells; LDL, low density lipoprotein.
In response to vascular injury, VSMCs convert from physiological contraction to pathological proliferation synthesis that through proliferation, migration, osteogenic transformation and macrophage-like transformation these play a significant role in the development and progression of AS plaque (103). The effect of glucose metabolism on VSMCs which mainly located in media is crucial. High glucose induced vasoconstriction in rat is highly sensitive that directly affects VSMCs contractile function (104). Previous studies shown that glucose concentration in diabetic patients up to 30.5 mmol/l, which can upregulate F-actin, α-SMA and cytoskeleton (105–107). Inflammatory factors such as TNF-α, IL-α, PDGF, Fibroblast growth factor 21 (FGF21) are produced at the site of high glucose injury (108), PI3K/Akt, NF-κB and other signal pathways also involved (109–113). Continuous high glucose environment in body increases the oxidation of vascular cells, making them in oxidative stress state over a long period of time, which is a key pathogenic factor for AS progression (82, 114–116). Severe vascular calcification in diabetic has also been confirmed to be related to VSMCs (117, 118). In addition, AGEs (119, 120), apoptosis (121, 122), oxidative stress (123, 124) and a variety of noncoding RNAs (125, 126) regulate AS under diabetic state. Recent studies about the effect of high glucose on VSMCs was listed in Table 3. Therefore, an important step for the prevention and treatment of diabetic AS is to inhibit the abnormal proliferation and migration of VSMCs.

Table 3 Mechanisms of high glucose in VSMCs function.
Cell senescence is a dynamic process of incremental development. Senescent VSMCs show a decrease in contractile protein expression. The senescent VSMCs in AS affect the size and stability of plaque (136). Senescent VSMCs secrete matrix degradation protease, which leads to collagen reduction and increases the risk of AS related complications (137). The combined treatment of Ang II and high glucose synergistically increases the proportion of aging regions in VSMCs, partly through autophagy, oxidative stress and p21-pRb pathway (138). Studies have shown that senescent VSMCs induced by proprotein invertase subtilisin/Kexin type 9 (PCSK9) is related to apoptosis pathway (139). Compared with control group, the burden of aortic plaque in diabetes mice was more serious, with fewer VSMCs, but the proportion of senescent cells in the plaque was larger (140). All above contents suggest that senescent VSMCs are closely related to the severity of atherosclerotic plaque in diabetes.
AGEs and AS
The heterogeneous molecules of AGEs were created in the classic Maillard reaction discovered in the early 20th century (127). And more than three decades ago, a theory about aging was proposed, which hypothesized that the slow and sustained accumulation of AGEs was a causal factor in aging, and the long-term accumulation of these compounds might alter the structure and function of proteins (141, 142). This process may also lead to the pathology of metabolic diseases, such as diabetes and AS, as well as oxidative stress and inflammation associated with neurodegenerative diseases of aging (127, 143). Maillard reaction, a classical generation pathway of endogenous AGEs, which consists of three main steps: first, reducing sugars react with proteins, lipids and nucleic acids to form unstable Schiff bases through non-enzymatic reactions, then undergo structural rearrangement to form stable and irreversible complex, and finally the complex is oxidized (glucose oxidation), dehydrated and degraded to the final product (144). Under physiological conditions, slow and complex glycosylation reactions between sugars form macromolecular toxic substances AGEs (145). The production pathways of AGEs can be divided into endogenous and exogenous pathways (146). Most of the exogenous pathways are derived from the western diet which is mainly thermal processing of food, especially frying, grilling, baking or barbecuing, which are the major sources of exogenous AGEs (147). At present, more than 20 kinds of AGEs have been identified, such as carboxymethyl-lysine (CML), carboxyethyl-lysine (CEL), pentosidine and so on (148).
In addition, smoke is also another important source of exogenous AGEs, and cigarettes contain generous glycosylation products (146). In healthy population, the intake and excretion of AGEs in a dynamic balance, but pathological states such as diabetes, aging and inflammation can break the balance and accelerate the non-enzymatic glycosylation (127, 145). AGEs are metabolized by binding with corresponding receptors located on wide variety types of cells surface, which then are released by degrading into a small soluble peptide (149). Kidney is an indispensable organ in the metabolic process of AGEs. About 30% intake of dietary AGEs eliminated by kidney in healthy body (150). If kidney function is impaired, this percentage will be lower.
A growing number of studies have testified that AGEs produce a marked effect in the above-mentioned effects on the function and phenotype of VSMCs. Under the condition of hyperglycemia, AGEs formation was accelerated. Accumulated AGEs have been associated with number of diseases, including AS and diabetes (151). AGEs are a group of glycated macromolecular protein with strong resistance to protease hydrolysis that are formed irreversibly through a chain of nonenzymatic chemical reactions (152). Previous studies have shown that the levels of AGEs in diabetic patients are significantly higher than non-diabetes patients, which are bound up with the severity of vascular complications (25, 153). At the same time, the accumulation of AGEs was detected in macrophages and VSMCs of atherosclerotic vessel walls (25). The function of AGEs in AS was summarized in Table 4.

Table 4 AGEs and AS.
AGEs and RAGE
As described above, most notably pathogenic mechanism is the specific binding of RAGE to AGEs, see Figure 2. RAGE is a member of the immunoglobulin superfamily and owns multiple ligands. RAGE exists on the surface of vascular cells membrane, most in VSMCs (163). In physiological condition, the expression level of RAGE is minimal, disease can upregulate its expression to an activatable state (164). Recent studies have identified four forms of RAGE in mammals, full length cell RAGE, N-truncated RAGE, and two C-truncated RAGE which has two isoforms, cleaved RAGE (c-RAGE), and endogenous secretory RAGE (esRAGE) (165). RAGE was treated differently to form c-RAGE and esRAGE, the former was formed by proteolytic cleavage, and the latter was the product of selective mRNA splicing (166, 167). It has been found that RAGE contains complete three domains that allow ligands to function properly in their biology. sRAGE consists of c-RAGE and esRAGE (168), which are nonfunctional because neither of them do not have a complete signal transduction transmembrane domain. Both sRAGE and esRAGE can competitively bind RAGE ligands, thus antagonize RAGE mediated pathological effects (168).
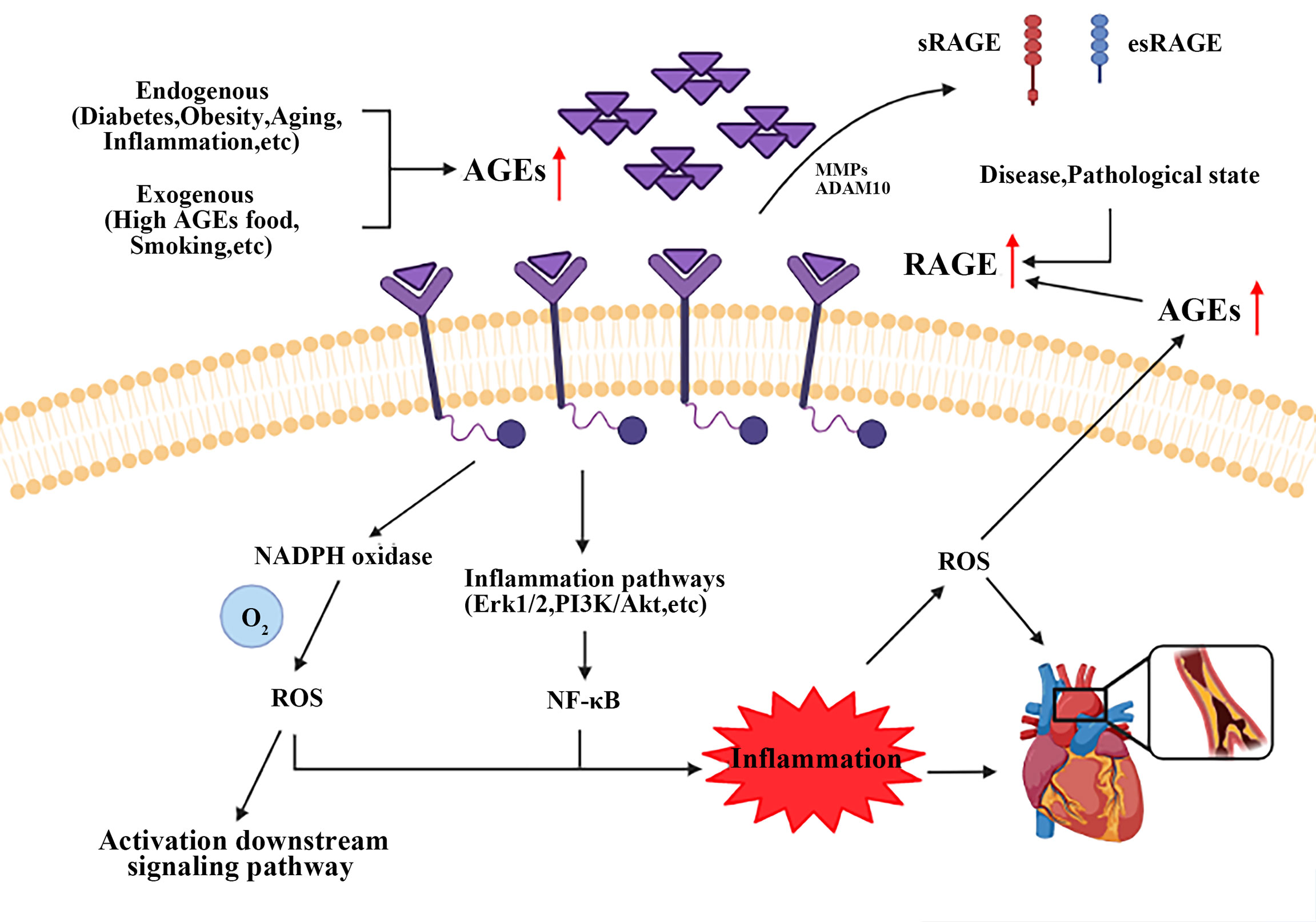
Figure 2 Brief mechanism of the interaction between AGEs and RAGE on VSMCs. Endogenous and exogenous pathways increase AGEs content, while disease and pathological states upregulate the expression of RAGE. AGEs-RAGE combination leads to the increase of ROS and activates inflammatory signaling pathways, such as Erk1/2, PI3K/Akt and NF-κB. All these events form a positive feedback loop, and ultimately promote AS progress. AGEs, advanced glycation end products; RAGE, receptor for advanced glycation end products. ROS, reactive oxygen species.
AGEs and VSMCs
VSMCs play important roles in AS, the contractile-synthetic phenotypic of VSMCs conversion was critical in atherosclerotic plaque formation and development (30). Abnormally AGEs (resulted from hyperglycemia) lead to the differentiation of contractile VSMCs in vessel medium into synthetic VSMCs (169). The mechanism of AGEs on VSMCs was summarized in Figure 3.
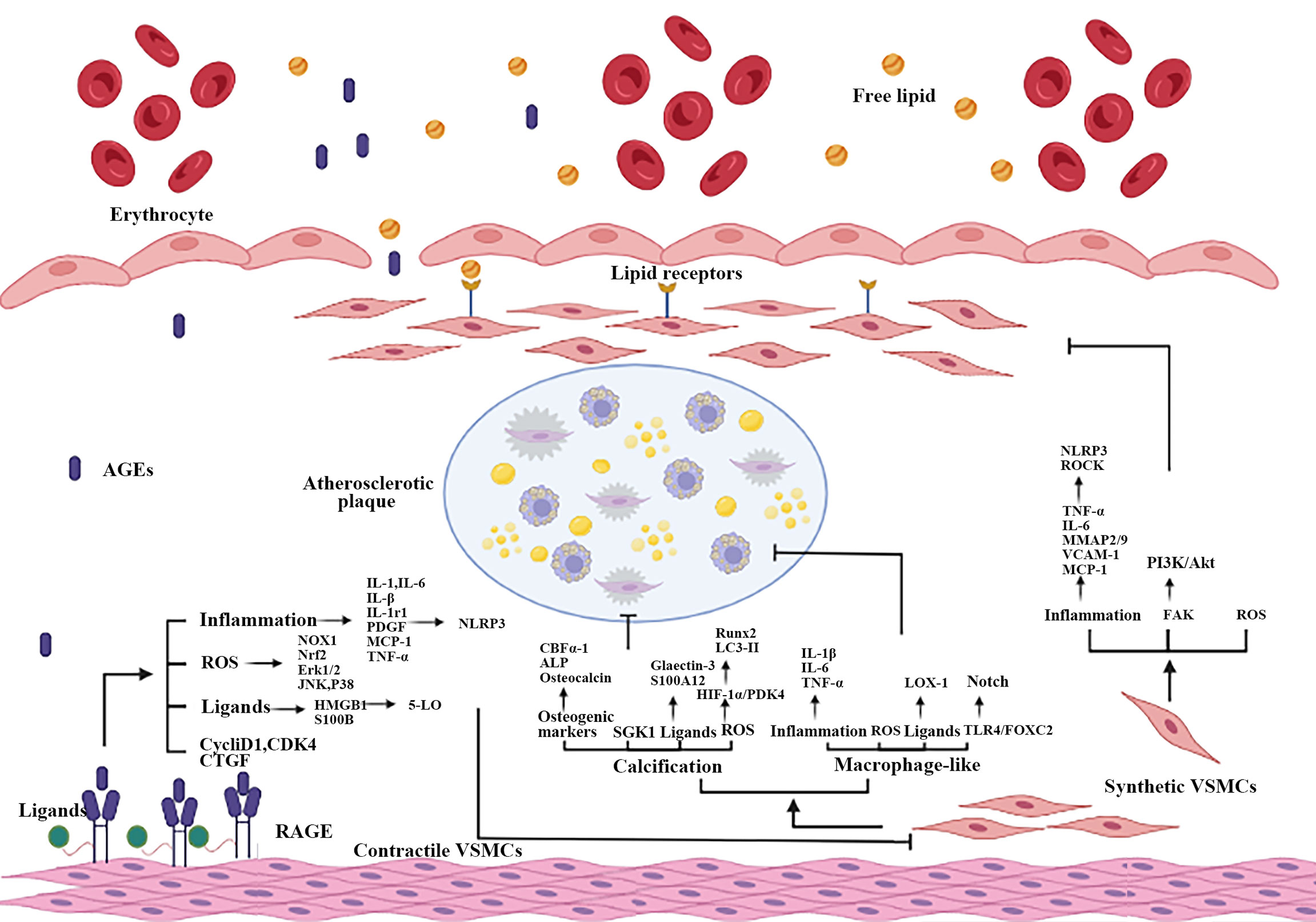
Figure 3 Part mechanisms of the interaction between AGEs and RAGE on atherosclerotic VSMCs. When vascular ECs are impaired, AGEs-RAGE interaction causes phenotypic transformation and VSMCs dysfunction, including proliferation, migration, calcification and the increase of macrophage-like VSMCs. Inflammation, oxidative stress, RAGE multiple ligands and other mechanisms are participated in AGEs promote atherosclerotic process by damaging VSMCs. ECs, endothelial cells.
Proliferation
VSMCs in the normal state are differentiated, quiescent and contracted, whereas VSMCs in the damaged state are dedifferentiated, proliferated and synthetic phenotypes (170). A growing body of evidences suggest that VSMCs exposed to AGEs exhibited a phenotype with proliferation. VSMCs proliferation has different dual roles during atherosclerotic lesion progression, VSMCs proliferation was needed in successful plaque repair and plaque stability; however, unregulated proliferation accelerates the progression of diabetic vasculopathy (171, 172). Excessive proliferation of medial VSMCs results the increased aortic stiffness and decreased aortic compliance. Blood serum levels of AGEs are elevated in CAD patients with type 2 diabetes as compared with CAD patients without diabetes (173). AGEs/RAGE interaction induced the proliferation of different species VSMCs, via increased ROS production and downstream pathway activation subsequently (174–176). RAGE expression was found to be significantly increased in human carotid AS (177), associated with elevated oxidative stressors locally (178). HMGB1 induces VSMCs to produce leukotrienes, increases 5-LO expression, and subsequently upregulates RAGE mRNA and protein expression in rapidly proliferating VSMCs, indirectly affecting vascular remodeling (179). Furthermore, VSMCs proliferation is suppressed in homozygous RAGE-null mice, relative to wild-type littermates. Increased ROS in VSMCs is a result of NADPH oxidase catalytic subunit differentially regulated by AGEs, which stimulates Nox1 transcription in young age, but Nox4 in aging under hyperlipidemic conditions (180). Glycated serum albumin promotes VSMCs proliferation by inducing the expression of inflammatory factors MCP-1 and IL-6 (181), which is inhibited by antioxidant N-acetylcysteine. Statins upregulate the expression of Nrf2-related antioxidant genes NQO1 and HO-1 in VSMCs through the ERK5-dependent Nrf2 pathway and inhibit the effect of AGEs-induced VSMCs proliferation (182). AGEs/ROS/ERK1/2 signaling pathway also induced VSMCs proliferation (183). Collectively, these results showed that inflammation and oxidative stress play a central role in the proliferation of VSMCs and inhibit the proliferation of VSMCs could be a useful method for AS treatment.
Migration
In fact, VSMCs migration plays different roles in atherosclerotic plaques at different stages. In early plaques, VSMCs migration contributes to stabilize plaques through forming fibrous caps, while in late plaques, the death of migrated VSMCs accelerates plaque rupture and corresponding clinical symptoms (184). AGEs promote VSMCs migration by stimulating inflammatory cytokine production and activating multiple pathways, leading to arterial disease (161, 162). Focal adhesion kinase (FAK) is expressed selectively in VSMCs, and links extracellular matrix/integrin and growth factors, inhibited FAK/PI3K/Akt pathways could promote VSMCs migration (185, 186). It is suggested that RAGE silence by lentivirus transfection leads to reduce the phosphorylation of Akt, thus reducing the expression of migration-related proteins (187). Crocetin pretreated VSMCs downregulated TNF-α, IL-6, MMP-2 and MMP-9, at same time, significantly inhibit the migration of VSMCs and the expression of RAGE protein (188). AGEs-induced VSMCs migration is due to increased production of PDGF which mediated activation of ROCK1 via the JNK pathway (189). High glucose treatment significantly increased the ROS production and VSMCs migration. Cilostazol reverses these phenomena in a dose-dependent manner. The protection effect of cilostazol on AS has been considered to inhibit superoxide, resulting in attenuation of NF-κB activation, vascular cell adhesion molecule 1 (VCAM-1)/MCP-1 expression and monocyte recruitment (190). In addition, phosphorylated Erk1/2 upregulates the number of K (CA) 3.1 channels, which is a necessary condition for RAGE mediated VSMCs migration (191, 192). The migration rate of VSMCs treated with AGEs increased significantly, increasing Bcl-2-associated athanogene 3 (BAG3) and ROS are involved (193). RAGE-NADPH oxidase-ROS pathway is unique way to increase AGEs-induced expression of LCN2 (194). In diabetic apoE−/− mice, activation of the ROCK1 (a branch of the TGF-β pathway) contributed to the RAGE-induced progress of AS (195).
It is reported the silencing of circWDR77 led to slower mobility in VSMCs, which was depend on miR-124 that targeting S100 calcium-binding protein A4 (S100A4) and fibroblast growth factor 2 (FGF-2) (196, 197). LncRNA-SMILR targeted miR-141 to activate RhoA/ROCK signaling, while promoting VSMCs migration (198). LncRNA LINC00281/ANXA2/NF-кB p65 signaling pathway, related to the pro-inflammatory response, promotes the phosphorylation of p65 and co-translocates with p65 into the nucleus, resulting in to VSMCs migration (199). Multiple RNAs regulate the process of AS by targeting HMGB1, a substance that plays an important role in VSMCs migration. LncRNA OIP5-AS1 regulated miR-141-3p/HMGB1 axis to promote the migration of VSMCs (200). Regulating the migration ability of VSMCs in different periods slowed the progression of AS.
Calcification and osteogenic differentiation
For a long time, vascular calcification was considered the end stage of AS. Research results show that vascular wall calcification is not a static and random process, but an active and strictly regulated process (201). AGEs-induced VSMCs convert to osteogenic phenotype and overexpressing osteogenic markers (CBFα-1, ALP and osteocalcin) expression, resulting in to unbalance of bone metabolism and calcification of vascular wall (120, 202, 203). AGEs increased the serum- and glucocorticoid-inducible kinase 1 (SGK1) expression in VSMCs. SGK1 knockdown restrained the high glucose-induced osteogenic trans-differentiation, which required NF-κB activation (204). AGEs stimulate the secretion of inflammatory response factors and increase CBFα-1 expression through Smad signaling pathway (120). AGEs increase the expression of NADPH oxidase in apoE-/- mice, induce endoplasmic reticulum oxidative stress, and promote late calcification of atherosclerotic lesions and calcification inside the aortic arch (205). One study clarified that metabolism related enzymes were closely related to ROS and VSMCs calcification (123). STZ-induced diabetes mice showed increased HMGB-1 translocation and expression, endoplasmic reticulum stress, mineralization and osteogenic gene expression (206). AGEs-treated VSMCs also exhibited a calcified phenotype (207). CML activates PDK4 to promote VSMCs calcification and glucose metabolism, which increases the expression of PDK4 by using the elevated level of ROS as a signal transduction intermediate (123). Another result showed that AGEs partially activated HIF-1α/PDK4 pathway in VSMCs to upregulate the expression level of Runx2 and aggravate the calcification of AS in vivo (121, 208). Organ culture of rat thoracic aorta also confirmed that AGEs promoted vascular calcification in a time-dependent manner (119). The activation of L-type calcium channels in VSMCs by AGEs is related to its osteogenic transformation, which may become a new research direction (161).
The stability of calcified plaque is related to two different AGEs receptors (galectin-3 and RAGE) (209). In carotid plaques of patients undergoing carotid endarterectomy, RAGE and inflammatory cells were only expressed in unstable inflammatory infiltrating areas with “microcalcification”, while galectin-3 and alkaline phosphatase were only expressed in fibrous areas and areas near “big calcification” (209). The targeted expression of RAGE ligand S100A12 in VSMCs isolated from apoE-/- mice will aggravate the characteristics of vascular calcification and plaque instability (210). Therefore, the regulation of AGEs receptors and ligands can be used as a treatment to enhance plaque stability. The role of autophagy in VSMCs calcification is receiving increasing attention. AGEs inhibit autophagy in a time-dependent manner by simultaneously downregulating p-AMPKα and upregulating the expression of p-mTOR, increasing the osteogenic differentiation and vascular wall calcification of VSMCs (119). AGEs via HIF-1α/PDK4 signaling pathway increased the expression of LC3-II protein, decreased the level of p62 protein and enhanced the autophagy ability of VSMCs (121). Statins inhibit TGF-β1 by activating autophagy to induce calcification, thereby achieving vascular benefit (211). The osteogenic effect of AGEs on VSMCs increases the hardness of blood vessels and weakens the elasticity. Calcified plaques may cause adverse clinical consequences. In addition, the role of different AGEs receptors in the calcification process can provide new ideas for stabilizing calcified plaque.
Macrophagic differentiation
The previous view considered monocytes recruited to the damaged site of blood vessels, then monocytes transformed into macrophages to phagocytize modified lipoproteins, which were the main source of foam cells in atherosclerotic plaques (212, 213). Several research results showed that VSMCs-based foam cells account for 45% to 90% approximately after exposure to lipids (such as cholesterol or lipoprotein), rather than macrophages, further highlights the important role of VSMCs in arterial disease (41, 214, 215). The level of serum CML and plaque RAGE in diabetic patients were higher than healthy people, which was significantly correlated with the number of macrophage-like VSMCs (216). AGEs downregulate VSMCs specific contractile markers (α-SMA, MYH11), decompose F-actin and increase collagen I, induce VSMCs transform to macrophage phenotype, thus losing the typical spindle appearance and lipid accumulation in VSMCs (217). Human diabetes-related atherosclerotic factors (hyperglycemia, ox-LDL, AGEs) can enhance the expression of LOX-1, suggesting that LOX-1 is involved in the interaction between CML and RAGE. LOX-1 expression has been detected in endothelial cells of early carotid AS and VSMCs of late carotid AS (114, 218). A variety of inflammatory factors (IL-1β, IL-6 and TNF-α, etc.) in the low-grade inflammatory environment of atherosclerotic vascular wall and long-term ROS stimulation accelerate the transformation from VSMCs to macrophages (219). VSMCs generate cyclic GMP in response to external stimulation and increase intravascular blood flow. Through cell fate mapping technology, PRKG1, a circulating GMP dependent protein kinase 1, is involved in the transformation of VSMCs into macrophage-like VSMCs in plaques. At the same time, the study also found that macrophage-like VSMCs came from mature VSMCs that migrated to plaque (220). VSMCs isolated from diabetes mice showed the same phenomenon (221). Indeed, whether macrophage-like VSMCs have macrophage function remains to be further studied. Weakening the effect of AGEs on promoting the transformation of VSMCs into macrophage-like VSMCs may reduce the number of foam cells in plaque.
Conclusion
VSMCs, as the key cell of blood vessels, mainly affect AS progression through functional changes and phenotypic transformation. VSMCs dysfunction, as one of the risk factors of cardiac metabolic diseases, plays an important role in AS pathogenesis. A growing number of studies have linked AGEs to AS. However, due to the complexity and multiple factors in AGEs -induced VSMCs dysfunction, it is not yet possible to prevent or treat atherosclerosis. AGEs accelerate AS progression by promoting the main aspects of functional changes (proliferation and migration) of VSMCs. Firstly, changing lifestyle and reducing AGEs intake should be widely advocated. Secondly, high blood glucose fluctuation can promote AGEs formation. Therefore, studying the role of AGEs in AS is helpful to solve AS in diabetes and may become a therapeutic target in the future. In a word, profound study of the effect and mechanism of AGEs on VSMCs in AS should more accurately focus on the key targets and alleviate or reverse the cardiovascular complications of diabetes.
Author contributions
LM and RY contributed equally to this work, LY and DZ contributed to the manuscript design, discussion and revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (8217020350) and Beijing Natural Science Foundation (7212055).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: Update from the GBD 2019 study. J Am Coll Cardiol (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
2. Safiri S, Karamzad N, Singh K, Carson-Chahhoud K, Adams C, Nejadghaderi SA, et al. Burden of ischemic heart disease and its attributable risk factors in 204 countries and territories, 1990-2019. Eur J Prev Cardiol (2022) 29:420–31. doi: 10.1093/eurjpc/zwab213
3. Saigusa R, Winkels H, Ley K. T Cell subsets and functions in atherosclerosis. Nat Rev Cardiol (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
4. Bays HE, Kulkarni A, German C, Satish P, Iluyomade A, Dudum R, et al. Ten things to know about ten cardiovascular disease risk factors. Am J Prev Cardiol (2022) 10:100342. doi: 10.1016/j.ajpc.2022.100342
5. Edgar L, Akbar N, Braithwaite AT, Krausgruber T, Gallart-Ayala H, Bailey J, et al. Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis. Circulation (2021) 144:961–82. doi: 10.1161/CIRCULATIONAHA.120.046464
6. Chen T, Huang W, Qian J, Luo W, Shan P, Cai Y, et al. Macrophage-derived myeloid differentiation protein 2 plays an essential role in ox-LDL-induced inflammation and atherosclerosis. EBioMedicine (2020) 53:102706. doi: 10.1016/j.ebiom.2020.102706
7. Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun (2020) 11:2127. doi: 10.1038/s41467-020-15983-6
8. Buglak NE, Jiang W, Bahnson ESM. Cinnamic aldehyde inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia in zucker diabetic fatty rats. Redox Biol (2018) 19:166–78. doi: 10.1016/j.redox.2018.08.013
9. Borghetti G, von Lewinski D, Eaton DM, Sourij H, Houser SR, Wallner M. Diabetic cardiomyopathy: Current and future therapies. beyond glycemic control. Front Physiol (2018) 9:1514. doi: 10.3389/fphys.2018.01514
10. Banovic M, Athithan L and McCann GP. Aortic stenosis and diabetes mellitus: An ominous combination. Diabetes Vasc Dis Res (2019) 16:310–23. doi: 10.1177/1479164118820657
11. Planas A, Simo-Servat O, Hernandez C, Simo R. Advanced glycations end products in the skin as biomarkers of cardiovascular risk in type 2 diabetes. Int J Mol Sci (2022) 23:6234. doi: 10.3390/ijms23116234
12. Yahagi K, Kolodgie FD, Otsuka F, Finn AV, Davis HR, Joner M, et al. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat Rev Cardiol (2016) 13:79–98. doi: 10.1038/nrcardio.2015.164
13. Grootaert MOJ, Bennett MR. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc Res (2021) 117:2326–39. doi: 10.1093/cvr/cvab046
14. Zhu Y, Qu J, He L, Zhang F, Zhou Z, Yang S, et al. Calcium in vascular smooth muscle cell elasticity and adhesion: Novel insights into the mechanism of action. Front Physiol (2019) 10:852. doi: 10.3389/fphys.2019.00852
15. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev (2004) 84:767–801. doi: 10.1152/physrev.00041.2003
16. Zhang F, Guo X, Xia Y, Mao L. An update on the phenotypic switching of vascular smooth muscle cells in the pathogenesis of atherosclerosis. Cell Mol Life Sci (2021) 79:6. doi: 10.1007/s00018-021-04079-z
17. Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
18. Miano JM, Fisher EA, Majesky MW. Fate and state of vascular smooth muscle cells in atherosclerosis. Circulation (2021) 143:2110–6. doi: 10.1161/CIRCULATIONAHA.120.049922
19. Yuan T, Yang T, Chen H, Fu D, Hu Y, Wang J, et al. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol (2019) 20:247–60. doi: 10.1016/j.redox.2018.09.025
20. Park W, Chawla A, O'Reilly EM. Pancreatic cancer: A review. JAMA (2021) 326:851–62. doi: 10.1001/jama.2021.13027
21. Sun B, Luo Z, Zhou J. Comprehensive elaboration of glycemic variability in diabetic macrovascular and microvascular complications. Cardiovasc Diabetol (2021) 20:9. doi: 10.1186/s12933-020-01200-7
22. Senior PA. Glucose as a modifiable cause of atherosclerotic cardiovascular disease: Insights from type 1 diabetes and transplantation. Atherosclerosis (2021) 335:16–22. doi: 10.1016/j.atherosclerosis.2021.09.001
23. Writing Group for the DERG. Coprogression of cardiovascular risk factors in type 1 diabetes during 30 years of follow-up in the DCCT/EDIC study. Diabetes Care (2016) 39:1621–30. doi: 10.2337/dc16-0502
24. Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med (2005) 353:2643–53. doi: 10.1056/NEJMoa052187
25. Yamagishi S, Nakamura K, Imaizumi T. Advanced glycation end products (AGEs) and diabetic vascular complications. Curr Diabetes Rev (2005) 1:93–106. doi: 10.2174/1573399052952631
26. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res (2010) 107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545
27. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation (2006) 114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854
28. Shi L, Ji Y, Liu D, Liu Y, Xu Y, Cao Y, et al. Sitagliptin attenuates high glucose-induced alterations in migration, proliferation, calcification and apoptosis of vascular smooth muscle cells through ERK1/2 signal pathway. Oncotarget (2017) 8:77168–80. doi: 10.18632/oncotarget.20417
29. Su SC, Hung YJ, Huang CL, Shieh YS, Chien CY, Chiang CF, et al. Cilostazol inhibits hyperglucose-induced vascular smooth muscle cell dysfunction by modulating the RAGE/ERK/NF-kappaB signaling pathways. J BioMed Sci (2019) 26:68. doi: 10.1186/s12929-019-0550-9
30. Xing Y, Pan S, Zhu L, Cui Q, Tang Z, Liu Z, et al. Advanced glycation end products induce atherosclerosis via RAGE/TLR4 signaling mediated-M1 macrophage polarization-dependent vascular smooth muscle cell phenotypic conversion. Oxid Med Cell Longev (2022) 2022:9763377. doi: 10.1155/2022/9763377
31. Defeudis G, Mazzilli R, Tenuta M, Rossini G, Zamponi V, Olana S, et al. Erectile dysfunction and diabetes: A melting pot of circumstances and treatments. Diabetes Metab Res Rev (2022) 38:e3494. doi: 10.1002/dmrr.3494
32. Isidori AM, Giammusso B, Corona G, Verze P. Diagnostic and therapeutic workup of erectile dysfunction: Results from a Delphi consensus of andrology experts. Sex Med (2019) 7:292–302. doi: 10.1016/j.esxm.2019.04.001
33. Wan L, Bai X, Zhou Q, Chen C, Wang H, Liu T, et al. The advanced glycation end-products (AGEs)/ROS/NLRP3 inflammasome axis contributes to delayed diabetic corneal wound healing and nerve regeneration. Int J Biol Sci (2022) 18:809–25. doi: 10.7150/ijbs.63219
34. Defeudis G, Gianfrilli D, Di Emidio C, Pofi R, Tuccinardi D, Palermo A, et al. Erectile dysfunction and its management in patients with diabetes mellitus. Rev Endocr Metab Disord (2015) 16(3)213–31. doi: 10.1007/s11154-015-9321-4
35. Ahuja P, Waris A, Siddiqui SS, Mukherjee A. Single nucleotide variants of receptor for advanced glycation end-products (AGER) gene: Is it a new opening in the risk assessment of diabetic retinopathy?-a review. J Genet Eng Biotechnol (2022) 20:17. doi: 10.1186/s43141-022-00297-5
36. Mahmoud AD, Ballantyne MD, Miscianinov V, Pinel K, Hung J, Scanlon JP, et al. The human-specific and smooth muscle cell-enriched LncRNA SMILR promotes proliferation by regulating mitotic CENPF mRNA and drives cell-cycle progression which can be targeted to limit vascular remodeling. Circ Res (2019) 125:535–51. doi: 10.1161/CIRCRESAHA.119.314876
37. Ikari Y, McManus BM, Kenyon J, Schwartz SM. Neonatal intima formation in the human coronary artery. Arterioscler Thromb Vasc Biol (1999) 19:2036–40. doi: 10.1161/01.atv.19.9.2036
38. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol (2000) 20:1262–75. doi: 10.1161/01.atv.20.5.1262
39. Aikawa M, Sivam PN, Kuro-o M, Kimura K, Nakahara K, Takewaki S, et al. Human smooth muscle myosin heavy chain isoforms as molecular markers for vascular development and atherosclerosis. Circ Res (1993) 73:1000–12. doi: 10.1161/01.res.73.6.1000
40. Andreeva ER, Pugach IM, Orekhov AN. Collagen-synthesizing cells in initial and advanced atherosclerotic lesions of human aorta. Atherosclerosis (1997) 130:133–42. doi: 10.1016/s0021-9150(96)06056-x
41. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation (2014) 129:1551–9. doi: 10.1161/CIRCULATIONAHA.113.005015
42. Campbell JH, Popadynec L, Nestel PJ, Campbell GR. Lipid accumulation in arterial smooth muscle cells. influence of phenotype. Atherosclerosis (1983) 47:279–95. doi: 10.1016/0021-9150(83)90059-x
43. Campbell JH, Reardon MF, Campbell GR, Nestel PJ. Metabolism of atherogenic lipoproteins by smooth muscle cells of different phenotype in culture. Arteriosclerosis (1985) 5:318–28. doi: 10.1161/01.atv.5.4.318
44. Little PJ, Tannock L, Olin KL, Chait A, Wight TN. Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to LDLs. Arterioscler Thromb Vasc Biol (2002) 22:55–60. doi: 10.1161/hq0102.101100
45. Langley SR, Willeit K, Didangelos A, Matic LP, Skroblin P, Barallobre-Barreiro J, et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J Clin Invest. (2017) 127:1546–60. doi: 10.1172/JCI86924
46. Chang MY, Potter-Perigo S, Tsoi C, Chait A, Wight TN. Oxidized low density lipoproteins regulate synthesis of monkey aortic smooth muscle cell proteoglycans that have enhanced native low density lipoprotein binding properties. J Biol Chem (2000) 275:4766–73. doi: 10.1074/jbc.275.7.4766
47. Misra A, Feng Z, Chandran RR, Kabir I, Rotllan N, Aryal B, et al. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun (2018) 9:2073. doi: 10.1038/s41467-018-04447-7
48. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med (2015) 21:628–37. doi: 10.1038/nm.3866
49. Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation (1998) 97:2307–15. doi: 10.1161/01.cir.97.23.2307
50. Okura Y, Brink M, Itabe H, Scheidegger KJ, Kalangos A, Delafontaine P. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation (2000) 102:2680–6. doi: 10.1161/01.cir.102.22.2680
51. Andreeva ER, Pugach IM, Orekhov AN. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis (1997) 135:19–27. doi: 10.1016/s0021-9150(97)00136-6
52. Imai H, Lee KT, Pastori S, Panlilio E, Florentin R, Thomas WA. Atherosclerosis in rabbits. architectural and subcellular alterations of smooth muscle cells of aortas in response to hyperlipemia. Exp Mol Pathol (1966) 5:273–310. doi: 10.1016/0014-4800(66)90036-0
53. Nahrendorf M. Myeloid cell contributions to cardiovascular health and disease. Nat Med (2018) 24:711–20. doi: 10.1038/s41591-018-0064-0
54. Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med (2013) 19:1166–72. doi: 10.1038/nm.3258
55. Nelken NA, Coughlin SR, Gordon D, Wilcox JN. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. (1991) 88:1121–7. doi: 10.1172/JCI115411
56. Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, et al. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci U S A (1990) 87:5134–8. doi: 10.1073/pnas.87.13.5134
57. Ross R, Masuda J, Raines EW, Gown AM, Katsuda S, Sasahara M, et al. Localization of PDGF-b protein in macrophages in all phases of atherogenesis. Science (1990) 248:1009–12. doi: 10.1126/science.2343305
58. Campbell JH, Rennick RE, Kalevitch SG, Campbell GR. Heparan sulfate-degrading enzymes induce modulation of smooth muscle phenotype. Exp Cell Res (1992) 200:156–67. doi: 10.1016/s0014-4827(05)80084-9
59. Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol (2008) 28:1429–31. doi: 10.1161/ATVBAHA.108.169078
60. Clarke MC, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: Effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res (2010) 106:363–72. doi: 10.1161/CIRCRESAHA.109.208389
61. Shaw PX, Horkko S, Tsimikas S, Chang MK, Palinski W, Silverman GJ, et al. Human-derived anti-oxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler Thromb Vasc Biol (2001) 21:1333–9. doi: 10.1161/hq0801.093587
62. Back M, Yurdagul A Jr., Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol (2019) 16:389–406. doi: 10.1038/s41569-019-0169-2
63. Li S, Sun Y, Liang CP, Thorp EB, Han S, Jehle AW, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res (2009) 105:1072–82. doi: 10.1161/CIRCRESAHA.109.199570
64. Bentzon JF, Sondergaard CS, Kassem M, Falk E. Smooth muscle cells healing atherosclerotic plaque disruptions are of local, not blood, origin in apolipoprotein e knockout mice. Circulation (2007) 116:2053–61. doi: 10.1161/CIRCULATIONAHA.107.722355
65. Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, Takakura N, et al. Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein e-deficient mice. Circulation (2001) 103:2955–60. doi: 10.1161/01.cir.103.24.2955
66. Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res (2015) 116:1312–23. doi: 10.1161/CIRCRESAHA.116.305012
67. Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater (2016) 15:335–43. doi: 10.1038/nmat4519
68. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ Res (2000) 87:1055–62. doi: 10.1161/01.res.87.11.1055
69. Rattazzi M, Bennett BJ, Bea F, Kirk EA, Ricks JL, Speer M, et al. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arterioscler Thromb Vasc Biol (2005) 25:1420–5. doi: 10.1161/01.ATV.0000166600.58468.1b
70. Pasterkamp G, den Ruijter HM, Libby P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat Rev Cardiol (2017) 14:21–9. doi: 10.1038/nrcardio.2016.166
71. Falk E. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br Heart J (1983) 50:127–34. doi: 10.1136/hrt.50.2.127
72. van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation (1994) 89:36–44. doi: 10.1161/01.cir.89.1.36
73. Davies MJ, Richardson PD, Woolf N, Katz DR, Mann J. Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J (1993) 69:377–81. doi: 10.1136/hrt.69.5.377
74. He M, Wang C, Sun JH, Liu Y, Wang H, Zhao JS, et al. Roscovitine attenuates intimal hyperplasia via inhibiting NF-kappaB and STAT3 activation induced by TNF-alpha in vascular smooth muscle cells. Biochem Pharmacol (2017) 137:51–60. doi: 10.1016/j.bcp.2017.04.018
75. Li H, Cheng Y, Simoncini T, Xu S. 17beta-estradiol inhibits TNF-alpha-induced proliferation and migration of vascular smooth muscle cells via suppression of TRAIL. Gynecol. Endocrinol (2016) 32:581–6. doi: 10.3109/09513590.2016.1141882
76. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol (2003) 23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3
77. Chi H, Messas E, Levine RA, Graves DT, Amar S. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein e heterozygote model: Pharmacotherapeutic implications. Circulation (2004) 110:1678–85. doi: 10.1161/01.CIR.0000142085.39015.31
78. Isoda K, Sawada S, Ishigami N, Matsuki T, Miyazaki K, Kusuhara M, et al. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol (2004) 24:1068–73. doi: 10.1161/01.ATV.0000127025.48140.a3
79. Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H, et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in apolipoprotein e-deficient mice. Atherosclerosis (2011) 216:313–20. doi: 10.1016/j.atherosclerosis.2011.02.026
80. Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao L, Zhang Y, et al. The landscape of circular RNA in cancer. Cell (2019) 176:869–881.e813. doi: 10.1016/j.cell.2018.12.021
81. Pu Z, Lu J, Yang X. Emerging roles of circular RNAs in vascular smooth muscle cell dysfunction. Front Genet (2021) 12:749296. doi: 10.3389/fgene.2021.749296
82. Zhang J, Cai W, Fan Z, Yang C, Wang W, Xiong M, et al. MicroRNA-24 inhibits the oxidative stress induced by vascular injury by activating the Nrf2/Ho-1 signaling pathway. Atherosclerosis (2019) 290:9–18. doi: 10.1016/j.atherosclerosis.2019.08.023
83. Gomez D, Baylis RA, Durgin BG, Newman AAC, Alencar GF, Mahan S, et al. Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med (2018) 24:1418–29. doi: 10.1038/s41591-018-0124-5
84. Ha YM, Lee DH, Kim M, Kang YJ. High glucose induces connective tissue growth factor expression and extracellular matrix accumulation in rat aorta vascular smooth muscle cells via extracellular signal-regulated kinase 1/2. Korean J Physiol Pharmacol (2013) 17:307–14. doi: 10.4196/kjpp.2013.17.4.307
85. Hwang AR, Nam JO, Kang YJ. Fluvastatin inhibits advanced glycation end products-induced proliferation, migration, and extracellular matrix accumulation in vascular smooth muscle cells by targeting connective tissue growth factor. Korean J Physiol Pharmacol (2018) 22:193–201. doi: 10.4196/kjpp.2018.22.2.193
86. Zhou S, Sun L, Cao C, Wu P, Li M, Sun G, et al. Hypoxia-induced microRNA-26b inhibition contributes to hypoxic pulmonary hypertension via CTGF. J Cell Biochem (2018) 119:1942–52. doi: 10.1002/jcb.26355
87. Kim EJ, Park SY, Baek SE, Jang MA, Lee WS, Bae SS, et al. HMGB1 increases IL-1beta production in vascular smooth muscle cells via NLRP3 inflammasome. Front Physiol (2018) 9:313. doi: 10.3389/fphys.2018.00313
88. Jeon EY, Baek SE, Kim JO, Choi JM, Jang EJ, Kim CD. A pivotal role for AP-1-Mediated osteopontin expression in the increased migration of vascular smooth muscle cells stimulated with HMGB1. Front Physiol (2021) 12:775464. doi: 10.3389/fphys.2021.775464
89. Cao T, Zhang L, Yao LL, Zheng F, Wang L, Yang JY, et al. S100B promotes injury-induced vascular remodeling through modulating smooth muscle phenotype. Biochim Biophys Acta Mol Basis Dis (2017) 1863:2772–82. doi: 10.1016/j.bbadis.2017.07.002
90. Mao C, Ma Z, Jia Y, Li W, Xie N, Zhao G, et al. Nidogen-2 maintains the contractile phenotype of vascular smooth muscle cells and prevents neointima formation via bridging Jagged1-Notch3 signaling. Circulation (2021) 144:1244–61. doi: 10.1161/CIRCULATIONAHA.120.053361
91. Rodriguez AI, Csanyi G, Ranayhossaini DJ, Feck DM, Blose KJ, Assatourian L, et al. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol (2015) 35:430–8. doi: 10.1161/ATVBAHA.114.304936
92. Zhang WB, Feng SY, Xiao ZX, Qi YF, Zeng ZF, Chen H. Down-regulating of MFN2 promotes vascular calcification via regulating RAS-RAF-ERK1/2 pathway. Int J Cardiol (2022) S0167-5273(22):00931-7. doi: 10.1016/j.ijcard.2022.06.033
93. Zhou W, Feng Q, Cheng M, Zhang D, Jin J, Zhang S, et al. LncRNA H19 sponges miR-103-3p to promote the high phosphorus-induced osteoblast phenotypic transition of vascular smooth muscle cells by upregulating Runx2. Cell Signal (2022) 91:110220. doi: 10.1016/j.cellsig.2021.110220
94. Edsfeldt A, Stenstrom KE, Sun J, Dias N, Skog G, Singh P, et al. Human atherosclerotic plaque progression is dependent on apoptosis according to bomb-pulse (14)C dating. JACC Basic Transl Sci (2021) 6:734–45. doi: 10.1016/j.jacbts.2021.08.005
95. Sun Z, Li L, Zhang L, Yan J, Shao C, Bao Z, et al. Macrophage galectin-3 enhances intimal translocation of vascular calcification in diabetes mellitus. Am J Physiol Heart Circ Physiol (2020) 318:H1068–79. doi: 10.1152/ajpheart.00690.2019
96. Ren K, Xu XD, Yu XH, Li MQ, Shi MW, Liu QX, et al. LncRNA-modulated autophagy in plaque cells: A new paradigm of gene regulation in atherosclerosis? Aging (Albany NY). (2020) 12:22335–49. doi: 10.18632/aging.103786
97. Chen MY, Ke JF, Zhang ZH, Li MF, Wang JW, Lu JX, et al. Deletion of Fam172a accelerates advanced atherosclerosis and induces plaque instability. Atherosclerosis (2021) 333:39–47. doi: 10.1016/j.atherosclerosis.2021.08.023
98. Pan Y, Liu T, Wang X, Sun J. Research progress of coumarins and their derivatives in the treatment of diabetes. J Enzyme Inhib Med Chem (2022) 37:616–28. doi: 10.1080/14756366.2021.2024526
99. Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri V, Orekhov AN. The diabetes mellitus-atherosclerosis connection: The role of lipid and glucose metabolism and chronic inflammation. Int J Mol Sci (2020) 21:1835. doi: 10.3390/ijms21051835
100. Sun HJ, Ren XS, Xiong XQ, Chen YZ, Zhao MX, Wang JJ, et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis (2017) 8:e3074. doi: 10.1038/cddis.2017.470
101. Samara VA, Das S, Reddy MA, Tanwar VS, Stapleton K, Leung A, et al. Angiotensin II-induced long non-coding RNA alivec regulates chondrogenesis in vascular smooth muscle cells. Cell (2021) 10:(10)2696. doi: 10.3390/cells10102696
102. Zhao D, Li J, Xue C, Feng K, Liu L, Zeng P, et al. TL1A inhibits atherosclerosis in apoE-deficient mice by regulating the phenotype of vascular smooth muscle cells. J Biol Chem (2020) 295:16314–27. doi: 10.1074/jbc.RA120.015486
103. Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res (2021) 128:474–91. doi: 10.1161/CIRCRESAHA.120.318353
104. Li T, Yang GM, Zhu Y, Wu Y, Chen XY, Lan D, et al. Diabetes and hyperlipidemia induce dysfunction of VSMCs: Contribution of the metabolic inflammation/miRNA pathway. Am J Physiol Endocrinol Metab (2015) 308:E257–269. doi: 10.1152/ajpendo.00348.2014
105. Soares RN, Ramirez-Perez FI, Cabral-Amador FJ, Morales-Quinones M, Foote CA, Ghiarone T, et al. SGLT2 inhibition attenuates arterial dysfunction and decreases vascular f-actin content and expression of proteins associated with oxidative stress in aged mice. Geroscience (2022) 44:1657–75. doi: 10.1007/s11357-022-00563-x
106. An XR, Li X, Wei W, Li XX, Xu M. Prostaglandin E1 inhibited diabetes-induced phenotypic switching of vascular smooth muscle cells through activating autophagy. Cell Physiol Biochem (2018) 50:745–56. doi: 10.1159/000494240
107. Madonna R, Geng YJ, Shelat H, Ferdinandy P, De Caterina R. High glucose-induced hyperosmolarity impacts proliferation, cytoskeleton remodeling and migration of human induced pluripotent stem cells via aquaporin-1. Biochim Biophys Acta (2014) 1842:2266–75. doi: 10.1016/j.bbadis.2014.07.030
108. Kim HJ, Park KG, Yoo EK, Kim YH, Kim YN, Kim HS, et al. Effects of PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid. Redox Signal (2007) 9:301–7. doi: 10.1089/ars.2006.1456
109. Yan Y, Li T, Li Z, He M, Wang D, Xu Y, et al. Metformin suppresses the progress of diabetes-accelerated atherosclerosis by inhibition of vascular smooth muscle cell migration through AMPK-Pdlim5 pathway. Front Cardiovasc Med (2021) 8:690627. doi: 10.3389/fcvm.2021.690627
110. Li H, Peng W, Zhuang J, Lu Y, Jian W, Wei Y, et al. Vaspin attenuates high glucose-induced vascular smooth muscle cells proliferation and chemokinesis by inhibiting the MAPK, PI3K/Akt, and NF-kappaB signaling pathways. Atherosclerosis (2013) 228:61–8. doi: 10.1016/j.atherosclerosis.2013.02.013
111. Zhang Q, Chen L, Zhao Z, Wu Y, Zhong J, Wen G, et al. HMGA1 mediated high-Glucose-Induced vascular smooth muscle cell proliferation in diabetes mellitus: Association between PI3K/Akt signaling and HMGA1 expression. DNA Cell Biol (2018) 37:389–97. doi: 10.1089/dna.2017.3957
112. Fan Z, Guo C, Zhang Y, Yao J, Liao L, Dong J. Hongjingtian injection inhibits proliferation and migration and promotes apoptosis in high glucose-induced vascular smooth muscle cells. Drug Des Devel Ther (2019) 13:4115–26. doi: 10.2147/DDDT.S220719
113. Wei W, Li XX, Xu M. Inhibition of vascular neointima hyperplasia by FGF21 associated with FGFR1/Syk/NLRP3 inflammasome pathway in diabetic mice. Atherosclerosis (2019) 289:132–42. doi: 10.1016/j.atherosclerosis.2019.08.017
114. Kattoor AJ, Goel A, Mehta JL. LOX-1: Regulation, signaling and its role in atherosclerosis. Antioxid. (Basel). (2019) 8(7):218. doi: 10.3390/antiox8070218
115. Zeya B, Chandra NC. LOX-1: Its cytotopographical variance and disease stress. J Biochem Mol Toxicol (2019) 33:e22375. doi: 10.1002/jbt.22375
116. Wang F, Bao Y, Shen X, Zengin G, Lyu Y, Xiao J, et al. Niazirin from moringa oleifera lam. attenuates high glucose-induced oxidative stress through PKCzeta/Nox4 pathway. Phytomedicine (2021) 86:153066. doi: 10.1016/j.phymed.2019.153066
117. Li W, Feng W, Su X, Luo D, Li Z, Zhou Y, et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via Runx2 in chronic kidney disease. J Clin Invest. (2022) 132(1):e150051. doi: 10.1172/JCI150051
118. Xu S, Ye F, Li L, Yan J, Bao Z, Sun Z, et al. Ghrelin attenuates vascular calcification in diabetic patients with amputation. BioMed Pharmacother. (2017) 91:1053–64. doi: 10.1016/j.biopha.2017.05.031
119. Liu Y, Li J, Han Y, Chen Y, Liu L, Lang J, et al. Advanced glycation end-products suppress autophagy by AMPK/mTOR signaling pathway to promote vascular calcification. Mol Cell Biochem (2020) 471:91–100. doi: 10.1007/s11010-020-03769-9
120. Son M, Oh S, Jang JT, Park CH, Son KH and Byun K. Attenuating effects of pyrogallol-Phloroglucinol-6,6-Bieckol on vascular smooth muscle cell phenotype changes to osteoblastic cells and vascular calcification induced by high fat diet. Nutrients (2020) 12(9):2777. doi: 10.3390/nu12092777
121. Yang R, Zhu Y, Wang Y, Ma W, Han X, Wang X, et al. HIF-1alpha/PDK4/autophagy pathway protects against advanced glycation end-products induced vascular smooth muscle cell calcification. Biochem Biophys Res Commun (2019) 517:470–6. doi: 10.1016/j.bbrc.2019.07.102
122. Cao J, Chen C, Chen Q, Gao Y, Zhao Z, Yuan Q, et al. Extracellular vesicle miR-32 derived from macrophage promotes arterial calcification in mice with type 2 diabetes via inhibiting VSMC autophagy. J Transl Med (2022) 20:307. doi: 10.1186/s12967-022-03502-8
123. Ma WQ, Han XQ, Wang Y, Wang X, Zhu Y, Liu NF. Nepsilon-carboxymethyl-lysine promotes calcium deposition in VSMCs via intracellular oxidative stress-induced PDK4 activation and alters glucose metabolism. Oncotarget (2017) 8:112841–54. doi: 10.18632/oncotarget.22835
124. Chen Z, Li R, Pei LG, Wei ZH, Xie J, Wu H, et al. High-mobility group box-1 promotes vascular calcification in diabetic mice via endoplasmic reticulum stress. J Cell Mol Med (2021) 25:3724–34. doi: 10.1111/jcmm.16075
125. Li Y, Xi Z, Yu Z, Yang C, Tan C. LincRNA-EPS increases TGF-beta expression to inhibit the wnt/beta-catenin pathway, VSMC osteoblastic differentiation and vascular calcification in diabetic mice. Exp Ther Med (2022) 23:425. doi: 10.3892/etm.2022.11352
126. Zhong JY, Cui XJ, Zhan JK, Wang YJ, Li S, Lin X, et al. LncRNA-ES3 inhibition by Bhlhe40 is involved in high glucose-induced calcification/senescence of vascular smooth muscle cells. Ann N Y Acad Sci (2020) 1474:61–72. doi: 10.1111/nyas.14381
127. Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, Bose N, et al. The role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab (2018) 28:337–52. doi: 10.1016/j.cmet.2018.08.014
128. Chuang WY, Yu MH, Yang TY, Chan KC, Wang CJ. Acarbose attenuates migration/proliferation via targeting microRNA-143 in vascular smooth muscle cells under diabetic conditions. J Food Drug Anal (2020) 28:461–74. doi: 10.38212/2224-6614.1241
129. Wu Y, Zhang M, Xu C, Chai D, Peng F, Lin J. Anti-diabetic atherosclerosis by inhibiting high glucose-induced vascular smooth muscle cell proliferation via Pin1/BRD4 pathway. Oxid Med Cell Longev (2020) 2020:4196482. doi: 10.1155/2020/4196482
130. Wang Y, Zhang Y, Gao X, Qian J, Yang J, Sun W, et al. Resistin-like molecule beta augments phenotypic modulation of human aortic smooth muscle cell triggered by high glucose. Endocr J (2021) 68:461–8. doi: 10.1507/endocrj.EJ20-0343
131. Zhou W, Wang W, Yuan XJ, Xiao CC, Xing Y, Ye SD, et al. The effects of RBP4 and vitamin d on the proliferation and migration of vascular smooth muscle cells via the JAK2/STAT3 signaling pathway. Oxid Med Cell Longev (2022) 2022:3046777. doi: 10.1155/2022/3046777
132. Henze LA, Estepa M, Pieske B, Lang F, Eckardt KU, Alesutan I, et al. Zinc ameliorates the osteogenic effects of high glucose in vascular smooth muscle cells. Cell (2021) 10(11):3083. doi: 10.3390/cells10113083
133. Shi LL, Hao M, Jin ZY, Peng GF, Tang YY, Kuang HY. Liraglutide alleviates diabetic atherosclerosis through regulating calcification of vascular smooth muscle cells. Dis Markers. (2022) 2022:5013622. doi: 10.1155/2022/5013622
134. Zhang P, Wang AP, Yang HP, Ai L, Zhang HJ, Wang YM, et al. Apelin-13 attenuates high glucose-induced calcification of MOVAS cells by regulating MAPKs and PI3K/AKT pathways and ROS-mediated signals. BioMed Pharmacother (2020) 128:110271. doi: 10.1016/j.biopha.2020.110271
135. Hu BA, Sai WW, Yuan J, Lan HT, Qi J, Wang D, et al. PGF2alpha-FP receptor ameliorates senescence of VSMCs in vascular remodeling by Src/PAI-1 signal pathway. Oxid Med Cell Longev (2022) 2022:2908261. doi: 10.1155/2022/2908261
136. Zha Y, Zhuang W, Yang Y, Zhou Y, Li H, Liang J. Senescence in vascular smooth muscle cells and atherosclerosis. Front Cardiovasc Med (2022) 9:910580. doi: 10.3389/fcvm.2022.910580
137. Gong C, Qi Y, Xu Y, Tang X, Liang F, Chen L. Parecoxib improves atherosclerotic plaque stability by suppressing inflammation and inhibiting matrix metalloproteinases production. BioMed Pharmacother (2021) 138:111423. doi: 10.1016/j.biopha.2021.111423
138. Bai HY, Li H, Zhou X, Gu HB, Shan BS. AT2 receptor stimulation inhibits vascular smooth muscle cell senescence induced by angiotensin II and hyperglycemia. Am J Hypertens (2022) hpac083. doi: 10.1093/ajh/hpac083
139. Guo Y, Tang Z, Yan B, Yin H, Tai S, Peng J, et al. PCSK9 (Proprotein convertase Subtilisin/Kexin type 9) triggers vascular smooth muscle cell senescence and apoptosis: Implication of its direct role in degenerative vascular disease. Arterioscler Thromb Vasc Biol (2022) 42:67–86. doi: 10.1161/ATVBAHA.121.316902
140. Li Y, Qin R, Yan H, Wang F, Huang S, Zhang Y, et al. Inhibition of vascular smooth muscle cells premature senescence with rutin attenuates and stabilizes diabetic atherosclerosis. J Nutr Biochem (2018) 51:91–8. doi: 10.1016/j.jnutbio.2017.09.012
141. Bjorksten J. The crosslinkage theory of aging. J Am Geriatr Soc (1968) 16:408–27. doi: 10.1111/j.1532-5415.1968.tb02821.x
143. Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology (2005) 15:16R–28R. doi: 10.1093/glycob/cwi053
144. Perez-Burillo S, Rufian-Henares JA, Pastoriza S. Effect of home cooking on the antioxidant capacity of vegetables: Relationship with maillard reaction indicators. Food Res Int (2019) 121:514–23. doi: 10.1016/j.foodres.2018.12.007
145. Fishman SL, Sonmez H, Basman C, Singh V, Poretsky L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: a review. Mol Med (2018) 24:59. doi: 10.1186/s10020-018-0060-3
146. Garay-Sevilla ME, Rojas A, Portero-Otin M, Uribarri J. Dietary AGEs as exogenous boosters of inflammation. Nutrients (2021) 13(8):2802. doi: 10.3390/nu13082802
147. Snelson M, Coughlan MT. Dietary advanced glycation end products: Digestion, metabolism and modulation of gut microbial ecology. Nutrients (2019) 22(12):6491. doi: 10.3390/nu11020215
148. Perer J, Jandova J, Fimbres J, Jennings EQ, Galligan JJ, Hua A, et al. The sunless tanning agent dihydroxyacetone induces stress response gene expression and signaling in cultured human keratinocytes and reconstructed epidermis. Redox Biol (2020) 36:101594. doi: 10.1016/j.redox.2020.101594
149. Chen JH, Lin X, Bu C, Zhang X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr Metab (Lond) (2018) 15:72. doi: 10.1186/s12986-018-0306-7
150. Tessier FJ, Boulanger E, Howsam M. Metabolic transit of dietary advanced glycation end-products - the case of N(E)-carboxymethyllysine. Glycoconj J (2021) 38:311–7. doi: 10.1007/s10719-020-09950-y
151. Wolf AR, Wesener DA, Cheng J, Houston-Ludlam AN, Beller ZW, Hibberd MC, et al. Bioremediation of a common product of food processing by a human gut bacterium. Cell Host Microbe (2019) 26:463–477.e468. doi: 10.1016/j.chom.2019.09.001
152. Indyk D, Bronowicka-Szydelko A, Gamian A, Kuzan A. Advanced glycation end products and their receptors in serum of patients with type 2 diabetes. Sci Rep (2021) 11:13264. doi: 10.1038/s41598-021-92630-0
153. Nin JW, Jorsal A, Ferreira I, Schalkwijk CG, Prins MH, Parving HH, et al. Higher plasma levels of advanced glycation end products are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: a 12-year follow-up study. Diabetes Care (2011) 34:442–7. doi: 10.2337/dc10-1087
154. Singh S, Siva BV, Ravichandiran V. Advanced glycation end products: key player of the pathogenesis of atherosclerosis. Glycoconj J (2022) 39(4):547–63. doi: 10.1007/s10719-022-10063-x
155. Li Z, Zhong Q, Yang T, Xie X, Chen M. The role of profilin-1 in endothelial cell injury induced by advanced glycation end products (AGEs). Cardiovasc Diabetol (2013) 12:141. doi: 10.1186/1475-2840-12-141
156. Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol (2009) 20:742–52. doi: 10.1681/ASN.2008050514
157. Zhou Q, Cheng KW, Gong J, Li ETS, Wang M. Apigenin and its methylglyoxal-adduct inhibit advanced glycation end products-induced oxidative stress and inflammation in endothelial cells. Biochem Pharmacol (2019) 166:231–41. doi: 10.1016/j.bcp.2019.05.027
158. Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest (2009) 119:136–45. doi: 10.1172/JCI35535
159. Wang ZQ, Jing LL, Yan JC, Sun Z, Bao ZY, Shao C, et al. Role of AGEs in the progression and regression of atherosclerotic plaques. Glycoconj J (2018) 35:443–50. doi: 10.1007/s10719-018-9831-x
160. Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J Jr., Pomerantzeff PM, et al. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol (2008) 28:463–70. doi: 10.1161/ATVBAHA.107.156745
161. Molinuevo MS, Fernandez JM, Cortizo AM, McCarthy AD, Schurman L, Sedlinsky C. Advanced glycation end products and strontium ranelate promote osteogenic differentiation of vascular smooth muscle cells in vitro: Preventive role of vitamin d. Mol Cell Endocrinol (2017) 450:94–104. doi: 10.1016/j.mce.2017.04.022
162. Takahashi M. NLRP3 inflammasome as a key driver of vascular disease. Cardiovasc Res (2022) 118:372–85. doi: 10.1093/cvr/cvab010
163. Barlovic DP, Soro-Paavonen A, Jandeleit-Dahm KA. RAGE biology, atherosclerosis and diabetes. Clin Sci (Lond) (2011) 121:43–55. doi: 10.1042/CS20100501
164. Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med (Berl). (2009) 87:235–47. doi: 10.1007/s00109-009-0439-2
165. Prasad K. AGE-RAGE stress and coronary artery disease. Int J Angiol (2021) 30:4–14. doi: 10.1055/s-0040-1721813
166. Tam XH, Shiu SW, Leng L, Bucala R, Betteridge DJ, Tan KC. Enhanced expression of receptor for advanced glycation end-products is associated with low circulating soluble isoforms of the receptor in type 2 diabetes. Clin Sci (Lond) (2011) 120:81–9. doi: 10.1042/CS20100256
167. Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J (2003) 370:1097–109. doi: 10.1042/BJ20021371
168. Prasad K, Dhar I, Zhou Q, Elmoselhi H, Shoker M, Shoker A. AGEs/sRAGE, a novel risk factor in the pathogenesis of end-stage renal disease. Mol Cell Biochem (2016) 423:105–14. doi: 10.1007/s11010-016-2829-4
169. Meloche J, Paulin R, Courboulin A, Lambert C, Barrier M, Bonnet P, et al. RAGE-dependent activation of the oncoprotein Pim1 plays a critical role in systemic vascular remodeling processes. Arterioscler Thromb Vasc Biol (2011) 31:2114–24. doi: 10.1161/ATVBAHA.111.230573
170. Fang Q, Tian M, Wang F, Zhang Z, Du T, Wang W, et al. Amlodipine induces vasodilation via Akt2/Sp1-activated miR-21 in smooth muscle cells. Br J Pharmacol (2019) 176:2306–20. doi: 10.1111/bph.14679
171. Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, et al. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res (2016) 119:1313–23. doi: 10.1161/CIRCRESAHA.116.309799
172. Zhang L, Cheng H, Yue Y, Li S, Zhang D, He R. H19 knockdown suppresses proliferation and induces apoptosis by regulating miR-148b/WNT/beta-catenin in ox-LDL -stimulated vascular smooth muscle cells. J BioMed Sci (2018) 25:11. doi: 10.1186/s12929-018-0418-4
173. Kilhovd BK, Berg TJ, Birkeland KI, Thorsby P, Hanssen KF. Serum levels of advanced glycation end products are increased in patients with type 2 diabetes and coronary heart disease. Diabetes Care (1999) 22:1543–8. doi: 10.2337/diacare.22.9.1543
174. Tian X, Zhang Q, Huang Y, Chen S, Tang C, Sun Y, et al. Endothelin-1 downregulates sulfur Dioxide/Aspartate aminotransferase pathway via reactive oxygen species to promote the proliferation and migration of vascular smooth muscle cells. Oxid Med Cell Longev (2020) 2020:9367673. doi: 10.1155/2020/9367673
175. You B, Liu Y, Chen J, Huang X, Peng H, Liu Z, et al. Vascular peroxidase 1 mediates hypoxia-induced pulmonary artery smooth muscle cell proliferation, apoptosis resistance and migration. Cardiovasc Res (2018) 114:188–99. doi: 10.1093/cvr/cvx234
176. Furmanik M, Chatrou M, van Gorp R, Akbulut A, Willems B, Schmidt H, et al. Reactive oxygen-forming Nox5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle-mediated vascular calcification. Circ Res (2020) 127:911–27. doi: 10.1161/CIRCRESAHA.119.316159
177. Koska J, Saremi A, Howell S, Bahn G, De Courten B, Ginsberg H, et al. Advanced glycation end products, oxidation products, and incident cardiovascular events in patients with type 2 diabetes. Diabetes Care (2018) 41:570–6. doi: 10.2337/dc17-1740
178. Fukami K, Yamagishi S, Okuda S. Role of AGEs-RAGE system in cardiovascular disease. Curr Pharm Des (2014) 20:2395–402. doi: 10.2174/13816128113199990475
179. Jang EJ, Baek SE, Kim EJ, Park SY, Kim CD. HMGB1 enhances AGE-mediated VSMC proliferation via an increase in 5-LO-linked RAGE expression. Vascul. Pharmacol (2019) 118-119:106559. doi: 10.1016/j.vph.2019.04.001
180. Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, et al. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal (2015) 23:1389–409. doi: 10.1089/ars.2014.6221
181. Hattori Y, Suzuki M, Hattori S, Kasai K. Vascular smooth muscle cell activation by glycated albumin (Amadori adducts). Hypertension (2002) 39:22–8. doi: 10.1161/hy1201.097300
182. Hwang AR, Han JH, Lim JH, Kang YJ, Woo CH. Fluvastatin inhibits AGE-induced cell proliferation and migration via an ERK5-dependent Nrf2 pathway in vascular smooth muscle cells. PloS One (2017) 12:e0178278. doi: 10.1371/journal.pone.0178278
183. Yuan X, Zhang Z, Gong K, Zhao P, Qin J, Liu N. Inhibition of reactive oxygen species/extracellular signal-regulated kinases pathway by pioglitazone attenuates advanced glycation end products-induced proliferation of vascular smooth muscle cells in rats. Biol Pharm Bull (2011) 34:618–23. doi: 10.1248/bpb.34.618
184. Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol (2015) 209:13–22. doi: 10.1083/jcb.201412052
185. Soleimani AA, Ghasmpour G, Mohammadi A, Gholizadeh M, Abkenar BR, Najafi M. Focal adhesion kinase-related pathways may be suppressed by metformin in vascular smooth muscle cells in high glucose conditions. Endocrinol Diabetes Metab (2022) 5(4):e351. doi: 10.1002/edm2.351
186. Wu X, Bian F, Hu H, Zhu T, Li C, Zhou Q. Effects of kindlin-2 on proliferation and migration of VSMC and integrinbeta1 andbeta3 activity via FAK-PI3K signaling pathway. PloS One (2020) 15:e0225173. doi: 10.1371/journal.pone.0225173
187. Yuan G, Si G, Hou Q, Li Z, Xu K, Wang Y, et al. Advanced glycation end products induce proliferation and migration of human aortic smooth muscle cells through PI3K/AKT pathway. BioMed Res Int (2020) 2020:8607418. doi: 10.1155/2020/8607418
188. Xiang M, Yang R, Zhang Y, Wu P, Wang L, Gao Z, et al. Effect of crocetin on vascular smooth muscle cells migration induced by advanced glycosylation end products. Microvasc Res (2017) 112:30–6. doi: 10.1016/j.mvr.2017.02.004
189. Tang L, Dai F, Liu Y, Yu X, Huang C, Wang Y, et al. RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol Res (2018) 133:201–12. doi: 10.1016/j.phrs.2018.05.011
190. Kim JE, Sung JY, Woo CH, Kang YJ, Lee KY, Kim HS, et al. Cilostazol inhibits vascular smooth muscle cell proliferation and reactive oxygen species production through activation of AMP-activated protein kinase induced by heme oxygenase-1. Korean J Physiol Pharmacol (2011) 15:203–10. doi: 10.4196/kjpp.2011.15.4.203
191. Spiekerkoetter E, Guignabert C, de Jesus Perez V, Alastalo TP, Powers JM, Wang L, et al. S100A4 and bone morphogenetic protein-2 codependently induce vascular smooth muscle cell migration via phospho-extracellular signal-regulated kinase and chloride intracellular channel 4. Circ Res (2009) 105:639–47:613. doi: 10.1161/CIRCRESAHA.109.205120
192. Zhao LM, Su XL, Wang Y, Li GR, Deng XL. KCa3.1 channels mediate the increase of cell migration and proliferation by advanced glycation endproducts in cultured rat vascular smooth muscle cells. Lab Invest (2013) 93:159–67. doi: 10.1038/labinvest.2012.163
193. Li C, Chang Y, Li Y, Chen S, Chen Y, Ye N, et al. Advanced glycation end products promote the proliferation and migration of primary rat vascular smooth muscle cells via the upregulation of BAG3. Int J Mol Med (2017) 39:1242–54. doi: 10.3892/ijmm.2017.2938
194. Chung TW, Choi HJ, Kim CH, Jeong HS, Ha KT. Lipocalin-2 elicited by advanced glycation end-products promotes the migration of vascular smooth muscle cells. Biochim Biophys Acta (2013) 1833:3386–95. doi: 10.1016/j.bbamcr.2013.10.011
195. Bu DX, Rai V, Shen X, Rosario R, Lu Y, D'Agati V, et al. Activation of the ROCK1 branch of the transforming growth factor-beta pathway contributes to RAGE-dependent acceleration of atherosclerosis in diabetic ApoE-null mice. Circ Res (2010) 106:1040–51. doi: 10.1161/CIRCRESAHA.109.201103
196. Chen J, Cui L, Yuan J, Zhang Y, Sang H. Circular RNA WDR77 target FGF-2 to regulate vascular smooth muscle cells proliferation and migration by sponging miR-124. Biochem Biophys Res Commun (2017) 494:126–32. doi: 10.1016/j.bbrc.2017.10.068
197. Choe N, Kwon DH, Shin S, Kim YS, Kim YK, Kim J, et al. The microRNA miR-124 inhibits vascular smooth muscle cell proliferation by targeting S100 calcium-binding protein A4 (S100A4). FEBS Lett (2017) 591:1041–52. doi: 10.1002/1873-3468.12606
198. Lei S, Peng F, Li ML, Duan WB, Peng CQ, Wu SJ. LncRNA-SMILR modulates RhoA/ROCK signaling by targeting miR-141 to regulate vascular remodeling in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol (2020) 319:H377–91. doi: 10.1152/ajpheart.00717.2019
199. Cheng L, Wang H, Maboh R, Mao G, Wu X, Chen H. LncRNA LINC00281/Annexin A2 regulates vascular smooth muscle cell phenotype switching via the nuclear factor-kappa b signaling pathway. J Cardiovasc Transl Res (2022). doi: 10.1007/s12265-022-10242-z
200. Dong H, Jiang G, Zhang J, Kang Y. LncRNA OIP5-AS1 promotes the proliferation and migration of vascular smooth muscle cells via regulating miR-141-3p/HMGB1 pathway. Am J Med Sci (2022) 363:538–47. doi: 10.1016/j.amjms.2022.02.012
201. Chinetti G, Neels JG. Roles of nuclear receptors in vascular calcification. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22126491
202. Xu Z, Liu X, Wang Z, Tao J, Han Z, Gu M, et al. Effect of sirolimus on arteriosclerosis induced by advanced glycation end products via inhibition of the ILK/mTOR pathway in kidney transplantation recipients. Eur J Pharmacol (2017) 813:1–9. doi: 10.1016/j.ejphar.2017.06.038
203. Tada Y, Yano S, Yamaguchi T, Okazaki K, Ogawa N, Morita M, et al. Advanced glycation end products-induced vascular calcification is mediated by oxidative stress: functional roles of NAD(P)H-oxidase. Horm Metab Res (2013) 45:267–72. doi: 10.1055/s-0032-1329965
204. Poetsch F, Henze LA, Estepa M, Moser B, Pieske B, Lang F, et al. Role of SGK1 in the osteogenic transdifferentiation and calcification of vascular smooth muscle cells promoted by hyperglycemic conditions. Int J Mol Sci (2020) 21(19):7207. doi: 10.3390/ijms21197207
205. Li Z, Yang P, Feng B. Effect of atorvastatin on AGEs-induced injury of cerebral cortex via inhibiting NADPH oxidase -NF-kappaB pathway in ApoE(-/-) mice. Mol Biol Rep (2020) 47:9479–88. doi: 10.1007/s11033-020-05998-z
206. Thinschmidt JS, Colon-Perez LM, Febo M, Caballero S, King MA, White FA, et al. Depressed basal hypothalamic neuronal activity in type-1 diabetic mice is correlated with proinflammatory secretion of HMBG1. Neurosci Lett (2016) 615:21–7. doi: 10.1016/j.neulet.2016.01.014
207. Kennon AM, Stewart JA Jr.RAGE differentially altered in vitro responses in vascular smooth muscle cells and adventitial fibroblasts in diabetes-induced vascular calcification. Front Physiol (2021) 12:676727. doi: 10.3389/fphys.2021.676727
208. Zhu Y, Ma WQ, Han XQ, Wang Y, Wang X, Liu NF. Advanced glycation end products accelerate calcification in VSMCs through HIF-1alpha/PDK4 activation and suppress glucose metabolism. Sci Rep (2018) 8:13730. doi: 10.1038/s41598-018-31877-6
209. Sun Z, Wang Z, Li L, Yan J, Shao C, Bao Z, et al. RAGE/galectin-3 yields intraplaque calcification transformation via sortilin. Acta Diabetol (2019) 56:457–72. doi: 10.1007/s00592-018-1273-1
210. Hofmann Bowman MA, Gawdzik J, Bukhari U, Husain AN, Toth PT, Kim G, et al. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein e-null mice by activating an osteogenic gene regulatory program. Arterioscler Thromb Vasc Biol (2011) 31:337–44. doi: 10.1161/ATVBAHA.110.217745
211. Liu D, Cui W, Liu B, Hu H, Liu J, Xie R, et al. Atorvastatin protects vascular smooth muscle cells from TGF-beta1-stimulated calcification by inducing autophagy via suppression of the beta-catenin pathway. Cell Physiol Biochem (2014) 33:129–41. doi: 10.1159/000356656
212. McLaren JE, Michael DR, Ashlin TG, Ramji DP. Cytokines, macrophage lipid metabolism and foam cells: Implications for cardiovascular disease therapy. Prog Lipid Res (2011) 50:331–47. doi: 10.1016/j.plipres.2011.04.002
213. Jinnouchi H, Guo L, Sakamoto A, Torii S, Sato Y, Cornelissen A, et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell Mol Life Sci (2020) 77:1919–32. doi: 10.1007/s00018-019-03371-3
214. Dubland JA, Francis GA. So much cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr Opin Lipidol (2016) 27:155–61. doi: 10.1097/MOL.0000000000000279
215. Fisher EA, Miano JM. Don't judge books by their covers: vascular smooth muscle cells in arterial pathologies. Circulation (2014) 129:1545–7. doi: 10.1161/CIRCULATIONAHA.114.009075
216. Bao Z, Li L, Geng Y, Yan J, Dai Z, Shao C, et al. Advanced glycation end products induce vascular smooth muscle cell-derived foam cell formation and transdifferentiate to a macrophage-like state. Mediators Inflamm (2020) 2020:6850187. doi: 10.1155/2020/6850187
217. Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res (2018) 114:529–39. doi: 10.1093/cvr/cvy023
218. Singh S, Gautam AS. Upregulated LOX-1 receptor: Key player of the pathogenesis of atherosclerosis. Curr Atheroscler Rep (2019) 21:38. doi: 10.1007/s11883-019-0801-y
219. Vendrov AE, Sumida A, Canugovi C, Lozhkin A, Hayami T, Madamanchi NR, et al. NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol (2019) 21:101063. doi: 10.1016/j.redox.2018.11.021
220. Lehners M, Dobrowinski H, Feil S, Feil R. cGMP signaling and vascular smooth muscle cell plasticity. J Cardiovasc Dev Dis (2018) 5(2):20. doi: 10.3390/jcdd5020020
Keywords: atherosclerosis, diabetes mellitus, advanced glycation end products, vascular smooth muscle cells, receptor for advanced glycation end products
Citation: Mao L, Yin R, Yang L and Zhao D (2022) Role of advanced glycation end products on vascular smooth muscle cells under diabetic atherosclerosis. Front. Endocrinol. 13:983723. doi: 10.3389/fendo.2022.983723
Received: 01 July 2022; Accepted: 11 August 2022;
Published: 31 August 2022.
Edited by:
Annunziata Nusca, Campus Bio-Medico University, ItalyReviewed by:
Giuseppe Defeudis, Università Campus Bio-Medico, ItalyLihua Li, Affiliated Hospital of Jiangsu University, China
Federico Bernardini, Policlinico Universitario Campus Bio-Medico, Italy
Copyright © 2022 Mao, Yin, Yang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Longyan Yang, lyyang15@ccmu.edu.cn; Dong Zhao, zhaodong@ccmu.edu.cn
†These authors have contributed equally to this work