Targeting a cancer-specific LYPD3 glycoform for tumor therapy
 Theresa Neumann† Evelyn Hartung†
Theresa Neumann† Evelyn Hartung†  Johanna Gellert* Lisa Weiß Manon Weiske Naomi Kast Stephanie Gurka Sophie Marinoff
Johanna Gellert* Lisa Weiß Manon Weiske Naomi Kast Stephanie Gurka Sophie Marinoff  Anika Jäkel Antje Danielczyk
Anika Jäkel Antje Danielczyk  Patrik Kehler
Patrik Kehler- Glycotope GmbH, Berlin, Germany
Introduction: One of the most drastic changes in cancer is the altered glycosylation of proteins and lipids, giving rise to truncated O-glycans like the Thomsen Friedenreich (TF) or Thomsen nouvelle (Tn) antigen, which are almost absent on normal cells. Combined protein-carbohydrate epitopes comprising these specific glycans are ideal candidates for potent targeted therapies given their excellent tumor specificity and broad cancer expression.
Methods and results: We have generated GT-002, a monoclonal antibody specifically targeting the epithelial glycoprotein LYPD3 only in the presence of a TF glycosylation. It does not cross-react with non-glycosylated LYPD3 or TF on other glycoproteins in ELISA and flow cytometry. GT-002 binds to various tumor cell lines and stains tumor tissues of different cancer indications including squamous cell carcinoma of the head and neck. The remarkable tumor specificity was confirmed in an immunohistochemistry study on a normal human tissue panel including several LYPD3-positive organs, where GT-002 elicited almost completely abolished normal tissue binding. Consequently, we observed markedly reduced binding of GT-002 to normal human tissues compared to Lupartumab, a conventional anti-LYPD3 antibody previously in clinical development as antibody-drug conjugate (BAY1129980). Neuraminidase treatment of healthy tissues, resulting in cleavage of sialic acid residues, re-established binding of GT-002 comparable to Lupartumab, showing that the GT-002 epitope is masked by sialic acid in normal cells.
Discussion: We believe that GT-002 is a promising candidate for development of antibody-drug- and radio-conjugates as well as bispecific molecules and chimeric antigen receptor therapeutics and highlights the powerful potential of antibodies against combined protein-carbohydrate epitopes to reduce on-target/off-tumor cytotoxicity.
1 Introduction
Targeted therapies including chimeric antigen receptor (CAR) T cells and antibody-drug conjugates (ADCs) are promising strategies for cancer treatment, but they substantially rely on the tumor specificity of the target structure. This has proven a major challenge, particularly in solid tumors, which, unlike hematological malignancies, lack lineage-specific markers. Thus, severe on-target/off-tumor toxicities have been observed in clinical trials targeting tumor-associated rather than tumor-specific antigens (Lamers et al., 2006; Morgan et al., 2010; Schoeffski et al., 2021).
Glycosylation is one of the most common post-translational modifications of lipids and proteins, regulating diverse biological processes like cell signaling, protein folding and extracellular scaffolding (Varki et al., 2022). It is therefore not surprising that changes in glycosylation can drastically impact the biology of the cell, significantly contributing to the development of cancer and various other pathologies (Munkley and Elliott, 2016; Reily et al., 2019). Tumor-associated alterations of the complex glycan biosynthesis pathway can provoke abnormal neosynthesis of blood group antigens, enhancement of N-glycan β1,6GlcNAc-branching, increased sialylation or incomplete O-glycan synthesis yielding short, truncated O-glycans (Kudelka et al., 2015; Pinho and Reis, 2015).
Given that healthy cells express mature elongated and often branched O-glycans, short O-glycans like the Thomsen Friedenreich (TF, Galβ1-3GalNAcα1-O-Ser/Thr), the Thomsen nouvelle antigen (Tn, GalNAcα1-O-Ser/Thr) and the sialylated form sTn (Neu5Acα2-6GalNAcα1-O-Ser/Thr) elicit superior tumor specificity, becoming de novo exposed on the cell surface of different types of cancer (Springer, 1984; Itzkowitz et al., 1989; Cao et al., 1995; Fu et al., 2016). Furthermore, they have been shown to promote oncogenic features like immune escape, tumor growth and metastasis (Yu et al., 2007; Cazet et al., 2010; Radhakrishnan et al., 2014; Dong et al., 2018). In particular, the TF antigen, also called CD176, T antigen or core-1, has been detected on the surface of cancers including breast, lung, head and neck and ovary as well as hematological malignancies (Therkildsen et al., 1995; Baldus et al., 1999; Takanami, 1999; Schindlbeck et al., 2006; Cao et al., 2008; Wiest et al., 2012; Karacosta et al., 2018), but is virtually absent from normal tissues (Cao et al., 1995). Several TF carrier proteins have been identified which comprise known cancer stem cell markers as well as widely utilized biomarkers in cancer diagnostics (Lin et al., 2010; Karsten and Goletz, 2013; Saldova et al., 2013; Li et al., 2019; Hernaez et al., 2022).
Truncated O-glycans expressed on tumor cells are thus ideal candidates for targeted therapies given their remarkable specificity and broad application opportunities (Steentoft et al., 2018; Mereiter et al., 2019). The generally low intrinsic affinities of antibodies targeting pure glycan epitopes can be overcome by developing therapeutic antibodies against combined protein-carbohydrate epitopes, which elicit high affinities while still bearing the exceptional tumor specificity dictated by the glycan (Danielczyk et al., 2006; Lavrsen et al., 2012; Haji-Ghassemi et al., 2015; Hernaez et al., 2022). Still, only one antibody targeting a glycan epitope has reached approval for cancer treatment (Dhillon, 2015; Ladenstein et al., 2020; Balaguer et al., 2022), underlining the technical and immunological difficulties of successfully leveraging the cancer glycome.
Here, we report the identification of GT-002, a monoclonal antibody (mAb) targeting a highly tumor-specific glycosylated epitope of LY6/PLAUR Domain Containing 3 (LYPD3) as first case study for purposive generation of mAbs against protein carbohydrate mix epitopes.
LYPD3, also named C4.4A, is a glycosylphosphatidylinositol (GPI)-anchored cell surface glycoprotein that was first described as a metastasis-associated protein in rats (Claas et al., 1996; Roesel et al., 1998; Hansen et al., 2004). Since then, it has been linked to carcinogenesis in different human cancers and is reported to be overexpressed in squamous cell carcinoma (SCC) of the skin, head and neck and esophagus, colorectal cancer and other indications (Konishi et al., 2010; Gu et al., 2011; Kriegbaum et al., 2014; Liu et al., 2015). LYPD3 is also highly expressed on healthy epithelia of the proximal digestive tract, female reproductive organs, tonsillar squamous epithelium as well as skin keratinocytes, suggesting potential for on-target/off-tumor toxicities (Hansen et al., 2004; Hansen et al., 2007; Jacobsen et al., 2017). Of note, LYPD3 is also known to carry several O-glycans on its extracellular domain, encouraging the targeting of a tumor-specific glycosylated epitope on LYPD3 for cancer therapy.
Lupartumab-amadotin, an anti-LYPD3 ADC directed against a conventional protein epitope (BAY1129980, Willuda et al., 2017) has been tested in a phase 1 study for the treatment of non-small-cell lung cancer (NSCLC), but clinical trial results have not been published (NCT02134197).
We demonstrate that GT-002, due to its glycosylation-dependent recognition of LYPD3, elicits excellent tumor specificity and drastically reduced normal tissue binding compared to Lupartumab, providing a highly promising candidate for the development of potent ADCs, bispecifics or CAR-based therapeutics.
2 Materials and methods
2.1 Antigen production and purification
The soluble ECD of LYPD3, its STP-rich domain and irrelevant off-target proteins were recombinantly expressed in the human myeloid leukemia cell lines NM-F9 (F9) and NM-H9D8 (H9D8) (Glycotope) to carry aberrant O-glycans without (TF/Tn) or with sialic acids (sialyl-TF/-Tn), respectively. Proteins were purified from supernatants via His- or Twin-Strep-tag® by cOmplete His-Tag column (Roche) or Strep-Tactin® XT 4Flow column (IBA Lifesciences). Additionally, to generate non-sialylated control antigen, LYPD3 purified from transfected H9D8 was incubated with neuraminidase (Sialidase A, Agilent) according to manufacturer’s instructions. For the generation of de-galactosylated LYPD3 (Tn), 1 µg protein purified from transfected F9 cells was incubated with 10 units GalactEXO (Genovis) in a mix of PBS pH 7.4 and 20 mM Tris pH 6.8 at 37°C for 24 h. For LYPD3 lacking O-glycans (deOglyc), the protein was incubated with a 1:1 mixture of immobilized GalactEXO (Genovis) and immobilized GalNAcEXO (Genovis) for 4–7 d at room temperature (RT).
2.2 Cell culture and cell lines
All cell culture media and supplements were purchased from Biowest and Serana. MDA-MB-231 (DSMZ) were cultured in Leibovitz’s L15 medium supplemented with 1 mM glutamine and 10% fetal bovine serum (FBS). CaOV-3 (ATCC) were cultivated in Dulbecco’s Modified Eagle Medium supplemented with 10% FBS and 2 mM glutamine. ZR-75-1 (ATCC) were cultured in RPMI1640 medium with 10% FBS and 1 mM glutamine. The human F9 cell line was used for flow cytometry binding experiments either as untransfected cell line or transfected with the complete human LYPD3 coding sequence (LYPD3-F9). F9 and H9D8 cells were cultivated under standard conditions with Glycotope medium +0.002% human serum albumin (HSA). Human embryonic kidney 293T (HEK293T) cells with GALE and GALK2 deficiency unable to generate mucin type O-linked glycans (Termini et al., 2017) were used for the recombinant expression of non-glycosylated LYPD3 (LYPD3-HEK-OglycKO). These cells were cultivated in Dulbecco’s Modified Eagle Medium supplemented with 4 mM glutamine, 10% FBS and 1 μg/mL puromycin.
2.3 Anti-LYPD3 antibody discovery and mAb production
Antibody discovery was performed by Icosagen (Estonia). Hybrifree technology (Kivi et al., 2016) was used to isolate LYPD3-specific antibodies from splenocytes of rabbits and chicken immunized with purified LYPD3-ECD from transfected F9 cells. Splenic B cells with antigen specificity for the aberrantly O-glycosylated STP-rich domain of LYPD3 were enriched by negative and positive selection using off- and on-target proteins. cDNA of antibody variable domains was amplified and cloned into an expression vector library in order to produce chimeric IgG1 antibodies with a human backbone in Chinese Hamster Ovary (CHO) cells. Supernatants of single clones with unique VH/VL sequences were screened for glycosylation-dependent LYPD3 binding in ELISA and flow cytometry. GT-002 was selected as lead candidate due to its unique binding properties.
To generate material for further characterization, anti-LYPD3 antibodies GT-002 and Lupartumab (BAY 111262) were recombinantly expressed in CHO cells as human and murine IgG1. Supernatants were produced in 50–700 mL batch culture and purified by Protein A chromatography.
2.4 Glycan-directed control antibodies
Anti-TF NM-TF2 (murine hybridoma IgM, Glycotope, Goletz et al., 2003) or its humanized derivative GatoMab (recombinant hIgM, Glycotope) as well as anti-Tn 5F4 (recombinant mIgM, Glycotope, sequence kindly provided by H. Clausen) served as glycan-directed control antibodies to characterize O-glycosylation of antigens and cell lines in ELISA and flow cytometry studies.
2.5 O-glycan profiling
The 2-AB labelling was performed using the EZGlyco® O-Glycan Prep Kit (S-BIO) according to the manufacturer’s instructions. Identification and quantification of O-glycan structures were performed on an ultra-performance liquid chromatography (UPLC) system (Waters, Acquity I-Class) with a fluorescence detector (FLR) (Waters, Acquity FLR) coupled to a quadrupole time-of-flight (QTOF) tandem mass spectrometer (Bruker, Compact). The ions were generated with electrospray ionization (ESI). UPLC-ESI-QTOF MS/MS was operated using HyStar Software (Bruker). The FLR settings were: λex = 330 nm and λem = 420 nm, 20 Hz. The 2-AB labeled glycans were separated by hydrophilic interaction chromatography (HILIC) using ACQUITY UPLC Glycan BEH Amide Precolumn (Waters) and ACQUITY UPLC Glycan BEH Amide (Waters). Acetonitrile and 100 mM ammonium formiate (pH 4.5) were used as mobile phases A and B, respectively. A gradient of 85% A to 50% A in 15 min was applied at a flow rate of 0.25 mL/min. The column temperature was 45°C and the injected amount was proportional to 2 µg of initial sample. The MS device was operated in ESI + mode with a capillary voltage of 4.5 kV, a desolvation temperature of 220°C and an end plate offset of 500 V. Full scan acquisition was performed with AutoMS2 over a mass range of 200–2,200 m/z with a scan rate of 2 Hz. O-glycans were identified based on the retention times of the glycans of a bovine fetuin standard and using intact and fragment masses. The evaluation of the FLR chromatograms and the extracted ion chromatograms were performed with DataAnalysis (version 5.2, Bruker). The relative quantification of the O-glycan structures was based on the peak areas of the fluorescence signals. For unknown retention times and co-eluting structures, additional identification and quantification via mass spectrometry was employed. The peeling product peak was high only in de-galactosylated samples due to free galactose in solution (8.5% for LYPD3 from NM-F9, 36.4% for de-galactosylated LYPD3 and 4.5% for LYPD3 from NM-H9D8). They were excluded from glycan composition calculations of all samples.
2.6 Specificity analysis by ELISA
To test binding to different glycoforms of LYPD3 or to irrelevant glycoproteins, MaxiSorp plates (Thermo Scientific) were coated with 35 nM of the indicated proteins dissolved in PBS overnight at 4°C. After washing with PBS +0.05% Tween-20 and blocking with PBS +2% BSA, anti-LYPD3 GT-002 and Lupartumab, anti-TF GatoMab and anti-Tn 5F4 were added as primary antibodies at the indicated concentrations diluted in PBS +1% BSA for 1.5 h at RT. After washing, anti-LYPD3 antibodies were incubated for 1 h at RT with anti-human IgG (hIgG)-HRP or anti-mouse IgG (mIgG)-HRP (Jackson ImmunoResearch), anti-TF with anti-human-IgM-HRP (JIR) and anti-Tn with anti-mouse IgM-HRP (JIR) as secondary antibodies. Substrate reaction was performed with TMB (tebu-bio) and after stopping with 2.5 N H2SO4, fluorescence was measured at 460 nm/620 nm with a Spark plate reader (TECAN). EC50 values of GT-002 and Lupartumab were calculated based on six individual ELISA experiments on O-glycosylated LYPD3 from NM-F9 using Graphpad Prism software version 5.04.
2.7 Flow cytometric analysis
Cells were harvested and, if indicated, treated with 5 mU/mL neuraminidase (from Arthrobacter ureafaciens, Roche) in RPMI1640 for 30 min at 37°C. They were washed with PBS +0.2% BSA, then seeded at a density of 5 × 104 cells in 50 µL. Cells were then incubated with primary antibodies for 30 min at 4°C and washed with PBS +0.2% BSA. Appropriate secondary antibodies were incubated for 30 min at 4°C and again washed with PBS +0.2% BSA. For exclusion of dead cells, DAPI (Merck) was added directly before measurement at a concentration of 3 µM. Cells were measured on a FACS Canto II (BD) or Attune Nxt (ThermoFisher) flow cytometer and data analyzed using FlowJo software.
2.8 Immunohistochemistry
Tissue microarrays (TMA) were purchased from TissueArray (formerly US Biomax). The following TMA were used: ES243, HN804, MC632, OR301a, OR601c, RE441, SK801d, OR481a, FDA999y2, LC242b, and LC726b. Further FFPE sections of rectal and NSCLC SCC were obtained from PrecisionMed. Frozen optimal cutting temperature compound-embedded healthy tissues of esophagus, tonsil and skin were ordered from Amsbio. PDX and CDX tumor FFPE sections were provided by EPO GmbH.
FFPE tissue sections and tissue microarrays were deparaffinated and heat-induced epitope retrieval was performed using citrate buffer (pH 6). To block endogenous peroxidase, sections were incubated with 3% H2O2 for 10 min at RT, then washed.
Cryostat sections were fixed in acetone for 10 min and washed with PBS. Endogenous peroxidase was inhibited by a blocking solution (PBS + 1 mM NaN3 + 10 mM glucose +1 U/mL glucose oxidase) for 1 h at 37°C, then washed.
BlockAid solution (Invitrogen) was applied to FFPE and cryo-sections for 60 min to block unspecific binding. While for ELISA and flow cytometry GT-002 and Lupartumab were deployed with human IgG1 backbones, in IHC both antibodies were used with murine IgG1 backbones to overcome background problems of human IgG on human tissue. Specificities of murine and human IgG were analyzed beforehand and showed comparable binding patterns (Supplementary Data; Supplementary Figure S1). Protein-specific anti-LYPD3 polyclonal antibodies HPA041529 (Atlas Antibodies) or AF5428 (R&D Systems) served as positive control. To confirm specificity of staining, mouse IgG1a (Biolegend), rabbit IgG (RbNP15, Invitrogen) and sheep IgG (Jackson ImmunoResearch) were used as irrelevant isotype controls. After 1 h incubation at RT, tissue sections were washed and incubated for 30 min with HRP-conjugated secondary reagents Envision Flex anti-mouse Ig-HRP (Dako), CRF Anti-Polyvalent HRP polymer (DAB) Stain Kit (ScyTek) or anti-sheep Ig-HRP (Jackson ImmunoResearch), followed by 3,3-diaminobenzidine (DAB) substrate reaction and counterstaining with Mayer’s Haematoxylin (epredia). Sections were mounted in Entellan (Merck). Images were analyzed by a slide scanner microscope (Olympus, SLIDEVIEW VS200). Positive tumor cases were defined by membranous binding of test antibodies to ≥10% of cancerous cells.
2.9 Neuraminidase treatment of tissue sections
Cryostat sections were treated as described above. After blocking of endogenous peroxidase, sections were incubated with 0.02 U/mL neuraminidase (from A. ureafaciens, Roche) for 60 min at RT to remove sialic acids. Unspecific binding was blocked with BlockAid solution (Invitrogen) and the protocol continued as described above, except that primary antibodies were incubated overnight.
2.10 Internalization assay
Cells were harvested and seeded to 5 × 103 cells per well of a 96-flat bottom plate (TPP) in medium. The respective antibody was incubated with a 3-fold molar excess of anti-human Fabfluor-pH antibody labeling dye (Sartorius) for 15 min at RT and then added to the cells. Phase contrast and red fluorescence was monitored over time in an Incucyte S3 (Sartorius) at 37°C. Percentage of lysosomal routing was expressed as the Red Area (Red positive cells)/Phase Area (Total cell confluency).
3 Results
3.1 GT-002 specifically binds to an O-glycosylated epitope on LYPD3
We have generated antibodies against the serine-threonine-proline (STP)-rich region of LYPD3 carrying tumor-specific truncated O-glycans. Antibodies were screened for glycosylation-dependent binding of LYPD3 via ELISA and flow cytometry using different on- and off-target proteins and LYPD3-expressing cell lines with varying glycosylation patterns. We identified GT-002 as lead candidate due to its outstanding specificity towards tumor-glycosylated LYPD3.
The glycosylation-dependent binding of GT-002 was analyzed by ELISA using proteins comprising the extracellular domain (ECD) of LYPD3 with or without truncated O-glycans. GT-002 bound only to O-glycosylated LYPD3 but not to the protein lacking O-glycans (Figure 1A). In contrast, Lupartumab bound both LYPD3 glycoforms, as expected. Full dose-response curves of GT-002 and Lupartumab revealed comparable EC50 values of 5.5 ± 1.2 ng/mL and 3.5 ± 0.9 ng/mL, respectively (Figure 1B). Glyco-dependent binding of GT-002 to LYPD3 was further confirmed by flow cytometry using recombinant cell lines expressing glycosylated or non-glycosylated LYPD3 on their surface (Figure 1C). LYPD3-F9 and F9 wildtype cells express mostly TF- and some Tn-glycosylated proteins, whereas O-glycans are absent due to knock-out (KO) of UDP-galactose-4-epimerase (GALE) and galactokinase 2 (GALK2) in LYPD3-HEK-OglycKO cells. O-glycan and LYPD3 expression of cell lines was confirmed using an anti-TF control antibody and Lupartumab, respectively. GT-002 only recognized LYPD3-F9 but not LYPD3-HEK-OglycKO cells, which was in accordance with ELISA results. Both anti-LYPD3 antibodies bound weakly to F9 wild type cells, which express only low amounts of endogenous, glycosylated LYPD3.
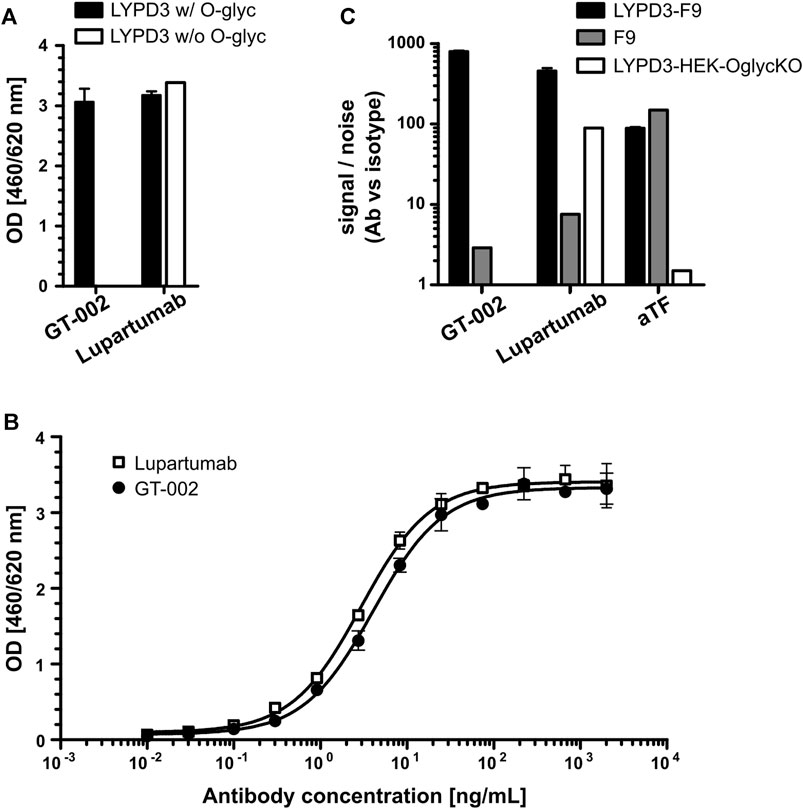
FIGURE 1. Glycosylation-dependent recognition of LYPD3 by GT-002. (A) LYPD3 proteins carrying truncated O-glycans (w/O-glyc) or no O-glycans (w/o O-glyc) were coated onto ELISA plates and tested for binding of 250 ng/mL GT-002 and Lupartumab, both expressed with human IgG1 backbones. (B) Dose-response curves (exemplary experiment measured in duplicates) of GT-002 (black circles) and Lupartumab (white squares) were determined by ELISA on LYPD3 carrying truncated O-glycans. (C) Flow cytometric analysis of cells overexpressing LYPD3 carrying truncated O-glycans (LYPD3-F9), no O-glycans (LYPD3-HEK-OglycKO) or low endogenous LYPD3 levels (F9) using 10 μg/mL GT-002. Lupartumab (1 μg/mL) and aTF mAb (0.1 μg/mL) served as expression controls. Signal/noise ratios were calculated as quotient of MFI (mean fluorescence intensity) values of mAb and appropriate isotype control staining. Error bars indicate standard deviation (SD) of duplicates.
3.2 The epitope of GT-002 comprises a combined TF-LYPD3 glycopeptide epitope
To further characterize the glyco-epitope recognized by GT-002, we analyzed binding to different LYPD3-ECD glycoforms. Glycosylation patterns of the different LYPD3 variants were analyzed by O-glycan profiling (Table 1; Figure 2A). LYPD3 expressed in F9 cells mostly carried TF (97%) and only minimal amounts of Tn. LYPD3 isolated from H9D8 cells comprised 59.1% sialylated O-glycans comprising 19.8% 3’sTF (Neu5Acα2-3Galβ1-3GalNAcα1-O-Ser/Thr), 18.8% 6’sTF (Neu5Acα2-6Galβ1-3GalNAcα1-O-Ser/Thr), 13.7% 3,6’sTF (Neu5Acα2-3(Neu5Acα2-6)Galβ1-3GalNAcα1-O-Ser/Thr) and 6.8% sTn. Furthermore, 38.9% non-sialylated TF and 2% Tn were detected, respectively. GT-002 bound strongly to TF-carrying LYPD3 purified from F9 cells and much weaker to sialylated LYPD3 from H9D8 (Figure 2B). Neuraminidase treatment of LYPD3 from NM-H9D8, releasing 91.1% TF and 8.9% Tn, re-established binding of GT-002, suggesting that GT-002 does not tolerate α2-nor α6-sialylation in its epitope. We also tested recognition of de-galactosylated LYPD3 from F9 cells with an O-glycan composition of 93% Tn and 7% remaining TF (F9, de-gal.). We observed no binding of GT-002 showing that GalNAc (Tn) is not sufficient but the disaccharide Galβ1-3GalNAcα (TF) must be present on LYPD3 to be recognized by GT-002. Lupartumab served as anti-protein control and bound to all LYPD3 glycoforms (Figure 2B).
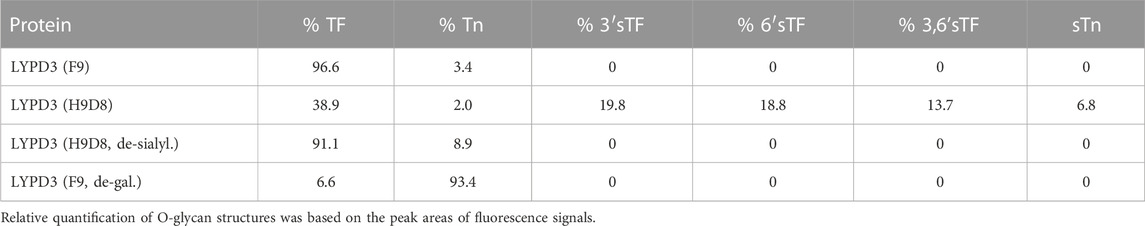
TABLE 1. O-glycan profiles of LYPD3 glycoforms used for specificity analysis of GT-002.
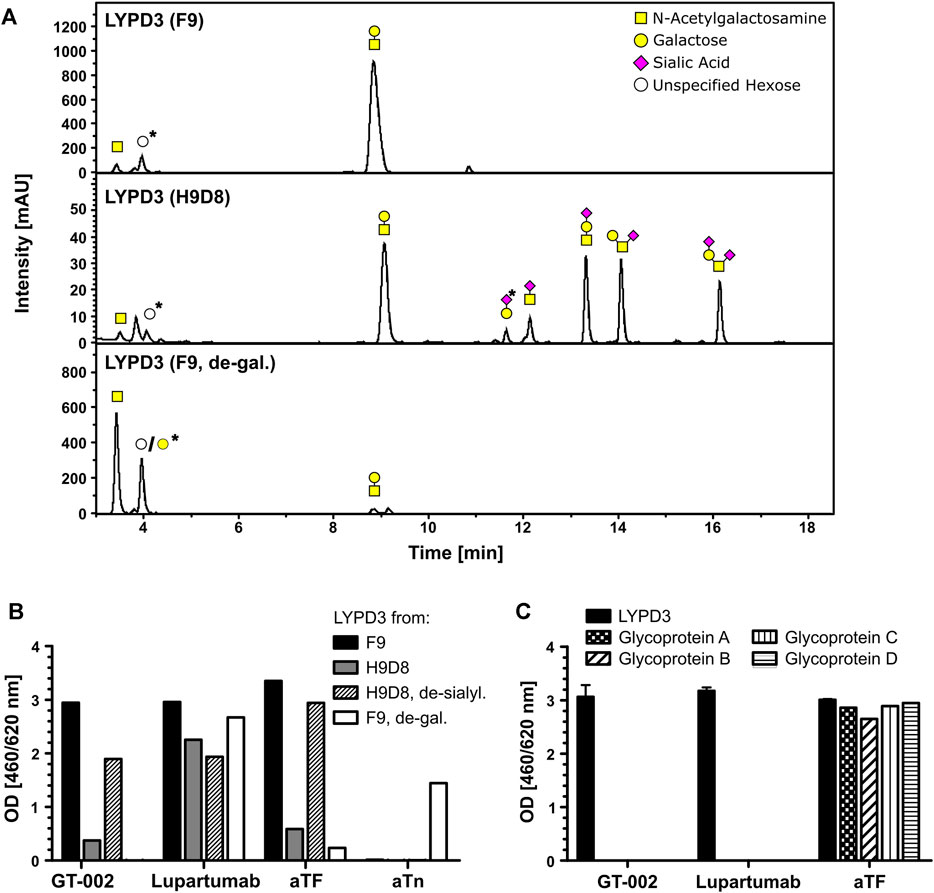
FIGURE 2. Fine-specificity of GT-002 in ELISA. (A) O-glycan profiling of LYPD3 glycoforms used for binding studies. Monosaccharide symbols follow the CFG recommended symbol nomenclature (Varki et al., 2015). Open circles: unspecified hexose; peaks marked with an asterisk are methodically-derived peeling products. (B) Recognition of indicated LYPD3 glycoforms by GT-002 (12.5 ng/mL) was analyzed by ELISA. LYPD3 purified from different cell lines was treated with neuraminidase (de-sialyl.) or with galactosidase (de-gal.), where indicated. Lupartumab (10 ng/mL), aTF mAb (1 μg/mL) and aTn mAb (5 μg/mL) served as controls. (C) Binding of GT-002 (250 ng/mL) to irrelevant TF-carrying glycoproteins produced in F9 cells was tested by ELISA. Lupartumab (250 ng/mL) and aTF mAb (1 μg/mL) served as controls. Error bars indicate standard deviation (SD) of duplicates.
Specificity towards the LYPD3 protein was analyzed by ELISA using other TF-carrying glycoproteins produced in F9 cells (Figure 2C). We found that GT-002 only binds to TF-LYPD3, but not to any of the other glycoproteins tested, strongly suggesting that GT-002 specifically recognizes TF in the context of the LYPD3 backbone.
From these data, we conclude that the epitope of GT-002 comprises a combined TF-LYPD3 glycopeptide epitope.
3.3 GT-002 binds to LYPD3-expressing tumor cell lines
Binding of GT-002 to tumor cell lines was analyzed in flow cytometry using LYPD3-positive cell lines (CaOV-3 and ZR-75-1) as well as the LYPD3-negative cell line MDA-MB-231. Cell lines were tested with or without prior enzymatic removal of sialic acids by neuraminidase due to hypersialylation by tumor cell lines under cell culture conditions, which has also been reported by others (Steentoft et al., 2019; Nishida et al., 2022). Staining with an anti-TF antibody showed moderate TF expression on untreated cells, which was strongly enhanced after neuraminidase treatment (Figure 3A). LYPD3 expression of CaOV-3 and ZR-75-1 with and without pre-treatment was confirmed using Lupartumab. GT-002 bound to CaOV-3 and ZR-75-1 only after removal of sialic acid, which was in accordance with ELISA results. LYPD3-negative MDA-MB-231 were not recognized by GT-002 or Lupartumab. Signal to noise ratios of GT-002 were slightly lower compared to Lupartumab, possibly reflecting the presence of some LYPD3 glycoforms not bound by GT-002 or different affinities of the antibodies to LYPD3 on the cell surface. In contrast to the flow cytometry experiments, cell line-derived xenograft (CDX) sections of CaOV-3 grown in mice were bound by GT-002 in immunohistochemistry (IHC) without prior removal of sialic acids (Figure 3B). GT-002 was able to bind these tumors with 20% positive cells and a weak to moderate intensity, suggesting that the sialylation of its epitope by tumor cell lines in vitro might not reflect the glycosylation pattern of LYPD3 in vivo.
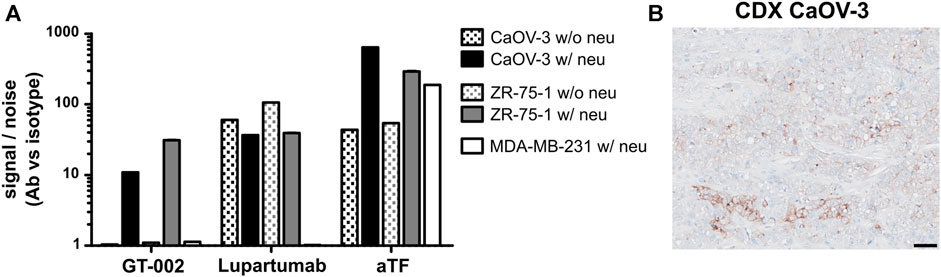
FIGURE 3. Binding of GT-002 to tumor cells. (A) LYPD3-positive (CaOV-3 and ZR-75-1) and -negative (MDA-MB-231) human tumor cell lines were treated with neuraminidase (w/neu) or not treated (w/o neu) and stained with 10 μg/mL GT-002. Lupartumab (1 μg/mL) and aTF (10 μg/mL) served as expression controls. Signal to noise ratios were calculated as the quotient of MFI values of mAb and appropriate isotype control stainings. (B) Immunohistochemical staining of a human tumor CDX section derived from the CaOV-3 cell line with 10 μg/mL GT-002. Scale bars indicate 50 µm. Error bars indicate SD of duplicates.
3.4 GT-002 recognizes a cancer-specific epitope
LYPD3 is described to be overexpressed in tumor indications like SCC of the skin, head and neck and esophagus as well as colorectal cancer. Using an anti-LYPD3 polyclonal antibody for IHC staining of formalin-fixed, paraffin-embedded (FFPE) tumor tissue arrays, strong LYPD3 expression was observed in SCC of head and neck, especially lip, oral cavity and nose, as well as of the vulva, penis and anal area/rectum, but not in NSCLC (Supplementary Data, Supplementary Figure S2; Supplementary Table S1). GT-002 was reactive with all of these indications with up to 80% positive cells per core and varying staining intensities (Figure 4A), demonstrating the presence of its TF-glycosylated epitope of LYPD3 in cancer tissues. Most cases were stained by GT-002 in SCC of anal area/rectum (71.4%), lip (60.9%) and vulva (60.0%) (Table 2). Also, patient-derived xenograft tumors (PDX) from head and neck SCC were stained by GT-002 with % positive cells similar to the CDX sections (Figures 3B, 4B).
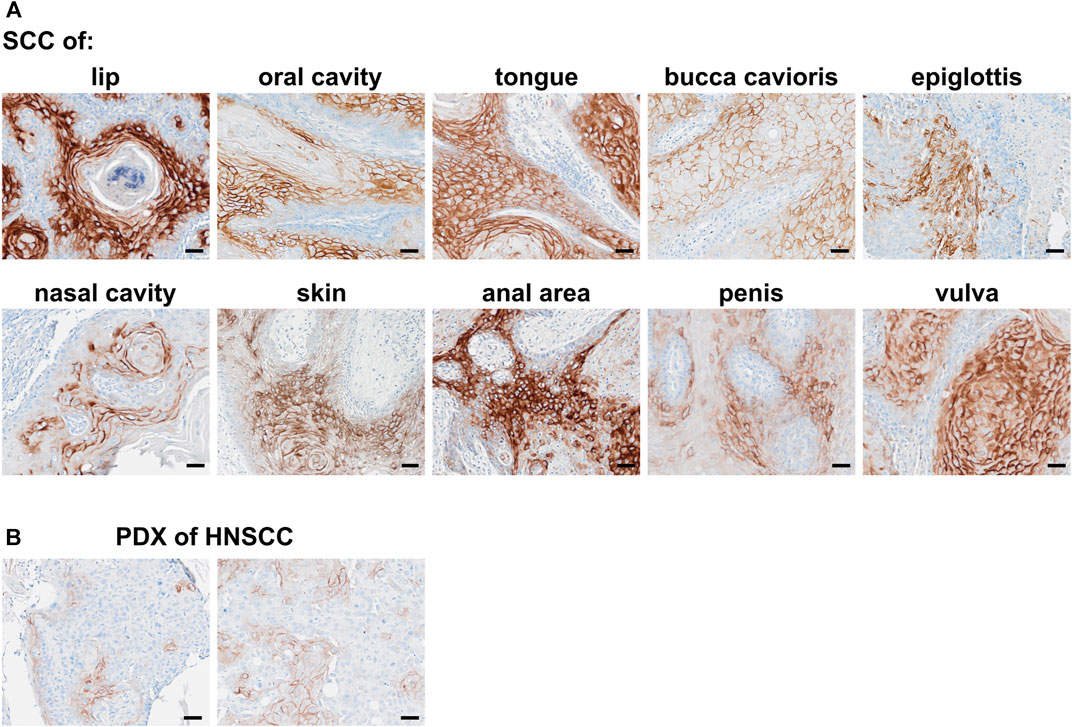
FIGURE 4. Immunohistochemical staining of squamous cell carcinomas (SCC) with GT-002. Human FFPE tumor tissues were stained with 10 μg/mL GT-002. (A) SCC primary tumors of indicated organs. (B) PDX tumor sections from head and neck SCC (HNSCC). Scale bars indicate 50 µm.
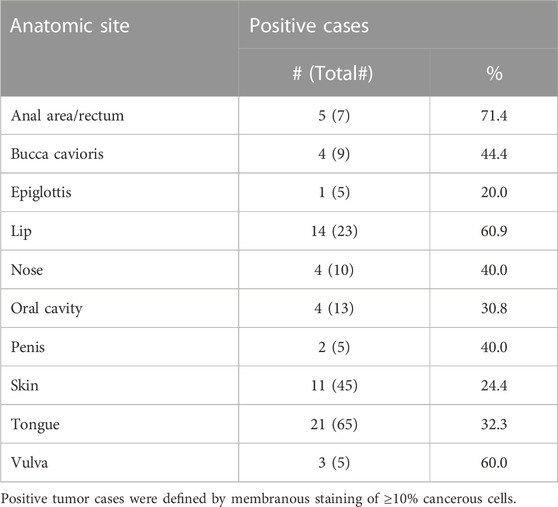
TABLE 2. Summary of immunohistochemistry with GT-002 on different TMAs comprising SCC tumor sections.
Cancer specificity of GT-002 was further analyzed by an IHC study using an FFPE normal tissue microarray comprising 33 organs with up to three donors per organ. Membranous expression of LYPD3 protein detected by an anti-LYPD3 polyclonal antibody was found on normal skin, esophagus, tonsil and ectocervix, which was in accordance with previously published expression data (Hansen et al., 2004; Hansen et al., 2007; Jacobsen et al., 2017). In contrast, GT-002 only bound to the epidermal layer of normal skin, whereas all other tissues were negative (Table 3). We verified these results on cryo-preserved normal tissue sections of esophagus, tonsil and skin and compared GT-002 to Lupartumab, which is not compatible with FFPE but with frozen tissues. While LYPD3 expression was evident in all three tissues given the strong membranous staining by Lupartumab, GT-002 again did not bind to squamous epithelium of esophagus and tonsil, but only to the epidermis (Figure 5A).
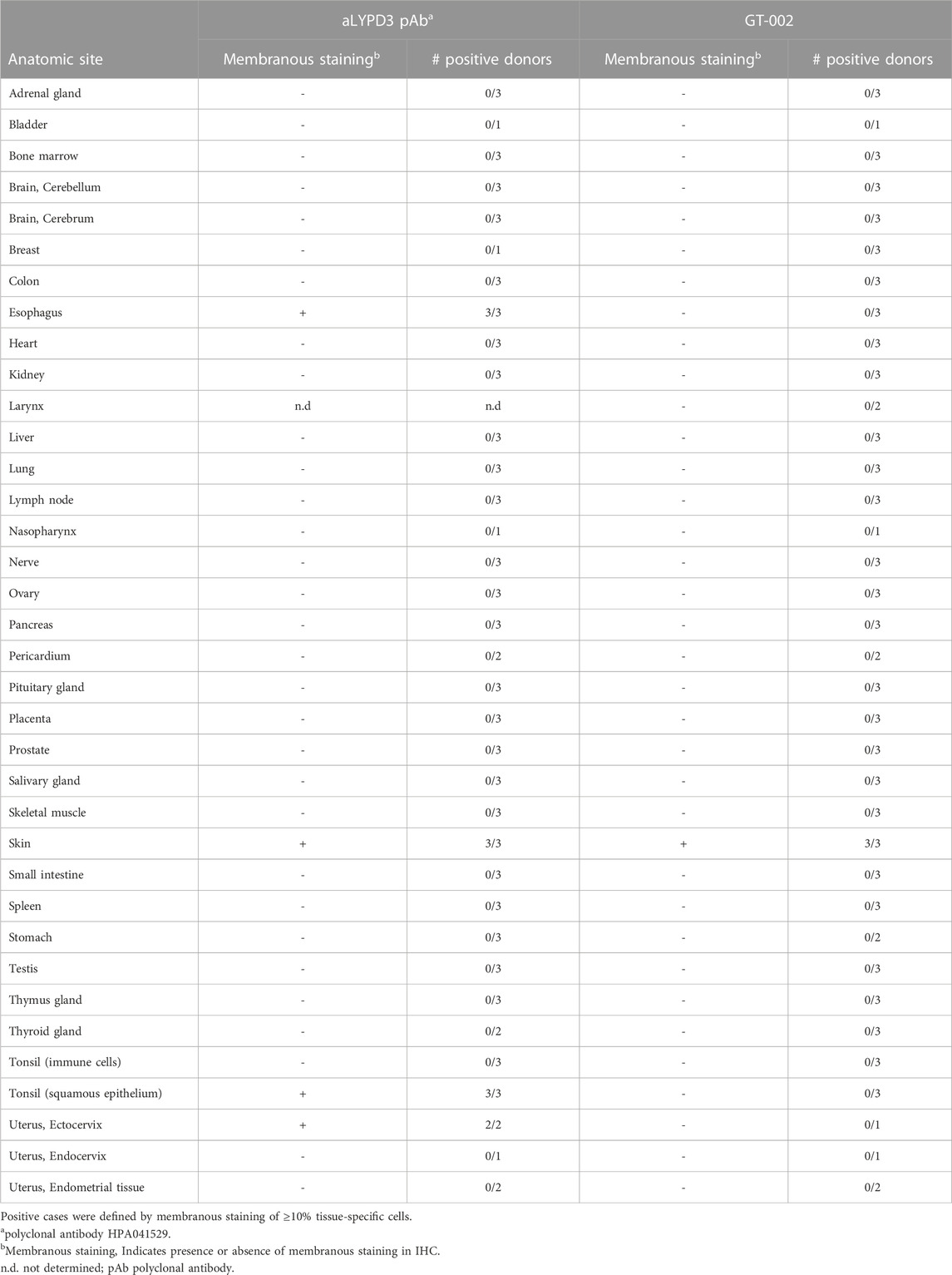
TABLE 3. Summary of immunohistochemistry with GT-002 on normal tissue sections.
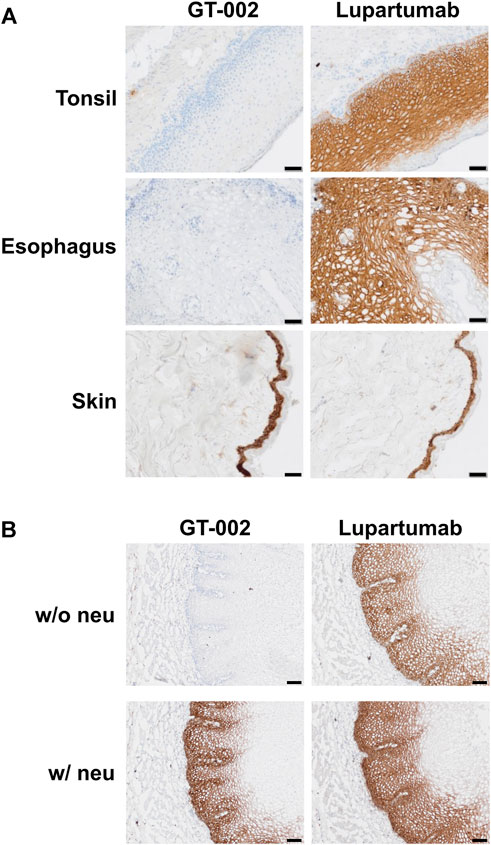
FIGURE 5. Immunohistochemical staining of cryo-preserved healthy human tissue sections by GT-002. (A) Tonsil, esophagus and skin sections were stained with GT-002 and Lupartumab (both 10 μg/mL). (B) Esophagus tissue sections with (w/neu) or without (w/o neu) prior neuraminidase treatment were stained with GT-002 and Lupartumab (both 10 μg/mL). Scale bars indicate 50 µm.
To check whether binding of GT-002 to normal tissues is absent due to sialylation of its epitope, normal esophagus tissue was treated with neuraminidase before staining with GT-002 or Lupartumab. Staining of Lupartumab was unaffected by neuraminidase treatment, whereas pre-treatment resulted in an intense staining of normal esophageal epithelium by GT-002 similar to Lupartumab (Figure 5B), confirming expression of sialylated-TF glycoforms of LYPD3 in normal epithelia.
3.5 GT-002 is effectively internalized indicating potential as ADC
Given its high tumor specificity and the known internalization property of its protein target (Willuda et al., 2017), we investigated whether GT-002 has potential as ADC. For this purpose, we performed an internalization assay with LYPD3-F9 cells, in which GT-002 was labeled with an anti-human Fab fragment conjugated pH-sensitive fluorophore. After internalization and entering the acidic lysosomes, red fluorescence is emitted and can therefore be used to quantify internalization. As shown in Figure 6A, GT-002 and Lupartumab were very quickly internalized into LYPD3-F9 cells within only 1 h incubation time and have entered a maximum of 83% and 91% of cells after 23 h, respectively, whereas the isotype control measured only 11.5% at the final timepoint. Both antibodies and the isotype control were only minimally internalized (<10%) into the untransfected control cell line F9 (Figure 6B). These results indicate effective internalization of GT-002 after binding to LYPD3, which is the basis for development of a toxin conjugate.
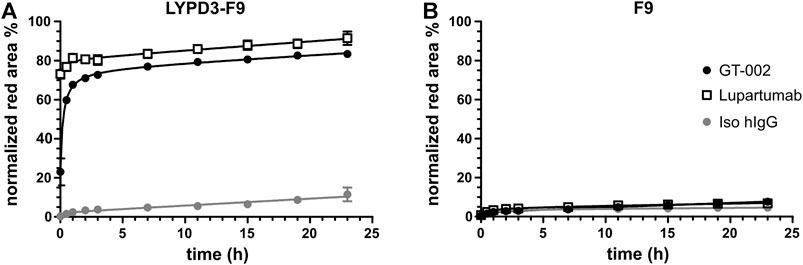
FIGURE 6. Internalization of GT-002 into tumor cell line. GT-002 (black circles), Lupartumab (white squares) or an isotype control (grey circle) where labeled with anti-human Fabfluor-pH antibody labeling dye and then added to LYPD3-F9 cells (A) or F9 cells (B). Internalization, measured as red fluorescence induced by low acidic pH after antibody uptake, was monitored for 24 h. Percentage of lysosomal routing was expressed as the Red Area (Red positive cells)/Phase Area (Total cell confluency). Error bars indicate SD of duplicates.
4 Discussion
One of the most striking characteristics of tumor cells is their altered glycosylation, resulting in de novo exposure of truncated O-glycans like TF, Tn, and sTn, which are highly tumor-specific and constitute excellent candidates for targeted therapy. Especially antibodies against mixed epitopes, comprising these truncated glycans in context of a specific peptide stretch, combine high-affinity antigen recognition with strongly reduced on-target/off-tumor binding compared to conventional protein-targeting antibodies.
LYPD3 is a highly glycosylated cell surface protein expressed in different types of cancers and has been clinically evaluated as a therapeutic target in a phase I study of the protein-directed ADC Lupartumab-amadotin (NCT02134197). We identified LYPD3 as a carrier of truncated O-glycans and developed an antibody, GT-002, which recognizes a tumor-specific glycosylated epitope in the STP-rich region of LYPD3.
Our results revealed that GT-002 strongly recognizes TF-carrying LYPD3, but neither closely related LYPD3 glycoforms comprising Tn and sialylated TF/Tn, nor other O-glycosylated proteins. Consequently, tumor cell binding of GT-002 analyzed by flow cytometry was strictly dependent on LYPD3 expression and only detected on LYPD3-positive tumor cell lines after exposure of TF by neuraminidase treatment. Tumor cell lines per se expressed only moderate levels of TF under normal cell culture conditions due to masking by sialic acid, however, TF and the epitope of GT-002 were released upon neuraminidase treatment. Although increased sialylation has been recognized as a hallmark of cancer (Vajaria and Patel, 2016), it does not generally apply to all glycosylation sites or glycoproteins. Rather, it describes the overall high expression of sialylated glycans like sTn, sialylated Lewis structures, gangliosides as well as sialylated N-glycans in tumors alongside with non-sialylated, aberrant O-glycans (Munkley, 2022). In addition, we generally observed high levels of sialylation by tumor cell lines in culture, which has been described by others (Steentoft et al., 2019; Nishida et al., 2022) and does not necessarily reflect the degree of sialylation in vivo.
These discrepancies in sialylation between cell lines in vitro and tumor cells in vivo were notable in our experiments when comparing flow cytometry results with immunohistochemical staining of CDX tumor sections from mice. Although in flow cytometry, GT-002 cell binding to CaOV-3 cells was only detected after neuraminidase treatment, CaOV-3 CDX sections elicited 20% GT-002-positive cells without prior enzymatic release of sialic acids. Most importantly, GT-002 showed considerable binding to various tissue sections from PDX models and cancer patients, further underlining the broad expression of its non-sialylated epitope in cancer.
GT-002 was able to stain SCC tumor tissues of several indications, mirroring the squamous epithelial expression of LYPD3 in normal tissues combined with the de novo exposure of TF in tumors. Variable staining results between different tumor indications are evident, ranging from 24.4% positive cases in SCC of the skin up to 71.4% positive cases in SCC of the anal area and rectum (Table 2). Although staining results of the anti-LYPD3 pAb control also vary between tumor indications, divergent staining results are not uncommon especially for antibodies recognizing glycopeptide- or glyco-epitopes, given that expression of these epitopes is dependent on the very complex and heterogenous pathway of O-glycosylation (Loureiro et al., 2018; Steentoft et al., 2019; Rømer et al., 2021). Consequently, a considerable degree of protein glycoform heterogeneity is observed between different tumors, even individual tumor cells, which is reflected by the divergent numbers of GT-002-positive tumor cases. Importantly, these mechanisms strongly suggest a fluctuation of truncated O-glycan expression of tumor cells, rather than that some tumor cells do not express these tumor glycans at all (Goletz et al., 2003). Thus, it can be speculated that GT-002 can reach the majority of LYPD3-positive tumor cells over time. Furthermore, apart from LYPD3 glycoform heterogeneity in the tumor, these staining differences might be caused by divergent affinities. Technical differences in the staining procedure due to the polyclonal nature of the control antibody might also have an influence on the observed staining patterns. Nonetheless, this slight reduction in tumor binding is a good compromise for the high tumor selectivity elicited by GT-002 and can be compensated by the optimal choice of conjugate, therapeutic modality and the respective dosage. In order to identify patients who will benefit most from GT-002-derived therapeutical constructs, an IHC-based screening to identify high expression of the GT-002 epitope might be indicated before therapy.
Interestingly, we could not confirm strong membranous expression of LYPD3 in NSCLC SCC, which was chosen as the therapeutical indication for Lupartumab-amadotin (NCT02134197, Willuda et al., 2017). Staining of several NSCLC SCC sections with a polyclonal control antibody revealed only low membranous LYPD3 expression levels in our hands (Supplementary Data, Supplementary Table S1), hence NSCLC was not further analyzed with GT-002. Instead, highest binding of GT-002 was observed in SCC of the head and neck (HNSCC), which about 800,000 people worldwide were diagnosed with in 2020 (Sung et al., 2021). Apart from the one targeted therapy option with anti-EGFR antibody Cetuximab, the current first line treatment for HNSCC includes checkpoint inhibitors like Pembrolizumab (Cramer et al., 2019; Harrington et al., 2022). Especially for recurrent or metastatic disease however, the overall survival remains poor and only a subset of patients responds to checkpoint inhibition therapy (Burtness et al., 2019). GT-002 offers significant promise as a valuable treatment option for these patients due to its broad staining of several HNSCC sub-indications.
Tumor selectivity of GT-002 was confirmed by an IHC study using a normal human tissue array. The array consisted of 33 organs and showed staining of GT-002 only in the epidermis, while all other organs were completely negative. A comparison to Lupartumab on cryo-preserved healthy tonsil, skin and esophagus sections revealed strong staining of all three organs by Lupartumab. In contrast, GT-002 again only stained the epidermis but no other tissue, although binding was comparable between the two antibodies as shown by ELISA using O-glycosylated LYPD3 produced in F9 cells. The differences in normal tissue recognition thus highlight the advantage of targeting a cancer-specific glycosylated epitope on LYPD3. Staining of GT-002 on healthy esophagus tissue was re-established after treatment with neuraminidase, confirming that its epitope is masked by sialic acid in normal tissue. Similar results have been observed with an anti-TF-MUC21 antibody, which bound to healthy murine tissues only after treatment with neuraminidase, and are further supported by previous reports demonstrating the wide occurrence of sialylated TF in normal epithelia (Clausen et al., 1988; Cao et al., 1995; Nishida et al., 2022).
Considering these results, GT-002 could be an excellent candidate for CAR therapy against solid tumors. CARs targeting lineage-specific markers in hematological malignancies have demonstrated major successes in the clinic, but severe toxicities were observed with CARs targeting solid tumor antigens with considerable normal tissue expression (Lamers et al., 2006; Schoeffski et al., 2021). Protein-carbohydrate epitopes like TF-LYPD3 bound by GT-002 are very restricted in healthy tissues and thus ideal candidates for CAR development. To date, only few CARs directed against glyco-epitopes have been developed, but demonstrated promising cytotoxicity against solid tumor cell lines in vitro and in vivo combined with high tumor specificities (Posey et al., 2016; He et al., 2019). Moreover, a phase I study of Tn-MUC1 CAR-Ts for the treatment of solid tumors revealed no evidence of safety concerns or on-target/off-tumor toxicities (NCT04025216, Gutierrez et al., 2021).
Apart from possible cellular therapies, the effective internalization of GT-002 in vitro supports its potential as a therapeutic antibody-drug conjugate. Results of the phase I study of Lupartumab-amadotin have not been published, leaving the underlying reasons for the early termination in 2018 up to speculation. In any case, the almost abolished binding of GT-002 to normal tissues strongly suggests an improved toxicity profile and larger therapeutic window, enabling conjugation of a more toxic payload or higher tolerable dosage of a GT-002 ADC or radioimmunoconjugate.
The high tumor selectivity of GT-002 could also be exploited in bispecific antibodies against solid tumors to ensure an efficient accumulation of the construct at the tumor. Possible combinations are numerous and include other tumor-associated antigens as well as checkpoint inhibitors and immune cell engagers (Liguori et al., 2022).
Taken together, GT-002 serves as case study for successful development of mAbs against protein-carbohydrate epitopes with excellent tumor selectivity. The concept is applicable to other glycoproteins and hence widens the portfolio of treatment options to otherwise undruggable, normal tissue-expressed protein antigens.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
TN: Conceptualization, Data curation, Project administration, Supervision, Visualization, Writing–original draft. EH: Data curation, Formal Analysis, Visualization, Writing–original draft. JG: Conceptualization, Project administration, Writing–review and editing. LW: Data curation, Investigation, Writing–review and editing. MW: Data curation, Investigation, Writing–review and editing. NK: Data curation, Investigation, Writing–review and editing. SG: Methodology, Project administration, Supervision, Writing–review and editing. SM: Methodology, Project administration, Supervision, Writing–review and editing. AJ: Conceptualization, Methodology, Project administration, Supervision, Writing–review and editing. AD: Conceptualization, Writing–review and editing. PK: Conceptualization, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The studies were funded by Glycotope GmbH.
Acknowledgments
We would like to thank Monique Winzek, Klara Haas, Sigrid Wiese, Luisa Willmann, Antoinette Kranz, Carolin Zehmke-Lange and Monique Rönick for their technical support and Lisa Kalfhues for the assistance in editing the manuscript.
Conflict of interest
Authors TN, EH, JG, LW, MW, NK, SG, SM, AJ, AD, and PK were employed by Glycotope GmbH.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2023.1298916/full#supplementary-material
Abbreviations
ADC, antibody-drug conjugate; CAR, chimeric antigen receptor; CDX, cell line-derived xenograft; DAP, 3,3-diaminobenzidine; CHO, Chinese Hamster Ovary; ECD, extracellular domain; FFPE, formalin-fixed, paraffin-embedded; GALK2, galactokinase 2; GPI, glycophosphatidylinositol; HNSCC, squamous cell carcinoma of the head and neck; IHC, immunohistochemistry; KO, knock-out; LYPD3, LY6/PLAUR Domain Containing 3; mAb, monoclonal antibody; MS, mass spectrometry; MMAE, Monomethyl auristatin E; NSCLC, non-small-cell lung cancer; PDX, patient-derived xenograft; SCC, squamous cell carcinoma; 3’sTF, Neu5Acα2-3Galβ1-3GalNAcα1-O-Ser/Thr; 6’sTF, Neu5Acα2-6Galβ1-3GalNAcα1-O-Ser/Thr; 3,6’sTF, Neu5Acα2-3(Neu5Acα2-6)Galβ1-3GalNAcα1-O-Ser/Thr; sTn, NeuAcα2-6GalNAcα1-O-Ser/Thr; STP, serine-threonine-proline; TF, Galβ1-3GalNAcα1-OSer/Thr; TMA, tissue microarray; Tn, GalNAcα1-O-Ser/Thr; GALE, UDP-galactose-4- epimerase.
References
Balaguer, J., Hidalgo, L. G., Hladun, R., Vega, C. M., and Alonso, V. P. (2022). Recent evidence-based clinical guide for the use of dinutuximab beta in pediatric patients with neuroblastoma. Target. Oncol. 18 (1), 77–93. doi:10.1007/s11523-022-00930-w
Baldus, S. E., Hanisch, F. G., Monaca, E., Karsten, U. R., Zirbes, T. K., Thiele, J., et al. (1999). Immunoreactivity of Thomsen-Friedenreich (TF) antigen in human neoplasms: the importance of carrier-specific glycotope expression on MUC1. Histology Histopathol. 14, 1153–1158. doi:10.14670/HH-14.1153
Burtness, B., Harrington, K. J., Greil, R., Soulières, D., Tahara, M., de Castro, G., et al. (2019). Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet 394 (10212), 1915–1928. doi:10.1016/S0140-6736(19)32591-7
Cao, Y., Karsten, U. R., Liebrich, W., Haensch, W., Springer, G. F., and Schlag, P. M. (1995). Expression of Thomsen-Friedenreich-related antigens in primary and metastatic colorectal carcinomas. a reevaluation. Cancer 76 (10), 1700–1708. doi:10.1002/1097-0142(19951115)76:10<1700::aid-cncr2820761005>3.0.co;2-z
Cao, Y., Merling, A., Karsten, U., Goletz, S., Punzel, M., Kraft, R., et al. (2008). Expression of CD175 (tn), CD175s (sialosyl-tn) and CD176 (Thomsen-Friedenreich antigen) on malignant human hematopoietic cells. Int. J. Cancer 123 (1), 89–99. doi:10.1002/ijc.23493
Cazet, A., Julien, S., Bobowski, M., Krzewinski-Recchi, M.-A., Harduin-Lepers, A., Groux-Degroote, S., et al. (2010). Consequences of the expression of sialylated antigens in breast cancer. Carbohydr. Res. 345 (10), 1377–1383. doi:10.1016/j.carres.2010.01.024
Claas, C., Herrmann, K., Matzku, S., Moeller, P., and Zpeller, M. (1996). Developmentally regulated expression of metastasis-associated antigens in the rat. Cell Growth Differ. 7, 663–678.
Clausen, H., Stroud, M., Parker, J., Springer, G., and Sen-Itiroh, H. (1988). Monoclonal antibodies directed to the blood group a associated structure, galactosyl-a: specificity and relation to the Thomsen-Friedenreich antigen. Mol. Immunol. 25 (2), 199–204. doi:10.1016/0161-5890(88)90068-5
Cramer, J. D., Burtness, B., Le, Q. T., and Ferris, R. L. (2019). The changing therapeutic landscape of head and neck cancer. Nat. Rev. Clin. Oncol. 16 (11), 669–683. doi:10.1038/s41571-019-0227-z
Danielczyk, A., Stahn, R., Faulstich, D., Löffler, A., Märten, A., Karsten, U., et al. (2006). PankoMab: a potent new generation anti-tumour MUC1 antibody. Immunotherapy 55 (11), 1337–1347. doi:10.1007/s00262-006-0135-9
Dhillon, S. (2015). Dinutuximab: first global approval. Drugs 75 (8), 923–927. doi:10.1007/s40265-015-0399-5
Dong, X., Jiang, Y., Liu, J., Liu, Z., Gao, T., An, G., et al. (2018). T-synthase deficiency enhances oncogenic features in human colorectal cancer cells via activation of epithelial-mesenchymal transition. BioMed Res. Int. 2018, 9532389–9532397. doi:10.1155/2018/9532389
Fu, C., Zhao, H., Wang, Y., Cai, H., Xiao, Y., Zeng, Y., et al. (2016). Tumor-associated antigens: tn antigen, sTn antigen, and t antigen. HLA 88 (6), 275–286. doi:10.1111/tan.12900
Goletz, S., Cao, Y., Danielczyk, A., Ravn, P., Schoeber, U., and Karsten, U. (2003). Thomsen-Friedenreich antigen: the "hidden" tumor antigen. Adv. Exp. Med. Biol. 535, 147–162. doi:10.1007/978-1-4615-0065-0_10
Gu, Y., Lin, S., Li, J.-L., Nakagawa, H., Chen, Z., Jin, B., et al. (2011). Altered LKB1/CREB-regulated transcription co-activator (CRTC) signaling axis promotes esophageal cancer cell migration and invasion. Oncogene 31 (4), 469–479. doi:10.1038/onc.2011.247
Gutierrez, R., Shah, P. D., Hamid, O., Garfall, A. L., Posey, A., Bishop, M. R., et al. (2021). Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor t-cells in patients with advanced TnMUC1 positive solid tumors. J. Clin. Oncol. 39 (15_Suppl. l), e14513. doi:10.1200/jco.2021.39.15_suppl.e14513
Haji-Ghassemi, O., Blackler, R. J., Young, N. M., and Evans, S. V. (2015). Antibody recognition of carbohydrate epitopes. Glycobiology 25 (9), 920–952. doi:10.1093/glycob/cwv037
Hansen, L. V., Gårdsvoll, H., Nielsen, B. S., Lund, L. R., Danø, K., Jensen, O. N., et al. (2004). Structural analysis and tissue localization of human C4.4A: a protein homologue of the urokinase receptor. Biochem. J. 380 (3), 845–857. doi:10.1042/BJ20031478
Hansen, L. V., Lærum, O. D., Illemann, M., Nielsen, B. S., and Ploug, M. (2007). Altered expression of the urokinase receptor homologue, C4.4A, in invasive areas of human esophageal squamous cell carcinoma. Int. J. Cancer 122 (4), 734–741. doi:10.1002/ijc.23082
Harrington, K. J., Burtness, B., Greil, R., Soulières, D., Tahara, M., de Castro, G., et al. (2022). Pembrolizumab with or without chemotherapy in recurrent or metastatic head and neck squamous cell carcinoma: updated results of the phase III KEYNOTE-048 study. J. Clin. Oncol. 41 (4), 790–802. doi:10.1200/JCO.21.02508
He, Y., Schreiber, K., Wolf, S. P., Wen, F., Steentoft, C., Zerweck, J., et al. (2019). Multiple cancer-specific antigens are targeted by a chimeric antigen receptor on a single cancer cell. JCI Insight 4 (21), e130416. doi:10.1172/jci.insight.130416
Hernaez, D. C., Hughes, M. R., Li, Y., Rocca, I. M., Dean, P., Brassard, J., et al. (2022). Targeting a tumor-specific epitope on podocalyxin increases survival in human tumor preclinical models. Front. Oncol. 12, 856424. doi:10.3389/fonc.2022.856424
Itzkowitz, S. H., Yuan, M., Montgomery, C. K., Kjeldsen, T., Takahashi, H. K., Bigbee, W. L., et al. (1989). Expression of Tn, sialosyl-Tn, and T antigens in human colon cancer. Cancer Res. 49, 197–204.
Jacobsen, B., Larouche, D., Rochette-Drouin, O., Ploug, M., and Germain, L. (2017). Expression of C4.4A in an in vitro human tissue-engineered skin model. BioMed Res. Int. 2017, 2403072–2403079. doi:10.1155/2017/2403072
Karacosta, L. G., Fisk, J. C., Jessee, J., Tati, S., Turner, B., Ghazal, D., et al. (2018). Preclinical analysis of JAA-f11, a specific anti-Thomsen-Friedenreich antibody via immunohistochemistry and in vivo imaging. Transl. Oncol. 11 (2), 450–466. doi:10.1016/j.tranon.2018.01.008
Karsten, U., and Goletz, S. (2013). What makes cancer stem cell markers different? SpringerPlus 2 (1), 301. doi:10.1186/2193-1801-2-301
Kivi, G., Teesalu, K., Parik, J., Kontkar, E., Ustav, M., Noodla, L., et al. (2016). HybriFree: a robust and rapid method for the development of monoclonal antibodies from different host species. BMC biotechnology 16, 2. doi:10.1186/s12896-016-0232-6
Konishi, K., Yamamoto, H., Mimori, K., Takemasa, I., Mizushima, T., Ikeda, M., et al. (2010). Expression of C4.4A at the invasive front is a novel prognostic marker for disease recurrence of colorectal cancer. Cancer Sci. 101 (10), 2269–2277. doi:10.1111/j.1349-7006.2010.01674.x
Kriegbaum, M. C., Clausen, O. P. F., Lærum, O. D., and Ploug, M. (2014). Expression of the ly6/uPAR-domain proteins C4.4A and haldisin in non-invasive and invasive skin lesions. J. Histochem. & Cytochem. 63 (2), 142–154. doi:10.1369/0022155414563107
Kudelka, M. R., Ju, T., Heimburg-Molinaro, J., and Cummings, R. D. (2015). Simple sugars to complex disease: mucin-type O-glycans in cancer. Adv. Cancer Res. 126, 53–135. Elsevier. doi:10.1016/bs.acr.2014.11.002
Ladenstein, R., Poetschger, U., Valteau-Couanet, D., Luksch, R., Castel, V., Ash, S., et al. (2020). Investigation of the role of dinutuximab beta-based immunotherapy in the siopen high-risk neuroblastoma 1 trial (hr-nbl1). Cancers 12 (2), 309. doi:10.3390/cancers12020309
Lamers, C. H., Sleijfer, S., Vulto, A. G., Kruit, W. H., Kliffen, M., Debets, R., et al. (2006). Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol. 24 (13), e20–e22. doi:10.1200/JCO.2006.05.9964
Lavrsen, K., Madsen, C. B., Rasch, M. G., Woetmann, A., Ødum, N., Mandel, U., et al. (2012). Aberrantly glycosylated MUC1 is expressed on the surface of breast cancer cells and a target for antibody-dependent cell-mediated cytotoxicity. Glycoconj. J. 30 (3), 227–236. doi:10.1007/s10719-012-9437-7
Li, X., Xu, Y., and Zhang, L. (2019). Serum CA153 as biomarker for cancer and noncancer diseases. Prog. Mol. Biol. Transl. Sci. 162, 265–276. Elsevier. doi:10.1016/bs.pmbts.2019.01.005
Liguori, L., Polcaro, G., Nigro, A., Conti, V., Sellitto, C., Perri, F., et al. (2022). Bispecific antibodies: a novel approach for the treatment of solid tumors. Pharmaceutics 14 (11), 2442. doi:10.3390/pharmaceutics14112442
Lin, W.-M., Karsten, U., Goletz, S., Cheng, R.-C., and Cao, Y. (2010). Expression of CD176 (Thomsen-Friedenreich antigen) on lung, breast and liver cancer-initiating cells. Int. J. Exp. Pathology 92 (2), 97–105. doi:10.1111/j.1365-2613.2010.00747.x
Liu, J.-F., Mao, L., Bu, L.-L., Ma, S.-R., Huang, C.-F., Zhang, W.-F., et al. (2015). C4.4a as a biomarker of head and neck squamous cell carcinoma and correlated with epithelial mesenchymal transition. Am. J. cancer Res. 5, 3505–3515.
Loureiro, L. R., Sousa, D. P., Ferreira, D., Chai, W., Lima, L., Pereira, C., et al. (2018). Novel monoclonal antibody L2A5 specifically targeting sialyl-Tn and short glycans terminated by alpha-2-6 sialic acids. Sci. Rep. 8 (1), 12196. doi:10.1038/s41598-018-30421-w
Mereiter, S., Balmaña, M., Campos, D., Gomes, J., and Reis, C. A. (2019). Glycosylation in the era of cancer-targeted therapy: where are we heading? Cancer Cell 36 (1), 6–16. doi:10.1016/j.ccell.2019.06.006
Morgan, R. A., Yang, J. C., Kitano, M., Dudley, M. E., Laurencot, C. M., and Rosenberg, S. A. (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18 (4), 843–851. doi:10.1038/mt.2010.24
Munkley, J. (2022). Aberrant sialylation in cancer: therapeutic opportunities. Cancers 14 (17), 4248. doi:10.3390/cancers14174248
Munkley, J., and Elliott, D. J. (2016). Hallmarks of glycosylation in cancer. Oncotarget 7 (23), 35478–35489. doi:10.18632/oncotarget.8155
Nishida, J., Shichino, S., Tsukui, T., Hoshino, M., Okada, T., Okada, K., et al. (2022). Unique glycoform-dependent monoclonal antibodies for mouse mucin 21. Int. J. Mol. Sci. 23 (12), 6718. doi:10.3390/ijms23126718
Pinho, S. S., and Reis, C. A. (2015). Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15 (9), 540–555. doi:10.1038/nrc3982
Posey, A. D., Schwab, R. D., Boesteanu, A. C., Steentoft, C., Mandel, U., Engels, B., et al. (2016). Engineered CAR T cells targeting the cancer-associated tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 44 (6), 1444–1454. doi:10.1016/j.immuni.2016.05.014
Radhakrishnan, P., Dabelsteen, S., Madsen, F. B., Francavilla, C., Kopp, K. L., Steentoft, C., et al. (2014). Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc. Natl. Acad. Sci. 111 (39), E4066–E4075. doi:10.1073/pnas.1406619111
Reily, C., Stewart, T. J., Renfrow, M. B., and Novak, J. (2019). Glycosylation in health and disease. Nat. Rev. Nephrol. 15 (6), 346–366. doi:10.1038/s41581-019-0129-4
Roesel, M., Claas, C., Seiter, S., Herlevsen, M., and Zöller, M. (1998). Cloning and functional characterization of a new phosphatidyl-inositol anchored molecule of a metastasizing rat pancreatic tumor. Oncogene 17 (15), 1989–2002. doi:10.1038/sj.onc.1202079
Rømer, T. B., Aasted, M. K. M., Dabelsteen, S., Groen, A., Schnabel, J., Tan, E., et al. (2021). Mapping of truncated O-glycans in cancers of epithelial and non-epithelial origin. Br. J. Cancer 125 (9), 1239–1250. Epub 2021 Sep 15. PMID: 34526666; PMCID: PMC8548315. doi:10.1038/s41416-021-01530-7
Saldova, R., Struwe, W., Wynne, K., Elia, G., Duffy, M., and Rudd, P. (2013). Exploring the glycosylation of serum CA125. Int. J. Mol. Sci. 14 (8), 15636–15654. doi:10.3390/ijms140815636
Schindlbeck, C., Jeschke, U., Schulze, S., Karsten, U., Janni, W., Rack, B., et al. (2006). Prognostic impact of Thomsen-Friedenreich tumor antigen and disseminated tumor cells in the bone marrow of breast cancer patients. Breast Cancer Res. Treat. 101 (1), 17–25. doi:10.1007/s10549-006-9271-3
Schoeffski, P., Concin, N., Suarez, C., Subbiah, V., Ando, Y., Ruan, S., et al. (2021). A phase 1 study of a CDH6-targeting antibody-drug conjugate in patients with advanced solid tumors with evaluation of inflammatory and neurological adverse events. Oncol. Res. Treat. 44 (10), 547–556. doi:10.1159/000518549
Springer, G. F. (1984). T and Tn, general carcinoma autoantigens. Science 224 (4654), 1198–1206. doi:10.1126/science.6729450
Steentoft, C., Fuhrmann, M., Battisti, F., Coillie, J. V., Madsen, T. D., Campos, D., et al. (2019). A strategy for generating cancer-specific monoclonal antibodies to aberrant O-glycoproteins: identification of a novel dysadherin-Tn antibody. Glycobiology 29 (4), 307–319. doi:10.1093/glycob/cwz004
Steentoft, C., Migliorini, D., King, T. R., Mandel, U., June, C. H., and Posey, A. D. (2018). Glycan-directed CAR-t cells. Glycobiology 28 (9), 656–669. doi:10.1093/glycob/cwy008
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Takanami, I. (1999). Expression of Thomsen-Friedenreich antigen as a marker of poor prognosis in pulmonary adenocarcinoma. Oncol. Rep. 6 (2), 341–344. doi:10.3892/or.6.2.341
Termini, J. M., Silver, Z. A., Connor, B., Antonopoulos, A., Haslam, S. M., Dell, A., et al. (2017). HEK293t cell lines defective for o-linked glycosylation. PLOS ONE 12 (6), e0179949. doi:10.1371/journal.pone.0179949
Therkildsen, M., Mandel, U., Christensen, M., and Dabelsteen, E. (1995). The Thomsen-Friedenreich (T) simple mucin-type carbohydrate antigen in salivary gland carcinomas. Eur. J. Cancer Part B Oral Oncol. 31 (6), 361–367. doi:10.1016/0964-1955(95)00044-5
Vajaria, B. N., and Patel, P. S. (2016). Glycosylation: a hallmark of cancer? Glycoconj. J. 34 (2), 147–156. doi:10.1007/s10719-016-9755-2
Varki, A., Cummings, R. D., Aebi, M., Packer, N. H., Seeberger, P. H., Esko, J. D., et al. (2015). Symbol nomenclature for graphical representations of glycans. Glycobiology 25 (12), 1323–1324. doi:10.1093/glycob/cwv091
Varki, A., Cummings, R. D., Esko, J. D., Stanley, P., Hart, G. W., Aebi, M., et al. (2022). Essentials of glycobiology. Harbor (NY): Cold Spring Harbor Laboratory Press, 79–92.
Wiest, I., Alexiou, C., Kuhn, C., Schulze, S., Kunze, S., Mayr, D., et al. (2012). Expression of different carbohydrate tumour markers and galectins 1 and 3 in normal squamous and malignant epithelia of the upper aaerodigestive tract. Anticancer Res. 32, 2023–2029.
Willuda, J., Linden, L., Lerchen, H.-G., Kopitz, C., Stelte-Ludwig, B., Pena, C., et al. (2017). Preclinical antitumor efficacy of BAY 1129980a novel auristatin-based anti-C4.4A (LYPD3) antibody-drug conjugate for the treatment of non-small cell lung cancer. Mol. Cancer Ther. 16 (5), 893–904. doi:10.1158/1535-7163.MCT-16-0474
Keywords: cancer therapy, monoclonal antibody, protein-carbohydrate epitope, squamous cell carcinoma, truncated O-glycans
Citation: Neumann T, Hartung E, Gellert J, Weiß L, Weiske M, Kast N, Gurka S, Marinoff S, Jäkel A, Danielczyk A and Kehler P (2023) Targeting a cancer-specific LYPD3 glycoform for tumor therapy. Front. Drug Discov. 3:1298916. doi: 10.3389/fddsv.2023.1298916
Received: 22 September 2023; Accepted: 08 November 2023;
Published: 22 November 2023.
Edited by:
Sigrid A. Langhans, Nemours Children’s Health, United StatesReviewed by:
Andrea Medeiros, Universidad de la República, UruguayDoretta Cuffaro, University of Pisa, Italy
Copyright © 2023 Neumann, Hartung, Gellert, Weiß, Weiske, Kast, Gurka, Marinoff, Jäkel, Danielczyk and Kehler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johanna Gellert, johanna.gellert@glycotope.com
†These authors have contributed equally to this work