Possible molecular mechanisms underlying the development of atherosclerosis in cancer survivors
 Priyanka Banerjee1,†
Priyanka Banerjee1,†  Julia Enterría Rosales2,3
Julia Enterría Rosales2,3  Khanh Chau1
Khanh Chau1  Minh T. H. Nguyen1,4
Minh T. H. Nguyen1,4  Sivareddy Kotla2
Sivareddy Kotla2  Steven H. Lin2
Steven H. Lin2  Anita Deswal2
Anita Deswal2  Robert Dantzer5
Robert Dantzer5  Elizabeth A. Olmsted-Davis1
Elizabeth A. Olmsted-Davis1  Hung Nguyen6
Hung Nguyen6  Guangyu Wang1
Guangyu Wang1  John P. Cooke1
John P. Cooke1  Jun-ichi Abe2*
Jun-ichi Abe2*  Nhat-Tu Le1*†
Nhat-Tu Le1*†
- 1Center for Cardiovascular Regeneration, Department of Cardiovascular Sciences, Houston Methodist Research Institute, Houston, TX, United States
- 2Department of Cardiology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 3School of Medicine, Instituto Tecnológico de Monterrey, Guadalajara, Mexico
- 4Department of Life Science, University of Science and Technology of Hanoi, Vietnam Academy of Science and Technology, Hanoi, Vietnam
- 5Department of Symptom Research, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 6Cancer Division, Burnett School of Biomedical Science, College of Medicine, University of Central Florida, Orlando, FL, United States
Cancer survivors undergone treatment face an increased risk of developing atherosclerotic cardiovascular disease (CVD), yet the underlying mechanisms remain elusive. Recent studies have revealed that chemotherapy can drive senescent cancer cells to acquire a proliferative phenotype known as senescence-associated stemness (SAS). These SAS cells exhibit enhanced growth and resistance to cancer treatment, thereby contributing to disease progression. Endothelial cell (EC) senescence has been implicated in atherosclerosis and cancer, including among cancer survivors. Treatment modalities for cancer can induce EC senescence, leading to the development of SAS phenotype and subsequent atherosclerosis in cancer survivors. Consequently, targeting senescent ECs displaying the SAS phenotype hold promise as a therapeutic approach for managing atherosclerotic CVD in this population. This review aims to provide a mechanistic understanding of SAS induction in ECs and its contribution to atherosclerosis among cancer survivors. We delve into the mechanisms underlying EC senescence in response to disturbed flow and ionizing radiation, which play pivotal role in atherosclerosis and cancer. Key pathways, including p90RSK/TERF2IP, TGFβR1/SMAD, and BH4 signaling are explored as potential targets for cancer treatment. By comprehending the similarities and distinctions between different types of senescence and the associated pathways, we can pave the way for targeted interventions aim at enhancing the cardiovascular health of this vulnerable population. The insights gained from this review may facilitate the development of novel therapeutic strategies for managing atherosclerotic CVD in cancer survivors.
1. Introduction
As individual age, the prevalence of various disorders and diseases, including atherosclerotic CVD, increases in the population (1). Atherosclerotic CVD remains the leading cause of morbidity and mortality worldwide, particularly in those aged 65 and older (2, 3) (see Table 1). Various scoring systems, such as the Pooled cohort equations (PCE), Framingham risk score, and Reynolds Risk Score, have been utilized to assess the risk of atherosclerotic CVD. These scoring systems consistently indicate that age is a significant risk factor for atherosclerotic CVD, with both females and males having a greater risk of CVD as they age (see Table 2).
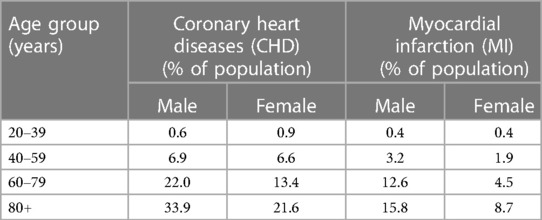
Table 1. The heart disease statistics in different age groups in United States (national health and nutrition examination survey) (4).
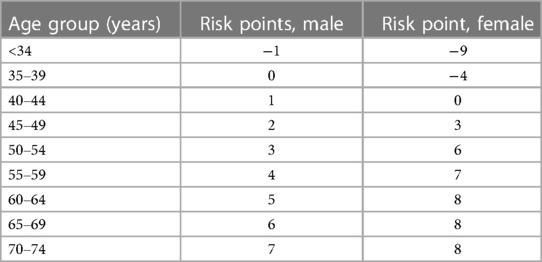
Table 2. Framingham risk score for risk of CVD with age and sex (5).
Cancer survivors who have undergone cancer treatment, including radiation therapy, face an elevated risk of developing atherosclerotic CVD, with a 1.3–3.6 fold increase in developing coronary artery disease (CAD) and a 1.7–18.5-fold increase in developing atherosclerotic risk factors, which may lead to potentially fatal consequences (6, 7). A cross-sectional study conducted by the National Health and Nutrition Examination Survey (NHANES) assessed the 10-year risk of atherosclerotic CVD using the Pooled Cohort Equations in both cancer survivors and non-cancer patients. The study found that the risk of atherosclerotic CVD was higher in cancer survivors compared to non-cancer patients (8) (see Table 3). This increased prevalence and mortality has been linked to cancer therapies, including IR and chemotherapeutic agents (2).
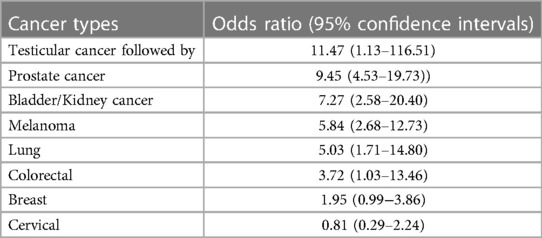
Table 3. The 10-year risk of atherosclerotic CVD in cancer survivors (8).
Cellular senescence, a hallmark of aging, occurs when cells cease to divide in response to various stress stimuli, both internal and external. These stimuli include d-flow, oncogene activation, DNA damage, mitochondrial dysfunction, reactive oxygen species (ROS), and cancer treatments including radiation therapy and chemotherapy. Recent study by Milanovic and colleagues has shown that chemotherapy may induce senescence in cancer cells, which can have negative effects on tissue functions due to the secretion of various factors that regulate vital biological processes, such as cellular metabolism, cell growth, and inflammatory signaling (9, 10). This secretion of factors known as senescence-associated secretory phenotype (SASP). These senescent cancer cells can also escape cell cycle arrest and apoptosis, leading to an increase in their clonogenic growth potential. This phenomenon is known as SAS (11–16).
Milanovic and colleagues' seminal observation revealed clear differences between replicative senescence (RS) and stress-induced premature senescence (SIPS) (11–16). SIPS is not necessarily associated with telomere shortening, unlike RS. Additionally, SIPS may promote cancer treatment resistance, tumorigenesis, and numerous age-related disorders, including atherosclerotic CVD in cancer survivors (13–20).
Cancer treatment can also lead to changes in the structure and function of the vasculature, including alterations in the morphology and function of ECs that can induce EC senescence (7). EC senescence can result in changes in the hemodynamics, structure, and function of the vasculature (21–23) and these alterations are driven by a complex interplay of molecular and cellular mechanisms. These mechanisms include DNA damage response (DDR), Shelterin disruption, aberrant post-translational modifications, signaling pathways, elevated generation of ROS, and sustained activation of pro-inflammatory and pro-fibrotic transcription factors (21, 24, 25), all of which can cause a positive feedback loop contributing to the sustained inflammation, which can explain the elevated risk of atherosclerotic CVD in cancer survivors.
In this review, we aim to provide a comprehensive understanding of the possible molecular mechanisms regulating the development of atherosclerotic CVD in cancer survivors to establish rational therapeutic directions based on a mechanistic understanding of their interactions and interplay between cancer therapy and hemodynamics. We will summarize the similarities and differences between RS and SIPS, the latter of which plays a critical role in the induction of the SAS phenotype. We will describe the importance of the regulatory mechanisms for telomere DNA protection, as telomere DNA damage and dysfunction are critical for the induction of SIPS. We will also examine the key pathways involved in cellular senescence. Finally, we will discuss potential therapeutic interventions that may benefit patients with atherosclerotic CVD, with a focus on the BH4 pathway.
Our goal is to provide current knowledge to help future studies understand how cellular senescence is triggered in ECs in response to cancer treatments and how a distinct type of SASP (SAS) in ECs may contribute to the development of atherosclerotic CVD in cancer survivors.
Additionally, we aim to highlight the potential of senolytic drugs in preventing, delaying, and alleviating various age-related disorders, including atherosclerotic CVD, in humans (26).
2. Cellular senescence has been implicated in the development of atherosclerosis and cancer
2.1. Atherosclerosis
Atherosclerosis is a chronic and progressive inflammatory disease that involves various risk factors, including d-flow and elevated level of oxidized low-density lipoprotein (oxLDL) (27). It can present in different clinical forms, such as CAD, ischemic heart disease, ischemic stroke, and peripheral arterial disease (PAD) (28). This disease typically begins in early life and has a long subclinical phase (2, 3). The aging process is a significant risk factor for atherosclerosis due to the deterioration of the balance between vasodilator and vasoconstriction factors secreted by ECs, which leads to vascular senescence and dysfunction. Cellular senescence have been shown to play a significant role in the development and progression of atherosclerosis (29). The accumulation of senescent cells during the aging process upregulates the expression of numerous molecules that fuel age-associated disorders, including atherosclerotic CVD (30–32).
Recent studies have also suggested that cellular senescence may play a role in plaque destabilization and rupture, leading to acute cardiovascular events. For instance, senescent vascular smooth muscle cells (VSMCs) within the plaque have been shown to have increased matrix metalloproteinase (MMP) activity, which can contribute to plaque rupture and thrombosis. Additionally, senescent cells can secrete SASP, characterized by the release of numerous molecules including proinflammatory cytokines and chemokines, such as interleukine 1β (IL1β), tumor necrosis factor α (TNFα), interferon γ (IFNγ), and transforming growth factor β (TGFβ). The SASP can lead to chronic inflammation by inducing the persistent pro-inflammatory senescence phenotype (PISP), promoting the recruitment and activation of immune cells within the plaque, impairing cholesterol efflux from macrophages, and leading to the accumulation of lipid-laden foam cells in the plaque (30–32).
2.2. Cancer
Cellular senescence can have a dual role in cancer, both suppressing and promoting its development and progression (33). On one hand, it can suppress cancer by halting the cell cycle of cells with DNA damage or mutations that could lead to cancer. On the other hand, senescent cells can also contribute to cancer development and progression by releasing SASP. The SASP can be beneficial by preventing cell division and promoting immune clearance of damaged cells, thus reducing the risk of tumor formation. However, the SASP can also promote inflammation, tissue remodeling, and angiogenesis, creating a microenvironment that is favorable for cancer cell growth and spread (34, 35).
Over time, the accumulate of senescent cells in tissues can lead to chronic inflammation, tissue dysfunction, and organ failure, all of which can increase the risk of cancer development (36). Several studies have shown that the presence of senescent cells in tissues is associated with a higher risk of cancer and poorer prognosis in cancer patients. Therefore, strategies aimed at eliminating senescent cells or modulating their SASP may have therapeutic potential for the prevention and treatment of cancer.
2.3. Cancer survivors
Milanovic and colleagues have demonstrated that chemotherapy can drive a specific subset of senescent cancer cells to reprogram and acquire a proliferative phenotype known as SAS. This unique state allows these cells to bypass cell cycle arrest and significantly enhances their growth and proliferation (9–16), independent of senescence-induced cell cycle arrest (17, 18) or cell death (13, 14). This adaptive mechanism may underlie the resistance of cancer cells to chemotherapy and radiation therapy, enabling their survival and proliferation even in the presence of treatment (13, 14, 19).
These findings highlight the significant role of cellular senescence in the development and progression of atherosclerotic CVD and cancer, including among cancer survivors. However, it is important to note that senescence is also associated with impaired angiogenesis (37). Studies have demonstrated that inhibiting telomerase in ECs can reduce angiogenesis in tumor (38) and animal model of hind limb ischemia (39). Therefore, targeting senescent cells could be a promising therapeutic strategy for managing these diseases. Additionally, while cancer treatment can induce EC senescence (40), it remains unclear whether the SAS phenotype in ECs expressing SASP contributes to the delayed onset of atherosclerotic CVD following cancer treatment. Further studies are needed to investigate this aspect and shed light on the interplay between EC senescence, SAS status, and the development of atherosclerosis in cancer survivors.
3. Cellular senescence
3.1. Cellular senescence is related to telomere shortening
Cellular senescence is a state of irreversible cell cycle arrest, first described by Hayflick and Moorhead in 1961 when they observed that human diploid fibroblasts underwent this process after serial passaging. Successive cell division can shorten telomeres to a critical length, known as the Hayflick limit (41–43), at which point they are unable to form a t-loop, causing cells to become senescent (44–47). This type of growth arrest was later named RS (48–50).
Telomeres are repetitive hexanucleotide 5′-TTAGGG-3′tandem sequences that cap the chromosome ends, responsible for maintaining genomic stability (41–43). On eukaryotic chromosomes, they span approximately 3–10 kb and end with a 3′-end single-stranded overhang (51). After each round of replication, the ends of chromosomes resemble damaged DNA, triggering DDR. This leads to the inability to fully replicate DNA strands, causing telomeres to shorten by about 50–200 base pairs after each cell division (44–47).
To prevent continuous shortening of telomeres, the telomerase protein, which is a telomere-specific ribonucleoprotein, adds single-stranded telomeric repeats to the chromosomal 3′ ends (52). However, many human somatic cells lack telomerase, including human dermal fibroblast cells. Therefore, during cell division, the telomere length progressively shortens. After a certain time, the telomeres reach a critical length, and a DDR signal triggers senescence in the cells. This can cause cell cycle arrest and cellular senescence by upregulating p53-mediated p21 transcription (53–55).
Cellular senescence is related to shortening of telomeres to the Hayflick limit and detected through upregulated expression of senescence associated β-galactosidase (SaβG) and cyclin-dependent kinase inhibitors p21Cip1 (or p21Waf1) and p16INK4a (56, 57). This process can lead to loss of tissue homeostasis and increased susceptibility to age-related disorders and diseases, including atherosclerotic CVD and cancer. Poorer survival rates have been reported in individuals over 60 years of age with shorter telomeres compared to their younger counterparts (58).
3.2. Cellular senescence can occur independently of telomere shortening
Telomere shortening has been linked to cellular senescence (59), but the relationship between these two is complex (60–62). Telomere shortening that occurs after successive cell divisions can lead to RS, which prevents further replication (44–47). However, cellular senescence can also occur independently of telomere shortening (52), indicating that telomere shortening alone may not be the sole cause of cellular senescence. This offers a significant opportunity for further studies. For instance, ventricular stiffness and impaired cardiac function can occur during aging due to cardiomyocyte hypertrophy and fibrosis, although the relationship between cardiac hypertrophy, fibrosis, and telomere length remains unclear (63). Cells with telomerase expression, such as human keratinocytes, epithelial cells, and rodent cells, can maintain long telomeres in culture but still undergo senescence (52, 64). Telomere attrition, but not the inherently short leukocyte telomere length at birth, has been found to be the main cause of leukocyte telomere shortening and correlates with telomere length in atherosclerotic CVD patients (65).
Additionally, studies have shown that cellular senescence is also a characteristic of chronic stress, which can provoke premature senescence, or so-called SIPS through internal and external stimuli (66–69). SIPS can occur independently of telomere shortening, although telomere shortening may be related to SIPS. For instance, SIPS can be induced in immortalized human foreskin fibroblasts expressing telomerase (hTERT-BJ1) exposed to hydrogen peroxide or Ultraviolet B (45). Another example is the induction of SIPS in renal tubular cells exposed to the urine of patients with calcium oxalate kidney stones, likely due to oxidative stress induced by oxalate and calcium oxalate monohydrate (70).
RS and SIPS are induced at different time frames and regulated by different mechanisms as reviewed elsewhere (44–47, 71). Figure 1 provides a summary of the mechanisms involved.
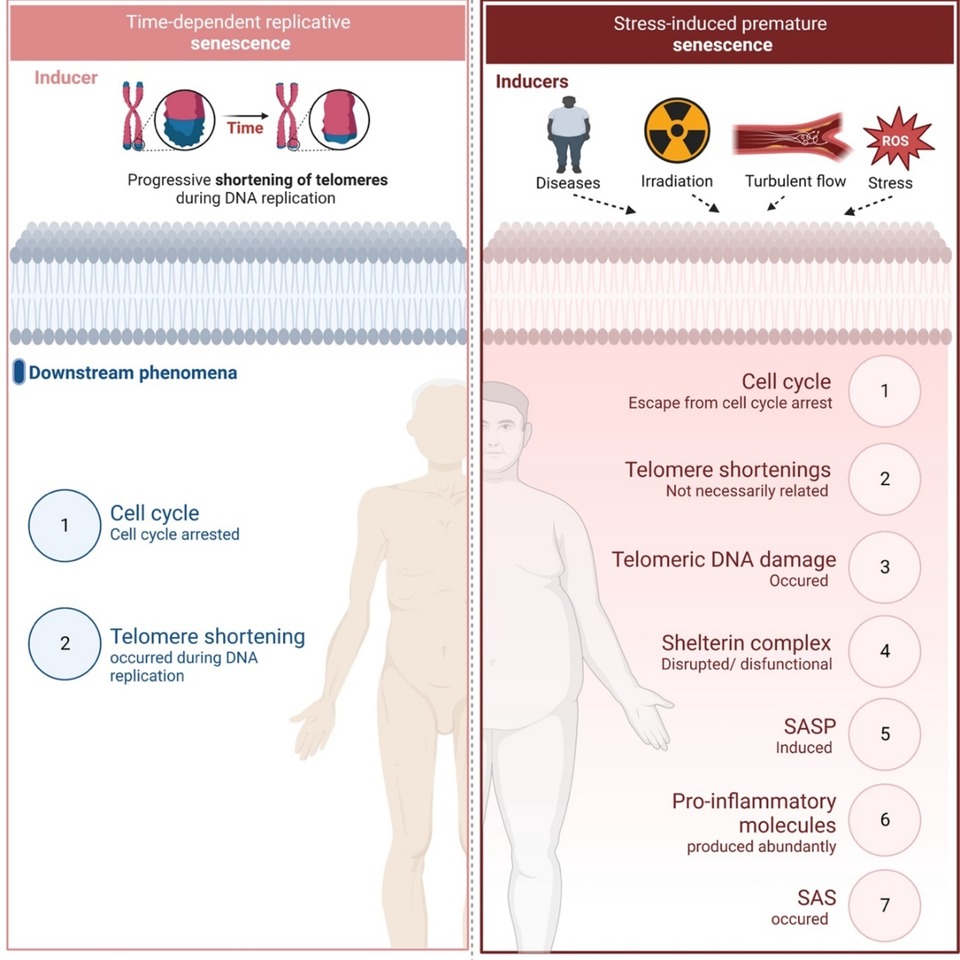
Figure 1. Replicative senescence vs stress-induced premature senescence. Different inducers and down-stream effects of two types of senescence (All the figures were made in Biorender.com).
3.3. Telomeric DNA damage can induce PISP
DNA damage, as detected by the formation of g-H2AX foci (g-foci), has been linked to cellular senescence and aging at the organismal level. Both telomeric and non-telomeric DNA damage have been implicated in cellular senescence. Nakamura and colleagues have shown that g-foci related to senescence can be found at uncapped telomeres or non-telomeric DNA damage sites on chromosomes in both humans and mice (13). Telomeric DNA is vulnerable to oxidation due to the low redox potential of guanine (72). However, repair of telomeric DNA damage is less effective and much slower than repair of genomic DNA damage, which occurs within 24 h (73–75). This leads to long-lasting telomeric DNA damage signaling that can induce the delay and persistent DDR at telomeres for months (76), resulting in the formation of telomere-associated DDR foci (TAF) (63). This process occurs in aging post-mitotic cells, such as cardiomyocytes and neurons, even though these cells have relatively longer telomeres (73). These observations suggest that stress-induced telomeric DNA damage and the persistent DDR and subsequent TAF formation, not telomere shortening, can induce PISP (77–79).
3.4. The SASP has been implicated in both atherosclerosis and cancer
Senescent cells, including both RS and SIPS, are known to secrete a variety of molecules that contribute to pro-inflammatory and pro-tumorigenic environment, collectively referred to as SASP (10, 13, 15, 80–84). These molecules include pro-inflammatory cytokines, chemokines, growth factors, pro-angiogenic factors, small molecules, lipids, ROS, and proteases (69, 80, 85). The SASP is reversible, as suggested by Coppé and colleagues (17). Unlike apoptotic and quiescent cells, SASP cells remain metabolically active and have been found to express significantly higher levels of glycolytic pathway enzymes such as hexokinase, phosphoglycerate kinase, and phosphoglycerate mutase, as well as higher glycolytic activity compared to young cells (77, 86, 87). Furthermore, mitochondrial ROS (mtROS) production and succinate induction are upregulated in SASP cells, even when both oxidative phosphorylation (OXPHOS) and glycolysis are inhibited by low dose IR without necrosis or apoptosis (88).
In addition to their effects of neighboring cells, SASP cells can also communicate via extracellular vesicles (EVs) (89). Activated senescent cells can generate more functional Evs than non-senescent cells by upregulating p53 expression, and these Evs can upregulate ROS induction and promote senescence in neighboring cells (90, 91).
3.5. SAS, a distinct type of SASP, has been found to contribute to the lasting effects of chemotherapy
Milanovic and colleagues reported that cancer therapy-induced senescence (TIS) can trigger the SAS proliferative phenotype in cancer cells, allowing them to evade senescence-induced cell cycle arrest and exhibit enhanced clonogenic growth potential (15, 16, 20). SAS is regulated independently of cell cycle arrest (17, 18) and cell death (13, 14, 92) and is considered a critical mechanism in the development of resistance to cancer therapy (13, 14, 19, 92, 93).
In addition to TIS, oncogene-induced senescence (OIS) is another phenomenon in which oncogenes are activated in non-tumor cells, resulting in a stable cell cycle arrest (85). Leon and colleagues have found that OIS and overexpression of the oncogene HRASG12V in IMR90 cells increase active histone H3K79 di- and tri-methylation (H3K79me2/3) at the ILIA gene locus (94). The histone methyltransferase disruptor of telomeric silencing 1-like (DOT1l) regulates the increase in H3K79me2/3 occupancy at the IL1A gene locus, which is critical for IL1A expression and gene expression regulation during OIS. However, while DOT1l is an epigenetic regulator of the SASP, its depletion does not affect OIS-induced cell cycle arrest, suggesting that it is not involved in SAS. Although epigenetic changes may lead to irreversible SASP in OIS cells (95, 96), their role in SAS remains unknown, and additional stressors that can result in both SASP and SAS necessitate further study.
It is important to note that, although the findings observed by Milanovic and colleagues were made on cancer cells, it is possible that a similar phenomenon might also occur in vascular cells, including ECs. Both senescence and macrophage proliferation contribute to CVD (97), and therefore, the SAS phenotype in ECs may also play a role in CVD. Therefore, further investigation on the role of SAS in ECs is necessary to fully understand its contribution to atherosclerotic CVD.
3.6. DDR is a link between atherosclerosis and resistance to cancer therapy
As we discussed above, cellular senescence has been implicated in the development of both atherosclerosis and cancer including in cancer survivors. At the molecular level, cellular senescence is associated with both telomeric and non-telomeric DNA damage in cells of the vessel wall, including ECs, triggering the DDR pathway (98–100). DDR maintains genetic stability and cell integrity when exposed to DNA damaging agents, such as IR and chemotherapeutic drugs used in cancer treatments. DDR induces cell cycle arrest for DNA repair and promote apoptosis and senescence to prevent propagation of damaged DNA. However, dysregulation of DDR can lead to resistance of cells to cancer treatments, highlighting its potential as a target to enhance sensitivity to cancer therapies (101, 102).
The major DDR pathways are regulated by the ataxia-telangiectasia mutated and ataxia-telangiectasia and Rad3 related (ATM/ATR) pathways, which phosphorylate proteins at DNA damage sites, including histone H2A (H2AX), to form phosphorylated gH2AX. Additionally, ATM/ATR activate CHK2 and CHK1, respectively (103), which prevent cells with damaged DNA from entering mitosis, especially when cells have a defective G1 checkpoint. However, a defective checkpoint is common in cancer cells due to p53 mutations. CHK1 is a potential therapeutic target, and CHK1 inhibitors are being developed and used as single agents or in combination with IR or genotoxic chemotherapies in preclinical and clinical studies (104). As such, these pathways can be targeted for further study.
4. Mechanisms involved in the regulation of telomere protection pathways
Cellular senescence can occur independently of telomere shortening, highlighting the importance of understanding the mechanisms that protect telomeres from DNA damage and dysfunction to mitigate senescence-associated disorders and diseases. In this section, we will explore the mechanisms involved in the regulation of telomere protection pathways.
Chromosome ends must be shielded from damage to prevent the acceleration of cellular senescence, which would lead to activation of DDR and DNA damage repair machinery. This protection is accomplished by specific factors associated with telomeres, including telomerase, non-coding telomeric repeat-containing RNAs (TERRA), and Shelterin (55, 105, 106).
4.1. Telomerase
Telomerase, which is comprised of a ribonucleoprotein with an RNA subunit TERC and a reverse transcriptase enzymatic subunit TERT, catalyzes the addition of repetitive hexanucleotide 5′-TTAGGG-3′sequences to chromosome ends. This process known as telomere elongation (107–113). In addition to this primary function, telomerase can also inhibit immune cell apoptosis, protect neurons from oxidative stress, and regulate inflammatory responses (114–119).
Furthermore, TERRA, a class of long noncoding RNAs transcribed at telomeres, is also suggested to participate in the protection of chromosome ends and telomeres. However, the mechanisms of telomere protection regulated by TERRA are yet to be fully elucidated (120). Additionally, the Shelterin can shape, safeguard, and protect telomeres during proliferation.
4.2. Shelterin
In addition to telomerase and TERRA, the Shelterin complex can shape, safeguard, and protect telomeres during proliferation. The Shelterin complex is comprised of six proteins: TERF1, TERF2, POT1, TIN2, TPP1, and TERF2IP, as illustrated in Figure 2. Its primary function is to safeguard telomeres by preserving their structure and function, ensuring genomic stability (121, 122). In the following sections, we will examine the specific roles of each Shelterin protein, and explore recent studies on the emerging functions of post-translational modifications of TERF2IP in the context of telomere protection, cellular senescence, and potential implications in atherosclerotic CVD.
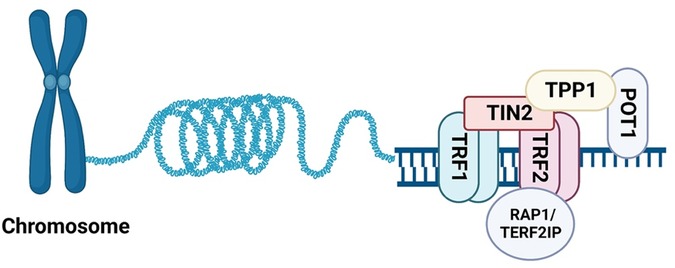
Figure 2. Assembling of Shelterin. The six known components of Shelterin: TRF1, TRF2, TERF2IP (RAP1), TIN2, TPP1, and POT1. TRF1 and TRF2 bind to telomeric DNA duplexes, while POT1 binds to single-stranded DNA in the 3′ overhang region. TERF2IP binds TRF2 and does not directly bind DNA (All the figures were made in Biorender.com).
Telomere repeat-binding factor 1 and 2 (TRF1 and TRF2) are two key components of vertebrate telomeres that share the TRF homology domain (TRFH) and bind double-stranded telomeric DNA as homodimers. TRFH on TRF1 and TRF2 is critical for TRF1-TRF2 dimerization, DNA binding, telomere localization, and modulation of TRF1-TRF2 interaction with other Shelterin proteins (123). To study how TRF1 and TRF2 locate TTAGGG repeats on DNA tightropes and assemble the Shelterin complex, Lin and colleagues used single-molecule fluorescence imaging to observe the dynamics of quantum dot (QD)-labeled TRF1 and TRF2 on λDNA and DNA substrates containing alternating regions of telomeric and non-telomeric sequences. They observed that TRF1 directly binds telomeric sequences with little 1D searching on non-telomeric DNA, while TRF2 extensive 1D searches on non-telomeric DNA through 1D sliding to find protein partners for assembling the Shelterin complex and stabilizing their interaction with specific telomeric DNA (124).
Besides their central role in telomere capping, elevated TRF2 expression is frequently found in tumors. El Mai and colleagues have demonstrated that TRF2 is expressed in the vasculature of most human cancers, where it colocalizes with the Wilms' tumor suppressor (WT1). TRF2 acts as a transcriptional target of WT1 and plays an essential role in EC proliferation, migration, and tube formation. Mechanistically, TRF2 binds and transactivates the promoter of angiogenic tyrosine kinase platelet-derived growth factor receptor β (PDGFRβ) through a mechanism distinct from that of telomere capping (125).
TRF1-interacting protein 2 (TIN2) is a key protein in the Shelterin complex, acting as a central mediator for TRF1 function. Using TRF1 as bait for interaction cloning, Kim and colleagues demonstrated that TIN2 interacts with TRF1 and co-localizes with it in the nucleus during metaphase chromosomes. TIN2 also binds telomere protection protein 1 (TPP1)/protection telomeres 1 (POT1) to regulate telomere length, telomeric capping, and telomerase activity (122, 126). POT1 (or POT1a and POT1b in rodents) is a single-stranded-DNA binding protein that plays a crucial role in telomeric capping and protection (122). In addition to modulating TRF1-TRF2 dimerization, TIN2 also acts as an adaptor protein that links TPP1/POT1 to TRF1-TRF2 on double-stranded telomeric DNA (127).
TTP1/POT1 are single-stranded telomeric DNA binding proteins that play essential roles in preventing the activation of the ATM/ATR pathways at telomeres (128–131). Although POT1a can block the binding of replication protein A (RPA) to telomeres, the binding affinities and abundance of TPP1/POT1a and RPA suggests that TPP1/POT1a is unlikely to exclude RPA, the major protein that binds single-stranded DNA. Takai and colleagues demonstrated that TIN2 deletion triggers the loss of telomere TPP1/POT1a, accumulation of RPA, and ATR activation, accompanied by all phenotypes provoked by POT1a/b deletion. While TIN2 has a minor role in TRF2-induced inhibition of ATM pathways (but not TRF2-induced inhibition of telomere fusions), it has a key role in TTP1/POT1-dependent inhibition of ATR pathways via stabilizing TPP1/POT1a on single-stranded telomeric DNA. This stabilization allows for effective exclusion of RPA and thus prevents ATR activation (128, 130, 131). Together, these observations indicate that TIN2 inhibits ATM/ATR, the two major DNA damage repair pathways, by stabilizing TRF2 on double-stranded telomeric DNA and stabilizing TPP1/POT1 on single-stranded-DNA overhang, respectively (130–133).
Of the six Shelterin proteins, TRF1-TRF2 bind double-stranded telomeric DNA through the C-terminal Myb domain and recruit TERF2IP, TIN2, TPP1, and POT1 for assembling the Shelterin complex (41). Among the six Shelterin proteins, TERF2IP, also known as repressor activator protein 1 (RAP1), is one of the most conserved proteins and directly binds TRF2 (55, 106, 122, 134).
The Shelterin complex is a promising target for chemotherapeutics. Table 4 provides a summary of the chemotherapeutics that have been used to target the Shelterin complex (135) (see Table 4).
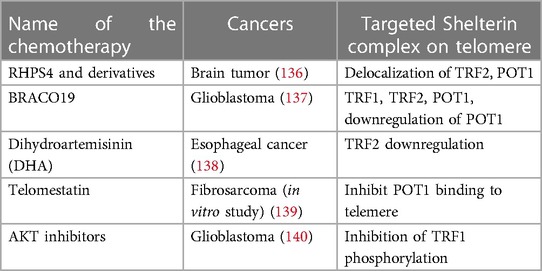
Table 4. Chemotherapeutics targeting Shelterin complex (135).
4.2.1. TRF1 and TRF2
To bind double-stranded telomeric DNA, TRF1 and TRF2 associate with TIN2, which stabilizes the telomeric localization of TRF1 and TRF2. Although TRF1 and TRF2 share similarities in structure and form TRF1-TRF2 dimers, their key functions are distinct from each other (141, 142). TRF1 negatively regulates telomerase-mediated telomere elongation to maintain telomeric homeostasis (143, 144). TRF1 depletion revealed TRF1 is a regulator of Shelterin localization on telomeres and maintenance of telomere functional structures. Martínez and colleagues have shown that conditional deletion of TRF1 in mouse embryonic fibroblasts is sufficient to induce severe telomeric damage without telomere shortening. These cells, in the absence of TRF1, rapidly underwent cellular senescence and had increased telomeric γH2AX foci and activation of the cell-cycle checkpoint kinase 2 and 1 (CHK2 and CHK1), which are known downstream targets for the ATM/ATR pathways, which is known as the major DDR pathways (145). Chromosomes with longer telomeric ends allow more TRF1 proteins to bind to telomeres, thereby inhibiting telomerase activity and suppressing telomere elongation. After multiple rounds of DNA replication, telomeric ends become shorter, which decreases TRF1 binding to telomeric ends and allows telomerase to activate telomere elongation. In aged ECs, both TRF1 mRNA and protein expression are decreased compared to that of young ECs (146–148).
TRF2, on the other hand, functions as a telomere capping protein and is involved in protecting chromosome ends from being recognized as DNA damage site, which can lead to genomic instability. In addition to its role in telomere attachment to nuclear membrane (149), TRF2 also plays a critical role in preventing telomere fusion and recombination. TRF2 depletion evokes telomere uncapping, leading to the activation of telomere dysfunctional DDR and induction of cellular senescence and oxidative stress, independent of telomerase activity (150). Specifically, TRF2 depletion rapidly induces end-to-end fusion, cellular senescence (147, 148), and cell death by activating the ATM/ATR pathways (145, 151, 152). In patients with heart failure, low TRF2 expression levels are associated with telomere shortening in cardiomycytes (153). These observations all together have demonstrated critical roles for both TRF1 and TRF2 in senescence and genomic instability (122, 154).
4.2.2. TIN2
TIN2 serves as an adaptor protein for TRF1 and helps in stabilizing the telomeric localization of TRF1 and TRF2 (126). Additionally, TIN2 interacts with TPP1 through its TPP1 interacting domain, which connects TIN2 to POT1, facilitating the telomeric localization of the TPP1-POT1 complex (122). TIN2 contains a mitochondrial localization domain at its N-terminus (155, 156), allowing it to translocate to mitochondria, where it undergoes post-translational modifications and regulates mitochondrial OXPHOS (155, 156). Depletion of TIN2 using short hairpin RNA (shRNA) prevents glycolysis and enhances OXPHOS, leading to a decrease in ROS synthesis (155). Conversely, TIN2 accumulation in mitochondria leads to an increase in ROS synthesis, promoting cellular senescence. Lee and colleagues have demonstrated that the RNA binding protein Human antigen R (HuR) binds to the 3′-untranslated region of TIN2 mRNA and inhibits TIN2 protein synthesis (157). During RS, diminished expression of HuR leads to increased stabilization of TIN2 mRNA, resulting in enhanced TIN2 protein synthesis, mitochondrial translocation, and acceleration of ROS synthesis and cellular senescence (157). Given the critical roles of mitochondrial metabolism and respiration in age- and stress-induced senescence (158, 159), the involvement of TNI2 in this process is of great interest and deserves further investigation.
4.2.3. TPP1
TTP1 binds TIN2, which tethers TIN2 to POT1 (122) and enhances POT1's binding affinity to single-stranded telomeric DNA (160–162). POT1 then recruits telomerase to telomeric ends by interacting with the telomerase N-terminal OB domain and TERT, the telomerase catalytic subunit (162). Sirtuin 1, a nicotinamide dinucleotide (NAD+)-dependent deacetylase, have been shown by Chen and colleagues to protect mesenchymal stem cells from senescence by mediating TPP1 expression, suggesting TPP1's involvement in cellular senescence (163). Additionally, Min and colleagues suggested that suppression of TPP1 expression leads to mitochondrial dysfunction and deregulated mitochondrial-ribosome function, which leads to telomere deprotection and RS in human diploid fibroblasts (164).
4.2.4. POT1
POT1 is considered a terminal transducer of TRF1-mediated telomere length regulation because it directly binds the 3′ single stranded G-overhang of telomeres, preventing telomerase access to telomeres (see Figure 2) (165). In addition to its telomere-protective functions, POT1 also binds to TPP1 and double-stranded DNA damage sites to suppress non-homologous end joining (NHEJ) and protect DNA from double-stranded breaks (166). Human POT1 has been shown to protect telomeric ends and regulate telomerase activity by displacing/replacing the G-quadruplex structure (167). POT1 mutations have been detected in various cancers including chronic lymphocytic leukemia, glioma, melanoma, angiosarcoma, and colorectal cancer (168). The most common mutations are found in the OB-fold domains at the N-terminus, which disrupts POT1 binding to telomeric ends (168). A recent study by Kelich and colleagues showed that a POT1 heterozygous mutation p(L259S) in a patient with idiopathic pulmonary fibrosis can drive telomere loss, telomere DNA damage, and pre-mature senescence (169). In mice, there are two homologues of human POT1, POT1a and POT1b, and knock-out studies suggested that POT1 is involved in protecting telomere single-stranded DNA from end-to-end fusion.
4.2.5. TERF2IP
TERF2IP, also known as RAP1, plays a crucial role in several biological processes, including telomere protection, homology-directed repair (HDR) regulation, inflammation, and metabolism (170, 171). TERF2IP binds telomeres through its association with TRF2, which enhances TRF2 binding to telomeres (172, 173). The TRF2-TERF2IP complex prevents the telomeric localization of Poly [ADP-ribose] polymerase 1 (PARP1) and structure-specific endonuclease subunit (SLX4) and thereby inhibits homologous recombination (HR)-triggered telomere attrition. Deletion of TERF2IP and TRF2 leads to the recruitment of PARP1 and SLX4 to telomeres, resulting in the recruitment of additional HR factors such as DNA repair protein RAD51, Exonuclease I (EXOI), and the MRN complex to telomeres (172). The role of TERF2IP in the development of premature senescence has been investigated in progeroid mice (77, 174). TERF2IP protects telomeres from NHEJ in both yeast and humans (175, 176), independently of its association with TRF2 (176). In senescent human cells, TERF2IP prevents fusion of critically short telomeres, thereby protecting telomeres (177) and regulates sub-telomeric transcription. In senescent cells with critically short telomeres, TERF2IP re-localizes to sub-telomeric regions and binds to the promoters of a group of genes called new Rap1 targets at senescence (NRTS), promoting loss of histones and activating a cascade of other genes in NRTS (178, 179).
4.3. Nitric oxide (NO) activates telomerase and delays EC senescence and the role of tetrahydrobiopterin (BH4)
4.3.1. NO signaling
NO has a crucial role in various physiological and pathological processes in the human body, including protecting telomeres from shortening, damage, and dysfunction by regulating telomerase activity, thus protecting cells from senescence. Additionally, NO can prevent DNA damage and oxidative stress, which can also contribute to telomere shortening and dysfunction. Exogenous NO has been demonstrated to delay EC senescence in culture, suggesting that telomere shortening can be modulated beyond the number of cellular divisions. The mechanism by which NO stimulates telomerase activity is not yet fully understood, but it may react with tissue-derived oxygen radicals, thereby reducing oxidative stress, which has been shown to accelerate EC senescence. Alternatively, NO may upregulate telomerase activity via transcriptional and/or posttranscriptional mechanisms (180).
NO has important cardioprotective and anti-inflammatory effects, as it inhibits vascular wall apoptosis and lipid oxidation, prevents VSMC growth, and inhibits white blood cell adhesion and platelet aggregation. Additionally, NO induces blood vessel dilation, which is a vital mediator of vascular tone (181, 182). The integrity of EC function, including their capability to proliferate and migrate, is essential for angiogenesis. Therefore, EC senescence and the subsequent reduction in their proliferative ability may contribute to compromised angiogenesis associated with age. Additionally, senescent ECs express adhesion molecules that promote neutrophil adhesion and inflammation, enhancing the chronic inflammatory process that contributes to the progression of atherosclerosis.
Conversely, endothelial NO is essential for angiogenesis and protects against atherosclerosis. However, the bioavailability of endothelial-derived NO is impaired with aging, which may accelerate EC senescence, impairing EC function and contributing to impaired angiogenesis and atherosclerotic progression (180, 183–186).
4.3.2. The impact of BH4 levels in anti-cancer treatment and their association with atherosclerotic CVD and the potential clinical applications
BH4 is an essential cofactor involved in several important enzymatic processes. It plays a vital role in the synthesis of neurotransmitters such as adrenaline, noradrenaline, serotonin, and dopamine, and participate in the degradation of phenylalanine (187). Additionally, BH4 serves as a cofactor for NOS3, which is responsible for the production of NO through the oxidation of L-Arginine to L-citrulline (188, 189). Supplementation with BH4 has been shown to have beneficial effects in improving conditions like hypertension and cardiac dysfunctions. By supporting NOS3 function, BH4 promotes the production of NO, thereby improving cardiovascular health (190).
However, under UV radiation or infrared exposure, BH4 can be oxidized to dihydrobiopterin (BH2), which can stimulate the production of higher levels of superoxide by eNOS. This oxidative process can lead to increased oxidative stress and potentially impact EC function (188). ECs have two pathways for synthesizing BH4: de novo synthesis from guanosine triphosphate (GTP) and a salvage pathway that involves recycling BH2 and quinoid-dihydrobiopterin (181, 191). These pathways are crucial for maintaining BH4 levels in ECs, which are essential for physiological processes and NO production, regulating EC function and vascular homeostasis.
Age-related studies have shown a decrease in vascular BH4 levels in animal models (192, 193), while in humans, aging has been observed to increase platelet and plasma BH2 levels without affecting BH4 levels or the BH4:BH2 ratio (182). This suggests increased oxidation of BH4 into BH2 with aging due to oxidative processes. Inhibiting the enzyme responsible for BH4 to BH2 conversion, such as with methotrexate treatment, leads to increased BH2 plasma levels, highlighting the importance of the BH4 recovery pathway for maintaining BH4 homeostasis (187, 194–196). Alterations in BH4 levels have been found to correlate with vascular dysfunction in ECs (188). In the context of tumor progression, BH4 promotes angiogenesis by activating eNOS for NO production (197). Conversely, inhibiting BH4 has been shown to attenuate tumor angiogenesis in a mouse model of hepatocellular carcinoma (198) (Figure 3). The changes in BH4 levels resulting from anti-cancer therapy and their potential impact on atherosclerotic CVD are summarized in Figure 3. These findings suggest that BH4 levels play a significant role in various physiological and pathological processes, including cancer progression and cardiovascular health. Further research is needed to fully understand the complex interactions and potential therapeutic implications of BH4 in cancer treatment and CVD.
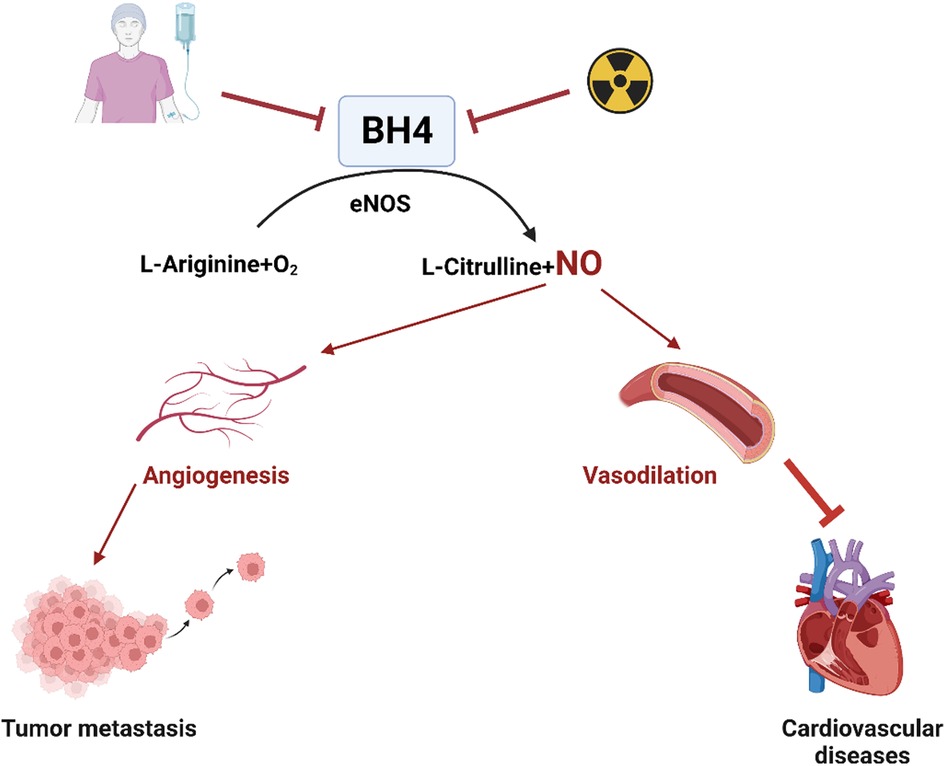
Figure 3. Chemotherapy or radiation therapy regulates BH4 levels and their association with atherosclerotic CVD (All the figures were made in Biorender.com).
5. The critical role of EC senescence in the development of atherosclerosis in cancer survivors
It is noteworthy that senescent ECs have been identified in human atherosclerotic plaques and have impaired function, such as reduced NO production and increased adhesion molecule expression, which can contribute to EC dysfunction and plaque formation (36, 174). The vascular system is crucial for maintaining health and survival (199, 200), and ECs plays a vital role in regulating vascular homeostasis. ECs line the luminal surface of blood vessels and dynamically respond to hemodynamic fluctuations to modulate blood flow. They also mediate the interaction of circulating blood components with the vessel wall and facilitate gas and nutrient exchange between the blood and tissues (71, 201). However, various stress stimuli, such as d-flow, increased production of ROS, DNA damage, mitochondrial dysfunction, exposure to inflammatory cytokines, cancer therapies, and activation of oncogenes, can induce EC senescence, adversely affecting these homeostatic functions, disrupting vascular integrity, and impairing their function (202).
The impairment of EC functions is an initial step in the progression of atherosclerotic CVD, which is linked to the aging process and a senescent phenotype in these cells. This can lead to disrupted permeability and pathological signaling cues, as evidenced by numerous studies (71, 203–205). Cancer therapy is a stress state that can induce EC senescence and subsequent vascular dysfunction, increasing the risk of atherosclerotic CVD (206). In ECs, therapy-induced senescence (TIS) is associated with chronic inflammation and the development of atherosclerotic lesions (11), which tend to occur in regions of the vasculature with d-flow (207). Notably, d-flow induces endothelial-to-mesenchymal transition (endoMT) through the transforming growth factor β (TGFß) signaling pathway (208), and this transition becomes pro-atherosclerotic through the deposition of fibronectin, increased expression of adhesion proteins, and recruitment of inflammatory cells (208).
Moreover, it is crucial to note that radiation therapy, a commonly cancer treatment, directly induces endoMT (209), which is accelerated by oxLDL and ultimately leads to atherosclerotic plaque formation (209). This endoMT not only leads to cardiovascular complications in cancer patient but also contributes to the development and progression of cancer-associated fibroblasts (CAFs) (210). It has been well-established that cancer and its therapies can induce or accelerate the aging process, which may explain the association between atherosclerotic CVD and cancer. Therefore, understanding the molecular mechanisms underlying EC senescence in atherosclerosis in cancer survivors is crucial to develop effective therapies for these conditions.
5.1. EndoMT
ECs can undergo a process called endoMT, which leads to their trans-differentiation into mesenchymal cells (211, 212). During this process, ECs lose their characteristic features and acquire a mesenchymal phenotype. EC-specific genes such as Cluster of Differentiation 31 [CD31 or Platelet Endothelial Cell Adhesion Molecule 1 (PECAM1)], Vascular Endothelial Cadherin (VE-Cadherin or CDH5), Von Willebrand Factor (vWF), Tyrosine Kinase with Immunoglobulin-like and EGF-like domains 1 (TIE1), and TEK Receptor Tyrosine Kinase (TIE2) are downregulated (27, 211, 213), while mesenchymal cell-specific genes such as α-smooth muscle actin [αSMA or Smooth Muscle 22α (SM22α)], Extra Domain A (EDA) of Fibronectin, N-cadherin, Vimentin, Fibroblast Specific Protein 1 (FSP1), Fibroblast Activating Protein (FAP), and Calponin are upregulated (27, 213) (see Figure 4).
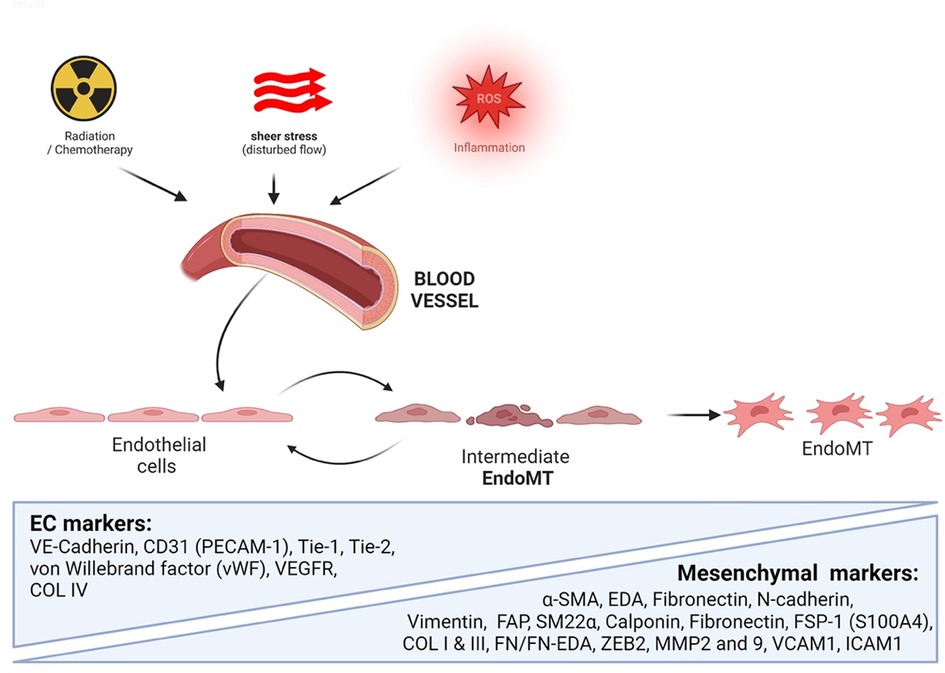
Figure 4. Extracellular stimuli induce endoMT. External stimuli such as radiotherapy, chemotherapy, d-flow, and inflammatory signals can trigger the transformation of ECs into mesenchymal cells, characterized by the enhanced expression of mesenchymal cell-specific markers (All the figures were made in Biorender.com).
Inflammatory mediators such as IL1β, TNFα, and NFκB cause EC dysfunction, leading to endoMT, which is the link between inflammation and EC inflammation-associated disorders (212). Recent studies have shown that ECs can also undergo a transient stage called partial or intermediate endoMT (see Figure 4) before being completely transformed into mesenchymal cells (214, 215). Tombor and colleagues showed that ECs are transiently transformed into mesenchymal-like cells with mesenchymal gene expression three days after myocardial infarction but return to their baseline phenotype within 14 days (214). “Transitioning” cells detected in human plaques co-expressed EC and mesenchymal genes involved in endoMT (216).
5.1.1. EndoMT contributes to atherosclerosis
EndoMT is essential during embryonic cardiac development and wound healing. However, dysregulated endoMT has been shown to contribute to atherogenesis (208, 216–219), which can cause flow-limiting lesions and ischemia in various organs, such as the heart, brain, and limbs, leading to angina, transient ischemic attacks and intermittent claudication. Plaque rupture, which can lead to acute ischemic syndromes such as myocardial infarction and cerebrovascular attack, is a common mechanism underlying atherosclerosis and driven by thinning of the fibrous cap induced by MMPs (220, 221). EndoMT-derived fibroblast-like cells are commonly present in atherosclerotic plaques and have been associated with plaque stability and rupture, as shown by Evrard and colleagues using EC-specific lineage tracing (212, 221–224). In vitro studies have revealed that various processes contributing to atherosclerosis, including TGFβ signaling, oxidative stress, and hypoxia, can activate endoMT (216).
5.1.2. EndoMT contributes to the development of CAFs
Myofibroblasts and perivascular mesenchymal cells, such as pericytes, have been implicated in cancer development and progression by promoting growth, metastasis, and chemotherapy resistance (210). CAFs in the tumor microenvironment (TME) secrete a variety of growth factors (EGF, HGF, IGF1, SDF1), cytokines (IL1, IL6, IL8, IL11, LIF), chemokines, and proangiogenic factors (VEGFA, SDF1, FGF2, IL8, PDGFC), which can cause a rewiring of cellular metabolism, provoke a SASP status in senescent cancer cells, and ultimately promote cancer progression by transforming the TME into a dense and fibrotic structure (210).
Zeisberg and colleagues have shown that TGFβ1 can promote endoMT in proliferating ECs, leading to upregulation of the mesenchymal marker FSP1 and downregulation of CD31/PECAM1. Furthermore, up to 40% of CAFs in murine models of melanoma and pancreatic carcinoma are derived from ECs via endoMT (225, 226). Anti-angiogenic therapy has also been shown to have a direct effect on reducing myofibroblasts and potentially hindering cancer progression (213, 225).
In atherosclerotic CVD, endoMT promotes the accumulation of mesenchymal cells in the sub-EC spaces, which contribute to the development of fibrous plaques. Similarly, in cancer, endoMT can promote tumor invasion and metastasis by enhancing cancer cell migration and invasion and increasing the number of cancer stem cells. Furthermore, endoMT has been implicated in cancer therapy resistance. Therefore, understanding the molecular mechanisms that regulate endoMT in atherosclerotic CVD and cancer survivors could lead to the development of novel therapies to prevent both diseases (220, 221).
5.2. Mechanisms of endoMT-induced atherosclerosis and cancer
There are various shared pathways in cancer and atherosclerosis development. These include TGFß signaling (227, 228), bone morphogenetic proteins (BMPs) (229), and NOTCH (230). SUMOylation appears to regulate the TGFß (231) and SMAD (232) pathways and contribute to endoMT as well as both cancer and atherosclerotic CVD development and progression.
EndoMT-induced atherosclerotic CVD and cancer share several mechanisms that contribute to disease progression. Both diseases involve the accumulation of fibroblast-like cells that contribute to disease development and progression (233–239). In atherosclerosis, endoMT-derived fibroblast-like cells contribute to plaque instability and rupture (220, 221, 233–238), while in cancer, CAFs promote tumor growth, angiogenesis, and chemotherapy resistance (240, 241). Both diseases are associated with chronic inflammation, which can promote endoMT in atherosclerosis and CAF activation in cancer (242–245). Both diseases involve alterations in cellular metabolism, with endoMT and CAF contribute to the rewiring of cellular metabolism to promote disease progression (246–248). Finally, both diseases involve dysregulation of signaling pathways such as TGFβ, BMPs, NOTCH, which promotes endoMT in atherosclerosis and is a key regulator of CAF activation in cancer (211, 227, 228).
5.2.1. TGFβ
Numerous types of cancer, including melanoma, esophageal carcinoma, and colon carcinoma secrete the soluble factor TGFβ (211, 227, 228). Inflammatory cytokines and d-flow inhibit FGFR1 expression in ECs, leading to activation of the TGFβ pathway, as reported by Chen and colleagues (208). Activated TGFβ signaling upregulates the expression of transcription factors (SNAIL, SLUG, and ZEB1), which, in turn, upregulates the expression of mesenchymal genes (αSMA orSM22α, calponin, vimentin, type I collagen, fibronectin, FSP1, N-cadherin, and MMP2/9) (212). TGFβ ligands secreted by TGFβ-producing cells bind the transmembrane TGFβRs, which are comprised of TGFβR1 (or ALK5) and TGFβR2 (213). This ligand-receptor association leads to the formation of the TGFβR1/2 heterodimer and the subsequent TGFβR2 auto-phosphorylation, which trans-phosphorylates TGFβR1 at specific sites, forming the active TGFβR1/2 complex. Activated TGFβR1/2 phosphorylates cytoplasmic small mothers against decapentaplegic SMAD2/3, promoting the nuclear translocation of SMAD2/3, where it binds the SMAD binding element of TGFβ target genes (such as SNAIL, SLUG, ZEB, TWIST) to upregulate their transcription (see Figure 5) (213, 249). In a SMAD-independent pathway, activated TGFβ signaling leads to the activation of the mitogen-activated protein kinase (MAPK) family of serine/threonine-specific protein kinases (ERK1/2, p38MAPK, JNK, and ERK5) (213). Medici and colleagues reported that TGFβ inhibition leads to the activation of SMAD, MEK (MAPK/ERK kinase), phosphoinositide 3-kinase (PI3K) and p38MAPK pathways, suppressing TGFβ-mediated endoMT via downregulating SNAIL expression (250). These findings suggest that activated TGFβ signaling plays a critical role in atherosclerotic CVD, cancer, and other fibrotic diseases by activating endoMT and fibroproliferative pathways (213).
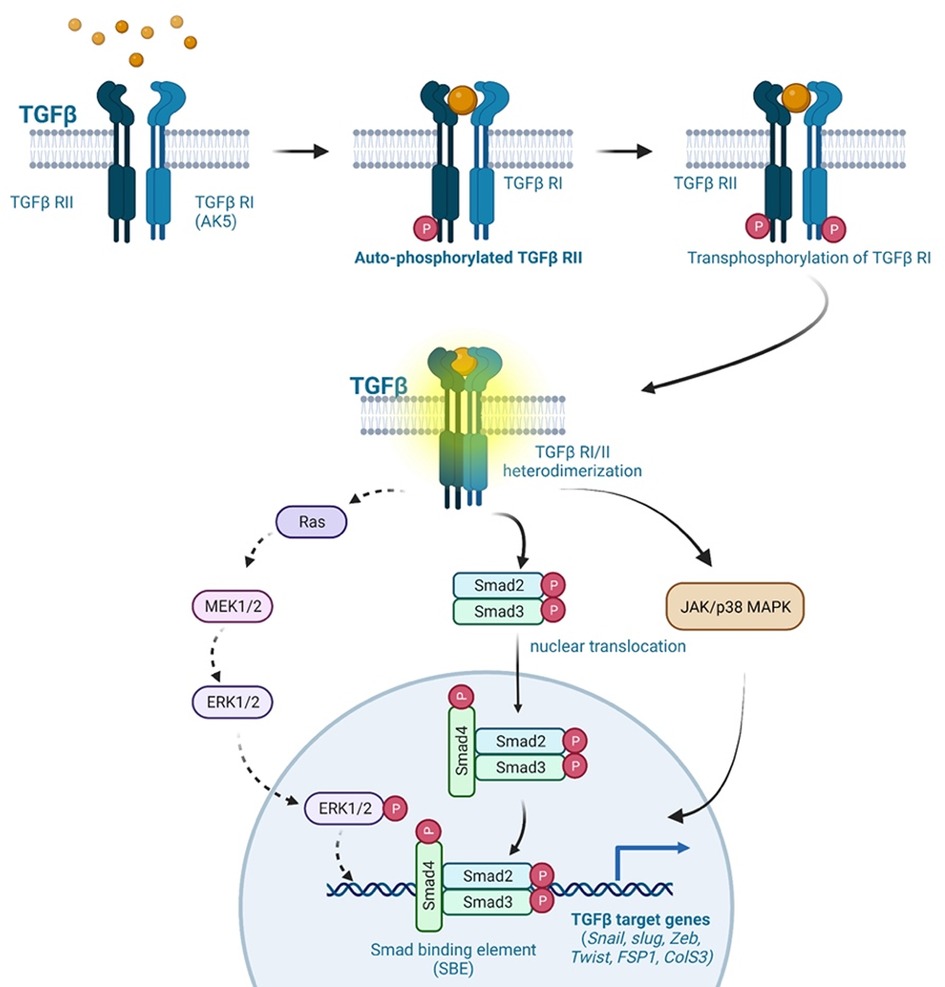
Figure 5. TGFβ signaling in endoMT. The TGFβ signaling pathway plays a crucial role in endoMT. Cancer cells secrete TGFβ ligands, which bind to TGFβR1 (ALK5) and TGFβR2 receptors present on ECs. This ligand-receptor binding initiates the formation of TGFβR1-TGFβR2 heterodimers, leading to the autophosphorylation of TGFβR2. Phosphorylated TGFβR2, in turn, phosphorylates TGFβR1, resulting in the activation of the TGFβR1/2 complex. The activated TGFβR1/2 complex transmits signals through both SMAD-dependent and SMAD-independent pathways. In the SMAD-dependent pathway, cytoplasmic SMAD2/3 proteins are phosphorylated by the activated TGFβR1/2 complex. Phosphorylated SMAD2/3 proteins then translocate to the nucleus, where they bind to specific SMAD binding elements (SNAIL, SLUG, ZEB, TWIST, FSP1, COLS3). This binding regulates the expression of genes involved in endoMT. In the SMAD-independent pathway, the activated TGFβR1/2 complex can also propagate signals through the activation of ERK, p38MAPK, and JNK signaling pathways. These pathways contribute to TGFβ-mediated effects on endoMT, CVD, fibrosis, and cancer progression. Overall, TGFβ signaling activation induces endoMT and is involved in various pathological processes, including CVD, fibrosis, and cancer progression (All the figures were made in Biorender.com).
5.2.2. BMPs
More than a dozen members of the BMP family, which belong to the TGFβ superfamily, have been identified. The balance between BMP and TGFβ is critical for maintaining tissue homeostasis. BMP expression is altered in various types of cancer, including hepatocellular, renal, colorectal, lung, breast, prostate, endometrial, and head and neck cancers (251). BMP binds BMP cell surface receptor type 1/2 (BMPR1/2) and activates serine/threonine kinases, playing multifunctional roles in different cell types (251, 252). Following ligand-receptor binding, BMPR2 oligomerizes with BMPR1(ALK1) and phosphorylates SMAD1/5/8. In ECs, BMP binds BMPR2 and suppresses TGFβ-induced endoMT. In patients with pulmonary arterial hypertension, loss of EC BMPR2 leads to the formation of a mixed heterodimeric BMPR1/TGFβR1/TGFβR2 complex, which activates downstream TGFβ-SMAD1/5 signaling. Activated TGFβ induces SMAD2/3 signaling by a high-affinity binding to the TGFβR2-TGFβR1(ALK5) heterodimer. Additionally, TGFβ activates a lateral SMAD1/5 signaling by complexing with TGFβR2/TGFβR1/BMPR1 (ALK1) and inducing endoMT (229, 253, 254).
5.2.3. NOTCH
NOTCH signaling plays important roles in both the development and pathology of the cardiac system. Mammals possess four receptors (NOTCH1/2/3/4) and five ligands (delta-like ligand 1/3/4 (DLL1/3/4) and Jagged 1/2 (JAG1/2)) (255). Upon ligand-receptor binding, enzymatic cleavages by ADAM10 and γ-secretase generate the NOTCH intracellular domain, which translocates to the nucleus, binds to the transcription factor CSL, and activates the transcription of NOTCH target genes (256). Recent studies have suggested the involvement of NOTCH signaling in endoMT (230). Noseda and colleagues have reported that EC NOTCH activation downregulates the expression of EC-specific genes (VE-cad, Tie1, Tie2, PECAM1, eNOS) and upregulates the expression of mesenchymal-specific genes (αSMA, fibronectin, PDGFRs), suggesting its role in endoMT development. This finding is supported by another study by Niessen and colleagues, demonstrating that EC NOTCH activation induces SLUG expression and endoMT (257). Additionally, NOTCH signaling has been implicated in atherosclerosis (258). TGFβ and NOTCH signaling act in a synergistic fashion during endoMT development, with TGFβ upregulating expression of NOTCH target genes such as JAG1, the receptor NOTCH1, N1ICD, and recombination signal binding protein J kappa (RBPJK)) (27, 259).
5.3. SUMOylation regulates endoMT-associated signaling
SUMOylation is involved in regulating many essential cellular processes, including those in the nucleus such as transcriptional activity, chromatin remodeling, and DDR (231, 260–263). It also occurs in the cytoplasmic compartment, particularly at intracellular membranes (264). One example of this is the regulation of the dynamin related GTPase DRP1, which mediates mitochondrial fission upon recruitment to the outer mitochondrial membrane (265, 266). Dysregulation of DRP1 SUMOylation affects mitochondrial division and has been associated with brain ischemia (267). Another important substrate of SUMOylation is the cystic fibrosis transmembrane conductance regulator (CFTR) is. Normally, CFTR resides in the plasma membrane, but its most common mutant form associated with cystic fibrosis contains a destabilizing phenylalanine deletion at position 508 (ΔF508) that causes protein degradation at the endoplasmic reticulum membrane (268). The degradation of ΔF508 is mediated by the ubiquitin-proteasome pathway but also involves SUMOylation (269). SUMOylation modulates the activity of multiple ion channels, including Kv7 potassium channels in hippocampal neurons linked to epilepsy and sudden death (270). Additionally, studies have shown SUMOylation regulates endoMT-associated signaling pathways, which are involved in the pathological progression of several diseases, including atherosclerosis and cancer.
5.3.1. TGFβ
TGFβ signaling is regulated at various levels, including TGFβR activation and post-translation modifications (271). TGFβ SUMOylation plays a critical role in epithelial to mesenchymal transition, which is similar to endoMT (231). As studies have shown that flow exerts profound effects on SUMOylation in atherosclerotic CVD (262, 272), we discuss the potential contributions of TGFβ SUMOylation to endoMT under flow conditions.
The TGFβ signaling cascade begins with TGFβR1/2 dimerization followed by TGFβ activation. Activated TGFβR1/2 promotes TGFβR1 K389 SUMOylation, which enhances TGFβR-SMAD association and SMAD2/3 phosphorylation (273). TGFβR SUMOylation also increases the metastasis and invasiveness of tumor cells. SENP2 inhibits TGFβR1 SUMOylation, thus suppressing TGFβ-induced epithelial to mesenchymal transition in bladder cancer (274). SENP2 overexpression in invasive bladder cancer cells, T24, suppresses TGFβ-mediated SMAD2/3 phosphorylation, downregulates the expression of mesenchymal markers N-cadherin and fibronectin, and inhibits epithelial to mesenchymal transition (274). Although we have reported the role of d-flow-induced SENP2 nuclear export on SUMOylation of ERK5 and p53 (263), there are no reports on flow effects on SENP2-regulated TGFβ/SMAD signaling. This area can be a subject of future studies.
5.3.2. SMAD
The SMAD family of proteins is divided into three subfamilies: receptor-activated SMADs (r-SMADs, including SMAD1/2/3/5/8), common mediator SMADs (Co-SMADs), and inhibitory SMADs (iSMADs including SMAD6/7) (275). R-SMADs are activated by specific ligand-receptor binding, such as TGFβR, and play a crucial role in transmitting signals from the receptor to the nucleus. iSMADs act as negative regulators of TGFβ signaling by inhibiting r-SMAD activation (276). rSMAD SUMOylation can modulate their function and activity by altering their subcellular localization, stability, and interaction with other proteins. For example, SUMOylation of SMAD4 at K113 and K159 increases its transcriptional activity by promoting interaction with histone acetyltransferase p300. SENP1/2 regulate TGFβ-induced epithelial to mesenchymal transition by inhibiting SMAD4 SUMOylation (277, 278). SMAD3 SUMOylation inhibits its transcriptional activity by promoting degradation. iSMAD SUMOylation can also modulate their function and stability; for instance, SMAD7 SUMOylation increases its inhibitory activity by promoting its stability and preventing proteasome-mediated degradation, while SMAD6 SUMOylation promotes its degradation and reduce its inhibitory activity (see Figure 6).
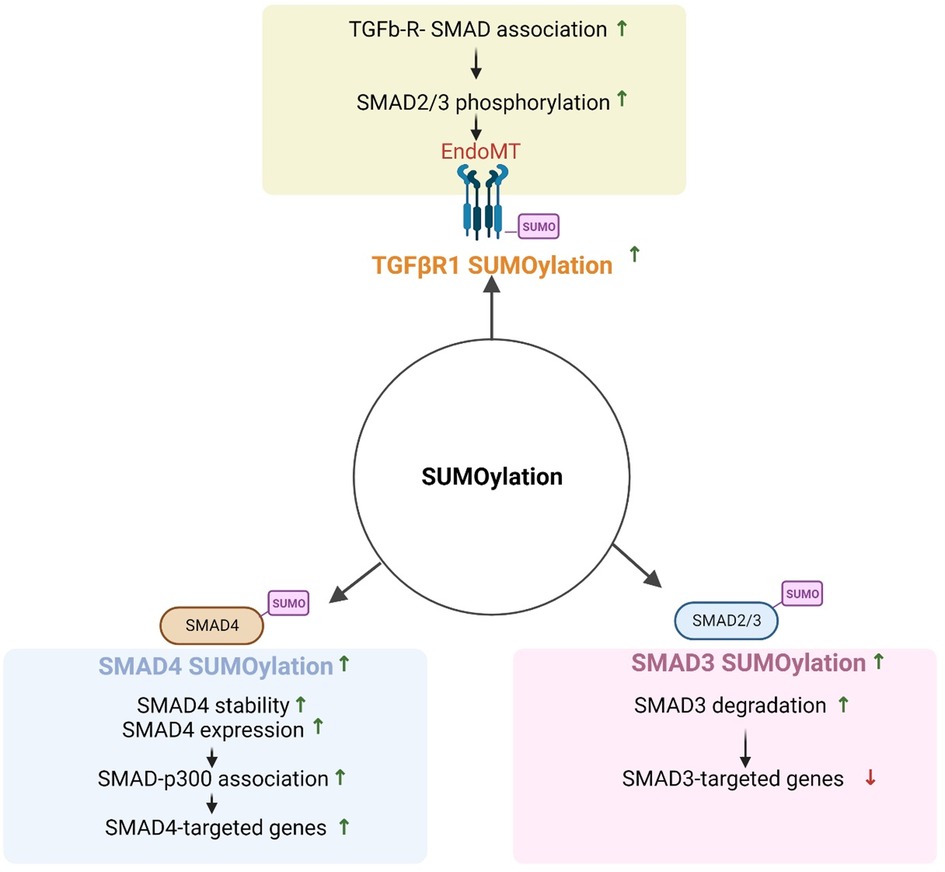
Figure 6. Regulation of TGFβ signaling by SUMOylation (All the figures were made in Biorender.com).
6. The role of blood flow in cellular senescence and the development of atherosclerosis in cancer survivors
ECs play a crucial role as transducers between blood flow mechanics and cellular signaling pathways in both physiological and pathological conditions. Mechano-sensors on the surface of ECs sense changes in blood flow and transduce these mechanical forces into biochemical signals, subsequently activating cellular responses that regulate downstream events (279). Hemodynamic shear stress, which is the frictional force generated by intraluminal blood flow on the surface of ECs, influences the formation of atherosclerotic plaque. Hemodynamic stress is expressed as a force per wall area (280, 281), but different patterns of shear stress can occur depending on the morphology of blood vessels. These patterns can be categorized as either linear or laminar flow (l-flow, also referred to as s-flow or u-flow in our previous publications) (282, 283) or d-flow, which is characterized by the recirculation of blood in vessel bends, branches, and bifurcations. These distinct flow patterns can have different effects on the secretory function and morphology of ECs. L-flow and d-flow have opposite effects on ECs and have been implicated in various cellular processes (71, 213, 284–286).
6.1. Blood flow in atherosclerosis
6.1.1. L-flow inhibits atherosclerosis
In regions of l-flow, ECs aligned longitudinally in the direction of blood flow and secrete factors for vasodilation and anticoagulation. L-flow promotes the generation of NO and expression of downstream molecules that improve vascular injury and suppress inflammation, making it an atheroprotective mechanical force that maintain EC homeostasis and function (191). This is accomplished in part through the activation of extracellular-signal regulated kinase 5 (ERK5), which increases endothelial nitric oxide synthase (eNOS) expression and inhibits EC inflammation (272, 287).
Studies on cultured human aortic ECs have shown that l-flow downregulates proliferation- and inflammation-related genes and upregulates survival-, angiogenesis- (e.g., tyrosine-protein kinase receptor Tie2 and vascular endothelial growth factor receptor Flk1) and vascular remodeling- (e.g., MMP1) related genes (288). This suggests that l-flow maintains a non-proliferative and non-inflammatory EC gene expression profile.
L-flow increases NO levels immediately through stimulation of eNOS, and long-term exposure can lead to increased mRNA and protein expression of eNOS. Acute eNOS activation could result from a shear-stress-derived intracellular calcium increase, which enhances calmodulin binding to eNOS. Nonetheless, eNOS phosphorylation by various phosphorylases, including PI3K/AKT, adenylate cyclase, and protein kinase A (PKA) have also been involved in the rapid response to l-flow (289). Long-term l-flow-dependent eNOS activation has been linked to SIRT1, a histone deacetylase, and its interaction with AMPK, as well as to various transcription factors that ultimately upregulate eNOS expression, such as KLF2 (290).
Furthermore, the resulting high shear stress has been found to increase BH4 levels by stimulating the first and rate-limiting step of its synthesis, GCH1. L-flow resulted in a ∼30-fold increase in GCH1 activity produced through phosphorylation of serine 81 by the α' subunit of casein kinase 2 (CK2), but did not produce increased protein expression, indicating BH4's short-term role in l-flow-associated NO synthesis. This, in turn, increases levels of NO and promotes an overall cardioprotective effect (291).
6.1.2. D-flow promotes atherosclerosis
In regions of d-flow, ECs undergo several changes including becoming polygonal and poorly aligned and undergo senescence and apoptosis. Moreover, they also secrete factors that lead to vasoconstriction and coagulation, promoting the generation of ROS, platelet aggregation, EC activation, senescence, inflammation, dysfunction, permeability, and endoMT as depicted in Figure 7 (207, 292, 293). This disruption of EC homeostasis (294) and proatherogenic signaling can worsen atherosclerotic CVD (207, 292). D-flow is a well-known stress stimulus that can cause vascular senescence, and several mechanisms such as endoMT (208), NO synthesis, telomere shortening (295, 296), oxidative stress (188), and chronic inflammation (297) have been proposed to explain how d-flow mediate atherosclerotic plaque formation (293, 298–302).
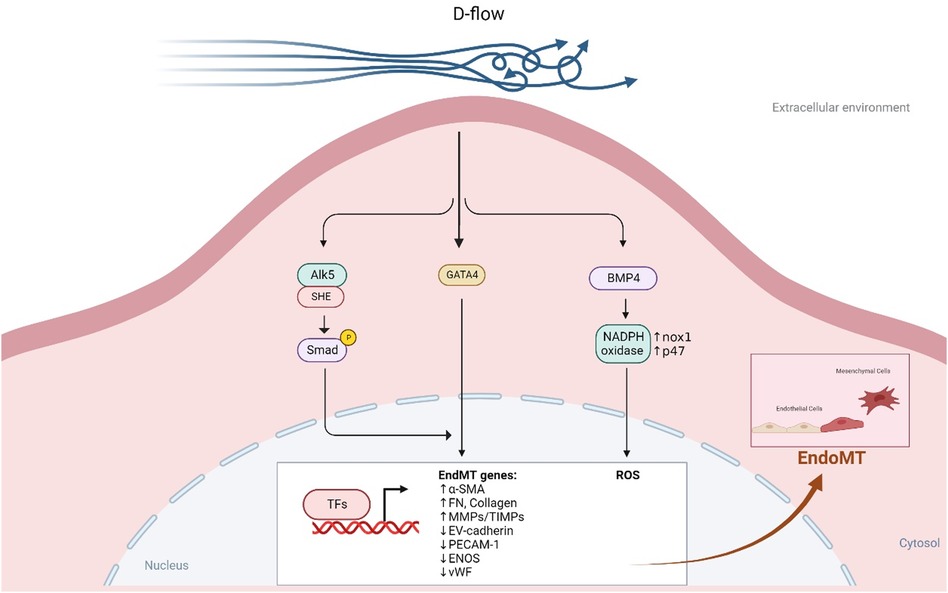
Figure 7. D-flow promotes endoMT (All the figures were made in Biorender.com).
6.1.3. D-flow induces endoMT
D-flow and l-flow have different effects on the accessibility of transcription factor binding motifs and cis-regulatory elements, leading to differential regulation of EC-specific gene expression at both genomic and epigenomic levels. Downstream factors, such as kruppel-like factors 2/4 (KLF2/4), are enriched in ECs exposed to l-flow, while known transcription factor binding motifs (RELA, Fos/Jun, and TEAD1) and novel transcription factor binding motifs (TEF, ETV3, and STAT1) are enriched in ECs exposed to d-flow (286).
Research using single-cell RNA-seq and single-cell assay for transposase accessible chromatin sequencing (ATACseq) in mice after partial carotid ligation showed that d-flow can reprogram ECs from athero-protective cells to atherogenic proinflammatory cells, endoMT cells, hematopoietic stem cells, endothelial progenitor cells, and cells with immune cell-like phenotypes (286). Mechanistically, d-flow can activate the TGFβR1 (ALK5) mechano-sensor in ECs, leading to downstream activation of molecules involved in d-flow-mediated endoMT, including TGFβR1-associated Src homology and collagen (Shc) (279). D-flow also promotes BMP4 production through Nox1-based NADPH oxidase, subsequently increasing ROS synthesis, a potential activator of endoMT (213, 303, 304) (see Figure 7). By contrast, l-flow prevents endoMT by upregulating the expression of extracellular matrix protein tenascin-X through KLF4 in mouse and human ECs, which binds TGFβ and hinders TGFβ-TGFβRs binding, thereby suppressing TGFβ-induced endoMT and atherogenesis (305).
Through a series of in vivo and in vitro studies, Mahmoud and colleagues have demonstrated that d-flow upregulates the expression of twist family bHLH transcriptional factor 1 (TWIST1) via a mechanism regulated by the developmental transcription factor GATA4. Microarray data revealed that both GATA4 and TWIST1 expression were enriched in ECs exposed to d-flow regions in the porcine aorta in vivo. In vitro studies using controlled flow systems also showed that GATA4 and TWIST1 expression were enhanced in cultured ECs exposed to d-flow compared to non-d-flow conditions (306, 307). Activation of the GATA4-TWIST1 pathway by d-flow upregulated SNAIL, a downstream transcription factor of TWIST1, and a positive regulator of endoMT markers such as SLUG, N-cadherin, and αSMA. These events subsequently induce proliferation, inflammation, permeability, and endoMT in ECs exposed to d-flow, leading to atherogenesis (308).
Chen and colleagues have found that d-flow, along with soluble inflammatory factors (IFNγ, TNFα, and IL1β), can decrease the expression of fibroblast growth factor receptor 1 (FGFR1) in ECs, leading to activation of downstream TGFβ signaling and subsequent endoMT activation (208). Studies in patients with coronary artery diseases have shown that disease severity correlates with FGFR1 expression, TGFβ signaling activation, and the extent of endoMT activation. EndoMT activation enhances atherosclerotic plaque formation by modulating fibronectin deposition and upregulating VCAM1 and ICAM1, which in turn recruit circulatory monocytes and leukocytes while generating new mesenchymal cells (208, 309). The extent of endoMT activation is positively correlated with the unstable phenotype of plaques, which can be driven by increased synthesis of collagen-MMPs by endoMT-derived fibroblast-like cells in atherosclerotic plaques (216).
Zhao and colleagues used a three-dimensional micro-engineered human coronary artery-on-a-chip model to demonstrate that d-flow drives ECs towards a proinflammatory endoMT phenotype, accompanied by elevated expression of VCAM1 and ICAM1, which can mediate recruitment of monocytes, leukocytes, and macrophages to plaque sites (310, 311). Activated macrophages and T lymphocytes release cytokines, which further increase MMP production (312), leading to degradation of the extracellular matrix and ultimately causing plaque ruptures. Macrophages can also directly induce apoptosis of vascular smooth muscle cells (313–315), which are responsible for collagen I and III production, critical factors for plaque stability and repair (312, 316). All these processes work together to prime atherosclerotic plaques for rupture (312, 317).
6.1.4. D-flow alters NO signaling
Altered synthesis or activity of NO and oxidative stress play a crucial role in d-flow-induced EC dysfunction (318). The levels of BH4 is reduced by d-flow (291) and aging (181, 188), leading to the production of superoxide (O2–), a potent ROS, and pro-atherosclerotic vascular dysfunction (191). In the absence of BH4, NO synthesis can result in the production of superoxide from the ferrous-dioxygen complex, leading to eNOS uncoupling, which could explain why d-flow produces ROS but decreases NOS (191).
6.2. Blood flow in cancer metastasis
A high-resolution model was developed to simulate the spread of circulating tumor cells (CTCs) through the bloodstream. The model includes stochastic adhesion events and uses a realistic model of global blood circulation to simulate cancer cell trajectories. The authors compared the model's predictions with data from thousands of human autopsies for seven different solid tumors, including lung, prostate, pancreatic, and colorectal cancers. They found that, on average, 40% of the variation in the metastatic distribution could be attributed to blood circulation (319–328).
6.3. D-flow promotes endoMT through SUMOylation
Activation of the transcription factor NFκB is critical for initiating an inflammatory genetic program critical that contributes to atherogenesis, which is triggered by d-flow (263, 272, 283, 329, 330). Ganguli and colleagues investigated the mechanisms underlying NFκB activation and found that d-flow regulates NFκB by modulating Ras-GTPase (331). Furthermore, Mabb and colleagues demonstrated that in response to genotoxic stress stimuli, the signal transducer and activator of transcription (STAT) activates NFkB through SUMOylating the NFkB essential modulator (NEMO) (332).
D-flow induces various posttranslational modifications that promote inflammatory signaling, including SUMOylation (262). SUMOylation is a reversible and dynamic process that occurs in approximately 20% of proteins and is associated with various diseases such as atherosclerosis and cancer (262, 263, 333–336). This process requires the activity of sentrin-specific proteases (SENPs) that catalyze the covalent attachment and detachment of SUMO to and from Lysine (K) residues on substrates (337). Six SENP isoforms have been identified (SENP1/2/3/5/6/7), each with substrate specificities and subcellular localizations. Among these isoforms, SENP2 binds NEMO, inhibits NEMO SUMOylation (338), and suppresses DNA damage-induced NFkB activation (339).
Knockout of SENP2 in mice leads to developmental defects in trophoblast stem cell niches and lineages due to dysregulation of the Mdm2-p53 signaling pathway in the placenta (340). In human hepatocellular carcinoma, SENP2 acts as a tumor suppressor by modulating β-catenin stability (341). Similarity, in osteosarcoma cells, SENP2 negatively regulates proliferation, migration, and invasion (342). It is important to note that NEMO is not the only substrate of SENP2, given its broad deSUMOylation activity (343). SENP2 contains multiple nuclear localization and export signals, allowing it to shuttle between the nucleus and cytoplasm (344), a process regulated by posttranslational modifications (263, 334). Depletion of SENP2 increases SUMOylation of ERK5 and p53 (338), leading to EC inflammation and apoptosis, respectively (263).
7. Possible molecular mechanisms for the development of atherosclerosis in cancer survivors
7.1. Both d-flow and cancer treatment can accelerate premature senescence
Atherogenesis predominantly occurs in regions of vascular ECs exposed to d-flow but not l-flow (287, 336, 337, 345–349). ECs exposed to d-flow exhibit accelerated generation of ROS, telomere shortening, and telomere dysfunction, leading to SIPS (71, 295–297, 350–352). During atherogenesis, senescent cells accumulate in d-flow regions as shown by Warboys and colleagues. They demonstrated that d-flow induces premature senescence via activation of the p53-p21 signaling pathway, which is attenuated by pharmacological activation of sirtuin 1 (SIRT1) (352).
Similarly, many cancer treatments, including radiation therapy and chemotherapy, can induce cellular senescence, a phenomenon known as TIS (206, 353, 354). While non-senescent inflammatory cells secret cytokines that activate a temporary inflammatory stage in ECs, subsets of cancer treatment-induced senescent cells secrete proinflammatory cytokines, chemokines, growth factors pro-angiogenic factors, and ROS, known as SASP, that promote inflammation and aberrant cell growth (10–12, 68, 355–357). SASP can be induced by both cancer treatment and d-flow and can produce long lasting inflammation (PISP) that instigates atherogenesis. Therefore, SASP may be an important target for mitigation strategies (79, 354, 358).
7.2. Both d-flow and cancer treatment can reduce BH4
Similar to d-flow (291), cancer therapy (188) reduce the levels of BH4 (181). Radiation creates an oxidative environment, which promotes the oxidation of BH4 into BH2 and other oxidized byproducts, causing the uncoupling of eNOS and leading to an increase in superoxide synthesis. BH4 deficiency, eNOS uncoupling, and vascular dysfunction can occur in irradiated cells due to the downregulation of GCH1, the enzyme that catalyzes the second step in BH4 de novo biosynthesis (359). The decreased BH4:BH2 ratio resulting from d-flow produces pro-atherosclerotic vascular dysfunction (181, 360, 361). EC dysfunction resulting from changes in the BH4:BH2 ratio and subsequent eNOS uncoupling is a characteristic feature of various diseases, including atherosclerosis.
7.3. Both d-flow and cancer treatment can activate p90RSK
Different flow patterns activate distinct kinase pathways that affect EC morphology and function. L-flow transiently activates both AMP-activated protein kinase (AMPK) and protein kinase B (PKB, or AKT), while d-flow activates only PKB (362, 363). In addition, our studies have shown that p90RSK, a redox sensitive kinase, is specifically activated not only d-flow, but not l-flow. Moreover, we have also demonstrated that p90RSK is activated not only by d-flow but also by chemotherapy and IR, suggesting its potential role on mediating cellular response to these stimuli (88, 287, 335, 364).
Radiation therapy is a common treatment modality for solid tumors (365), with over 30% of cancer patients in the United States receiving radiation therapy as part of their treatment plan, often combined with chemotherapy, immunotherapy, or surgery as first-line therapy (366). However, thoracic radiotherapy can result in delayed CVD development (367). Studies have shown that radiation can cause endoMT, which contributes to the development of atherosclerotic CVD and CAFs in cancer (209, 368–370), as discussed above.
To investigate the molecular mechanism underlying atherosclerotic CVD development after thoracic radiotherapy, Kim and colleagues exposed human aortic ECs to 5Gy of IR for 24 h and observed an increased expression of fibroblast markers αSMA and FSP1 accompanied by a decreased expression of EC markers CD31 and VE-cad. The authors also noted that oxLDL increases IR-induced endoMT, which contributes to atherosclerotic plaque formation (209). Similarly, irradiation of human pulmonary artery ECs induces endoMT with an increased expression of hypoxia inducible factor 1α (HIF1α) and activation of TGFβR1/SMAD signaling (368).
Recent research by Choi and colleagues reported that in human umbilical vein ECs exposed to radiation, TRP53 expression increases while TGFβ2 decreases endoMT. siRNA-mediated TRP53 silencing abrogated radiation-induced endoMT, resulting in a decreased expression of endoMT-related transcription factors such as SNAIL1/2 and zinc finger E-box-binding homeobox 2 (ZEB2) (370). These findings highlight the complex interplay between radiation-induced signaling pathways and the development of atherosclerotic CVD in cancer survivors. Furthermore, as 30hosphory earlier, p90RSK activation plays a potential role in mediating cellular response to radiation and chemotherapy, further underscoring its potential involvement in the development of atherosclerotic CVD in cancer survivors.
7.4. p90RSK activation regulates SUMOylation by driving SENP2 phosphorylation
We have found that d-flow activates p90RSK (287, 335), which phosphorylates SENP2 T368 (335). This leads to the export of SENP2 from the nucleus, causing a loss of its deSUMOylation activity within the nucleus. As a result, there is an increase in the SUMOylation of ERK5 and p53, ultimately leading to atherosclerosis. These findings help explain why atherosclerotic plaques mainly develop in vessel bends or bifurcations, where d-flow is generated (191, 291, 371, 372). Despite the growing understanding of the effects of flow on ECs (349), the mechanisms by which l-flow inhibits EC turnover, maintains EC homeostasis, and prevents atherogenesis remain incompletely understood.
Furthermore, we have discovered that cytoplasmic SENP2 plays a role in inhibiting the SUMOylation of the membrane-associated protein MAGI1, which is a tight and adherent junction protein with a newly identified role in EC function and atherosclerosis (334). Recently, we also reported that SENP2 regulates the SUMOylation of focal adhesion kinase (FAK) at K152 through a mechanism that is independent of T368 phosphorylation (373, 374). These findings suggest that the regulation of SUMOylation by SENP2 through post-translational modifications is more complex than previously thought and requires further investigation. For example, it is important to understand how the SUMOylation machinery is targeted to cell membranes and how it regulates post-modifications. Mass-spectrometry-based phospho-proteomics has revealed that SENP2 can be phosphorylated at S32, S333, and S344 residues (375, 376). However, the mechanisms underlying the31hosphorylateon of these residues by kinases activated during altered flow, as well as the associated regulatory mechanisms and biological consequences, remain to be investigated.
Figure 8 illustrates the plausible mechanism of endoMT development by d-flow, chemotherapy, or radiation through the activation of p90RSK.
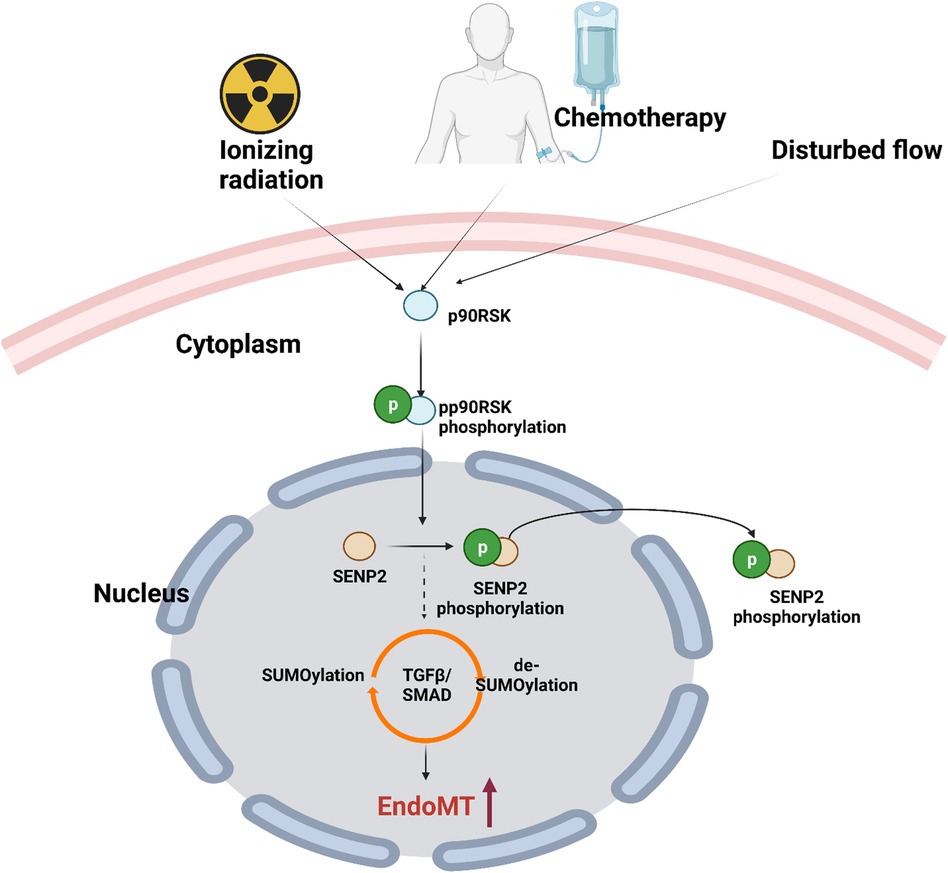
Figure 8. Endomt activation by d-flow, chemotherapy, or radiation therapy through the activation of p90RSK (All the figures were made in Biorender.com).
7.5. p90RSK activation accelerates SIPS by driving TERF2IP posttranslational modifications
Post-translational modifications are crucial for regulating the cellular localization and function of TERF2IP, which is necessary for protecting telomeres (295, 377). Our recent studies have revealed that activation of p90RSK leads to phosphorylation of TERF2IP S205. This phosphorylation event promotes TERF2IP nuclear export, which leads to the loss of TERF2IP telomeric protection and TRF2 removal from telomeric DNA, ultimately accelerating telomere shortening and cellular senescence (see Figure 9) (295). Additionally, TERF2IP can be modified by Small Ubiquitin-related Modifier (SUMO), which disrupts the TERF2IP-TRF2 association and leads to nuclear export (377). Both scenarios result in cytoplasmic TERF2IP-mediated activation of nuclear factor kappa B (NFkB) p65 subunit, ultimately leading to cellular senescence, inflammation, and atherosclerosis (295, 377–381).
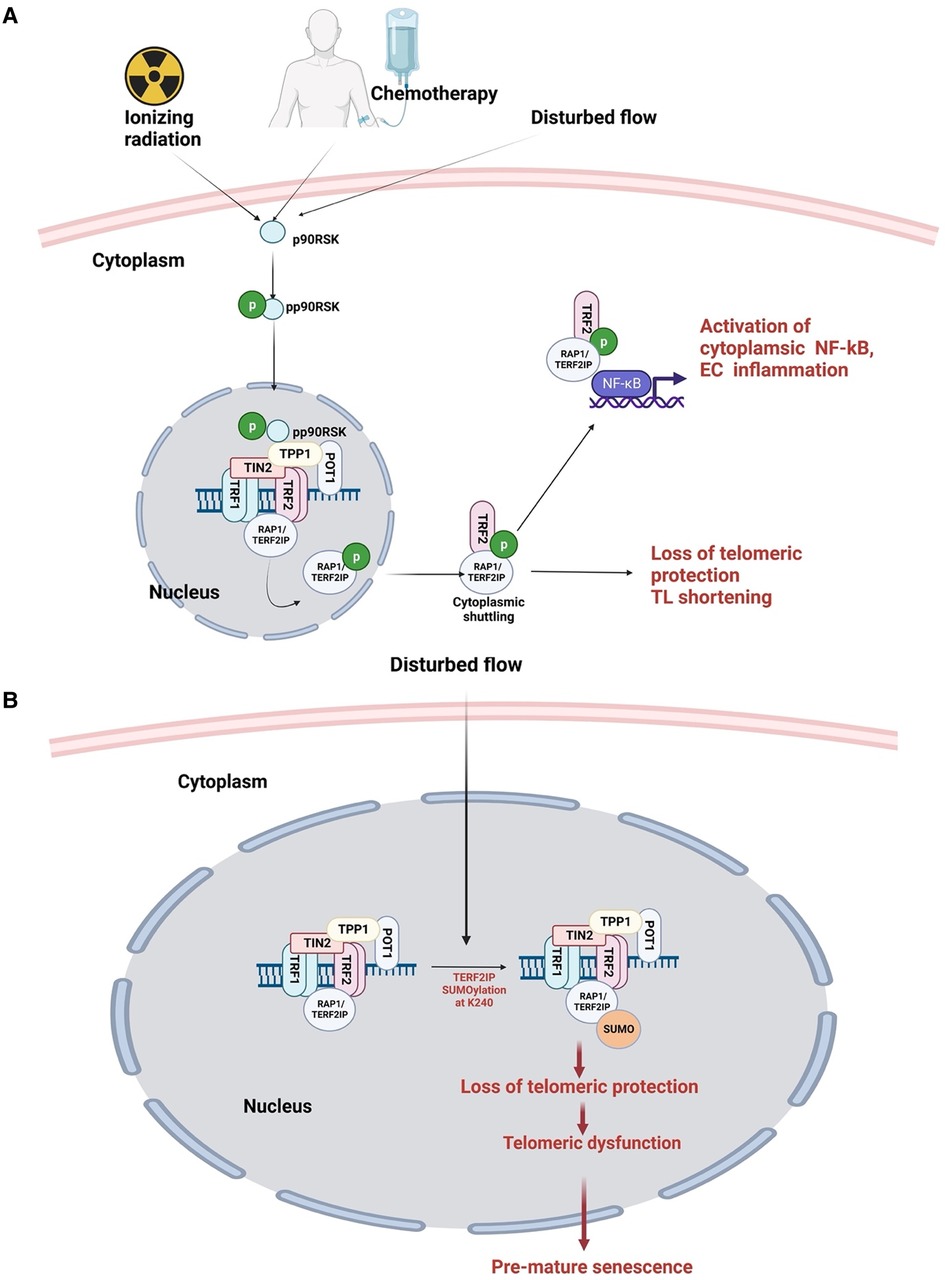
Figure 9. Posttranslational modifications of TERF2IP induced by d-flow and cancer treatment disrupt Shelterin and induce premature senescence. (A) External stimuli, such as radiation, chemotherapeutic agents, and d-flow, activate p90RSK through phosphorylation. Activated p90RSK translocates to the nucleus and phosphorylates TERF2IP (RAP1), leading to TERF2IP nuclear export and loss of telomeric protection. Unprotected telomere becomes susceptible to stress-induced damage, resulting in telomere shortening. Cytoplasmic TERF2IP activates p65 NFκB, triggering EC inflammation. (B) D-flow induces SUMOylation of TERF2IP at K240, causing loss of telomeric protection, telomeric dysfunction, and premature senescence (All the figures were made in Biorender.com).
Notably, TERF2IP expression is increased in d-flow regions of atherosclerotic plaques and has been linked to various cancers, such as breast cancers, gastric carcinoma, non-small cell lung cancer, and mantle cell lymphoma (106, 382–384). In metastatic colon cancer, TERF2IP activates NFkB, leading to the phosphatase of regenerating liver 3 (PRL3) activation and increased TERF2IP expression and nuclear export, ultimately promoting cancer cell invasiveness and metastasis (378–381, 385).
In a study using mice that received 2 Gy whole-body γ-radiation, it was found that VCAM1 expression is upregulated in the d-flow, further implicating the role of p90RSK-mediated TERF2IP S205 phosphorylation in the progression of atherosclerotic CVD progression induced by d-flow and chemoradiation therapies (295, 377).
8. Senotherapies for the treatment of age-related diseases
The accumulation of senescent cells during RS or SIPS is one of the challenges in the treatment of atherosclerotic CVD and other age-related diseases. Therefore, eliminating senescent cells using senolytic drugs or attenuating the SASP without inducing apoptosis of senescent cells using senomorphic drugs is considered a potential treatment strategy nowadays. Table 5 lists senotherapies commonly used in clinical or preclinical settings.
Table 5. List of senotherapies used for age related diseases (386).
9. Discussion
Our aim with this review is to offer a comprehensive overview of the interactions between multiple key components that contribute to SIPS and the development of atherosclerotic CVD in cancer survivors. We intend to identify potential therapeutic targets for the prevention and treatment of atherosclerotic CVD and provide new insights into the molecular mechanisms underlying atherosclerosis development in cancer survivors. We believe that the information presented in this review will be beneficial to a diverse audience of researchers and clinicians in the fields of cardiology, oncology, and molecular biology, as well as to patients and their families who may be concerned about the long-term health effects of cancer treatment. Finally, we emphasize the need for further research, particularly in ECs, to gain a better understanding of the mechanisms involved in the development of atherosclerosis in cancer survivors after receiving cancer therapy.
A potential link between BH4 and eNOS uncoupling has been proposed, but the precise mechanism underlying this process remains unclear (188). Preventing this oxidative process may limit the impact of aging on vascular function (182). Studies investigating the acute BH4 supplementation on vascular function have shown positive short-term effects, likely by preventing eNOS uncoupling and ROS formation, leading to overall cardioprotective NO synthesis. However, chronic administration of BH4 appears to have little long-term effect, possibly due to rapid oxidation into its inactivated form, BH2 (181). A recycling pathway that converts BH2 back to BH4 through DHFR exists, and reduced DHFR levels lead to altered BH4:BH2 ratios and eNOS uncoupling-derived EC dysfunction (181). Deficiencies in NO bioavailability, in which BH4 plays a critical role, are the primary cause of age-related reduction in endothelium-dependent dilation (389). This vascular impairment is a hallmark of the physiological aging process and is also involved in the pathophysiology of age-related conditions, as well as the deleterious effects of cancer treatment on the vasculature.
The impact of altered hemodynamic shear stress, either alone and in combination with cancer treatment, on DDR signaling pathways and SIPS in cancer survivors is not well understood and requires further investigation. However, it is possible that d-flow and cancer therapy accelerate premature senescence through p90RSK-driven posttranslational modifications of TERF2IP and disruption of the Shelterin complex, which is formed by TRF1, TRF2, TIN2, TPP1, POT1, and TERF2IP/RAP1 (121, 122). Alterations in any of these Shelterin proteins cause aberrant telomere protection and often lead to a pro-senescent stress-induced phenotype associated with disease states (122).
Author contributions
JA and NL were responsible for the manuscript's structure design. PB, JR, and NL contributed to writing and editing the manuscript. PB and KC created all the figures in Biorender.com. All authors contributed to the article and approved the submitted version.
Funding
This study was partially supported by funding from the National Institutes of Health (NIH) to JA, NL, and JC (HL149303 and HL163857), JA (AI156921), JC (HL148338 and HL157790), and NL (HL-134740 and HL157790).
Acknowledgments
All authors would like to thank James M. Kasper (Houston Methodist Research Institute) for a critical reading of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ATM, ataxia-telangiectasia-mutated; ATR, ataxia-telangiectasia and Rad3-related; BH4, Tetrahydrobiopterin (THB, INN); CAFs, cancer-associated fibroblasts, CD31, Cluster of Differentiation 31; CDH5, vascular endothelial cadherin; CHK, cell-cycle checkpoint kinase; CVD, cardiovascular disease; D-flow, disturbed flow; DDR, DNA damage response; ECs, endothelial cells; EDA, extra domain A; endoMT, endothelial-to-mesenchymal transition; eNOS, endothelial nitric oxide synthase; ERK, extracellular-signal regulated kinase; FAP, fibroblast activating protein; FSP1, fibroblast specific protein 1; IL-1β, interleukin β; IR, ionizing radiation; L-flow, laminar flow; NEMO, NF-kB essential modulator; NFκB, nuclear factor kappa B; NO, nitric oxide; NRTS, new Rap1 targets at senescence; PECAM-1, platelet endothelial cell adhesion molecule-1; POT1, Protection telomeres 1; ROS, reactive oxygen species; RPA, replication protein A; TERF2IP (RAP1), TERF2 interacting protein; TGFβ-R1, transforming growth factor beta receptor 1; TIE1, tyrosine kinase with immunoglobulin-like and EGF-like domains 1; TIE2, TEK receptor tyrosine kinase; TIN2, TRF1-interacting protein 2; TRF1/2, Telomere Repeat Binding Factor 1/2; TPP1, Telomere Protection Protein 1; TME, tumor microenvironment; TNF-α, tumor necrosis factor-alpha. SASP, senescence-associated secretory phenotype; SaβG, senescence associated
References
1. Luu J, Palczewski K. Human aging and disease: lessons from age-related macular degeneration. Proc Natl Acad Sci U S A. (2018) 115:2866–72. doi: 10.1073/pnas.1721033115
2. Armenian SH, Armstrong GT, Aune G, Chow EJ, Ehrhardt MJ, Ky B, et al. Cardiovascular disease in survivors of childhood cancer: insights into epidemiology, pathophysiology, and prevention. J Clin Oncol. (2018) 36:2135–44. doi: 10.1200/JCO.2017.76.3920
3. Kirkham AA, Davis MK. Exercise prevention of cardiovascular disease in breast cancer survivors. J Oncol. (2015) 2015:917606. doi: 10.1155/2015/917606
4. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics-2022 update: a report from the American heart association. Circulation. (2022) 145:e153–639. doi: 10.1161/cir.0000000000001052
5. Grundy SM, Pasternak R, Greenland P, Smith S Jr, Fuster V. Assessment of cardiovascular risk by use of multiple-risk-factor assessment equations: a statement for healthcare professionals from the American heart association and the American college of cardiology. Circulation. (1999) 100:1481–92. doi: 10.1161/01.CIR.100.13.1481
6. Velusamy R, Nolan M, Murphy A, Thavendiranathan P, Marwick TH. Screening for coronary artery disease in cancer survivors: JACC: cardioOncology state-of-the-art review. JACC CardioOncol. (2023) 5:22–38. doi: 10.1016/j.jaccao.2022.12.007
7. Florido R, Daya NR, Ndumele CE, Koton S, Russell SD, Prizment A, et al. Cardiovascular disease risk among cancer survivors: the atherosclerosis risk in communities (ARIC) study. J Am Coll Cardiol. (2022) 80:22–32. doi: 10.1016/j.jacc.2022.04.042
8. Zhang X, Pawlikowski M, Olivo-Marston S, Williams KP, Bower JK, Felix AS. Ten-year cardiovascular risk among cancer survivors: the national health and nutrition examination survey. PLoS One. (2021) 16:e0247919. doi: 10.1371/journal.pone.0247919
9. Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. (2011) 25:1245–61. doi: 10.1101/gad.625811
10. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
11. Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. (2012) 23:2240–52. doi: 10.1091/mbc.e11-11-0926
12. Gong YN, Crawford JC, Heckmann BL, Green DR. To the edge of cell death and back. FEBS J. (2019) 286:430–40. doi: 10.1111/febs.14714
13. Saleh T, Tyutynuk-Massey L, Cudjoe EK, Idowu MO, Landry JW, Gewirtz DA. Non-cell autonomous effects of the senescence-associated secretory phenotype in cancer therapy. Front Oncol. (2018) 8:164. doi: 10.3389/fonc.2018.00164
14. Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, et al. Tumor cell escape from therapy-induced senescence. Biochem Pharmacol. (2019) 162:202–12. doi: 10.1016/j.bcp.2018.12.013
15. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. (2018) 553:96–100. doi: 10.1038/nature25167
16. Milanovic M, Yu Y, Schmitt CA. The senescence-stemness alliance—a cancer-hijacked regeneration principle. Trends Cell Biol. (2018) 28:1049–61. doi: 10.1016/j.tcb.2018.09.001
17. Coppé J-P, Rodier F, Patil CK, Freund A, Desprez P-Y, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. (2011) 286:36396–403. doi: 10.1074/jbc.M111.257071
18. Ferrand M, Kirsh O, Griveau A, Vindrieux D, Martin N, Defossez P-A, et al. Screening of a kinase library reveals novel pro-senescence kinases and their common NF-kappaB-dependent transcriptional program. Aging (Albany NY). (2015) 7:986–1003. doi: 10.18632/aging.100845
19. Muñoz DP, Yannone SM, Daemen A, Sun Y, Vakar-Lopez F, Kawahara M, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight. (2019) 5(14):e124716. doi: 10.1172/jci.insight.124716
20. Dou Z, Berger SL. Senescence elicits stemness: a surprising mechanism for cancer relapse. Cell Metab. (2018) 27:710–1. doi: 10.1016/j.cmet.2018.03.009
21. Toda N. Age-related changes in endothelial function and blood flow regulation. Pharmacol Ther. (2012) 133:159–76. doi: 10.1016/j.pharmthera.2011.10.004
22. Vyssoulis GP, Karpanou EA, Liakos CI, Kyvelou S-MG, Tzamou VE, Michaelides AP, et al. Cardiovascular risk factor(s) prevalence in Greek hypertensives. Effect of gender and age. J Hum Hypertens. (2012) 26:443–51. doi: 10.1038/jhh.2011.55
23. Tarumi T, Zhang R. Cerebral blood flow in normal aging adults: cardiovascular determinants, clinical implications, and aerobic fitness. J Neurochem. (2018) 144:595–608. doi: 10.1111/jnc.14234
24. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res. (2018) 123:825–48. doi: 10.1161/CIRCRESAHA.118.312563
25. Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. Oxidative stress, aging, and diseases. Clin Interv Aging. (2018) 13:757–72. doi: 10.2147/CIA.S158513
26. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. (2020) 288:518–36. doi: 10.1111/joim.13141
27. Huang Q, Gan Y, Yu Z, Wu H, Zhong Z. Endothelial to mesenchymal transition: an insight in atherosclerosis. Front Cardiovasc Med. (2021) 8:734550. doi: 10.3389/fcvm.2021.734550
28. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. (2016) 118:535–46. doi: 10.1161/CIRCRESAHA.115.307611
29. Wu CM, Zheng L, Wang Q, Hu YW. The emerging role of cell senescence in atherosclerosis. Clin Chem Lab Med. (2020) 59:27–38. doi: 10.1515/cclm-2020-0601
30. Dominic A, Banerjee P, Hamilton DJ, Le NT, Abe JI. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. (2020) 37:101614. doi: 10.1016/j.redox.2020.101614
31. Morgan RG, Ives SJ, Lesniewski LA, Cawthon RM, Andtbacka RHI, Noyes RD, et al. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol. (2013) 305:H251–258. doi: 10.1152/ajpheart.00197.2013
32. Rossman MJ, Kaplon RE, Hill SD, McNamara MN, Santos-Parker JR, Pierce GL, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. (2017) 313:H890–5. doi: 10.1152/ajpheart.00416.2017
33. Schosserer M, Grillari J, Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. (2017) 7:278. doi: 10.3389/fonc.2017.00278
34. Yang J, Liu M, Hong D, Zeng M, Zhang X. The paradoxical role of cellular senescence in cancer. Front Cell Dev Biol. (2021) 9:722205. doi: 10.3389/fcell.2021.722205
35. Chambers CR, Ritchie S, Pereira BA, Timpson P. Overcoming the senescence-associated secretory phenotype (SASP): a complex mechanism of resistance in the treatment of cancer. Mol Oncol. (2021) 15:3242–55. doi: 10.1002/1878-0261.13042
36. Machado-Oliveira G, Ramos C, Marques ARA, Vieira OV. Cell senescence, multiple organelle dysfunction and atherosclerosis. Cells. (2020) 9(10):2146. doi: 10.3390/cells9102146
37. Moriya J, Minamino T. Angiogenesis, cancer, and vascular aging. Front Cardiovasc Med. (2017) 4:65. doi: 10.3389/fcvm.2017.00065
38. Falchetti ML, Mongiardi MP, Fiorenzo P, Petrucci G, Pierconti F, D’Agnano I, et al. Inhibition of telomerase in the endothelial cells disrupts tumor angiogenesis in glioblastoma xenografts. Int J Cancer. (2008) 122:1236–42. doi: 10.1002/ijc.23193
39. Zaccagnini G, Gaetano C, Della Pietra L, Nanni S, Grasselli A, Mangoni A, et al. Telomerase mediates vascular endothelial growth factor-dependent responsiveness in a rat model of hind limb ischemia. J Biol Chem. (2005) 280:14790–8. doi: 10.1074/jbc.M414644200
40. Prasanna PG, Citrin DE, Hildesheim J, Ahmed MM, Venkatachalam S, Riscuta G, et al. Therapy-induced senescence: opportunities to improve anticancer therapy. J Natl Cancer Inst. (2021) 113:1285–98. doi: 10.1093/jnci/djab064
41. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. (2005) 19:2100–10. doi: 10.1101/gad.1346005
42. Shammas MA. Telomeres, lifestyle, cancer, and aging. Curr Opin Clin Nutr Metab Care. (2011) 14:28–34. doi: 10.1097/MCO.0b013e32834121b1
43. Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. (1988) 85:6622–6. doi: 10.1073/pnas.85.18.6622
44. Chuenwisad K, More-krong P, Tubsaeng P, Chotechuang N, Srisa-Art M, Storer RJ, et al. Premature senescence and telomere shortening induced by oxidative stress from oxalate, calcium oxalate monohydrate, and urine from patients with calcium oxalate nephrolithiasis. Front Immunol. (2021) 12:696486. doi: 10.3389/fimmu.2021.696486
45. de Magalhaes JP, Chainiaux F, Remacle J, Toussaint O. Stress-induced premature senescence in BJ and hTERT-BJ1 human foreskin fibroblasts. FEBS Lett. (2002) 523:157–62. doi: 10.1016/S0014-5793(02)02973-3
46. Hewitt G, Jurk D, Marques FDM, Correia-Melo C, Hardy T, Gackowska A, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. (2012) 3:708. doi: 10.1038/ncomms1708
47. Naka K, Tachibana A, Ikeda K, Motoyama N. Stress-induced premature senescence in hTERT-expressing ataxia telangiectasia fibroblasts. J Biol Chem. (2004) 279:2030–7. doi: 10.1074/jbc.M309457200
48. Campisi J. The biology of replicative senescence. Eur J Cancer. (1997) 33:703–9. doi: 10.1016/S0959-8049(96)00058-5
49. Campisi J. Aging and cancer: the double-edged sword of replicative senescence. J Am Geriatr Soc. (1997) 45:482–8. doi: 10.1111/j.1532-5415.1997.tb05175.x
50. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
51. de Lange T. How telomeres solve the end-protection problem. Science. (2009) 326:948–52. doi: 10.1126/science.1170633
52. Mathon NF, Lloyd AC. Cell senescence and cancer. Nat Rev Cancer. (2001) 1:203–13. doi: 10.1038/35106045
53. Vera E, Bernardes de Jesus B, Foronda M, Flores JM, & Blasco MA. The rate of increase of short telomeres predicts longevity in mammals. Cell Rep. (2012) 2:732–7. doi: 10.1016/j.celrep.2012.08.023
54. Allsopp RC, Chang E, Kashefi-Aazam M, Rogaev EI, Piatyszek MA, Shay JW, et al. Telomere shortening is associated with cell division in vitro and in vivo. Exp Cell Res. (1995) 220:194–200. doi: 10.1006/excr.1995.1306
55. Zinder JC, Olinares PDB, Svetlov V, Bush MW, Nudler E, Chait BT, et al. Shelterin is a dimeric complex with extensive structural heterogeneity. Proc Natl Acad Sci U S A. (2022) 119:e2201662119. doi: 10.1073/pnas.2201662119
56. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
57. Gire V, Dulic V. Senescence from G2 arrest, revisited. Cell Cycle. (2015) 14:297–304. doi: 10.1080/15384101.2014.1000134
58. Cawthon RM, Smith KR, O'Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet. (2003) 361:393–5. doi: 10.1016/S0140-6736(03)12384-7
59. Marcotte R, Wang E. Replicative senescence revisited. J Gerontol A Biol Sci Med Sci. (2002) 57:B257–269. doi: 10.1093/gerona/57.7.B257
60. Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. Br Med J. (2014) 349:g4227. doi: 10.1136/bmj.g4227
61. Aviv A. Genetics of leukocyte telomere length and its role in atherosclerosis. Mutat Res. (2012) 730:68–74. doi: 10.1016/j.mrfmmm.2011.05.001
62. Aviv A, Levy D. Telomeres, atherosclerosis, and the hemothelium: the longer view. Annu Rev Med. (2012) 63:293–301. doi: 10.1146/annurev-med-050311-104846
63. Rossiello F, Jurk D, Passos JF, d'Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. (2022) 24:135–47. doi: 10.1038/s41556-022-00842-x
64. Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. (1998) 396:84–8. doi: 10.1038/23962
65. Benetos A, Toupance S, Gautier S, Labat C, Kimura M, Rossi PM, et al. Short leukocyte telomere length precedes clinical expression of atherosclerosis: the blood-and-muscle model. Circ Res. (2018) 122:616–23. doi: 10.1161/CIRCRESAHA.117.311751
66. Toussaint O, Medrano EE, von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol. (2000) 35:927–45. doi: 10.1016/S0531-5565(00)00180-7
67. Zhang S, Liu X, Bawa-Khalfe T, Lu L-S, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
68. Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. (2010) 29:273–83. doi: 10.1007/s10555-010-9220-9
69. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
70. Nakamura AJ, Chiang YJ, Hathcock KS, Horikawa I, Sedelnikova OA, Hodes RJ, et al. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin. (2008) 1:6. doi: 10.1186/1756-8935-1-6
71. Dominic A, Banerjee P, Hamilton DJ, Le NT, Abe JI. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. (2020) 37:101614. doi: 10.1016/j.redox.2020.101614
72. Fleming AM, Burrows CJ. G-quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem Res Toxicol. (2013) 26:593–607. doi: 10.1021/tx400028y
73. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
74. Gioia U, Francia S, Cabrini M, Brambillasca S, Michelini F, Jones-Weinert CW, et al. Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci Rep. (2019) 9:6460. doi: 10.1038/s41598-019-42892-6
75. Schumann S, Scherthan H, Pfestroff K, Schoof S, Pfestroff A, Hartrampf P, et al. DNA damage and repair in peripheral blood mononuclear cells after internal ex vivo irradiation of patient blood with (131)I. Eur J Nucl Med Mol Imaging. (2022) 49(5):1447–55. doi: 10.1007/s00259-021-05605-8
76. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. (2012) 14:355–65. doi: 10.1038/ncb2466
77. Barinda AJ, Ikeda K, Nugroho DB, Wardhana DA, Sasaki N, Honda S, et al. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat Commun. (2020) 11:481. doi: 10.1038/s41467-020-14387-w
78. Victorelli S, Lagnado A, Halim J, Moore W, Talbot D, Barrett K, et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. (2019) 38:e101982. doi: 10.15252/embj.2019101982
79. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. (2008) 133:1006–18. doi: 10.1016/j.cell.2008.03.038
80. Rodier F, Coppé J-P, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
81. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. (2019) 19:439–53. doi: 10.1038/s41568-019-0156-2
82. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. (2012) 111:245–59. doi: 10.1161/CIRCRESAHA.111.261388
83. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
84. Vaitiekus D, Muckiene G, Vaitiekiene A, Maciuliene D, Vaiciuliene D, Ambrazeviciute G, et al. Impact of arterial hypertension on doxorubicin-based chemotherapy-induced subclinical cardiac damage in breast cancer patients. Cardiovasc Toxicol. (2020) 20:321–7. doi: 10.1007/s12012-019-09556-3
85. Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. (2006) 281:29568–74. doi: 10.1074/jbc.M603307200
86. Sohn JY, Kwak HJ, Rhim JH, Yeo EJ. AMP-activated protein kinase-dependent nuclear localization of glyceraldehyde 3-phosphate dehydrogenase in senescent human diploid fibroblasts. Aging (Albany NY). (2022) 14:4–27. doi: 10.18632/aging.203825
87. Bittles AH, Harper N. Increased glycolysis in ageing cultured human diploid fibroblasts. Biosci Rep. (1984) 4:751–6. doi: 10.1007/BF01128816
88. Kotla S, Zhang A, Imanishi M, Ko KA, Lin SH, Gi YJ, et al. Nucleus-mitochondria positive feedback loop formed by ERK5 S496 phosphorylation-mediated poly (ADP-ribose) polymerase activation provokes persistent pro-inflammatory senescent phenotype and accelerates coronary atherosclerosis after chemo-radiation. Redox Biol. (2021) 47:102132. doi: 10.1016/j.redox.2021.102132
89. Simoncini S, Chateau A-L, Robert S, Todorova D, Yzydorzick C, Lacroix R, et al. Biogenesis of pro-senescent microparticles by endothelial colony forming cells from premature neonates is driven by SIRT1-dependent epigenetic regulation of MKK6. Sci Rep. (2017) 7:8277. doi: 10.1038/s41598-017-08883-1
90. Burger D, Kwart DG, Montezano AC, Read NC, Kennedy CRJ, Thompson CS, et al. Microparticles induce cell cycle arrest through redox-sensitive processes in endothelial cells: implications in vascular senescence. J Am Heart Assoc. (2012) 1:e001842. doi: 10.1161/JAHA.112.001842
91. Riquelme JA, Takov K, Santiago-Fernández C, Rossello X, Lavandero S, Yellon DM, et al. Increased production of functional small extracellular vesicles in senescent endothelial cells. J Cell Mol Med. (2020) 24:4871–6. doi: 10.1111/jcmm.15047
92. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 9:645593. doi: 10.3389/fcell.2021.645593
93. Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci. (2019) 76:1901–18. doi: 10.1007/s00018-019-03048-x
94. Leon KE, Buj R, Lesko E, Dahl ES, Chen C-W, Tangudu NK, et al. DOT1l modulates the senescence-associated secretory phenotype through epigenetic regulation of IL1A. J Cell Biol. (2021) 220(8):e202008101. doi: 10.1083/jcb.202008101
95. Zhu W, Zhang X, Yu M, Lin B, Yu C. Radiation-induced liver injury and hepatocyte senescence. Cell Death Discov. (2021) 7:244. doi: 10.1038/s41420-021-00634-6
96. Nacarelli T, Liu P, Zhang R. Epigenetic basis of cellular senescence and its implications in aging. Genes (Basel). (2017) 8(12):343. doi: 10.3390/genes8120343
97. Tang J, Lobatto ME, Hassing L, van der Staay S, van Rijs SM, Calcagno C, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. (2015) 1(3):e1400223. doi: 10.1126/sciadv.1400223
98. Cervelli T, Borghini A, Galli A, Andreassi MG. DNA damage and repair in atherosclerosis: current insights and future perspectives. Int J Mol Sci. (2012) 13:16929–44. doi: 10.3390/ijms131216929
99. Mahmoudi M, Mercer J, Bennett M. DNA damage and repair in atherosclerosis. Cardiovasc Res. (2006) 71:259–68. doi: 10.1016/j.cardiores.2006.03.002
100. Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res. (2001) 88:733–9. doi: 10.1161/hh0701.088684
101. Carrassa L, Damia G. DNA damage response inhibitors: mechanisms and potential applications in cancer therapy. Cancer Treat Rev. (2017) 60:139–51. doi: 10.1016/j.ctrv.2017.08.013
102. Desai A, Yan Y, Gerson SL. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair (Amst). (2018) 66–67:24–9. doi: 10.1016/j.dnarep.2018.04.004
103. Uryga A, Gray K, Bennett M. DNA damage and repair in vascular disease. Annu Rev Physiol. (2016) 78:45–66. doi: 10.1146/annurev-physiol-021115-105127
104. Qiu Z, Oleinick NL, Zhang J. ATR/CHK1 inhibitors and cancer therapy. Radiother Oncol. (2018) 126:450–64. doi: 10.1016/j.radonc.2017.09.043
105. de Lange T. Shelterin-mediated telomere protection. Annu Rev Genet. (2018) 5l2:223–47. doi: 10.1146/annurev-genet-032918-021921
106. Deregowska A, Wnuk M. RAP1/TERF2IP-A multifunctional player in cancer development. Cancers (Basel). (2021) 13(23):5970. doi: 10.3390/cancers13235970
107. Hockemeyer D, Collins K. Control of telomerase action at human telomeres. Nat Struct Mol Biol. (2015) 22:848–52. doi: 10.1038/nsmb.3083
108. Tong AS, Stern JL, Sfeir A, Kartawinata M, de Lange T, Zhu X-D, et al. ATM and ATR signaling regulate the recruitment of human telomerase to telomeres. Cell Rep. (2015) 13:1633–46. doi: 10.1016/j.celrep.2015.10.041
109. Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. (1996) 18:173–9. doi: 10.1002/(SICI)1520-6408(1996)18:2%3C173::AID-DVG10%3E3.0.CO;2-3
110. Hiyama K, Hirai Y, Kyoizumi S, Akiyama M, Hiyama E, Piatyszek MA, et al. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol. (1995) 155:3711–5. doi: 10.4049/jimmunol.155.8.3711
111. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PLC, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. (1994) 266:2011–5. doi: 10.1126/science.7605428
112. Feng J, Funk WD, Wang S-S, Weinrich SL, Avilion AA, Chiu C-P, et al. The RNA component of human telomerase. Science. (1995) 269:1236–41. doi: 10.1126/science.7544491
113. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. (2019) 20:299–309. doi: 10.1038/s41576-019-0099-1
114. Liu H, Yang Y, Ge Y, Liu J, Zhao Y. TERC promotes cellular inflammatory response independent of telomerase. Nucleic Acids Res. (2019) 47:8084–95. doi: 10.1093/nar/gkz584
115. Eitan E, Tamar A, Yossi G, Peleg R, Braiman A, Priel E. Expression of functional alternative telomerase RNA component gene in mouse brain and in motor neurons cells protects from oxidative stress. Oncotarget. (2016) 7:78297–309. doi: 10.18632/oncotarget.13049
116. Gazzaniga FS, Blackburn EH. An antiapoptotic role for telomerase RNA in human immune cells independent of telomere integrity or telomerase enzymatic activity. Blood. (2014) 124:3675–84. doi: 10.1182/blood-2014-06-582254
117. Kang HJ, Choi YS, Hong S-B, Kim K-W, Woo R-S, Won SJ, et al. Ectopic expression of the catalytic subunit of telomerase protects against brain injury resulting from ischemia and NMDA-induced neurotoxicity. J Neurosci. (2004) 24:1280–7. doi: 10.1523/JNEUROSCI.4082-03.2004
118. Richardson GD, Breault D, Horrocks G, Cormack S, Hole N, Owens WA. Telomerase expression in the mammalian heart. FASEB J. (2012) 26:4832–40. doi: 10.1096/fj.12-208843
119. Iannilli F, Zalfa F, Gartner A, Bagni C, Dotti CG. Cytoplasmic TERT associates to RNA granules in fully mature neurons: role in the translational control of the cell cycle inhibitor p15INK4B. PLoS One. (2013) 8:e66602. doi: 10.1371/journal.pone.0066602
120. Bettin N, Oss Pegorar C, Cusanelli E. The emerging roles of TERRA in telomere maintenance and genome stability. Cells. (2019) 8(3):246. doi: 10.3390/cells8030246
121. Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. (2011) 11:161–76. doi: 10.1038/nrc3025
122. Mir SM, Samavarchi Tehrani S, Goodarzi G, Jamalpoor Z, Jahanbakhsh A, Khelghati N, et al. Shelterin complex at telomeres: implications in ageing. Clin Interv Aging. (2020) 15:827–39. doi: 10.2147/CIA.S256425
123. Fairall L, Chapman L, Moss H, de Lange T, Rhodes D. Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol Cell. (2001) 8:351–61. doi: 10.1016/S1097-2765(01)00321-5
124. Lin J, Countryman P, Buncher N, Kaur PEL, Zhang Y, et al. TRF1 and TRF2 use different mechanisms to find telomeric DNA but share a novel mechanism to search for protein partners at telomeres. Nucleic Acids Res. (2014) 42:2493–504. doi: 10.1093/nar/gkt1132
125. El Maï M, Wagner K-D, Michiels J-F, Ambrosetti D, Borderie A, Destree S, et al. The telomeric protein TRF2 regulates angiogenesis by binding and activating the PDGFRbeta promoter. Cell Rep. (2014) 9:1047–60. doi: 10.1016/j.celrep.2014.09.038
126. Martinez P, Blasco MA. Role of shelterin in cancer and aging. Aging Cell. (2010) 9:653–66. doi: 10.1111/j.1474-9726.2010.00596.x
127. Kim SH, Kaminker P, Campisi J. TIN2, a new regulator of telomere length in human cells. Nat Genet. (1999) 23:405–12. doi: 10.1038/70508
128. Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol Cell. (2017) 67:162. doi: 10.1016/j.molcel.2017.05.033
129. Frescas D, de Lange T. Binding of TPP1 protein to TIN2 protein is required for POT1a,b protein-mediated telomere protection. J Biol Chem. (2014) 289:24180–7. doi: 10.1074/jbc.M114.592592
130. Frescas D, de Lange T. TRF2-tethered TIN2 can mediate telomere protection by TPP1/POT1. Mol Cell Biol. (2014) 34:1349–62. doi: 10.1128/MCB.01052-13
131. Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol Cell. (2011) 44:647–59. doi: 10.1016/j.molcel.2011.08.043
132. Hu C, Rai R, Huang C, Broton C, Long J, Xu Y, et al. Structural and functional analyses of the mammalian TIN2-TPP1-TRF2 telomeric complex. Cell Res. (2017) 27:1485–502. doi: 10.1038/cr.2017.144
133. Ye JZ-S, Donigian JR, van Overbeek M, Loayza D, Luo Y, Krutchinsky AN, et al. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem. (2004) 279:47264–71. doi: 10.1074/jbc.M409047200
134. Khattar E, Tergaonkar V. Role of Rap1 in DNA damage response: implications in stem cell homeostasis and cancer. Exp Hematol. (2020) 90:12–7. doi: 10.1016/j.exphem.2020.08.009
135. Vertecchi E, Rizzo A, Salvati E. Telomere targeting approaches in cancer: beyond length maintenance. Int J Mol Sci. (2022) 23(7):3784. doi: 10.3390/ijms23073784
136. Lagah S, Tan I-L, Radhakrishnan P, Hirst RA, Ward JH, O’Callaghan C, et al. RHPS4 G-quadruplex ligand induces anti-proliferative effects in brain tumor cells. PLoS One. (2014) 9:e86187. doi: 10.1371/journal.pone.0086187
137. Zhou G, Liu X, Li Y, Xu S, Ma C, Wu X, et al. Telomere targeting with a novel G-quadruplex-interactive ligand BRACO-19 induces T-loop disassembly and telomerase displacement in human glioblastoma cells. Oncotarget. (2016) 7:14925–39. doi: 10.18632/oncotarget.7483
138. Ma Q, Liao H, Xu L, Li Q, Zou J, Sun R, et al. Autophagy-dependent cell cycle arrest in esophageal cancer cells exposed to dihydroartemisinin. Chin Med. (2020) 15:37. doi: 10.1186/s13020-020-00318-w
139. Gomez D, Wenner T, Brassart B, Douarre C, O’Donohue M-F, El Khoury V, et al. Telomestatin-induced telomere uncapping is modulated by POT1 through G-overhang extension in HT1080 human tumor cells. J Biol Chem. (2006) 281:38721–9. doi: 10.1074/jbc.M605828200
140. Cherfils-Vicini J, Gilson E. Inhibiting TRF1 upstream signaling pathways to target telomeres in cancer cells. EMBO Mol Med. (2019) 11:e10845. doi: 10.15252/emmm.201910845
141. Okamoto K, Iwano T, Tachibana M, Shinkai Y. Distinct roles of TRF1 in the regulation of telomere structure and lengthening. J Biol Chem. (2008) 283:23981–8. doi: 10.1074/jbc.M802395200
142. Broccoli D, Smogorzewska A, Chong L, de Lange T. Human telomeres contain two distinct myb-related proteins, TRF1 and TRF2. Nat Genet. (1997) 17:231–5. doi: 10.1038/ng1097-231
143. van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. (1997) 385:740–3. doi: 10.1038/385740a0
144. Muñoz P, Blanco R, de Carcer G, Schoeftner S, Benetti R, Flores JM, et al. TRF1 controls telomere length and mitotic fidelity in epithelial homeostasis. Mol Cell Biol. (2009) 29:1608–25. doi: 10.1128/MCB.01339-08
145. Martínez P, Thanasoula M, Muñoz P, Liao C, Tejera A, McNees C, et al. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. (2009) 23:2060–75. doi: 10.1101/gad.543509
146. Hohensinner PJ, Kaun C, Buchberger E, Ebenbauer B, Demyanets S, Huk I, et al. Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochim Biophys Acta. (2016) 1863:360–7. doi: 10.1016/j.bbamcr.2015.11.034
147. Minamino T, Komuro I. Role of telomere in endothelial dysfunction in atherosclerosis. Curr Opin Lipidol. (2002) 13:537–43. doi: 10.1097/00041433-200210000-00010
148. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. (2002) 105:1541–4. doi: 10.1161/01.CIR.0000013836.85741.17
149. Ilicheva NV, Podgornaya OI, Voronin AP. Telomere repeat-binding factor 2 is responsible for the telomere attachment to the nuclear membrane. Adv Protein Chem Struct Biol. (2015) 101:67–96. doi: 10.1016/bs.apcsb.2015.06.009
150. Morgan RG, Walker AE, Trott DW, Machin DR, Henson GD, Reihl KD, et al. Induced Trf2 deletion leads to aging vascular phenotype in mice associated with arterial telomere uncapping, senescence signaling, and oxidative stress. J Mol Cell Cardiol. (2019) 127:74–82. doi: 10.1016/j.yjmcc.2018.11.014
151. Karlseder J, Kachatrian L, Takai H, Mercer K, Hingorani S, Jacks T, et al. Targeted deletion reveals an essential function for the telomere length regulator Trf1. Mol Cell Biol. (2003) 23:6533–41. doi: 10.1128/MCB.23.18.6533-6541.2003
152. Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. (2009) 138:90–103. doi: 10.1016/j.cell.2009.06.021
153. Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, et al. Telomere attrition and Chk2 activation in human heart failure. Proc Natl Acad Sci U S A. (2003) 100:5378–83. doi: 10.1073/pnas.0836098100
154. Derevyanko A, Whittemore K, Schneider RP, Jiménez V, Bosch F, Blasco MA. Gene therapy with the TRF1 telomere gene rescues decreased TRF1 levels with aging and prolongs mouse health span. Aging Cell. (2017) 16:1353–68. doi: 10.1111/acel.12677
155. Chen L-Y, Zhang Y, Zhang Q, Li H, Luo Z, Fang H, et al. Mitochondrial localization of telomeric protein TIN2 links telomere regulation to metabolic control. Mol Cell. (2012) 47:839–50. doi: 10.1016/j.molcel.2012.07.002
156. Sullivan LB, Santos JH, Chandel NS. Mitochondria and telomeres: the promiscuous roles of TIN2. Mol Cell. (2012) 47:823–4. doi: 10.1016/j.molcel.2012.09.006
157. Lee JH, Jung M, Hong J, Kim MK, Chung IK. Loss of RNA-binding protein HuR facilitates cellular senescence through posttranscriptional regulation of TIN2 mRNA. Nucleic Acids Res. (2018) 46:4271–85. doi: 10.1093/nar/gky223
158. Mistry Y, Poolman T, Williams B, Herbert KE. A role for mitochondrial oxidants in stress-induced premature senescence of human vascular smooth muscle cells. Redox Biol. (2013) 1:411–7. doi: 10.1016/j.redox.2013.08.004
159. Billard P, Poncet DA. Replication stress at telomeric and mitochondrial DNA: common origins and consequences on ageing. Int J Mol Sci. (2019) 20(19):4959. doi: 10.3390/ijms20194959
160. Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. (2007) 445:506–10. doi: 10.1038/nature05454
161. Ye JZ-S, Hockemeyer D, Krutchinsky AN, Loayza D, Hooper SM, Chait BT, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. (2004) 18:1649–54. doi: 10.1101/gad.1215404
162. Grill S, Padmanaban S, Friedman A, Perkey E, Allen F, Tesmer VM, et al. TPP1 mutagenesis screens unravel shelterin interfaces and functions in hematopoiesis. JCI Insight. (2021) 6(9):e138059. doi: 10.1172/jci.insight.138059
163. Chen H, Liu X, Zhu W, Chen H, Hu X, Jiang Z, et al. SIRT1 ameliorates age-related senescence of mesenchymal stem cells via modulating telomere shelterin. Front Aging Neurosci. (2014) 6:103. doi: 10.3389/fnagi.2014.00103
164. Min S, Kwon SM, Hong J, Lee Y-K, Park TJ, Lim SB, et al. Mitoribosomal deregulation drives senescence via TPP1-mediated telomere deprotection. Cells. (2022) 11. doi: 10.3390/cells11132079
165. Loayza D, De Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. (2003) 423:1013–8. doi: 10.1038/nature01688
166. Yu Y, Tan R, Ren Q, Gao B, Sheng Z, Zhang J, et al. POT1 inhibits the efficiency but promotes the fidelity of nonhomologous end joining at non-telomeric DNA regions. Aging (Albany NY). (2017) 9:2529–43. doi: 10.18632/aging.101339
167. Colgin LM, Baran K, Baumann P, Cech TR, Reddel RR. Human POT1 facilitates telomere elongation by telomerase. Curr Biol. (2003) 13:942–6. doi: 10.1016/S0960-9822(03)00339-7
168. Wu Y, Poulos RC, Reddel RR. Role of POT1 in human cancer. Cancers (Basel). (2020) 12(10):2739. doi: 10.3390/cancers12102739
169. Kelich J, Aramburu T, van der Vis JJ, Showe L, Kossenkov A, van der Smagt J, et al. Telomere dysfunction implicates POT1 in patients with idiopathic pulmonary fibrosis. J Exp Med. (2022) 219(5):e20211681. doi: 10.1084/jem.20211681
170. Yeung F, Ramírez CM, Mateos-Gomez PA, Pinzaru A, Ceccarini G, Kabir S, et al. Nontelomeric role for Rap1 in regulating metabolism and protecting against obesity. Cell Rep. (2013) 3:1847–56. doi: 10.1016/j.celrep.2013.05.032
171. Cai Y, Sukhova GK, Wong HK, Xu A, Tergaonkar V, Vanhoutte PM, et al. Rap1 induces cytokine production in pro-inflammatory macrophages through NFkappaB signaling and is highly expressed in human atherosclerotic lesions. Cell Cycle. (2015) 14:3580–92. doi: 10.1080/15384101.2015.1100771
172. Rai R, Chen Y, Lei M, Chang S. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat Commun. (2016) 7:10881. doi: 10.1038/ncomms10881
173. Janoušková E, Nečasová I, Pavloušková J, Zimmermann M, Hluchý M, Marini V, et al. Human Rap1 modulates TRF2 attraction to telomeric DNA. Nucleic Acids Res. (2015) 43:2691–700. doi: 10.1093/nar/gkv097
174. Honda S, Ikeda K, Urata R, Yamazaki E, Emoto N, Matoba S. Cellular senescence promotes endothelial activation through epigenetic alteration, and consequently accelerates atherosclerosis. Sci Rep. (2021) 11:14608. doi: 10.1038/s41598-021-94097-5
175. Pardo B, Marcand S. Rap1 prevents telomere fusions by nonhomologous end joining. EMBO J. (2005) 24:3117–27. doi: 10.1038/sj.emboj.7600778
176. Sarthy J, Bae NS, Scrafford J, Baumann P. Human RAP1 inhibits non-homologous end joining at telomeres. EMBO J. (2009) 28:3390–9. doi: 10.1038/emboj.2009.275
177. Lototska L, Yue J, Li J, Giraud-Panis M, Songyang Z, Royle NJ, et al. Human RAP1 specifically protects telomeres of senescent cells from DNA damage. EMBO Rep. (2020) 21:e49076. doi: 10.15252/embr.201949076
178. Song S, Perez JV, Svitko W, Ricketts MD, Dean E, Schultz D, et al. Rap1-mediated nucleosome displacement can regulate gene expression in senescent cells without impacting the pace of senescence. Aging Cell. (2020) 19:e13061. doi: 10.1111/acel.13061
179. Platt JM, Ryvkin P, Wanat JJ, Donahue G, Ricketts MD, Barrett SP, et al. Rap1 relocalization contributes to the chromatin-mediated gene expression profile and pace of cell senescence. Genes Dev. (2013) 27:1406–20. doi: 10.1101/gad.218776.113
180. Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, Fukatsu A, Funami J, Ding Q-F, et al. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci U S A. (2006) 103:17018–23. doi: 10.1073/pnas.0607873103
181. Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal. (2014) 20:3040–77. doi: 10.1089/ars.2013.5566
182. Bouly M, Bourguignon MP, Roesch S, Rigouin P, Gosgnach W, Bossard E, et al. Aging increases circulating BH(2) without modifying BH(4) levels and impairs peripheral vascular function in healthy adults. Transl Res. (2021) 238:36–48. doi: 10.1016/j.trsl.2021.07.004
183. Hayashi T, Yano K, Matsui-Hirai H, Yokoo H, Hattori Y, Iguchi A. Nitric oxide and endothelial cellular senescence. Pharmacol Ther. (2008) 120:333–9. doi: 10.1016/j.pharmthera.2008.09.002
184. Nanni S, Grasselli A, Benvenuti V, Aiello A, Pantisano V, Re A, et al. The role of nuclear endothelial nitric oxide synthase in the endothelial and prostate microenvironments. Horm Mol Biol Clin Investig. (2011) 5:91–6. doi: 10.1515/HMBCI.2011.011
185. Torella D, Leosco D, Indolfi C, Curcio A, Coppola C, Ellison GM, et al. Aging exacerbates negative remodeling and impairs endothelial regeneration after balloon injury. Am J Physiol Heart Circ Physiol. (2004) 287:H2850–2860. doi: 10.1152/ajpheart.01119.2003
186. Vasa M, Breitschopf K, Zeiher AM, Dimmeler S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res. (2000) 87:540–2. doi: 10.1161/01.RES.87.7.540
187. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. (2012) 33:829–37; 837a–837d. doi: 10.1093/eurheartj/ehr304
188. Feng Y, Feng Y, Gu L, Liu P, Cao J, Zhang S. The critical role of tetrahydrobiopterin (BH4) metabolism in modulating radiosensitivity: BH4/NOS axis as an angel or a devil. Front Oncol. (2021) 11:720632. doi: 10.3389/fonc.2021.720632
189. Goncalves DA, Jasiulionis MG, Melo FHM. The role of the BH4 cofactor in nitric oxide synthase activity and cancer progression: two sides of the same coin. Int J Mol Sci. (2021) 22(17):9546. doi: 10.3390/ijms22179546
190. Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol. (2006) 26:2439–44. doi: 10.1161/01.ATV.0000243924.00970.cb
191. Widder JD, Chen W, Li L, Dikalov S, Thöny B, Hatakeyama K, et al. Regulation of tetrahydrobiopterin biosynthesis by shear stress. Circ Res. (2007) 101:830–8. doi: 10.1161/CIRCRESAHA.107.153809
192. Perrier E, Fournet-Bourguignon M-P, Royere E, Molez S, Reure H, Lesage L, et al. Effect of uncoupling endothelial nitric oxide synthase on calcium homeostasis in aged porcine endothelial cells. Cardiovasc Res. (2009) 82:133–42. doi: 10.1093/cvr/cvp034
193. Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. (2009) 297:H1829–1836. doi: 10.1152/ajpheart.00230.2009
194. Spurlock CF, Aune ZT, Tossberg JT, Collins PL, Aune JP, Huston JW, et al. Increased sensitivity to apoptosis induced by methotrexate is mediated by JNK. Arthritis Rheum. (2011) 63:2606–16. doi: 10.1002/art.30457
195. Crabtree MJ, Hale AB, Channon KM. Dihydrofolate reductase protects endothelial nitric oxide synthase from uncoupling in tetrahydrobiopterin deficiency. Free Radic Biol Med. (2011) 50:1639–46. doi: 10.1016/j.freeradbiomed.2011.03.010
196. Yan H, Su R, Xue H, Gao C, Li X, Wang C. Pharmacomicrobiology of methotrexate in rheumatoid arthritis: gut microbiome as predictor of therapeutic response. Front Immunol. (2021) 12:789334. doi: 10.3389/fimmu.2021.789334
197. Chen L, Zeng X, Wang J, Briggs SS, O’Neill E, Li J, et al. Roles of tetrahydrobiopterin in promoting tumor angiogenesis. Am J Pathol. (2010) 177:2671–80. doi: 10.2353/ajpath.2010.100025
198. Dai Y, Cui J, Gan P, Li W. Downregulation of tetrahydrobiopterin inhibits tumor angiogenesis in BALB/c-nu mice with hepatocellular carcinoma. Oncol Rep. (2016) 36:669–75. doi: 10.3892/or.2016.4850
199. Clayton ZS, Craighead DH, Darvish S, Coppock M, Ludwig KR, Brunt VE, et al. Promoting healthy cardiovascular aging: emerging topics. J Cardiovasc Aging. (2022) 2:43. doi: 10.20517/jca.2022.27
200. Xu S, Lyu QR, Ilyas I, Tian XY, Weng J. Vascular homeostasis in atherosclerosis: a holistic overview. Front Immunol. (2022) 13:976722. doi: 10.3389/fimmu.2022.976722
201. Chang R, Mamun A, Dominic A, Le NT. SARS-CoV-2 mediated endothelial dysfunction: the potential role of chronic oxidative stress. Front Physiol. (2020) 11:605908. doi: 10.3389/fphys.2020.605908
202. Dominic A, Hamilton D, Abe JI. Mitochondria and chronic effects of cancer therapeutics: the clinical implications. J Thromb Thrombolysis. (2021) 51:884–9. doi: 10.1007/s11239-020-02313-2
203. Lin SH, Wang L, Myles B, Thall PF, Hofstetter WL, Swisher SG, et al. Propensity score-based comparison of long-term outcomes with 3-dimensional conformal radiotherapy vs intensity-modulated radiotherapy for esophageal cancer. Int J Radiat Oncol Biol Phys. (2012) 84:1078–85. doi: 10.1016/j.ijrobp.2012.02.015
204. Darby SC, Ewertz M, Hall P. Ischemic heart disease after breast cancer radiotherapy. N Engl J Med. (2013) 368:2527. doi: 10.1056/NEJMoa1209825
205. Darby SC, Ewertz M, McGale P, Bennet AM, Blom-Goldman U, Brønnum D, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med. (2013) 368:987–98. doi: 10.1056/NEJMoa1209825
206. Qin S, Schulte BA, Wang GY. Role of senescence induction in cancer treatment. World J Clin Oncol. (2018) 9:180–7. doi: 10.5306/wjco.v9.i8.180
207. Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal. (2011) 15:1405–14. doi: 10.1089/ars.2010.3679
208. Chen P-Y, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. (2015) 125:4514–28. doi: 10.1172/JCI82719
209. Kim M, Choi S-H, Jin YB, Lee H-J, Ji YH, Kim J, et al. The effect of oxidized low-density lipoprotein (ox-LDL) on radiation-induced endothelial-to-mesenchymal transition. Int J Radiat Biol. (2013) 89:356–63. doi: 10.3109/09553002.2013.763193
210. Czekay RP, Cheon DJ, Samarakoon R, Kutz SM, Higgins PJ. Cancer-associated fibroblasts: mechanisms of tumor progression and novel therapeutic targets. Cancers (Basel). (2022) 14(5):1231. doi: 10.3390/cancers14051231
211. Clere N, Renault S, Corre I. Endothelial-to-mesenchymal transition in cancer. Front Cell Dev Biol. (2020) 8:747. doi: 10.3389/fcell.2020.00747
212. Cho JG, Lee A, Chang W, Lee MS, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. (2018) 9:294. doi: 10.3389/fimmu.2018.00294
213. Piera-Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol Rev. (2019) 99:1281–324. doi: 10.1152/physrev.00021.2018
214. Tombor LS, John D, Glaser SF, Luxán G, Forte E, Furtado M, et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun. (2021) 12:681. doi: 10.1038/s41467-021-20905-1
215. Fang JS, Hultgren NW, Hughes CCW. Regulation of partial and reversible endothelial-to-mesenchymal transition in angiogenesis. Front Cell Dev Biol. (2021) 9:702021. doi: 10.3389/fcell.2021.702021
216. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman K-R, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. (2016) 7:11853. doi: 10.1038/ncomms11853
217. Markwald RR, Fitzharris TP, Smith WN. Sturctural analysis of endocardial cytodifferentiation. Dev Biol. (1975) 42:160–80. doi: 10.1016/0012-1606(75)90321-8
218. Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. (1977) 148:85–119. doi: 10.1002/aja.1001480108
219. Bostrom KI, Yao J, Guihard PJ, Blazquez-Medela AM, Yao Y. Endothelial-mesenchymal transition in atherosclerotic lesion calcification. Atherosclerosis. (2016) 253:124–7. doi: 10.1016/j.atherosclerosis.2016.08.046
220. Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists’ view. Eur Heart J. (2013) 34:719–28. doi: 10.1093/eurheartj/ehs411
221. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114:1852–66. doi: 10.1161/CIRCRESAHA.114.302721
222. Schneider F, Sukhova GK, Aikawa M, Canner J, Gerdes N, Tang S-MT, et al. Matrix-metalloproteinase-14 deficiency in bone-marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. (2008) 117:931–9. doi: 10.1161/CIRCULATIONAHA.107.707448
223. Furman C, Copin C, Kandoussi M, Davidson R, Moreau M, McTaggiart F, et al. Rosuvastatin reduces MMP-7 secretion by human monocyte-derived macrophages: potential relevance to atherosclerotic plaque stability. Atherosclerosis. (2004) 174:93–8. doi: 10.1016/j.atherosclerosis.2004.01.009
224. Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park D-H, Thum T. Transforming growth factor-beta-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler Thromb Vasc Biol. (2012) 32:361–9. doi: 10.1161/ATVBAHA.111.234286
225. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. (2007) 67:10123–8. doi: 10.1158/0008-5472.CAN-07-3127
226. Medici D, Kalluri R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin Cancer Biol. (2012) 22:379–84. doi: 10.1016/j.semcancer.2012.04.004
227. Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. TGF-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst. (2014) 106:djt369. doi: 10.1093/jnci/djt369
228. Garside VC, Chang AC, Karsan A, Hoodless PA. Co-ordinating notch, BMP, and TGF-beta signaling during heart valve development. Cell Mol Life Sci. (2013) 70:2899–917. doi: 10.1007/s00018-012-1197-9
229. Hiepen C, Jatzlau J, Hildebrandt S, Kampfrath B, Goktas M, Murgai A, et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFbeta responses and altered cell mechanics. PLoS Biol. (2019) 17:e3000557. doi: 10.1371/journal.pbio.3000557
230. Noseda M, McLean G, Niessen K, Chang L, Pollet I, Montpetit R, et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res. (2004) 94:910–7. doi: 10.1161/01.RES.0000124300.76171.C9
231. Chanda A, Sarkar A, Bonni S. The SUMO system and TGFbeta signaling interplay in regulation of epithelial-mesenchymal transition: implications for cancer progression. Cancers (Basel). (2018) 10(8):264. doi: 10.3390/cancers10080264
232. Lee PS, Chang C, Liu D, Derynck R. Sumoylation of Smad4, the common smad mediator of transforming growth factor-beta family signaling. J Biol Chem. (2003) 278:27853–63. doi: 10.1074/jbc.M301755200
233. Stein S, Weber J, Nusser-Stein S, Pahla J, Zhang HE, Mohammed SA, et al. Deletion of fibroblast activation protein provides atheroprotection. Cardiovasc Res. (2021) 117:1060–9. doi: 10.1093/cvr/cvaa142
234. Testai L, Brancaleone V, Flori L, Montanaro R, Calderone V. Modulation of EndMT by hydrogen sulfide in the prevention of cardiovascular fibrosis. Antioxidants (Basel). (2021) 10(6):910. doi: 10.3390/antiox10060910
235. Lu S, Jolly AJ, Strand KA, Dubner AM, Mutryn MF, Moulton KS, et al. Smooth muscle-derived progenitor cell myofibroblast differentiation through KLF4 downregulation promotes arterial remodeling and fibrosis. JCI Insight. (2020) 5(23):e139445. doi: 10.1172/jci.insight.139445
236. Kuwabara JT, Tallquist MD. Tracking adventitial fibroblast contribution to disease: a review of current methods to identify resident fibroblasts. Arterioscler Thromb Vasc Biol. (2017) 37:1598–607. doi: 10.1161/ATVBAHA.117.308199
237. Poduri A, Rateri DL, Howatt DA, Balakrishnan A, Moorleghen JJ, Cassis LA, et al. Fibroblast angiotensin II type 1a receptors contribute to angiotensin II-induced medial hyperplasia in the ascending aorta. Arterioscler Thromb Vasc Biol. (2015) 35:1995–2002. doi: 10.1161/ATVBAHA.115.305995
238. Fleenor BS, Bowles DK. Negligible contribution of coronary adventitial fibroblasts to neointimal formation following balloon angioplasty in swine. Am J Physiol Heart Circ Physiol. (2009) 296:H1532–1539. doi: 10.1152/ajpheart.00566.2008
239. LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech. (2018) 11(4):dmm029447. doi: 10.1242/dmm.029447
240. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol Lett. (2017) 14:2611–20. doi: 10.3892/ol.2017.6497
241. Joshi RS, Kanugula SS, Sudhir S, Pereira MP, Jain S, Aghi MK. The role of cancer-associated fibroblasts in tumor progression. Cancers (Basel). (2021) 13(6):1399. doi: 10.3390/cancers13061399
242. Yoshimatsu Y, Watabe T. Emerging roles of inflammation-mediated endothelial-mesenchymal transition in health and disease. Inflamm Regen. (2022) 42:9. doi: 10.1186/s41232-021-00186-3
243. Choi KJ, Nam JK, Kim JH, Choi SH, Lee YJ. Endothelial-to-mesenchymal transition in anticancer therapy and normal tissue damage. Exp Mol Med. (2020) 52:781–92. doi: 10.1038/s12276-020-0439-4
244. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. (2019) 12:86. doi: 10.1186/s13045-019-0770-1
245. Nguyen M, De Ninno A, Mencattini A, Mermet-Meillon F, Fornabaio G, Evans SS, et al. Dissecting effects of anti-cancer drugs and cancer-associated fibroblasts by on-chip reconstitution of immunocompetent tumor microenvironments. Cell Rep. (2018) 25:3884–3893.e3. doi: 10.1016/j.celrep.2018.12.015
246. Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, et al. A metabolic basis for endothelial-to-mesenchymal transition. Mol Cell. (2018) 69:689–698.e7. doi: 10.1016/j.molcel.2018.01.010
247. Strzyz P. Metabolism: a metabolic switch of fate. Nat Rev Mol Cell Biol. (2018) 19:211. doi: 10.1038/nrm.2018.14
248. Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics. (2021) 11:8322–36. doi: 10.7150/thno.62378
249. Ma J, Sanchez-Duffhues G, Goumans MJ, Ten Dijke P. TGF-beta-induced endothelial to mesenchymal transition in disease and tissue engineering. Front Cell Dev Biol. (2020) 8:260. doi: 10.3389/fcell.2020.00260
250. Medici D, Potenta S, Kalluri R. Transforming growth factor-beta2 promotes snail-mediated endothelial-mesenchymal transition through convergence of smad-dependent and smad-independent signalling. Biochem J. (2011) 437:515–20. doi: 10.1042/BJ20101500
251. Ehata S, Miyazono K. Bone morphogenetic protein signaling in cancer; some topics in the recent 10 years. Front Cell Dev Biol. (2022) 10:883523. doi: 10.3389/fcell.2022.883523
252. Gomez-Puerto MC, Iyengar PV, Garcia de Vinuesa A, Ten Dijke P, & Sanchez-Duffhues G. Bone morphogenetic protein receptor signal transduction in human disease. J Pathol. (2019) 247:9–20. doi: 10.1002/path.5170
253. Rol N, Kurakula KB, Happe C, Bogaard HJ, Goumans MJ. TGF-beta and BMPR2 signaling in PAH: two black sheep in one family. Int J Mol Sci. (2018) 19(9):2585. doi: 10.3390/ijms19092585
254. Hopper RK, Moonen J-RAJ, Diebold I, Cao A, Rhodes CJ, Tojais NF, et al. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation. (2016) 133:1783–94. doi: 10.1161/CIRCULATIONAHA.115.020617
255. Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu K, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther. (2022) 7:95. doi: 10.1038/s41392-022-00934-y
256. Shen W, Huang J, Wang Y. Biological significance of NOTCH signaling strength. Front Cell Dev Biol. (2021) 9:652273. doi: 10.3389/fcell.2021.652273
257. Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct notch target required for initiation of cardiac cushion cellularization. J Cell Biol. (2008) 182:315–25. doi: 10.1083/jcb.200710067
258. Lin QQ, Zhao J, Zheng CG, Chun J. Roles of notch signaling pathway and endothelial-mesenchymal transition in vascular endothelial dysfunction and atherosclerosis. Eur Rev Med Pharmacol Sci. (2018) 22:6485–91. doi: 10.26355/eurrev_201810_16062
259. Kluppel M, Wrana JL. Turning it up a notch: cross-talk between TGF beta and notch signaling. Bioessays. (2005) 27:115–8. doi: 10.1002/bies.20187
260. Hendriks IA, Vertegaal AC. A comprehensive compilation of SUMO proteomics. Nat Rev Mol Cell Biol. (2016) 17:581–95. doi: 10.1038/nrm.2016.81
261. Le NT, Martin JF, Fujiwara K, Abe JI. Sub-cellular localization specific SUMOylation in the heart. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:2041–55. doi: 10.1016/j.bbadis.2017.01.018
262. Heo KS, Berk BC, Abe J. Disturbed flow-induced endothelial proatherogenic signaling via regulating post-translational modifications and epigenetic events. Antioxid Redox Signal. (2016) 25:435–50. doi: 10.1089/ars.2015.6556
263. Heo K-S, Chang E, Le N-T, Cushman H, Yeh ETH, Fujiwara K, et al. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res. (2013) 112:911–23. doi: 10.1161/CIRCRESAHA.111.300179
264. Wasik U, Filipek A. Non-nuclear function of sumoylated proteins. Biochim Biophys Acta. (2014) 1843:2878–85. doi: 10.1016/j.bbamcr.2014.07.018
265. Choi S-G, Kim H, Jeong EI, Lee H-J, Park S, Lee S-Y, et al. SUMO-modified FADD recruits cytosolic Drp1 and caspase-10 to mitochondria for regulated necrosis. Mol Cell Biol. (2017) 37(2):e00254–16. doi: 10.1128/MCB.00254-16
266. Anderson CA, Blackstone C. SUMO wrestling with Drp1 at mitochondria. EMBO J. (2013) 32:1496–8. doi: 10.1038/emboj.2013.103
267. Fu J, Yu H-MI, Chiu S-Y, Mirando AJ, Maruyama EO, Cheng J-G, et al. Disruption of SUMO-specific protease 2 induces mitochondria mediated neurodegeneration. PLoS Genet. (2014) 10:e1004579. doi: 10.1371/journal.pgen.1004579
268. Meng X, Clews J, Kargas V, Wang X, Ford RC. The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability. Cell Mol Life Sci. (2017) 74:23–38. doi: 10.1007/s00018-016-2386-8
269. Gong X, Ahner A, Roldan A, Lukacs GL, Thibodeau PH, Frizzell RA. Non-native conformers of cystic fibrosis transmembrane conductance regulator NBD1 are recognized by Hsp27 and conjugated to SUMO-2 for degradation. J Biol Chem. (2016) 291:2004–17. doi: 10.1074/jbc.M115.685628
270. Qi Y, Wang J, Bomben VC, Li D-P, Chen S-R, Sun H, et al. Hyper-SUMOylation of the Kv7 potassium channel diminishes the M-current leading to seizures and sudden death. Neuron. (2014) 83:1159–71. doi: 10.1016/j.neuron.2014.07.042
271. Xu P, Liu J, Derynck R. Post-translational regulation of TGF-beta receptor and smad signaling. FEBS Lett. (2012) 586:1871–84. doi: 10.1016/j.febslet.2012.05.010
272. Woo C-H, Shishido T, McClain C, Lim JH, Li J-D, Yang J, et al. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res. (2008) 102:538–45. doi: 10.1161/CIRCRESAHA.107.156877
273. Kang JS, Saunier EF, Akhurst RJ, Derynck R. The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat Cell Biol. (2008) 10:654–64. doi: 10.1038/ncb1728
274. Tan M, Zhang D, Zhang E, Xu D, Liu Z, Qiu J, et al. SENP2 suppresses epithelial-mesenchymal transition of bladder cancer cells through deSUMOylation of TGF-betaRI. Mol Carcinog. (2017) 56:2332–41. doi: 10.1002/mc.22687
275. Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. (2001) 114:4359–69. doi: 10.1242/jcs.114.24.4359
276. Hata A, Chen YG. TGF-beta signaling from receptors to smads. Cold Spring Harb Perspect Biol. (2016) 8(9):a022061. doi: 10.1101/cshperspect.a022061
277. Zhang X, Wang H, Wang H, Xiao F, Seth P, Xu W, et al. SUMO-specific cysteine protease 1 promotes epithelial mesenchymal transition of prostate cancer cells via regulating SMAD4 deSUMOylation. Int J Mol Sci. (2017) 18(4):808. doi: 10.3390/ijms18040808
278. Chang C-C, Huang Y-S, Lin Y-M, Lin C-J, Jeng J-C, Liu S-M, et al. The role of sentrin-specific protease 2 substrate recognition in TGF-beta-induced tumorigenesis. Sci Rep. (2018) 8:9786. doi: 10.1038/s41598-018-28103-8
279. Mehta V, Pang K-L, Givens CS, Chen Z, Huang J, Sweet DT, et al. Mechanical forces regulate endothelial-to-mesenchymal transition and atherosclerosis via an Alk5-shc mechanotransduction pathway. Sci Adv. (2021) 7(28):eabg5060. doi: 10.1126/sciadv.abg5060
280. Cichon N, Lach D, Dziedzic A, Bijak M, Saluk J. The inflammatory processes in atherogenesis. Pol Merkur Lekarski. (2017) 42:125–8.28333905
281. Ingber DE. Mechanobiology and diseases of mechanotransduction. Ann Med. (2003) 35:564–77. doi: 10.1080/07853890310016333
282. Abe J, Berk BC. Novel mechanisms of endothelial mechanotransduction. Arterioscler Thromb Vasc Biol. (2014) 34:2378–86. doi: 10.1161/ATVBAHA.114.303428
283. Le N-T, Sandhu UG, Quintana-Quezada RA, Hoang NM, Fujiwara K, Abe J. Flow signaling and atherosclerosis. Cell Mol Life Sci. (2017) 74:1835–58. doi: 10.1007/s00018-016-2442-4
284. Morgan RG, Donato AJ, Walker AE. Telomere uncapping and vascular aging. Am J Physiol Heart Circ Physiol. (2018) 315:H1–5. doi: 10.1152/ajpheart.00008.2018
285. Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol. (2005) 40:634–42. doi: 10.1016/j.exger.2005.04.010
286. Andueza A, Kumar S, Kim J, Kang D-W, Mumme HL, Perez JI, et al. Endothelial reprogramming by disturbed flow revealed by single-cell RNA and chromatin accessibility study. Cell Rep. (2020) 33:108491. doi: 10.1016/j.celrep.2020.108491
287. Le N-T, Heo K-S, Takei Y, Lee H, Woo C-H, Chang E, et al. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. (2013) 127:486–99. doi: 10.1161/CIRCULATIONAHA.112.116988
288. Chen BP, Li YS, Zhao Y, Chen KD, Li S, Lao J, et al. DNA microarray analysis of gene expression in endothelial cells in response to 24-h shear stress. Physiol Genomics. (2001) 7:55–63. doi: 10.1152/physiolgenomics.2001.7.1.55
289. Guo D, Chien S, Shyy JY. Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ Res. (2007) 100:564–71. doi: 10.1161/01.RES.0000259561.23876.c5
290. Turpaev KT. Transcription factor KLF2 and its role in the regulation of inflammatory processes. Biochemistry (Mosc). (2020) 85:54–67. doi: 10.1134/S0006297920010058
291. Hsieh HJ, Liu CA, Huang B, Tseng AH, Wang DL. Shear-induced endothelial mechanotransduction: the interplay between reactive oxygen species (ROS) and nitric oxide (NO) and the pathophysiological implications. J Biomed Sci. (2014) 21:3. doi: 10.1186/1423-0127-21-3
292. Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. (2011) 91:327–87. doi: 10.1152/physrev.00047.2009
293. Chien S. Effects of disturbed flow on endothelial cells. Ann Biomed Eng. (2008) 36:554–62. doi: 10.1007/s10439-007-9426-3
294. Yang C, Xiao X, Huang L, Zhou F, Chen L, Zhao Y-Y, et al. Role of kruppel-like factor 4 in atherosclerosis. Clin Chim Acta. (2021) 512:135–41. doi: 10.1016/j.cca.2020.11.002
295. Kotla S, Vu HT, Ko KA, Wang Y, Imanishi M, Heo K-S, et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight. (2019) 4(9):e124867. doi: 10.1172/jci.insight.124867
296. Sabatino L, Picano E, Andreassi MG. Telomere shortening and ionizing radiation: a possible role in vascular dysfunction? Int J Radiat Biol. (2012) 88:830–9. doi: 10.3109/09553002.2012.709307
297. de Almeida AJPO, de Almeida Rezende MS, Dantas SH, de Lima Silva S, de Oliveira JCPL, de Lourdes Assunção Araújo de Azevedo F, et al. Unveiling the role of inflammation and oxidative stress on age-related cardiovascular diseases. Oxid Med Cell Longev. (2020) 2020:1954398. doi: 10.1155/2020/1954398
298. Weinbaum S, Tzeghai G, Ganatos P, Pfeffer R, Chien S. Effect of cell turnover and leaky junctions on arterial macromolecular transport. Am J Physiol. (1985) 248:H945–960. doi: 10.1152/ajpheart.1985.248.6.H945
299. Chien S, Lin SJ, Weinbaum S, Lee MM, Jan KM. The role of arterial endothelial cell mitosis in macromolecular permeability. Adv Exp Med Biol. (1988) 242:59–73. doi: 10.1007/978-1-4684-8935-4_8
300. Tovar-Lopez F, Thurgood P, Gilliam C, Nguyen N, Pirogova E, Khoshmanesh K, et al. A microfluidic system for studying the effects of disturbed flow on endothelial cells. Front Bioeng Biotechnol. (2019) 7:81. doi: 10.3389/fbioe.2019.00081
301. Arslan N, Isik S, Uykan O. Steady and disturbed flow effects on human umbilical vein endothelial cells (HUVECs) in vascular system: an experimental study. Acta Bioeng Biomech. (2010) 12:3–9.21361250
302. Chiu JJ, Wang DL, Chien S, Skalak R, Usami S. Effects of disturbed flow on endothelial cells. J Biomech Eng. (1998) 120:2–8. doi: 10.1115/1.2834303
303. Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ Res. (2004) 95:773–9. doi: 10.1161/01.RES.0000145728.22878.45
304. Jo H, Song H, Mowbray A. Role of NADPH oxidases in disturbed flow- and BMP4- induced inflammation and atherosclerosis. Antioxid Redox Signal. (2006) 8:1609–19. doi: 10.1089/ars.2006.8.1609
305. Liang G, Wang S, Shao J, Jin Y-J, Xu L, Yan Y, et al. Tenascin-X mediates flow-induced suppression of EndMT and atherosclerosis. Circ Res. (2022) 130:1647–59. doi: 10.1161/CIRCRESAHA.121.320694
306. Mahmoud M, Souilhol C, Serbanovic-Canic J, Evans P. GATA4-Twist1 signalling in disturbed flow-induced atherosclerosis. Cardiovasc Drugs Ther. (2019) 33:231–7. doi: 10.1007/s10557-019-06863-3
307. Mahmoud MM, Kim HR, Xing R, Hsiao S, Mammoto A, Chen J, et al. TWIST1 integrates endothelial responses to flow in vascular dysfunction and atherosclerosis. Circ Res. (2016) 119:450–62. doi: 10.1161/CIRCRESAHA.116.308870
308. Mahmoud MM, Serbanovic-Canic J, Feng S, Souilhol C, Xing R, Hsiao S, et al. Shear stress induces endothelial-to-mesenchymal transition via the transcription factor snail. Sci Rep. (2017) 7:3375. doi: 10.1038/s41598-017-03532-z
309. Rohwedder I, Montanez E, Beckmann K, Bengtsson E, Dunér P, Nilsson J, et al. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol Med. (2012) 4:564–76. doi: 10.1002/emmm.201200237
310. Seneviratne A, Hulsmans M, Holvoet P, Monaco C. Biomechanical factors and macrophages in plaque stability. Cardiovasc Res. (2013) 99:284–93. doi: 10.1093/cvr/cvt097
311. Zhao P, Yao Q, Zhang P-J, The E, Zhai Y, Ao L, et al. Single-cell RNA-seq reveals a critical role of novel pro-inflammatory EndMT in mediating adverse remodeling in coronary artery-on-a-chip. Sci Adv. (2021) 7(34):eabg1694. doi: 10.1126/sciadv.abg1694
312. Braganza DM, Bennett MR. New insights into atherosclerotic plaque rupture. Postgrad Med J. (2001) 77:94–8. doi: 10.1136/pmj.77.904.94
313. Boyle JJ, Bowyer DE, Weissberg PL, Bennett MR. Human blood-derived macrophages induce apoptosis in human plaque-derived vascular smooth muscle cells by fas-ligand/fas interactions. Arterioscler Thromb Vasc Biol. (2001) 21:1402–7. doi: 10.1161/hq0901.094279
314. Imanishi T, Han DKM, Hofstra L, Hano T, Nishio I, Conrad Liles W, et al. Apoptosis of vascular smooth muscle cells is induced by fas ligand derived from monocytes/macrophage. Atherosclerosis. (2002) 161:143–51. doi: 10.1016/S0021-9150(01)00631-1
315. Beck-Joseph J, Lehoux S. Molecular interactions between vascular smooth muscle cells and macrophages in atherosclerosis. Front Cardiovasc Med. (2021) 8:737934. doi: 10.3389/fcvm.2021.737934
316. Barnes MJ, Farndale RW. Collagens and atherosclerosis. Exp Gerontol. (1999) 34:513–25. doi: 10.1016/S0531-5565(99)00038-8
317. Clarke MCH, Littlewood TD, Figg N, Maguire JJ, Davenport AP, Goddard M, et al. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. (2008) 102:1529–38. doi: 10.1161/CIRCRESAHA.108.175976
318. Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. (2001) 33:1897–918. doi: 10.1006/jmcc.2001.1462
319. Jin J, Tang K, Xin Y, Zhang T, Tan Y. Hemodynamic shear flow regulates biophysical characteristics and functions of circulating breast tumor cells reminiscent of brain metastasis. Soft Matter. (2018) 14:9528–33. doi: 10.1039/C8SM01781F
320. Krog BL, Henry MD. Biomechanics of the circulating tumor cell microenvironment. Adv Exp Med Biol. (2018) 1092:209–33. doi: 10.1007/978-3-319-95294-9_11
321. Landwehr GM, Kristof AJ, Rahman SM, Pettigrew JH, Coates R, Balhoff JB, et al. Biophysical analysis of fluid shear stress induced cellular deformation in a microfluidic device. Biomicrofluidics. (2018) 12:054109. doi: 10.1063/1.5063824
322. Li S, Chen Y, Zhang Y, Jiang X, Jiang Y, Qin X, et al. Shear stress promotes anoikis resistance of cancer cells via caveolin-1-dependent extrinsic and intrinsic apoptotic pathways. J Cell Physiol. (2019) 234:3730–43. doi: 10.1002/jcp.27149
323. Wang X, Zhang Y, Feng T, Su G, He J, Gao W, et al. Fluid shear stress promotes autophagy in hepatocellular carcinoma cells. Int J Biol Sci. (2018) 14:1277–90. doi: 10.7150/ijbs.27055
324. Huang Q, Hu X, He W, Zhao Y, Hao S, Wu Q, et al. Fluid shear stress and tumor metastasis. Am J Cancer Res. (2018) 8:763–77.29888101
325. Hyler AR, Baudoin NC, Brown MS, Stremler MA, Cimini D, Davalos RV, et al. Fluid shear stress impacts ovarian cancer cell viability, subcellular organization, and promotes genomic instability. PLoS One. (2018) 13:e0194170. doi: 10.1371/journal.pone.0194170
326. Liu B, Han S, Hedrick BP, Modarres-Sadeghi Y, Lynch ME. Perfusion applied to a 3D model of bone metastasis results in uniformly dispersed mechanical stimuli. Biotechnol Bioeng. (2018) 115:1076–85. doi: 10.1002/bit.26524
327. Lee YH, Yeh CH. Laminar shear stress inhibits high glucose-induced migration and invasion in human bladder cancer cells. In Vitro Cell Dev Biol Anim. (2018) 54:120–8. doi: 10.1007/s11626-017-0217-3
328. Font-Clos F, Zapperi S, La Porta CAM. Blood flow contributions to cancer metastasis. iScience. (2020) 23:101073. doi: 10.1016/j.isci.2020.101073
329. Liu Y-Z, Xiao X, Hu C-T, Dai Y, Qu S-L, Huang L, et al. SUMOylation in atherosclerosis. Clin Chim Acta. (2020) 508:228–33. doi: 10.1016/j.cca.2020.05.033
330. Talamillo A, Ajuria L, Grillo M, Barroso-Gomila O, Mayor U, Barrio R. SUMOylation in the control of cholesterol homeostasis. Open Biol. (2020) 10:200054. doi: 10.1098/rsob.200054
331. Ganguli A, Persson L, Palmer IR, Evans I, Yang L, Smallwood R, et al. Distinct NF-kappaB regulation by shear stress through ras-dependent IkappaBalpha oscillations: real-time analysis of flow-mediated activation in live cells. Circ Res. (2005) 96:626–34. doi: 10.1161/01.RES.0000160435.83210.95
332. Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol. (2006) 8:986–93. doi: 10.1038/ncb1458
333. Dehnavi S, Sadeghi M, Penson PE, Banach M, Jamialahmadi T, Sahebkar A. The role of protein SUMOylation in the pathogenesis of atherosclerosis. J Clin Med. (2019) 18(11):1856. doi: 10.3390/jcm8111856
334. Abe J, Ko KA, Kotla S, Wang Y, Paez-Mayorga J, Shin IJ, et al. MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight. (2019) 4(7):e125570. doi: 10.1172/jci.insight.125570
335. Heo K-S, Le N-T, Cushman HJ, Giancursio CJ, Chang E, Woo C-H, et al. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest. (2015) 125:1299–310. doi: 10.1172/JCI76453
336. Hendriks IA, Lyon D, Young C, Jensen LJ, Vertegaal ACO, Nielsen ML. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat Struct Mol Biol. (2017) 24:325–36. doi: 10.1038/nsmb.3366
337. Sarge KD, Park-Sarge OK. SUMO and its role in human diseases. Int Rev Cell Mol Biol. (2011) 288:167–83. doi: 10.1016/B978-0-12-386041-5.00004-2
338. Johnson ES. Protein modification by SUMO. Annu Rev Biochem. (2004) 73:355–82. doi: 10.1146/annurev.biochem.73.011303.074118
339. Lee MH, Mabb AM, Gill GB, Yeh ET, Miyamoto S. NF-kappaB induction of the SUMO protease SENP2: a negative feedback loop to attenuate cell survival response to genotoxic stress. Mol Cell. (2011) 43:180–91. doi: 10.1016/j.molcel.2011.06.017
340. Chiu SY, Asai N, Costantini F, Hsu W. SUMO-specific protease 2 is essential for modulating p53-Mdm2 in development of trophoblast stem cell niches and lineages. PLoS Biol. (2008) 6:e310. doi: 10.1371/journal.pbio.0060310
341. Jiang QF, Tian YW, Shen Q, Xue HZ, Li K. SENP2 regulated the stability of beta-catenin through WWOX in hepatocellular carcinoma cell. Tumour Biol. (2014) 35:9677–82. doi: 10.1007/s13277-014-2239-8
342. Pei H, Chen L, Liao Q, Wang K, Chen S, Liu Z, et al. SUMO-specific protease 2 (SENP2) functions as a tumor suppressor in osteosarcoma via SOX9 degradation. Exp Ther Med. (2018) 16:5359–65. doi: 10.3892/etm.2018.6838
343. Mikolajczyk J, Drag M, Békés M, Cao JT, Ronai Z, Salvesen GS. Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J Biol Chem. (2007) 282:26217–24. doi: 10.1074/jbc.M702444200
344. Itahana Y, Yeh ET, Zhang Y. Nucleocytoplasmic shuttling modulates activity and ubiquitination-dependent turnover of SUMO-specific protease 2. Mol Cell Biol. (2006) 26:4675–89. doi: 10.1128/MCB.01830-05
345. Zhang L, Yang TH, Li DW. Roles of SUMOylation in heart development and cardiovascular diseases. Curr Mol Med. (2017) 16:877–84. doi: 10.2174/1566524016666161223110407
346. Cohen P. The regulation of protein function by multisite phosphorylation–a 25 year update. Trends Biochem Sci. (2000) 25:596–601. doi: 10.1016/S0968-0004(00)01712-6
347. Stastna M, Van Eyk JE. Posttranslational modifications of lysine and evolving role in heart pathologies-recent developments. Proteomics. (2015) 15:1164–80. doi: 10.1002/pmic.201400312
348. Olvera Lopez E, Ballard BD, & Jan A. Cardiovascular disease. Treasure Island (FL): StatPearls (2020).
349. Le N-T, Sandhu UG, Quintana-Quezada RA, Hoang NM, Fujiwara K, Abe J. Flow signaling and atherosclerosis. Cell Mol Life Sci. (2017) 74(10):1835–58. doi: 10.1007/s00018-016-2442-4
350. Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. (2009) 73:595–601. doi: 10.1253/circj.CJ-08-1169
351. Sun X, Feinberg MW. Vascular endothelial senescence: pathobiological insights, emerging long noncoding RNA targets, challenges and therapeutic opportunities. Front Physiol. (2021) 12:693067. doi: 10.3389/fphys.2021.693067
352. Warboys CM, de Luca A, Amini N, Luong L, Duckles H, Hsiao S, et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol. (2014) 34:985–95. doi: 10.1161/ATVBAHA.114.303415
353. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. (2022) 22:340–55. doi: 10.1038/s41568-022-00450-9
354. Banerjee P, Kotla S, Reddy Velatooru L, Abe RJ, Davis EA, Cooke JP, et al. Senescence-associated secretory phenotype as a hinge between cardiovascular diseases and cancer. Front Cardiovasc Med. (2021) 8:763930. doi: 10.3389/fcvm.2021.763930
355. Tsolou A, Passos JF, Nelson G, Arai Y, Zglinicki T. ssDNA fragments induce cell senescence by telomere uncapping. Exp Gerontol. (2008) 43:892–9. doi: 10.1016/j.exger.2008.08.043
356. Birch J, Barnes PJ, Passos JF. Mitochondria, telomeres and cell senescence: implications for lung ageing and disease. Pharmacol Ther. (2018) 183:34–49. doi: 10.1016/j.pharmthera.2017.10.005
357. Birch J, Passos JF. Targeting the SASP to combat ageing: mitochondria as possible intracellular allies? Bioessays. (2017) 39(5). doi: 10.1002/bies.201600235
358. He S, Sharpless NE. Senescence in health and disease. Cell. (2017) 169:1000–11. doi: 10.1016/j.cell.2017.05.015
359. Yan T, Zhang T, Mu W, Qi Y, Guo S, Hu N, et al. Ionizing radiation induces BH(4) deficiency by downregulating GTP-cyclohydrolase 1, a novel target for preventing and treating radiation enteritis. Biochem Pharmacol. (2020) 180:114102. doi: 10.1016/j.bcp.2020.114102
360. Herranz B, Marquez S, Guijarro B, Aracil E, Aicart-Ramos C, Rodriguez-Crespo I, et al. Integrin-linked kinase regulates vasomotor function by preventing endothelial nitric oxide synthase uncoupling: role in atherosclerosis. Circ Res. (2012) 110:439–49. doi: 10.1161/CIRCRESAHA.111.253948
361. Takeda M, Yamashita T, Shinohara M, Sasaki N, Takaya T, Nakajima K, et al. Plasma tetrahydrobiopterin/dihydrobiopterin ratio: a possible marker of endothelial dysfunction. Circ J. (2009) 73:955–62. doi: 10.1253/circj.CJ-08-0850
362. Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher AM. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res. (1998) 83:334–41. doi: 10.1161/01.RES.83.3.334
363. Kim S, Han J-H, Nam D-H, Kim G-Y, Lim JH, Kim J-R, et al. PAR-1 is a novel mechano-sensor transducing laminar flow-mediated endothelial signaling. Sci Rep. (2018) 8:15172. doi: 10.1038/s41598-018-33222-3
364. Vu HT, Kotla S, Ko KA, Fujii Y, Tao Y, Medina J, et al. Ionizing radiation induces endothelial inflammation and apoptosis via p90RSK-mediated ERK5 S496 phosphorylation. Front Cardiovasc Med. (2018) 5:23. doi: 10.3389/fcvm.2018.00023
365. Wang K, Tepper JE. Radiation therapy-associated toxicity: etiology, management, and prevention. CA Cancer J Clin. (2021) 71:437–54. doi: 10.3322/caac.21689
366. Royce TJ, Qureshi MM, Truong MT. Radiotherapy utilization and fractionation patterns during the first course of cancer treatment in the United States from 2004 to 2014. J Am Coll Radiol. (2018) 15:1558–64. doi: 10.1016/j.jacr.2018.04.032
367. Yang EH, Marmagkiolis K, Balanescu DV, Hakeem A, Donisan T, Finch W, et al. Radiation-induced vascular disease-A state-of-the-art review. Front Cardiovasc Med. (2021) 8:652761. doi: 10.3389/fcvm.2021.652761
368. Choi S-H, Hong Z-Y, Nam J-K, Lee H-J, Jang J, Yoo RJ, et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res. (2015) 21:3716–26. doi: 10.1158/1078-0432.CCR-14-3193
369. Choi S-H, Nam J-K, Kim B-Y, Jang J, Jin Y-B, Lee H-J, et al. HSPB1 inhibits the endothelial-to-mesenchymal transition to suppress pulmonary fibrosis and lung tumorigenesis. Cancer Res. (2016) 76:1019–30. doi: 10.1158/0008-5472.CAN-15-0952
370. Choi S-H, Kim A-R, Nam J-K, Kim J-M, Kim J-Y, Seo HR, et al. Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls CD44v6(+) cancer cell and macrophage polarization. Nat Commun. (2018) 9:5108. doi: 10.1038/s41467-018-07470-w
371. Paszkowiak JJ, Dardik A. Arterial wall shear stress: observations from the bench to the bedside. Vasc Endovascular Surg. (2003) 37:47–57. doi: 10.1177/153857440303700107
372. Heo KS, Fujiwara K, Abe J. Shear stress and atherosclerosis. Mol Cells. (2014) 37:435–40. doi: 10.14348/molcells.2014.0078
373. Zhou J, Yi Q, Tang L. The roles of nuclear focal adhesion kinase (FAK) on cancer: a focused review. J Exp Clin Cancer Res. (2019) 38:250. doi: 10.1186/s13046-019-1265-1
374. Velatooru LR, Abe RJ, Imanishi M, Gi YJ, Ko KA, Heo K-S, et al. Disturbed flow-induced FAK K152 SUMOylation initiates the formation of pro-inflammation positive feedback loop by inducing reactive oxygen species production in endothelial cells. Free Radic Biol Med. (2021) 177:404–18. doi: 10.1016/j.freeradbiomed.2021.09.023
375. Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJR, et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res. (2013) 12:260–71. doi: 10.1021/pr300630k
376. Bian Y, Song C, Cheng K, Dong M, Wang F, Huang J, et al. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J Proteomics. (2014) 96:253–62. doi: 10.1016/j.jprot.2013.11.014
377. Robinson NJ, Miyagi M, Scarborough JA, Scott JG, Taylor DJ, Schiemann WP. SLX4IP promotes RAP1 SUMOylation by PIAS1 to coordinate telomere maintenance through NF-kappaB and notch signaling. Sci Signal. (2021) 14(689):eabe9613. doi: 10.1126/scisignal.abe9613
378. Costantino S, Paneni F, Cosentino F. Ageing, metabolism and cardiovascular disease. J Physiol. (2016) 594:2061–73. doi: 10.1113/JP270538
379. Tabibzadeh S. Signaling pathways and effectors of aging. Front Biosci (Landmark Ed). (2021) 26:50–96. doi: 10.2741/4889
380. Donato AJ, Morgan RG, Walker AE, Lesniewski LA. Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol. (2015) 89:122–35. doi: 10.1016/j.yjmcc.2015.01.021
381. Gupta SC, Kunnumakkara AB, Aggarwal S, Aggarwal BB. Inflammation, a double-edge sword for cancer and other age-related diseases. Front Immunol. (2018) 9:2160. doi: 10.3389/fimmu.2018.02160
382. Matsutani N, Yokozaki H, Tahara E, Tahara H, Kuniyasu H, Kitadai Y, et al. Expression of MRE11 complex (MRE11, RAD50, NBS1) and hRap1 and its relation with telomere regulation, telomerase activity in human gastric carcinomas. Pathobiology. (2001) 69:219–24. doi: 10.1159/000055946
383. Xiao L, Lan X, Shi X, Zhao K, Wang D, Wang X, et al. Cytoplasmic RAP1 mediates cisplatin resistance of non-small cell lung cancer. Cell Death Dis. (2017) 8:e2803. doi: 10.1038/cddis.2017.210
384. Panero J, Alves-Paiva RM, Roisman A, Santana-Lemos BA, Falcão RP, Oliveira G, et al. Acquired TERT promoter mutations stimulate TERT transcription in mantle cell lymphoma. Am J Hematol. (2016) 91:481–5. doi: 10.1002/ajh.24324
385. Lian S, Meng L, Liu C, Xing X, Song Q, Dong B, et al. PRL-3 activates NF-kappaB signaling pathway by interacting with RAP1. Biochem Biophys Res Commun. (2013) 430:196–201. doi: 10.1016/j.bbrc.2012.11.036
386. Banerjee P, Gaddam N, Pandita TK, Chakraborty S. Cellular senescence as a brake or accelerator for oncogenic transformation and role in lymphatic metastasis. Int J Mol Sci. (2023) 24(3):2877. doi: 10.3390/ijms24032877
387. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
388. Triana-Martínez F, Picallos-Rabina P, Da Silva-álvarez S, Pietrocola F, Llanos S, Rodilla V, et al. Identification and characterization of cardiac glycosides as senolytic compounds. Nat Commun. (2019) 10:4731. doi: 10.1038/s41467-019-12888-x
Keywords: senescence, atherosclerosis, disturbed flow, sumoylation, endoMT
Citation: Banerjee P, Rosales JE, Chau K, Nguyen MTH, Kotla S, Lin SH, Deswal A, Dantzer R, Olmsted-Davis EA, Nguyen H, Wang G, Cooke JP, Abe J-i and Le N-T (2023) Possible molecular mechanisms underlying the development of atherosclerosis in cancer survivors. Front. Cardiovasc. Med. 10:1186679. doi: 10.3389/fcvm.2023.1186679
Received: 15 March 2023; Accepted: 17 May 2023;
Published: 2 June 2023.
Edited by:
Ippei Shimizu, Juntendo University, JapanReviewed by:
Agian Jeffilano Barinda, University of Indonesia, IndonesiaHyoung Kyu Kim, Inje University, Republic of Korea
© 2023 Banerjee, Rosales, Chau, Nguyen, Kotla, Lin, Deswal, Dantzer, Olmsted-Davis, Nguyen, Wang, Cooke, Abe, Le. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-ichi Abe jabe@mdanderson.org Nhat-Tu Le nhle@houstonmethodist.org
†These authors have contributed equally to this work