Inherited Thrombocytopenia: Update on Genes and Genetic Variants Which may be Associated With Bleeding
 Ibrahim Almazni
Ibrahim Almazni Rachel Stapley
Rachel Stapley Neil V. Morgan
Neil V. Morgan- Institute of Cardiovascular Sciences, College of Medical and Dental Sciences, University of Birmingham, Birmingham, United Kingdom
Inherited thrombocytopenia (IT) is comprised of a group of hereditary disorders characterized by a reduced platelet count as the main feature, and often with abnormal platelet function, which can subsequently lead to impaired haemostasis. Inherited thrombocytopenia results from genetic mutations in genes implicated in megakaryocyte differentiation and/or platelet formation and clearance. The identification of the underlying causative gene of IT is challenging given the high degree of heterogeneity, but important due to the presence of various clinical presentations and prognosis, where some defects can lead to hematological malignancies. Traditional platelet function tests, clinical manifestations, and hematological parameters allow for an initial diagnosis. However, employing Next-Generation Sequencing (NGS), such as Whole Genome and Whole Exome Sequencing (WES) can be an efficient method for discovering causal genetic variants in both known and novel genes not previously implicated in IT. To date, 40 genes and their mutations have been implicated to cause many different forms of inherited thrombocytopenia. Nevertheless, despite this advancement in the diagnosis of IT, the molecular mechanism underlying IT in some patients remains unexplained. In this review, we will discuss the genetics of thrombocytopenia summarizing the recent advancement in investigation and diagnosis of IT using phenotypic approaches, high-throughput sequencing, targeted gene panels, and bioinformatics tools.
Introduction
Platelets are small anucleate cells produced by megakaryocytes in the bone marrow (BM) where they circulate in the blood to protect the integrity of blood vessels. They play an important role in normal haemostasis to prevent excessive bleeding at the site of blood vessel injury (1, 2). Inherited Thrombocytopenias (ITs) are a heterogeneous group of disorders characterized by low platelet counts, often manifesting as bleeding diathesis which subsequently result in impaired haemostasis (3). In 1948, the disease inheritance pattern of one IT was initially discovered in a disorder called Bernard-Soulier syndrome (BSS) (4). Since then, the advancement in clinical and scientific research has led to an increased understanding of the molecular defects in patients with ITs. These defects are variable in severity, ranging from severe bleeding, which can be recognized within a few weeks after birth, to mild bleeding that may remain undiagnosed until incidental recognition during routine blood testing in adulthood (5). They manifest with different symptoms including epistaxis, easy bruising, petechiae, prolonged bleeding from cuts, gum bleeding, excessive bleeding after surgery, hematuria, and menorrhagia in women (6, 7). As bleeding is considered the main clinical complication for patients with IT, some patients with common ITs have the propensity to develop other disorders such as hematological malignancies and kidney failure (4, 8). Although there are other causes of thrombocytopenia, such as infections and immune disorders, IT is primarily caused by mutations in genes involved in megakaryocyte differentiation, maturation and platelet release (9). Since the last decade, next generation sequencing technologies, namely Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS) coupled with conventional Sanger sequencing and in-silico bioinformatic tools have been used in parallel to uncover novel genes with a pivotal role in megakaryocyte biology and platelet biogenesis (10, 11). To date, 40 genes have been reported to cause different forms of IT, which reflects the immense difficulty in identifying a single causative gene, particularly when accompanied by other hematological disorders [Table 1; Figure 1; (27, 28, 54)]. These genetic forms have various clinical manifestations, phenotypic presentations and sometimes associated with secondary qualitative defects in platelet function (7). Diverse platelet phenotypes mean there are several approaches in which they can be characterized. One such way is to classify genes based on their influence on megakaryocyte differentiation, platelet production, and removal (54), and will be discussed below. However, despite these advancements, nearly 50% of patients with IT of unknown genetic etiology still remain undiagnosed (6, 10).

Table 1. The direct genetic causes of inherited thrombocytopenia and their associated syndromes.
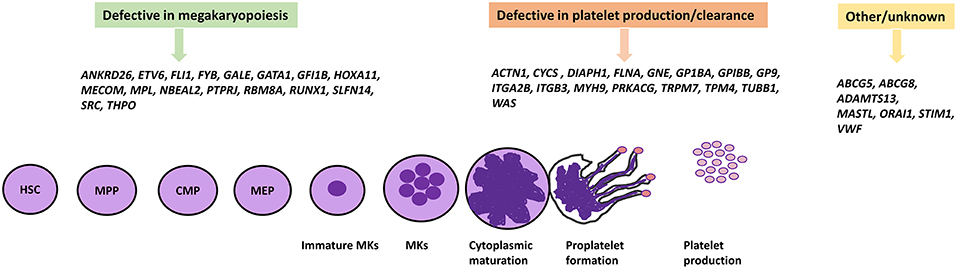
Figure 1. IT causative genes involved in megakaryopoiesis, platelet formation, and others. The differentiation of platelets from HSCs proceeds by multiple different cell lineages which involve many genes encoding a number of transcription factors and proteins. Genetic defects in these genes have been shown to cause IT. HSC, Hematopoietic stem cell; MPP, Multi-Potent Progenitor; CMP, Common myeloid progenitor; MEP, Megakaryocyte-erythroid progenitor.
IT Genes Associated with Megakaryocyte Differentiation and Maturation
The process of megakaryopoiesis and thrombopoiesis involves a complicated biological series of events. Megakaryocytes, like all blood cells, are derived from the hematopoietic stem cells (HSC) in the bone marrow during the lineage commitment stages. The hematopoietic stem cell differentiation process involves committed precursors that include the common myeloid progenitor (CMP) and the megakaryocyte-erythroid progenitor (MEP). The erythrocyte cells and megakaryocyte cells result from the MEP. Megakaryocyte precursors encompass a maturation process that results in mature polyploid megakaryocytes, and then leads to the formation of pro-platelets (54, 55). The process of megakaryopoiesis and thrombopoiesis involves multiple genes and transcriptions factors (detailed below) which play important roles in megakaryocyte differentiation, platelet formation, and release. IT can result from defects in these genes which present with variable phenotypic display and clinical presentation. As a result of the numerous clinical demonstrations of ITs, they can be characterized based on genes and their role during megakaryocyte differentiation, platelet production, and release (9, 56).
Some ITs result from defective changes from haemopoietic progenitor cells to MKs, leading to reduction or absence in the number of bone marrow MKs. Thrombopoietin (TPO), an acidic glycoprotein, is the main regulator of the megakarypoiesis and thrombopoiesis mechanism in humans, acting through its receptor c-Mpl. It is required for megakaryocytes to fulfill their developmental proliferation and for the subsequent maturation of platelets (57). Affected individuals from a large Micronesian family displayed idiopathic anemia and mild thrombocytopenia as a result of mutations in TPO and MPL genes (21, 58). The main defective mechanism in several forms of IT is a change in MK maturation which therefore leads to the production of immature and dysfunctional MKs. However, the differentiation and maturation of MKs is regulated by several transcription factors such as GATA1. GATA1 is highly expressed in the erythroid and megakaryocytic lineage, and plays a vital role in the maturation and development of erythroid cells and megakaryocytes (59). X-linked thrombocytopenia with thalassemia and X-linked thrombocytopenia with dyserythropoietic anemia are both caused by mutations in GATA1, resulting in impaired MK and erythroid cell maturation. As a consequence, GATA1-mutated patients are characterized with large platelets and reduced α-granule contents. They also display a variable degree of anemia and abnormal morphology of red blood cells (60). Additional transcription factors known to be involved in the maturation of megakaryocytes are RUNX1, ETV6, ANKRD26, FLI-1, and the transcriptional repressor GFI1B acting by binding to promoter regions in MK expressed genes. Thus, multiple mechanisms in MK and platelet maturation are affected as result of alterations in these genes (61, 62). A previous study identified a point mutation in the third helix of HOXA11 homeodomain causing an inherited syndrome of congenital amegakaryocytic thrombocytopenia and radio-ulnar synostosis (19). Thrombocytopenia absent radii (TAR) syndrome results from a combination of a microdeletion on Chromosome 1 including the RBM8A gene alongside a low frequency non-coding single nucleotide polymorphism (SNP) within the regulatory region of RBM8A (23). As a consequence, hematopoietic progenitors from patients with TAR syndrome fail to differentiate into MKs in vitro (63). Gray Platelet syndrome is characterized by a deficiency in α granule content which also results in a platelet function defect. It is associated with enlarged platelets and mild thrombocytopenia with moderate to severe bleeding as a result of biallelic mutations in NBEAL2, the gene encoding the neurobeachin-like-2 protein (22). Variants in the 5'UTR of ANKRD26 cause familial thrombocytopenia type-2 (THC2) with propensity to leukemia, which result in loss of RUNX1 and FLI1 binding and prevents gene silencing (12). Moreover, heterozygous variants specifically located in the promoter region between c.-134G and c.-113 region highly affect gene expression. Patients with THC2 are characterized by small MKs with hypolobulated nuclei as a result of dysmegakaryopoiesis (64). A mutation in the FYB1 gene has recently been identified to cause IT and although the exact mechanism of the mutation is still ambiguous, it has been suggested that thrombocytopenia arises from a reduction of mature MKs in the bone marrow and synthesis of small platelets (16).
Defects in Proplatelet Formation and Platelet Release
After megakaryopoesis, proplatelets form extensions which lead to “budding” at the tips and platelet release into the circulation. Mature MKs undergo essential processing by extending long branches called proplatelets via the bone marrow sinusoids, and subsequently release platelets into the blood circulation. These processes are underpinned by cytoskeletal changes and cellular signaling where most causative mutations of IT disrupt the pathway reducing the circulating platelet count (65, 66). Mature polyploid MKs cytoplasm extend long beaded cytoplasmic protrusions, as a result of microtubule sliding. The dimerisation of β1-tubulin with α-tubulin polymerizes into long microtubule bundles inside the MK cortex. A mixed polarity of microtubule bundles runs throughout the extension of proplatelets which are thought to provide fundamental force for microtubule sliding and proplatelet elongation (65). TUBB1 encodes for β1-tubulin and mutations within TUBB1 are associated with an autosomal dominant form of IT known as a congenital macrothrombocytopenia (43). WASp is a multidomain protein belonging to a family of actin nucleation-promoting factors (NPFs) which are specifically expressed in hematopoietic cells. WASp plays an important role in actin polymerization by transmission of surface signals via the actin-related protein (Arp)2/3 complex (44, 67). Mutations have been identified in the WAS gene which cause a rare X-linked disorder called Wiskott-Aldrich syndrome (WAS). Patients are characterized by micro-thrombocytopenia and immunodeficiency with predispostion to malignancies (68). The transmission of extracellular signals into the cytoskeleton is mediated via membrane bound receptors which have been associated with mutations in IT. One of the main membrane receptors in platelets/MKs is the GP1b-IX-V complex, which binds specifically to Von-Willebrand factor (VWF). This receptor is comprised of four subunits including GP1bα, GP1bβ, GPIX, and GPV. Binding of VWF with GP1bα leads to activation and signal transmission to form the extending proplatelet. Mutations in the encoding genes GP1BA, GP1BB, and GP9 cause monoallelic and biallelic forms of BSS (69, 70). Other receptors include the receptor for fibrinogen, integrin GPIIb-IIIa, which is encoded by the genes ITGA2B and ITGB3. Affected mutations in ITGA2B and ITGB3 have been identified to cause Glanzmann thrombasthenia (GT) (71), but patients have a normal platelet count.
Initial Diagnosis of Hereditary Thrombocytopenia
Identification of the genetic cause in patients with IT is challenging and many patients may be misdiagnosed with acquired thrombocytopenia such as immune thrombocytopenic purpura (ITP). IT can be recognized in patients when a low platelet count has been identified after birth, the presence of familial medical history with similar clinical presentations, evaluation of peripheral blood films, and physical examination. Moreover, the presence of a severe bleeding tendency (in combination with a low platelet count), a lifelong history of diathesis, and evidence of other clinical complications that are typically associated with thrombocytopenia in syndromic forms, and all help to diagnose IT (4, 72). Platelets are involved in other biological roles beyond hemostasis, such as immunity and inflammation (73–75) therefore, mutations in platelet specific genes may cause functional disruption in hemostasis, other biological pathways or both. Furthermore, some proteins are expressed in megakaryocytes and platelets and can be found in other cell types. GATA1 is a prominent example which involves megakaryopoiesis and erythropoiesis (76). MYH9 also has an important role in the platelet cytoskeleton and has been found expressed in kidney and inner ear cilia (77). Based on this, inherited bleeding disorders can be classified into three categories including disorders that (i) affect only platelets, (ii) disorders that are associated with syndromic or non-syndromic phenotypes, and (iii) disorders with increased risk of haematologic malignancies. This classification can be used for both diagnostic and prognostic purposes (4, 11).
Syndromic Disorders Associated With IT
The number of IT forms identified has increased over the last few years since the implementation of NGS. Consequently, it has been shown that the bleeding is not the only clinical phenotype with IT, but patients with some IT forms have propensity to develop more syndromic disorders as result of molecular defects in genes responsible for thrombocytopenia. For instance, hematological malignancies, bone marrow aplasia, skeletal malformation, liver and kidney malfunction, and deafness (Figure 2). The development of these diseases can be more severe for patients than the bleeding itself (4) however, it is still important to recognize if these manifestations are present in the relatives. Some syndromic phenotypes associated with ITs are variable between family members or can arise later in life. For example, development of deafness, kidney malfunction and/or cataract in patients with MYH9-RD occur only in adult individuals and it has been reported that patients of the same MYH9-RD pedigrees have variable clinical manifestations (78).
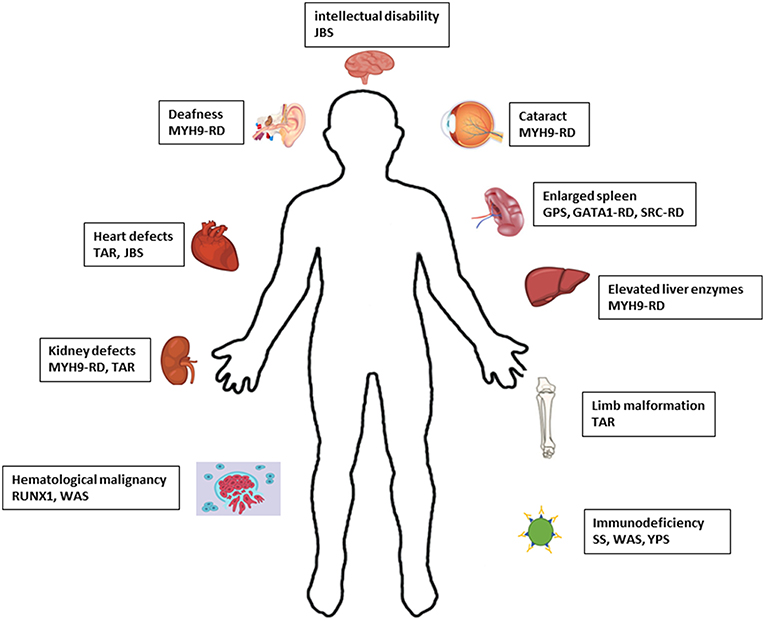
Figure 2. Schematic representation of the defects associated with syndromic IT. JBS, Jacobsen syndrome; RD, related disease; TAR, Thrombocytopenia-absent radius; GPS, Gray platelet syndrome; SS, Stormorken syndrome; WAS, Wiskott-Aldrich syndrome; YPS, York platelet syndrome.
The Genetic Diagnosis of IT
Some patients with a low platelet count may be falsely diagnosed and receive unnecessary treatments such as immunosuppression and splenectomies and therefore it is paramount that strong evidence must prove that the condition is truly genetic. Genetic diagnosis is a vital approach in providing patients with clinical benefits and prevent unnecessary treatments. Patients with genetic mutations in RUNX1 have a predisposition to develop hematological malignancies where the genetic information can be used to monitor the patients' hematological parameters very closely. This emphasizes the importance and need for definitive genetic diagnostic tools to provide quick and cost-effective diagnosis for screening patients with IT (6, 8). The molecular basis of ITs has been elucidated since the adoption of Sanger sequencing and linkage analysis in the 1990s. Recently, Sanger sequencing is considered a low throughput and time-consuming approach which can be used initially as a standard tool to investigate patients based on precise clinical findings and phenotype (4). A targeted thrombocytopenia gene specific panel is a useful approach which can be used as initial screening prior to WES. This targeted panel encompasses all known genes associated with IT and their related genes. The aim of using an IT gene specific panel is to filter out patients based on variants in known IT-causal genes and subsequently allowing for WES for patients with unknown genetic etiology (4, 6). The ThromboGenomics project provided a multi-gene high-throughput sequencing platform (HTS) for the diagnosis of heritable bleeding disorders (79). The HTS platform covers approximately 96 genes associated with inherited bleeding, thrombotic, coagulation, and platelet disorders. The panel showed high sensitivity in detecting causative variants in patients who had not been previously investigated at the molecular level. It has a high sensitivity to detect variants in the exonic region as well as many of exonic-intronic boundaries and untranslated regions (UTRs) (6, 79).
Next Generation Sequencing
Targeted NGS platforms can be efficiently applied to determine the causative genes of IT. As the molecular basis of ITs remain unknown in many patients, WGS or WES may be required which improves the knowledge of ITs at the molecular level. Several national and international consortia have adopted these approaches to identify disease-causing genes associated with IT. The genes SLFN14, FYB, STIM1, GFI1b, and ETV6 are some examples of causative genes detected by these approaches. The results obtained by HTS improves the understanding of the functional role in some causative genes, whose function in platelet production was previously unknown. These techniques will bring substantial benefits to improve our understanding of the molecular mechanisms in megakaryocyte and platelet biogenesis (14, 16, 24, 49, 80, 81). However, distinguishing pathogenic variants from non-pathogenic variants often requires complex functional and cell line studies to prove causality (7).
Bioinformatic Tools
Bioinformatic tools can be conducted to determine candidate variants from WES or WGS data. A wide range of variants, ~25,000–40,000 variants can be identified per single patient in WES. These variants can be filtered for novelty by direct comparison using a database from the 100,000 Genomes Project, Exon Variant Server (EVS), dbSNP versions, Exome Aggregation Consortium (ExAC) (gnomAD), and in-house databases of whole exomes and/or whole genomes. A database of known platelet-related genes and genes involved in platelet formation, function, lifespan, or death can be compared with the patient's genes in order to narrow the candidate genetic variants down. Variants with MAF (minor allele frequency) ≥0.01 are generally excluded given the rarity of most of these genetic defects in IT. Variants not known to change the amino acid or those that do not have a potential effect on protein, such as synonymous variants and intron variants can also be excluded. Splice site variants occurring >5 base pair away from the exome can be also excluded, although this can potentially result in splicing or regulatory mutations being missed. Comparisons with other affected and unaffected family members on the database can be used to select candidate variants. Also, pathogenicity prediction can be assessed by using different tools such PolyPhen2, Provean, SIFT, Mutation Taster, mRNA expression levels which predict the potential effect of amino acid changes on protein structure and function and also measure the conservation of amino acids among different species. Sapientia is a recently developed clinical diagnostic platform established by Congenica to help clinicians, clinical scientists, and researchers with genetic diagnosis and identification of disease-causing genes, by interrogating the human genome with multiple bioinformatic tools. It can help streamline the process of diagnosis, ensuring patients are receiving accurate information and treatments for their individual platelet or megakaryocyte defect.
Conclusions
The advances in NGS techniques improves our knowledge about the molecular mechanisms of IT. The major risk factor for patients with ITs is the development of additional syndromic disorders rather than bleeding itself. Due to the polygenic nature of ITs and disorders involved in hematopoiesis, identifying a singular causative gene for platelet and megakaryocyte function is particularly difficult. A combination of whole blood counting and platelet functional assays will highlight the platelet phenotype, however only familial studies and genetic sequencing will help to identify any genetic defect. With the introduction of HTS and various genome browser software such as Sapienta, the diagnosis process has been modernized to highlight candidate variants in known platelet affected genes and reveal variants in novel genes in which hemostatic input remains to be explored. Approaches such as these may be implemented within clinical settings in the future, however, bioinformatic pipelines are yet to be standardized across all facilities. Aside from bioinformatic training and the initial financial burden of installation of the software, there are clear reasons that updating current genetic analysis in hematological disorders benefits healthcare in the wider community. This will ensure IT families obtain a clear diagnosis and receive correct treatment based on their genetically influenced megakaryocyte or platelet defect.
Author Contributions
IA wrote the manuscript. IA, RS, and NM critically reviewed and edited the review.
Funding
The work in the author's laboratories is supported by the British Heart Foundation (PG/16/103/32650; FS/18/11/33443) and the Saudi Arabia Cultural Bureau in London.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. D'Andrea G, Chetta M, Margaglione M. Inherited platelet disorders: thrombocytopenias and thrombocytopathies. Blood Transfus. (2009) 7:278. doi: 10.2450/2009.0078-08
2. Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. (2007) 357:2482–94. doi: 10.1056/NEJMra071014
3. Balduini A, Raslova H, Di Buduo CA, Donada A, Ballmaier M, Germeshausen M, et al. Clinic, pathogenic mechanisms and drug testing of two inherited thrombocytopenias, ANKRD26-and MYH9-related diseases. Eur J Med Genet. (2018) 61:715–22. doi: 10.1016/j.ejmg.2018.01.014
4. Noris P, Pecci A. Hereditary thrombocytopenias: a growing list of disorders. Hematology. (2017) 2017:385–99. doi: 10.1182/asheducation-2017.1.385
5. Drachman JG. Review in translational hematology inherited thrombocytopenia : when a low platelet count does not mean ITP. Blood. (2004) 103:390–8. doi: 10.1182/blood-2003-05-1742
6. Johnson B, Doak R, Allsup D, Astwood E, Evans G, Grimley C, et al. A comprehensive targeted next-generation sequencing panel for genetic diagnosis of patients with suspected inherited thrombocytopenia. Res Pract Thromb Haemost. (2018) 2:640–52. doi: 10.1002/rth2.12151
7. Johnson B, Lowe GC, Futterer J, Lordkipanidzé M, MacDonald D, Simpson MA, et al. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica. (2016) 101:1170–9. doi: 10.3324/haematol.2016.146316
8. Morgan NV, Daly ME. Gene of the issue: RUNX1 mutations and inherited bleeding. Platelets. (2017) 28:208–10. doi: 10.1080/09537104.2017.1280151
9. Balduini CL, Melazzini F, Pecci A. Inherited thrombocytopenias—recent advances in clinical and molecular aspects. Platelets. (2017) 28:3–13. doi: 10.3109/09537104.2016.1171835
10. Savoia A. Molecular basis of inherited thrombocytopenias: an update. Curr Opin Hematol. (2016) 23:486–92. doi: 10.1097/MOH.0000000000000269
11. Greinacher A, Eekels JJM. Diagnosis of hereditary platelet disorders in the era of next-generation sequencing: “primum non nocere.” J Thromb Haemost. (2019) 17:551–4. doi: 10.1111/jth.14377
12. Bluteau D, Balduini A, Balayn N, Currao M, Nurden P, Deswarte C, et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J Clin Invest. (2014) 124:580–91. doi: 10.1172/JCI71861
13. Pippucci T, Savoia A, Perrotta S, Ria Pujol-Moix N, Noris P, Castegnaro G, et al. Mutations in the 5'UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet. (2011) 88:115–20. doi: 10.1016/j.ajhg.2010.12.006
14. Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. (2015) 47:535–8. doi: 10.1038/ng.3253
15. Stevenson WS, Rabbolini DJ, Beutler L, Chen Q, Gabrielli S, Mackay JP, et al. Paris-Trousseau thrombocytopenia is phenocopied by the autosomal recessive inheritance of a DNA-binding domain mutation in FLI1. Blood. (2015) 126:2027–30. doi: 10.1182/blood-2015-06-650887
16. Koren A, Khayat M, Hauschner H, Pretorius E, Rosenberg N, Zalman L, et al. Deleterious mutation in the FYB gene is associated with congenital autosomal recessive small-platelet thrombocytopenia. J Thromb Haemost. (2015) 13:1285–92. doi: 10.1111/jth.12966
17. Freson K, Wijgaerts A, Van Geet C. GATA1 gene variants associated with thrombocytopenia and anemia. Platelets. (2017) 28:731–4. doi: 10.1080/09537104.2017.1361525
18. Stevenson WS, Morel-Kopp MC, Chen Q, Liang HP, Bromhead CJ, Wright S, et al. GFI1B mutation causes a bleeding disorder with abnormal platelet function. J Thromb Haemost. (2013) 11:2039–47. doi: 10.1111/jth.12368
19. Horvat-Switzer RD, Thompson AA. HOXA11 mutation in amegakaryocytic thrombocytopenia with radio-ulnar synostosis syndrome inhibits megakaryocytic differentiation in vitro. Blood Cells Mol Dis. (2006) 37:55–63. doi: 10.1016/j.bcmd.2006.04.001
20. Germeshausen M, Ancliff P, Estrada J, Metzler M, Ponstingl E, Schwabe D, et al. MECOM-associated syndrome: a heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Adv. (2018) 2:586–96. doi: 10.1182/bloodadvances.2018016501
21. Ihara K, Ishii E, Eguchi M, Takada H, Suminoe A, Good RA, et al. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Med Sci. (1999) 96:3132–6. doi: 10.1073/pnas.96.6.3132
22. Pluthero FG, Di Paola J, Carcao MD, Kahr WHA. NBEAL2 mutations and bleeding in patients with gray platelet syndrome. Platelets. (2018) 29:632–5. doi: 10.1080/09537104.2018.1478405
23. Manukjan G, Bösing H, Schmugge M, Strauß G, Schulze H. Impact of genetic variants on haematopoiesis in patients with thrombocytopenia absent radii (TAR) syndrome. Br J Haematol. (2017) 179:606–17. doi: 10.1111/bjh.14913
24. Fletcher SJ, Johnson B, Lowe GC, Bem D, Drake S, Lordkipanidzé M, et al. SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J Clin Invest. (2015) 125:3600–5. doi: 10.1172/JCI80347
25. Turro E, Greene D, Wijgaerts A, Thys C, Lentaigne C, Bariana TK, et al. A dominant gain-of-function mutation in universal tyrosine kinase SRC causes thrombocytopenia, myelofibrosis, bleeding, and bone pathologies. Sci Transl Med. (2016) 8:328ra30. doi: 10.1126/scitranslmed.aad7666
26. Noris P, Marconi C, De Rocco D, Melazzini F, Pippucci T, Loffredo G, et al. A new form of inherited thrombocytopenia due to monoallelic loss of function mutation in the thrombopoietin gene. Br J Haematol. (2017) 181:698–701. doi: 10.1111/bjh.14694
27. Wen R, Wang D. PTPRJ: a novel inherited thrombocytopenia gene. Blood. (2019) 133:1272–4. doi: 10.1182/blood-2019-01-895102
28. Seo A, Gulsuner S, Pierce S, Ben-Harosh M, Shalev H, Walsh T, et al. Inherited thrombocytopenia associated with mutation of UDP-galactose-4-epimerase (GALE). Hum Mol Genet. (2019) 28:133–42. doi: 10.1093/hmg/ddy334
29. Kunishima S, Okuno Y, Yoshida K, Shiraishi Y, Sanada M, Muramatsu H, et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet. (2013) 92:431–8. doi: 10.1016/j.ajhg.2013.01.015
30. Ong L, Morison IM, Ledgerwood EC. Megakaryocytes from CYCS mutation-associated thrombocytopenia release platelets by both proplatelet-dependent and -independent processes. Br J Haematol. (2017) 176:268–79. doi: 10.1111/bjh.14421
31. Futterer J, Dalby A, Lowe GC, Johnson B, Simpson MA, Motwani J, et al. Mutation in GNE is associated with severe congenital thrombocytopenia. Blood. (2018) 132:1855–8. doi: 10.1182/blood-2018-04-847798
32. Revel-Vilk S, Shai E, Turro E, Jahshan N, Hi-Am E, Spectre G, et al. GNE variants causing autosomal recessive macrothrombocytopenia without associated muscle wasting. Blood. (2018) 132:1851–4. doi: 10.1182/blood-2018-04-845545
33. Izumi R, Niihori T, Suzuki N, Sasahara Y, Rikiishi T, Nishiyama A, et al. GNE myopathy associated with congenital thrombocytopenia: a report of two siblings. Neuromuscul Disord. (2014) 24:1068–72. doi: 10.1016/j.nmd.2014.07.008
34. Sivapalaratnam S, Westbury SK, Stephens JC, Greene D, Downes K, Kelly AM, et al. Rare variants in GP1BB are responsible for autosomal dominant macrothrombocytopenia. Blood. (2017) 129:520–4. doi: 10.1182/blood-2016-08-732248
35. Balduini A, Malara A, Balduini CL, Noris P. Megakaryocytes derived from patients with the classical form of Bernard-Soulier syndrome show no ability to extend proplatelets in vitro. Platelets. (2011) 22:308–11. doi: 10.3109/09537104.2010.547960
36. Wright SD, Michaelides K, Johnson DJD, West NC, Tuddenham EGD. Double heterozygosity for mutations in the platelet glycoprotein IX gene in three siblings with Bernard-Soulier syndrome. Blood. (1993) 81:2339–47.
37. Burk CD, Newman PJ, Lyman S, Gill J, Coller BS, Poncz M. A deletion in the gene for glycoprotein lib associated with Glanzmann's thrombasthenia. J Clin Invest. (1991) 87:270–6. doi: 10.1172/JCI114982
38. Nurden AT, Pillois X, Wilcox DA. Glanzmann Thrombasthenia: state of the art and future directions. Semin Thromb Hemost. (2013) 39:642. doi: 10.1055/s-0033-1353393
39. Balduini CL, Pecci A, Savoia A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br J Haematol. (2011) 154:161–74. doi: 10.1111/j.1365-2141.2011.08716.x
40. Manchev VT, Hilpert M, Berrou E, Elaib Z, Aouba A, Boukour S, et al. A new form of macrothrombocytopenia induced by a germ-line mutation in the PRKACG gene. Blood. (2014) 124:2554–63. doi: 10.1182/blood-2014-01-551820
41. Stritt S, Nurden P, Favier R, Favier M, Ferioli S, Gotru SK, et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg2+ homeostasis and cytoskeletal architecture. Nat Commun. (2016) 7:11097. doi: 10.1038/ncomms11097
42. Pleines I, Woods J, Chappaz S, Kew V, Foad N, Ballester-Beltrán J, et al. Mutations in tropomyosin 4 underlie a rare form of human macrothrombocytopenia. J Clin Invest. (2017) 127:814–29. doi: 10.1172/JCI86154
43. Kunishima S, Kobayashi R, Itoh TJ, Hamaguchi M, Saito H. Mutation of the β1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood. (2009) 113:458–61. doi: 10.1182/blood-2008-06-162610
44. Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. (2013) 1285:26–43. doi: 10.1111/nyas.12049
45. Nurden P, Debili N, Coupry I, Bryckaert M, Youlyouz-Marfak I, Solé G, et al. Thrombocytopenia resulting from mutations in filamin A can be expressed as an isolated syndrome. Blood. (2011) 118:5928–37. doi: 10.1182/blood-2011-07-365601
46. Stritt S, Nurden P, Turro E, Greene D, Jansen SB, Westbury SK, et al. A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood. (2016) 127:2903–14. doi: 10.1182/blood-2015-10-675629
47. Bastida JM, Benito R, Janusz K, Díez-Campelo M, Hernández-Sánchez JM, Marcellini S, et al. Two novel variants of the ABCG5 gene cause xanthelasmas and macrothrombocytopenia: a brief review of hematologic abnormalities of sitosterolemia. J Thromb Haemost. (2017) 15:1859–66. doi: 10.1111/jth.13777
48. Levy GG, Nichols WC, Lian EC, Foroud T, Mcclintick JN, Mcgee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura.Nature. (2001) 413:488–94. doi: 10.1038/35097008
49. Nesin V, Wiley G, Kousi M, Ong EC, Lehmann T, Nicholl DJ, et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc Natl Acad Sci USA. (2014) 111:4197–202. doi: 10.1073/pnas.1312520111
50. Markello T, Chen D, Kwan JY, Horkayne-Szakaly I, Morrison A, Simakova O, et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1 HHS public access. Mol Genet Metab. (2015) 114:474–82. doi: 10.1016/j.ymgme.2014.12.307
51. Cooney KA, Nichols WC, Bruck ME, Bahou WF, Shapiro AD, Bowie EJW, et al. The molecular defect in type IIB von Willebrand disease. J Clin Invest. (1991) 87:1227–33. doi: 10.1172/JCI115123
52. Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. (2015) 1356:45–79. doi: 10.1111/nyas.12938
53. Hurtado B, Trakala M, Ximénez-Embún P, El Bakkali A, Partida D, Sanz-Castillo B, et al. Thrombocytopenia-associated mutations in Ser/Thr kinase MASTL deregulate actin cytoskeletal dynamics in platelets. J Clin Invest. (2018) 128:5351–67. doi: 10.1172/JCI121876
54. Johnson B, Fletcher SJ, Morgan NV. Inherited thrombocytopenia: novel insights into megakaryocyte maturation, proplatelet formation and platelet lifespan. Platelets. (2016) 27:519–25. doi: 10.3109/09537104.2016.1148806
55. Schulze H, Shivdasani RA. Mechanisms of thrombopoiesis. J Thromb Haemost. (2005) 3:1717–24. doi: 10.1111/j.1538-7836.2005.01426.x
56. Ogawa M. Differentiation and proliferation of hematopoietic stem cells. Blood. (1993) 81:2844–53.
57. Drachman JG, Griffin JD, Kaushansky K. The c-Mpl ligand (thrombopoietin) stimulates tyrosine phosphorylation of Jak2, Shc, and c-Mpl. J Biol Chem. (1995) 270:4979–82. doi: 10.1074/jbc.270.10.4979
58. Dasouki MJ, Rafi SK, Olm-Shipman AJ, Wilson NR, Abhyankar S, Ganter B, et al. Exome sequencing reveals a thrombopoietin ligand mutation in a Micronesian family with autosomal recessive aplastic anemia. Blood. (2013) 122:3440–9. doi: 10.1182/blood-2012-12-473538
59. Geddis AE. Megakaryopoiesis. Semin Hematol. (2010) 47:212–9. doi: 10.1053/j.seminhematol.2010.03.001
60. Millikan PD, Balamohan SM, Raskind WH, Kacena MA. Inherited thrombocytopenia due to GATA-1 mutations. Semin Thromb Hemost. (2011) 37:682–9. doi: 10.1055/s-0031-1291378
61. Tijssen MR, Ghevaert C. Transcription factors in late megakaryopoiesis and related platelet disorders. J Thromb Haemost. (2013) 11:593–604. doi: 10.1111/jth.12131
62. Foudi A, Kramer DJ, Qin J, Ye D, Behlich A-S, Mordecai S, et al. Distinct, strict requirements for Gfi-1b in adult bone marrow red cell and platelet generation. J Exp Med. (2014) 211:909–27. doi: 10.1084/jem.20131065
63. Letestu R, Vitrat N, Massé A, Le Couedic J-P, Lazar V, Rameau P, et al. Existence of a differentiation blockage at the stage of a megakaryocyte precursor in the thrombocytopenia and absent radii (TAR) syndrome. Blood. (2000) 95:1633–41.
64. Loffredo G, Balduini CL, Notarangelo LD, Casale M, Perrotta S, Pujol-Moix N, et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood. (2011) 117:6673–80. doi: 10.1182/blood-2011-02-336537
65. Bender M, Thon JN, Ehrlicher AJ, Wu S, Mazutis L, Deschmann E, et al. Microtubule sliding drives proplatelet elongation and is dependent on cytoplasmic dynein. Blood. (2015) 125:860–8. doi: 10.1182/blood-2014-09-600858
66. Machlus KR, Italiano JE. The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. (2013) 201:785–96. doi: 10.1083/jcb.201304054
67. Snapper SB, Rosen FS. The Wiskott-Aldrich Syndrome Protein (WASP): roles in signaling and cytoskeletal organization. Annu. Rev. Immunol. (1999) 17:905–29. doi: 10.1146/annurev.immunol.17.1.905
68. Kirchhausen T, Rosen FS. Disease mechanism: unravelling Wiskott-Aldrich syndrome. Curr Biol. (1996) 6:676–8. doi: 10.1016/S0960-9822(09)00447-3
69. Savoia A, Kunishima S, De Rocco D, Zieger B, Rand ML, Pujol-Moix N, et al. Spectrum of the mutations in bernard-soulier syndrome. Hum Mutat. (2014) 35:1033–45. doi: 10.1002/humu.22607
70. Balduini A, Malara A, Pecci A, Badalucco S, Bozzi V, Pallotta I, et al. Proplatelet formation in heterozygous Bernard-Soulier syndrome type Bolzano. J Thromb Haemost. (2009) 7:478–84. doi: 10.1111/j.1538-7836.2008.03255.x
71. Kunishima S, Kashiwagi H, Otsu M, Takayama N, Eto K, Onodera M, et al. Heterozygous ITGA2B R995W mutation inducing constitutive activation of the IIb3 receptor affects proplatelet formation and causes congenital macrothrombocytopenia. Blood. (2011) 117:5479–84. doi: 10.1182/blood-2010-12-323691
72. Pecci A. Diagnosis and treatment of inherited thrombocytopenias. Clin Genet. (2016) 89:141–53. doi: 10.1111/cge.12603
73. Broadley SP, Plaumann A, Coletti R, Lehmann C, Wanisch A, Seidlmeier A, et al. Dual-track clearance of circulating bacteria balances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe. (2016) 20:36–48. doi: 10.1016/j.chom.2016.05.023
74. Kim S-J, Davis R, Jenne C. Platelets as modulators of inflammation. Semin Thromb Hemost. (2018) 44:91–101. doi: 10.1055/s-0037-1607432
75. Swinkles M, Rijkers M, Voorberg C, Vidarsson G, Leebeek FWG, Jansen AJG. Emerging concepts in immune thrombocytopenia. Front Immunol. (2018) 9:880. doi: 10.3389/fimmu.2018.00880
76. Leonard M, Brice M, Engel JD, Papayannopoulou T. Dynamics of GATA transcription factor expression during erythroid differentiation. Blood. (1993) 82:1071–9.
77. Marigo V, Nigro A, Pecci A, Montanaro D, Stazio M Di, Balduini CL, et al. Correlation between the clinical phenotype of MYH9-related disease and tissue distribution of class II nonmuscle myosin heavy chains. Genomics. (2004) 83:1125–33. doi: 10.1016/j.ygeno.2003.12.012
78. Pecci A, Klersy C, Gresele P, Lee KJD, De Rocco D, Bozzi V, et al. MYH9-related disease: a novel prognostic model to predict the clinical evolution of the disease based on genotype-phenotype correlations Europe PMC funders group. Hum Mutat. (2014) 35:236–47. doi: 10.1002/humu.22476
79. Simeoni I, Stephens JC, Hu F, Deevi SV, Megy K, Bariana TK, et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood. (2016) 127:2791–803. doi: 10.1182/blood-2015-12-688267
80. Kahr WH, Hinckley J, Li L, Schwertz H, Christensen H, Rowley JW, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. (2011) 43:738–40. doi: 10.1038/ng.884
Keywords: inherited thrombocytopenia, platelets, megakaryocytes, genes, mutations, bleeding
Citation: Almazni I, Stapley R and Morgan NV (2019) Inherited Thrombocytopenia: Update on Genes and Genetic Variants Which may be Associated With Bleeding. Front. Cardiovasc. Med. 6:80. doi: 10.3389/fcvm.2019.00080
Received: 24 April 2019; Accepted: 31 May 2019;
Published: 19 June 2019.
Edited by:
Vera Ignjatovic, Murdoch Childrens Research Institute, AustraliaReviewed by:
Patricia B. Maguire, University College Dublin, IrelandCécile Oury, University of Liège, Belgium
Copyright © 2019 Almazni, Stapley and Morgan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neil V. Morgan, n.v.morgan@bham.ac.uk