Trends in the Contribution of Genetic Susceptibility Loci to Hyperuricemia and Gout and Associated Novel Mechanisms
 Jianan Zhao
Jianan Zhao Shicheng Guo
Shicheng Guo Steven J. Schrodi
Steven J. Schrodi Dongyi He
Dongyi He- 1Department of Rheumatology, Shanghai Guanghua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Guanghua Clinical Medical College, Shanghai University of Traditional Chinese Medicine, Shanghai, Shanghai, China
- 3Computation and Informatics in Biology and Medicine, University of WI-Madison, Madison, WI, United States
- 4Department of Medical Genetics, School of Medicine and Public Health, University of WI-Madison, Madison, WI, United States
- 5Arthritis Institute of Integrated Traditional and Western Medicine, Shanghai Chinese Medicine Research Institute, Shanghai, China
- 6Institute of Arthritis Research in Integrative Medicine, Shanghai Academy of Traditional Chinese Medicine, Shanghai, China
Hyperuricemia and gout are complex diseases mediated by genetic, epigenetic, and environmental exposure interactions. The incidence and medical burden of gout, an inflammatory arthritis caused by hyperuricemia, increase every year, significantly increasing the disease burden. Genetic factors play an essential role in the development of hyperuricemia and gout. Currently, the search on disease-associated genetic variants through large-scale genome-wide scans has primarily improved our understanding of this disease. However, most genome-wide association studies (GWASs) still focus on the basic level, whereas the biological mechanisms underlying the association between genetic variants and the disease are still far from well understood. Therefore, we summarized the latest hyperuricemia- and gout-associated genetic loci identified in the Global Biobank Meta-analysis Initiative (GBMI) and elucidated the comprehensive potential molecular mechanisms underlying the effects of these gene variants in hyperuricemia and gout based on genetic perspectives, in terms of mechanisms affecting uric acid excretion and reabsorption, lipid metabolism, glucose metabolism, and nod-like receptor pyrin domain 3 (NLRP3) inflammasome and inflammatory pathways. Finally, we summarized the potential effect of genetic variants on disease prognosis and drug efficacy. In conclusion, we expect that this summary will increase our understanding of the pathogenesis of hyperuricemia and gout, provide a theoretical basis for the innovative development of new clinical treatment options, and enhance the capabilities of precision medicine for hyperuricemia and gout treatment.
Introduction
Gout is the leading cause of inflammatory arthritis in males. This is primarily due to multiple mechanisms resulting in the deposition of urate in the synovial fluid and other tissues to form monosodium urate crystals, which are further stimulated by inflammatory irritants, ultimately resulting in gout. The global prevalence of gout is approximately 0.1%–10%, and the incidence ranges from 0.3 to 6 cases per 1,000 person-years (Kuo et al., 2015; Liu et al., 2015; GBD, 2017). With a worldwide trend of an aging population, the medical disease burden of gout is increasing (Smith et al., 2014). Risk factors for hyperuricemia and gout include the use of medications (thiazides, cyclosporine, low-dose aspirin), insulin resistance, metabolic syndrome, obesity, renal insufficiency, abnormal blood pressure, purine-rich foods, alcohol, and sugary drinks (Neogi, 2011). The role of wine in gout may be contradictory, in any case, a retrospective study said individuals with established gout and pre-existing risk factors should limit all types of alcohol intake to prevent gout episodes (Nieradko-Iwanicka, 2021).
The main source of uric acid is the metabolism of purines and nucleotides in food produced in the liver and excreted by the intestines and kidneys (Köttgen et al., 2013a). Uric acid is reabsorbed and secreted into the proximal tubules of the kidneys, and urate transporter-1 (URAT1), glucose transporter 9 (GLUT9) (also known as GLUT9L), organic anion transporter 4 (OAT4), and organic anion transporter 4 (OAT10) are responsible for its reabsorption. TP-binding cassette superfamily G member 2 (ABCG2), adenosine triphosphate (ATP) binding cassette subfamily C member 4 (ABCC4), and organic anion transporter (NPT1 and NPT4) proteins mediate uric acid excretion. The balance between reabsorption and secretion is related to the homeostasis of uric acid. Otherwise, hyperuricemia and gout can occur. A high serum uric acid concentration is the primary risk factor for gout. Controlling the metabolism of uric acid in circulation at reasonable levels plays a vital role in preventing and improving gout (Chung and Kim, 2021). The progression from high blood uric acid levels to gout occurs in three main steps, hyperuricemia, the deposition of monosodium urate crystals, and inflammatory responses in the joints (Dalbeth et al., 2016). Toll-like receptor (TLR) and NLRP3 inflammasome activation and the associated inflammatory responses are critical factors in the progression of hyperuricemia to gout. This primarily involves activation of the downstream TLR4 and nuclear factor-κB (NF-kB) pathways, activation of the NLRP3 inflammasome, and production of interleukin (IL)-1β, which together regulate immune, metabolic, and inflammatory processes (Qing et al., 2013; McKinney et al., 2015; Rasheed et al., 2016).
Gene variants in related functional proteins can affect uric acid metabolism and inflammation in vivo (Reginato et al., 2012). Current research is focused on the heritability of uric acid-associated phenotypes, estimated to be around 40%–70%, implying a clear indication of the importance of its genetic role (Köttgen et al., 2013a). Commonly used drugs to treat acute gout attacks include nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine, and glucocorticoids (Terkeltaub, 2003). Several biologically targeted agents have also been developed, such as IL-1/1β antagonists, anakinra, rilonacept, canakinumab (So et al., 2007; Terkeltaub et al., 2009; Neogi, 2010; So et al., 2010). In addition, patients with gout require a combination of long-term treatments to lower uric acid levels, such as allopurin, probenecid, and sulfinpyrazone (Terkeltaub, 2003). Although existing gout treatment drugs have achieved some efficacy, the multiple side effects and even poor drug response in some patients suggest that we should focus, at least in part, on the genetic mechanisms underlying hyperuricemia and gout to identify other effective and well-tolerated clinical treatment options. Large-scale genome-wide association studies have identified many risk loci (Tin et al., 2019a); however, the biological mechanisms underlying hyperuricemia and gout remain unclear. A meta-analysis of the Global Biobank Meta-analysis Initiative improved the understanding of this disease, as well as risk prediction, by integrating GWAS results from six major ancestral groups (African ancestry from African or mixed-race immigrants, mixed-race Americans, Central and South Asians, East Asians, Europeans, and Middle Easterners), while also providing insight into the underlying biology of the traits being studied by integrating gene and protein expression data, enabling the identification of disease-related genes and drug candidates (Zhou et al., 2021). This review discusses the mechanisms through which gene variants affect hyperuricemia and gout by searching Pubmed and GBMI (https://www.globalbiobankmeta.org/and http://results.globalbiobankmeta.org/) database. This review further explores and discusses the relationship between multiple biological agents and genetic variants and how they potentially affect gout and hyperuricemia to provide a theoretical reference for further clinical treatment options.
Association Between Uric Acid Transporter-Related Gene Variants and Hyperuricemia and Gout
A decline in kidney function is a vital cause of hyperuricemia and gout. As the kidney is the main organ that excretes uric acid, when this occurs, uric acid excretion by this organ is also reduced. The increase in uric acid in the blood aggravates hyperuricemia, negatively affecting each other and forming a vicious cycle (Asgari and Hilton, 2021). The proximal tubule excretes most uric acid, and when specific lesions occur there, dysfunction leads to low uric acid excretion, which is often associated with genetic variants of specific uric acid transport proteins. Pleiotropy in genetic variation can underlie the regulation of renal function, hyperuricemia, and gout (García-Nieto et al., 2022). For example, Mendelian randomization analysis can be used to analyze causality in confounding situations. Using this approach to determine the relationship between genetic variants regulating blood uric acid excretion in the kidney and renal function, it was found that the uric acid transporter genetic risk score (mainly comprising solute carrier family 2 member 9 (SLC2A9), ABCG2, solute carrier family 22 member 11 (SLC22A11), solute carrier family 17 member 1 (SLC17A1), and solute carrier family 22 member 12 (SLC22A12)) was positively associated with improved renal function in European Caucasian males. The uric acid transporter protein genetic risk score was used as an instrumental variable. Mendelian randomization for renal function using the two-stage least squares method to assess the effect of urate on renal function quantitatively (Hughes et al., 2014). In conclusion, it was found that the variant with the strongest effect on the protection of renal function was located in SLC22A11 (Hughes et al., 2014). A meta-analysis further identified four gene loci (SLC2A9, ABCG2, SLC22A12, and MAF BZIP transcription factor (MAF)) associated with blood uric acid levels and renal function in an East Asian population (Okada et al., 2012). Another meta-analysis of GWASs on serum urea salt concentrations and gout in African Americans found genome-wide significance at three loci (SLC2A9, solute carrier family 2 member 12 (SLC2A12), and SLC22A12) (Tin et al., 2011). Previous genome-wide significant loci associated with serum urate levels, such as SLC2A12, were identified and validated in a meta-analysis by combining GWAS data from more than 14,000 individuals (Köttgen et al., 2013a). Similarly, ABCG2, SLC2A9, solute carrier family 16 member 9 (SLC16A9), glucokinase (hexokinase 4) regulator (GCKR), SLC22A11, SLC22A12, PDZ domain containing 1 (PDZK1), and SLC17A1 were found to be significantly associated with hyperuricemia and gout risk in Asian, native Hawaiian, and Pacific Islander populations estimated using the biospecimens repository at the University of Hawai’i (Alghubayshi et al., 2022). Most gout-related genetic studies have focused on this mechanism and have made some exciting discoveries, but there still could be a need to focus on this and explore it more extensively in the future (Figure 1).
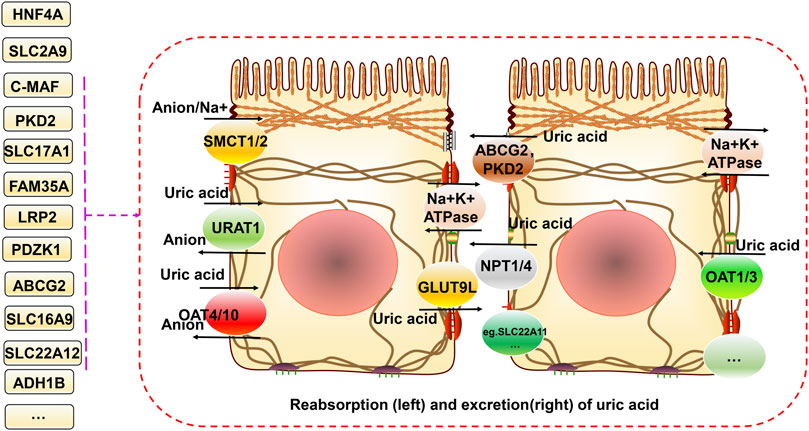
FIGURE 1. Relationship between gene variants and reabsorption and excretion of uric acid in hyperuricemia and gout. Uric acid is reabsorbed and secreted into the proximal tubules of the kidneys. URAT1, GLUT9 (also known as GLUT9L), OAT4, and OAT10 are responsible for its reabsorption. ABCG2, ABCC4, NPT1, and NPT4 mediate uric acid excretion. The balance between reabsorption and secretion is related to uric acid homeostasis. In addition, SMCT1/2 proteins can regulate ion concentrations both inside and outside the cell. Different gene variants have different effects on these processes.
Hepatocyte Nuclear Factor 4 Alpha (HNF4A), Hepatocyte Nuclear Factor 4 Gamma (HNF4G), and PDZK1
HNF4A encodes a nuclear transcription factor that binds DNA and modulates the transcription of multiple genes, mainly in the form of homodimers. A missense variant in HNF4A (rs1800961) is probably the most likely leading and causal variant resulting in better transactivation of the promoter of the urate transporter protein-encoding gene ABCG2 (Tin et al., 2019a). Additionally, HNF4A can also control gene expression in pancreatic islets, potentially further associating with uric acid and gout by affecting insulin secretion (Yoon et al., 2001). The inheritance and expression of different alleles of HNF4A might also have potential effects on renal function, but the exact mechanism remains unknown (Leask et al., 2020).
PDZK1 primarily encodes a scaffolding protein containing the PDZ structural domain. It mediates the localization of cell surface proteins and is linked to cholesterol metabolism through the regulation of multiple receptors. HNF4A can also directly regulate PDZK1. The T-allele of PDZK1 single-nucleotide polymorphism (SNP) (rs1967017) enhances HNF4A binding to the promoter of PDZK1, augmenting its expression, potentially increasing uric acid transport, and regulating uric acid homeostasis, as PDZK1 is a scaffolding protein for multiple transport proteins (Ketharnathan et al., 2018). A C-MAF BZIP transcription factor-encoding (C-MAF) SNP (rs889472) might also be associated with gout susceptibility by affecting uric acid metabolism (Higashino et al., 2018), and part of the mechanism could be related to regulation of the transcription factor HNF4A (Leask et al., 2018). MAF/c-MAF is mainly expressed in the proximal tubules of the kidney and is a critical factor for maintaining differentiation and functional integrity (Imaki et al., 2004; Tsuchiya et al., 2015). Its b-ZIP structure can form dimers with other b-ZIP proteins and bind to DNA as transcription factors to regulate the functions of various organs, such as the kidney and pancreas (Yang and Cvekl, 2007; Tsuchiya et al., 2015). Leask et al. summarized the genetic mechanisms underlying the detailed regulation of uric acid levels mediated by MAF variants, mainly involving the proximal signal cis-expression quantitative trait loci (cis-eQTL) of MAF (controls the expression of MAF transcriptional regulator RNA (MAFTRR)) and the distal signal cis-eQTL (controls the expression of LINC01229). The MAFTRR lncRNA region binds to the MAF promoter and recruits the histone imprint H3K27me3 to repress MAF transcription, whereas the removal of both LINC01229 and MAFTRR promotes MAF expression (Leask and Merriman, 2021).
HNF4G also encodes a transcription factor involved in the positive regulation of transcription by RNA polymerase II. It has a lower transcriptional activation potential than that of HNF4A. An HNF4G SNP (rs2941484) can increase gout susceptibility in the Chinese population and mainly affects serum uric acid concentration and gout risk in men (Dong et al., 2017). In Chinese Han men, the TT genotype of the HNF4G rs2941484 may represent a gender-specific genetic marker of hyperuricemia. The distribution frequency of TT and CC+CT alleles in hyperuricemic and normokalemic males differed considerably (p = 0.011) in the rs2941484 recessive model (Chen et al., 2017). miR-34a can regulate HNF4G to control the survival, proliferation, and invasion of bladder cancer cells (Sun et al., 2015) and might bind to endogenous fatty acids to regulate fatty acid metabolic pathways affecting gout (Wisely et al., 2002).
ABCG2 and Polycystin 2 (PKD2)
ABCG2 is a multispecific heterotrimeric and endogenous transporter protein expressed mainly in the kidney, liver, and gastrointestinal tract that affects drug metabolism and plays a key role in uric acid excretion. Its variants can lead to destabilization of the nucleotide-binding structural domain of ABCG2, resulting in its reduced expression and dysfunction, leading to the inadequate renal excretion of urate, causing hyperuricemia and gout (Wong et al., 2016). ABCG2 variants (rs2231142) are variants associated with gout and an increased frequency of erythema (Onuora, 2020). Individuals carrying the ABCG2 SNP (rs2231142) have a nearly 2-fold increased susceptibility to gout (Lee et al., 2019), and alcohol consumption independently increases the risk of gout stones in the Han Chinese population in Taiwan (Tu et al., 2014). The alpha kinase 1 variant in combination with the ABCG2 SNP (rs2231142), the SLC2A9 SNP (rs1014290), or the SLC22A12 SNP (rs475688 and rs3825016) is linked to gout in the recessive model (Tu et al., 2018). ABCG2 SNP (rs2231142) significantly increased the risk of gout in Asians (dominant model: OR = 2.64, 95% CI = 2.04–3.43, p = 0.02 for heterogeneity; recessive model: OR = 3.19, 95% CI = 2.56–3.97, p = 0.28 for heterogeneity; co-dominant model: OR = 1.37, 95% CI = 1.18–1.59, p = 0.09 for heterogeneity) as well as other populations (dominant model: OR = 1.85, 95% CI = 1.20–2.85, p < 0.0001 for heterogeneity; recessive model: OR = 3.78, 95% CI = 2.28–6.27, p = 0.19 for heterogeneity; co-dominant model: OR = 1.48, 95% CI = 1.26–1.74, p = 0.19 for heterogeneity) (Li et al., 2015a). The ABCG2 SNP (rs72552713) also significantly increased the risk of gout in Asians (dominant model: OR = 3.87, 95% CI = 2.07–7.24, p = 0.06 for heterogeneity) (Li et al., 2015a). ABCG2 and PKD2 were found to have epistatic interactions, and two SNP pairs (rs2728121:rs1481012 and rs2728121:rs2231137) were mainly identified as associated with the serum urate concentration or risk of hyperuricemia (Dong et al., 2020). ABCG2 variants might affect disease progression through inflammatory pathways, in addition to lowering uric acid excretion. The knockdown of ABCG2 in endothelial cells leads to higher IL-8 release, which further leads to inflammation (Chen et al., 2018). ABCG2 deficiency in hepatocytes leads to mitochondrial dysfunction and dynamics. Owing to increased intracellular protoporphyrin IX/DRP-1-mediated mitochondrial fission, abnormal protein function results in aggregate formation, leading to excessive reactive oxygen species activation of the NLRP3 inflammasome, which plays a role in the development of gout (Lin et al., 2013). Mitochondrial dysfunction can induce the NLRP3 inflammasome in gout to promote IL-1β and inflammation (Gosling et al., 2018). In addition, monosodium urate crystals also disrupt proteasomal degradation, leading to increased P62 expression, impaired cellular autophagy, and the inability to clear dysfunctional proteins, thus leading to aggregates formation. An ABCG2 SNP (rs2231142) enhances this autophagic impairment, diminishes the formation of neutrophil extracellular traps, and aggravates gout via the overactive release of the NLRP3 inflammasome and IL-1β. Neutrophil extracellular traps can degrade cytokines and chemokines to limit inflammation (Luciani et al., 2010; Shi et al., 2012; Choe et al., 2014; Schauer et al., 2014). PKD2 is localized near ABCG2 and encodes a urate transporter protein. PKD2 variants in autosomal dominant polycystic kidney disease result in PKD2 transporter dysfunction and elevated serum urate concentrations, which are associated with hyperuricemia and gout (Mejías et al., 1989; Puig et al., 1993). A transcript assay revealed that PDK2 and ABCG2 gene expression levels are positively correlated; thus, the regulators of PDK2 interact with ABCG2 to indirectly influence gout incidence (Dong et al., 2020).
SLC16A9, SLC17A1, and Shieldin Complex Subunit 2 (FAM35A)
A SLC16A9 SNP (rs2242206) can affect the function of its encoded monocarboxylate transporter 9 (MCT9) protein, resulting in inadequate urate excretion in the kidney (Kolz et al., 2009; Nakayama et al., 2013). SLC2A9 is expressed in the liver, kidney, and bone cells and transports various substances, including urates and sugars. The SNP rs734553 alters protein affinity to increase the risk of hyperuricemia and gout (Yi et al., 2018).
SLC17A1 encodes the NPT1 protein. The SNP rs1183201 appears to have a protective effect against diseases by enhancing urate excretion and transport (Kolz et al., 2009). A meta-analysis of GWASs on serum uric acid and gout in 28,283 Caucasian individuals found genome-wide significance for the SLC17A1 SNP with serum urate levels (Yang et al., 2010).
FAM35A variants are associated with gout and hyperuricemia via a mechanism that might involve a reduction in uric acid excretion during renal excretion (Nakayama et al., 2017). FAM35A encodes a DNA repair protein expressed mainly in the distal tubules of the kidney and has not been directly linked to uric acid metabolism in functional assays. Therefore, there might be other indirect mechanisms and the potential involvement of kidney function in the regulation of uric acid excretion (Nakayama et al., 2017; García-Nieto et al., 2022).
SLC22A12
SLC22A12 encodes the transporter protein URAT1, which is primarily responsible for urate reabsorption following urine filtration. Tin et al. identified 97 rare variants of SLC22A12, of which functional validation of p. Trp325, p. Cys405, and p. Met467 variants revealed that they cause loss of function of the encoded protein affecting serum uric acid levels. Individuals carrying SLC22A12 variants have a lower risk of developing gout (Tin et al., 2018). Linkage disequilibrium between SLC22A12 and SLC22A11 might be associated with uric acid in Caucasian individuals (Yang et al., 2010). Novel G65W variants of SLC22A12 (rs12800450) are characterized as functional alleles with an approximately 6–10-fold greater effect on uric acid than that observed for common variants in SLC22A12 (Tin et al., 2011). Existing drugs have been developed to target URAT1, such as probenecid and benzbromarone. In addition, a new URAT1 inhibitor for the treatment of chronic gout, lesinurad (Zurampic®; RDEA594), was approved in the United States and Europe in 2016 (Miner et al., 2016). However, lesinurad alone appears to impair renal function and should be used in combination with xanthine oxidase inhibitors, and recipients should be closely monitored for renal function (Narang and Dalbeth, 2018). The alcohol dehydrogenase 1B (Class I), beta polypeptide (ADH1B) SNP (rs129984) might increase the NADH/NAD ratio to promote lactate production by facilitating ethanol conversion to highly reactive acetaldehyde, thereby increasing uric acid reabsorption in synergy with the SLC22A12-encoded transporter protein URAT1 (Lieber et al., 1962; Edenberg, 2007; Macgregor et al., 2009; Sandoval-Plata et al., 2021).
Solute Carrier Family 22 Member 6 (SCL22A6)
SLC22A6 primarily encodes organic anion transporter 1 (OAT1) involved in eliminating endogenous and exogenous organic anions from the kidney. Tanner et al. identified multiple SNPs in SLC22A6 associated with hyperuricemia, including rs3017670, rs2276300, rs4149171, and rs4149170. Strong association studies with gout have been performed; however, there is potential evidence linking it to gout (Tanner et al., 2017). Granados et al. found altered tryptophan metabolite profiles in SLC22A6-knockout mice, including several gut microbiota metabolites that are thought to be deleterious for chronic kidney disease. Probenecid, a gout treatment drug, elevates the levels of circulating tryptophan metabolites. Different variants affect the ability of OAT1 to regulate tryptophan metabolism, thus potentially causing gout. Therefore, based on the relationship between OAT1 and tryptophan metabolism, it might be a potential future direction for targets of drug development (Granados et al., 2021). Liu et al. also demonstrated that OAT1 is associated with various metabolic processes, including the tricarboxylic acid cycle, tryptophan metabolism, and other amino acids, fatty acids, and prostaglandins (Liu et al., 2016).
SLC2A9
SLC2A9 mainly encodes the GLUT9 protein. The missense variants (rs16890979) of SLC2A9 showed an association with uric acid and gout (Dehghan et al., 2008). On the one hand, SLC2A9 is related to regulation by the transcription factor HNF4A. HNF4A overexpression enhances the activity of SLC2A9. The mRNA expression levels of HNF4A and SLC2A9 are significantly correlated, and there is an interaction between them (Prestin et al., 2014). The contribution of the coding sequence variants of SLC2A9 to overall uric acid metabolism is still unknown because of the presence of linkage disequilibrium and heterogeneity, but 24 annotated nonsynonymous variants have been identified (Reginato et al., 2012). The effects of variants in SLC2A9 (Val253Ile, and Arg265His) are also inconsistent based on studies on gout and hyperuricemia, and further studies are required (Hollis-Moffatt et al., 2009; Tu et al., 2010; Urano et al., 2010; Reginato et al., 2012). On the other hand, SLC2A9 can exchange uric acid with glucose and fructose, which are involved in gluconeogenesis. This may also have a potential impact on hyperuricemia and gout (Batt et al., 2014).
EFFECT OF GENE VARIANTS RELATED TO GLUCOLIPID METABOLISM ON HYPERURICEMIA AND GOUT
Multiple metabolic factors, including abnormalities in glucose regulation, lipid levels, obesity, and arterial hypertension, are associated with primary gout and hyperuricemia (González-Senac et al., 2014). The glycolytic pathway leads to increased serum uric acid levels through various mechanisms. In addition, insulin resistance and high blood glucose levels can directly affect uric acid clearance in the kidneys (Padova et al., 1964). Hyperinsulinemia increases urate reabsorption in the kidney and decreases renal uric acid and sodium excretion, and this effect can also occur at sites other than the proximal tubule (Quinones Galvan et al., 1995; Maaten et al., 1997). Studies have demonstrated that a high intake of fructose or other high-calorie foods can dramatically increase serum uric acid levels beyond what the body can typically handle, resulting in urate deposition with a severe disruption in hepatocyte metabolism. The rapid intake of fructose can also cause an increase in blood lactate, probably via a mechanism involving blockage of the gluconeogenic pathway caused by the inhibition of glucose-phosphate isomerase mediated by fructose 1-phosphate, leading to the excessive production of lactic acid in hepatocytes (Perheentupa and Raivio, 1967). In addition, fructose phosphorylation in the liver can increase serum uric acid levels by interacting with aldolase B, ATP, and adenosine monophosphate deaminase 2 (AMPD2) (Lanaspa et al., 2011). Dyslipidemia, insulin resistance, hyperuricemia, and gout are interrelated (Schmidt et al., 1996). Excessive alcohol intake, but not including wine, has been shown to increase serum uric acid levels in several studies (Choi and Curhan, 2004; Yu et al., 2008). Some alcohols, such as beer, contain high levels of purines, and excessive intake can increase uric acid synthesis, leading to hyperuricemia and resistance to some of the antioxidant components of the plasma (van der Gaag et al., 2000; Nishioka et al., 2002). The specific underlying mechanism might involve the degradation of adenosine triphosphate to monophosphate during alcohol metabolism, thereby increasing adenosine and uric acid synthesis. The oxidation of alcohol (ethanol) increases blood lactate, further decreases uric acid excretion, and potentially affects lipid metabolism, thereby increasing the risk of hyperuricemia and gout (Nakamura et al., 2012). Multiple gene variants are associated with glucose metabolism and are potentially associated with hyperuricemia and gout.
GCKR, MLX Interacting Protein (MLXIP), and MLX Interacting Protein-Like (MLXIPL)
GCKR encodes the GCKR subfamily of proteins that are regulatory proteins that inhibit glucokinase in the liver and pancreatic islet cells by binding noncovalently with the enzyme to form inactive complexes. GCKR variants (rs1260326) are missense variants that serve as possible candidate causal variants for which the leucine allele leads to increased glucokinase GCK activity, resulting in increased glycolytic flux, which facilitates hepatic glucose metabolism (Beer et al., 2009). A GCKR SNP (rs780094) is strongly associated with gout in Polynesian, European, Japanese, and Chinese populations (Wang et al., 2012; Köttgen et al., 2013a; Urano et al., 2013). In the recessive model, GCKR SNP (rs780094) was shown to be associated with the risk of hyperuricemia in men in the Uyghur population of Xinjiang in China (p = 0.015, OR = 1.311) (Wang et al., 2018). GCKR and NFAT5 are associated with glucose metabolism or the insulin response, and GCKR increases the metabolites that cause gout-related factors through glycolysis (Köttgen et al., 2013a; Rasheed et al., 2017). MLXIP encodes a protein that forms a heterodimer with MAX dimerization protein. It regulates the genes that moderate cellular glucose levels. MLXIPL encodes a Myc/Max/Mad superfamily basic helix-loop-helix leucine zipper transcription factor that forms a heterodimeric complex. That binds and activates the carbohydrate response element-binding protein motif within the triglyceride synthesis gene promoter in a glucose-dependent manner. MLXIP and MLXIPL variants can also correlate with serum urate concentrations (Boocock et al., 2020). MLXIPL is primarily associated with cellular carbohydrate metabolism and glycolytic processes. It is directly responsible for regulating glucose flux, interpreted as the pentose phosphate pathway producing ribose 5-phosphate, an essential precursor of de novo purine synthesis, and is involved in the production of uric acid (Köttgen et al., 2013a). In addition, the overproduction of lactate affects the transmembrane transport of urate, leading to impaired clearance of urate by the kidney (Luo et al., 2005; Tong et al., 2009; Levine and Puzio-Kuter, 2010).
Patatin-Like Phospholipase Domain-Containing 3 (PNPLA3) and Insulin Like Growth Factor 1 Receptor (IGF1R)
PNPLA3 encodes an active lipase that hydrolyzes various lipids and is associated with oxidative stress (Huang et al., 2011). A PNPLA3 SNP (rs738409) is associated with hyperuricemia in a Japanese population (Nakatochi et al., 2019a). The rs738409-G allele was found to be associated with a reduced risk of gout in phenome-wide association studies (Diogo et al., 2018). This study indicated that the PNPLA3 SNP (rs738409) enhances susceptibility to metabolism-related fatty liver disease (MAFLD) and is involved in the pathology of liver fibrosis (Kawaguchi et al., 2018; Namjou et al., 2019). In the recessive model, the PNPLA3 SNP (rs738409) was associated with NAFLD in different ethnic groups in China: Han (OR = 1.84, 95% CI: 1.03–3.27, p = 0.036), Uyghur (OR = 2.25, 95% CI: 1.23–4.09, p = 0.006) (Zhang et al., 2014). IGF1R encodes the insulin-like growth factor I receptor. This receptor binds insulin-like growth factor with a high affinity. It has tyrosine kinase activity. The insulin-like growth factor I receptor plays a critical role in transformation events. IGF1R SNPs (rs12908437, rs659854, rs1291127, and rs4966024) might correlate with blood uric acid levels by affecting the body mass index (BMI) (Park et al., 2021). An abnormal BMI is indicative of abnormal lipid metabolism, and plasma uric acid is a powerful antioxidant (Ames et al., 1981). Thus, PNPLA3 and IGF1R variants might be linked to hyperuricemia and gout by affecting lipid metabolism and oxidative stress.
APOBEC1 Complementation Factor (A1CF)
A1CF encodes a protein that may primarily act as an RNA binding subunit and be involved in RNA editing or processing. Rasheed et al. found that both a GCKR SNP (rs780094) and A1CF SNP (rs10821905) interact with alcohol exposure to increase the risk of gout in a European population under alcohol exposure conditions, suggesting that the involvement of GCKR and AICF in alcohol metabolism promotes the development of gout (Rasheed et al., 2017). The A1CF SNP has been previously associated with hyperuricemia (Köttgen et al., 2013b). Makoto et al. further investigated the association between the A1CF SNP (rs10821905) and gout in Japanese individuals. They found that it was significantly associated with elevated serum uric acid and gout via a mechanism that might involve the regulation of dyslipidemia and uric acid metabolism (Kawaguchi et al., 2021). Further investigation of the mechanism of interaction between alcohol and AICF could suggest that the metabolite acetate of alcohol (ethanol) leads to the increased production of diacylglycerol and further activates protein kinase C and AICF phosphorylation in the nucleus. This leads to the increased production of apolipoprotein B (ApoB)-48 and decreased production of ApoB-100 at the transcriptional level, further causing increased free fatty acid production from very-low-density lipoproteins/triglycerides, which stimulates downstream TLR, the NLRP3 inflammasome, and IL-1β to activate the inflammatory response and produce more monosodium urate crystals (Joosten et al., 2010; Jump et al., 2013; Rasheed et al., 2017).
Lipoprotein Receptor-Related Protein 2 (LRP2)
LRP2 encodes an endocytic receptor protein, low-density lipoprotein-related protein 2, which is associated with multiple ligands such as ApoB, lipoprotein lipase, and lactoferrin. It is expressed in numerous tissues, such as proximal renal tubules (Christensen and Birn, 2002). LRP2 SNPs (rs2390793, rs2544390, and rs16856823) are associated with blood uric acid (Kamatani et al., 2010; Kanai et al., 2018; Nakatochi et al., 2019b; Tin et al., 2019b) and increased gout susceptibility in Japanese (Akashi et al., 2020) and Chinese populations (Dong et al., 2015). However, its variants might lead to renal tubular dysfunction, affecting the renal reabsorption of uric acid (Kamatani et al., 2010; Kanai et al., 2018; Nakatochi et al., 2019b; Tin et al., 2019b). In contrast, rs2544390 was shown to have a non-additive interaction effect with alcohol consumption (beer or spirits), which can increase the risk of serum urate accumulation and gout in alcohol drinkers (Rasheed et al., 2013). However, there are additional contradictory results showing that LRP2 is not associated with gout susceptibility (Nakayama et al., 2014). LRP2 can also regulate the activity of lipoprotein lipase to modulate lipid metabolism, which is associated with uric acid metabolism (Rasheed et al., 2013). Additional experiments are needed to clarify the potential biological mechanisms and links between LRP2 and hyperuricemia and gout.
NLRP3 INFLAMMASOME AND INFLAMMATION-ASSOCIATED GENES PROMOTE THE PROGRESSION OF HYPERURICEMIA TO GOUT
Pyroptosis, involving the NLRP3 inflammasome, can lead to cell destruction and the release of the pro-inflammatory factors IL-18 and IL-1β, thus promoting inflammation, which has been discussed in rheumatoid arthritis and MAFLD. Both have similarities to gouty arthritis in terms of disease mechanisms (Zhao et al., 2021a; Zhao et al., 2021b). As mentioned earlier, the excessive deposition of uric acid leads to the appearance of monosodium urate crystals, which are stimulated by the NLRP3 inflammasome and inflammatory factors to progress further toward inflammation. Many genetic variants could be involved in this (Figure 2).
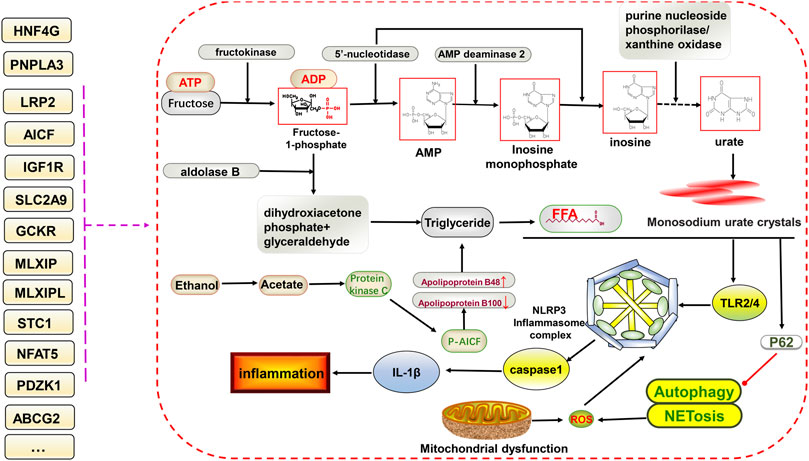
FIGURE 2. Potential relationship between gene variants and glucolipid metabolism and NLRP3 inflammasome-mediated inflammatory pathways in hyperuricemia and gout. The ingestion of fructose increases uric acid formation via the gluconeogenic pathway. In addition, triglycerides are also produced, increasing free fatty acid contents via the lipid metabolic pathway. Free fatty acids and monosodium urate crystals together stimulate downstream TLR2/4 and NLRP3 inflammasome formation, facilitate the of release IL-1β, and inhibit P62-mediated activation of autophagy and NETosis, ultimately promoting inflammation. Gene variants have different effects on different processes.
IGF1R
IGF1R might be associated with activation of the NLRP3 inflammasome in gout. Spadaro et al. found that macrophages lacking the IGF1R have reduced NLRP3 activation and a controlled inflammatory response (Spadaro et al., 2016). Liang et al. found that IGF1R primarily regulates vascular homeostasis and precise endothelial functions and that IGF1R deficiency impairs endothelial function in experimental mice and increases the degree of fibrosis in renal disease, which is associated with a poor wound healing response owing to repeated irritation from inflammation (Liang et al., 2015). Thus, both studies suggest that IGF1R variants might influence gout by regulating inflammation. In addition, a IGF1R SNP (rs7193778) and PDZK1 SNP (rs112129861) could interact with each other, further enriching our understanding of the genetic and biological mechanisms underlying uric acid accumulation and gout (Fernández-Torres et al., 2019).
Stanniocalcin 1 (STC1)
STC1 encodes stanniocalcin-1, a glycoprotein that plays a role in multiple biological responses, including bone development, angiogenesis, and inflammatory responses (Yeung et al., 2012). Studies have reported that STC1 is associated with elevated serum uric acid levels (Köttgen et al., 2013b). An STC1 SNP (rs17786744) might cause the crystalline precipitation of sodium urate to trigger the inflammatory process, further exacerbating cartilage damage and promoting knee osteoarthritis, which could be associated with the inflammatory response in gouty arthritis. In addition, an interaction between an STC1 SNP (rs17786744) and GCKR SNP (rs1260326) synergistically promotes crystalline precipitation with urate-promoting gout (Fernández-Torres et al., 2019).
The Association Between Genetic Variants Involved in Other Mechanisms and Hyperuricemia and Gout
Various factors, such as coffee intake, tryptophan metabolism, B-cell development and activation, and sex hormones, are interlinked with genetic variants that play a role in hyperuricemia and gout. Hutton et al. found a negative association between coffee intake and gout. ABCG2, GCKR, MLXIPL, and cytochrome P450 family 1 subfamily A member 2 (CYP1A2) are variants associated with coffee consumption habits, and GCKR and ABCG2 are associated with low coffee intake and a high gout risk. Coffee consumption habits indirectly affect the association between gene variants and gout. In contrast, the direct effect of these gene variants on gout is still possible through other mechanisms, as described previously herein (Hutton et al., 2018). Evidence from studies involving genetic variants associated with other mechanisms is relatively scarce, and further research is needed in the future. Therefore, in this section, we summarize briefly the association of other mechanisms with gout, including sleep rhythm, immune response and B-cell activation (hypocretin receptor 2 (HCRTR2), cytokine-dependent hematopoietic cell linker (CLNK), guanine nucleotide-binding protein a-stimulating polypeptide (GNAS)), sex hormones (breast cancer-amplified sequence 3 (BCAS3)).
HCRTR2, CLNK and GNAS
The protein encoded by HCRTR2 is a G protein-coupled receptor involved in the regulation of feeding. The encoded proteins bind to orexin A and orexin B. A HCRTR2 SNP (rs4715517), a variant associated with serum uric acid, appears to be specific to Asian populations with significantly higher allele frequencies than those in European populations. Differences in allele frequencies might contribute to interethnic differences in serum uric acid levels (Park et al., 2021). HCRTR2 is mainly involved in the sleep rhythm of the body (Lane et al., 2017; Dashti et al., 2019). With the accelerated pace of life in modern society, irregular sleep affects the immune system and the function of multiple organs, including the kidneys and liver. Therefore, genes regulating sleep rhythms might be potentially associated with hyperuricemia and gout. CLNK, a member of the SLP76 family, plays an essential role in integrating immunotyrosine-based activation motif-bearing receptors and integrins and is a positive regulator of immune response signaling (Yu et al., 2001; Wu and Koretzky, 2004). The allele “G" of CLNK SNP (rs2041215 and rs1686947) was identified as susceptibility genes for gout in the Chinese population by using dominant model (OR 1.66; 95% CI 1.04–2.63; p = 0.031) (OR 2.19; 95% CI 1.38–3.46; p = 0.001) and additive model (OR 1.39; 95% CI 1.00–1.93; p = 0.049) (OR 1.67; 95% CI 1.19–2.32; p = 0.003), respectively (Jin et al., 2015). A CLNK SNP (rs16869924) within the established SLC2A9 gout-associated locus was shown to increase the risk of gout in Polynesian and Chinese Tibetan individuals, genetically independent on the SLC2A9 association signal (Lan et al., 2016; Ji et al., 2021). It is hypothesized that CLNK mainly regulates B-cell development and activation and co-mediates the formation of immune complexes through the STAT signaling pathway to promote gout, as suggested be a combination of related studies (Siniachenko et al., 1984; Wang et al., 2002; Marrero et al., 2006). GNAS encodes GSa protein, which activates downstream cyclic AMP (cAMP) production and promotes signaling (Turan and Bastepe, 2015; Tafaj and Jüppner, 2017). GNAS variants predispose patients to an abnormal synovial environment and the deposition of uric acid crystals, promoting the formation of gout and related osteoarthritis (Rhyu and Bhat, 2021).
BCAS3
BCAS3 encodes proteins that are associated with several functions, such as angiogenesis, activation and recruitment of cell division cycle 42, reorganization of the actin cytoskeleton at the leading edge, regulation of cell polarity, the endothelial cell migration, filopodia formation, estrogen receptor response, and autophagy.
Rs11653176 in BCAS3 is significantly associated with uric acid levels and gout in Japanese and Chinese Han populations (Li et al., 2015b; Sakiyama et al., 2018). BCAS3 can activate estrogen receptor alpha (Sakiyama et al., 2018). Studies have shown that sex hormones can affect uric acid levels (Adamopoulos et al., 1977). Postmenopausal women might have elevated uric acid levels owing to a decrease in estrogen, especially estradiol, because estrogen is more effective in promoting urate clearance by the kidneys (Hak and Choi, 2008). Similarly, the effects of some gene variants on serum uric acid levels and gout appear to be sex-specific. For example, variants in SLC2A9 and ABCG2, which are associated with urate concentrations, are sex-specific (Köttgen et al., 2013a). An SLC16A9 SNP (rs12356193) was found to be weakly associated with gout but strongly associated with blood uric acid and showed a sex-specific difference (Köttgen et al., 2013a). It is thus possible that sex hormones primarily contribute to sex differences in disease or drug efficacy.
Gene Variants as Potential Diagnostic Markers of Drug Efficacy and Prognosis
A case-control association study of gout in Chinese populations revealed that CLNK SNPs (rs2041215 and rs1686947) are associated with various clinicopathological parameters and might have potential as diagnostic and prognostic markers for patients with gout (Jin et al., 2015). Patients with gout carrying an ABCG2 SNP (rs2231142) respond poorly to allopurine therapy (Wen et al., 2015; Roberts et al., 2017; Wallace et al., 2018). A GWAS and polygenic risk scores in patients with asymptomatic hyperuricemia and gout revealed that ABCG2 (rs2231142, rs13120400, and rs7672194), SLC2A9 (rs16890979 and rs16891234), SLC22A11 (rs2078267), GCKR (rs1260326), matrix extracellular phosphoglycoprotein (MEPE) (rs114580333), protein phosphatase, Mg2+/Mn2+ dependent 1 K-divergent transcript (PPM1K-DT) (rs4693211, rs28793136, and rs1545207), LOC105377323 (rs114791459), and alcohol dehydrogenase 1B (Class I), beta polypeptide (ADH1B) (rs1229984) SNPs can be used as markers of asymptomatic hyperuricemia to identify transition predictors (Sandoval-Plata et al., 2021). However, little research has been conducted on genetic variants as markers to predict disease progression and drug efficacy, as serum uric acid levels can effectively predict the risk of gout. However, this might provide more relevant results that could be uncovered through in-depth studies in the future.
Conclusion
Gout is a form of arthritis that damages patients’ physical and mental health and causes severe pain during acute attacks. Identifying individuals at risk in the early stages of the disease is essential to prevent and reduce hyperuricemia and gout and to provide pharmacological and lifestyle interventions to better treat patients with clinically diagnosed gout. The identification of genetic variants might help in disease prevention and intervention. Many GWASs have performed to uncover loci related to hyperuricemia and gout, mostly linking it to uric acid transporter proteins, such as the widely studied URAT1 and GLUT9. Some drugs have been used as targets for drug development (see Table 1). We also summarize the latest clinical trials of these genes, and some of these were conducted in the context of gout, which is certainly instructive. Although some trials were not investigated in the context of gout, they have some informative implications for the clinical management of gout, which urgently needs to be studied in depth in the context of gout in the future (see Table2). Currently, the most elucidated is the effect of variants in the uric acid transporter protein gene on hyperuricemia and gout. We aim to increase our understanding of the genetic mechanisms behind the disease by adding descriptions of other genes of potential clinical value. All of these genes are undoubtedly promising and essential. Associations between genetic variants and traits are often located in regions of strong linkage disequilibrium and aided by eQTL analysis and fine localization studies, these can be exploited to identify true causal variants of gout in complex genetic backgrounds. Genetic-related issues in multiple disease contexts still need attention and elucidation, such as disease-specific genetic variants in different ethnic backgrounds, genetic variants based on sex differences, rare and low frequency variants, functional polymorphisms in genetic susceptibility genes, and epigenetic mechanisms. With the rapid development of modern molecular biotechnologies and multi-omics techniques, these issues require further clarification. In addition, attention should be paid to the interconnection between hyperuricemia/gout and other diseases, such as metabolic syndrome and cardiovascular diseases, as well as the role of genetic factors in these diseases. Elucidating these genetic issues will contribute to the improvement of clinical outcomes and precision medicine.

TABLE 1. Gene variants associated with hyperuricemia and gout.
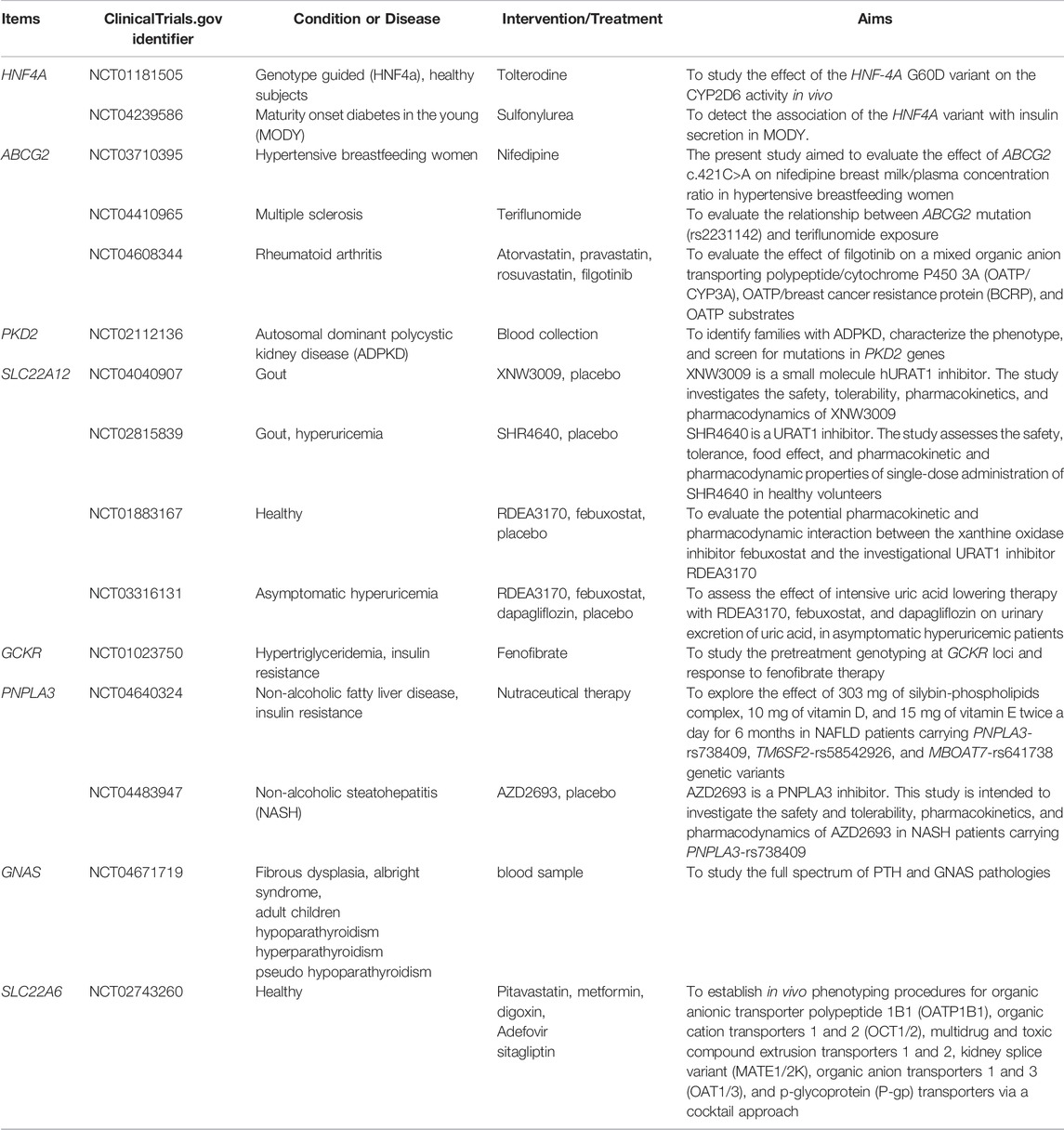
TABLE 2. Gene tests in clinical trials.
Author Contributions
JZ is responsible for the collection, collation, and writing of the original manuscript. SG, SS, and DH are responsible for the concept development, revision, and manuscript review. All authors reviewed and accepted the final version.
Funding
This work was funded by the National Natural Science Funds of China (82074234 and 82071756), National Key Research and Development Project (2018YFC1705200 and 2018YFC1705203), Shanghai Chinese Medicine Development Office, National Administration of Traditional Chinese Medicine, Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology, State Administration of Traditional Chinese Medicine, National TCM Evidence-Based Medicine Research and Construction Project, Basic TCM Evidence-Based Capacity Development Program, Shanghai Municipal Health Commission, and East China Region-based Chinese and Western Medicine Joint Disease Specialist Alliance.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adamopoulos, D., Vlassopoulos, C., Seitanides, B., Contoyiannis, P., and Vassilopoulos, P. (1977). The Relationship of Sex Steroids to Uric Acid Levels in Plasma and Urine. Eur. J. Endocrinol. 85 (1), 198–208. doi:10.1530/acta.0.0850198
Akashi, A., Nakayama, A., Kamatani, Y., Higashino, T., Shimizu, S., Kawamura, Y., et al. (2020). A Common Variant of LDL Receptor Related Protein 2 (LRP2) Gene Is Associated with Gout Susceptibility: a Meta-Analysis in a Japanese Population. Hum. Cell 33 (2), 303–307. doi:10.1007/s13577-019-00318-5
Alghubayshi, A., Edelman, A., Alrajeh, K., and Roman, Y. (2022). Genetic Assessment of Hyperuricemia and Gout in Asian, Native Hawaiian, and Pacific Islander Subgroups of Pregnant Women: Biospecimens Repository Cross-Sectional Study. BMC Rheumatol. 6 (1), 1. doi:10.1186/s41927-021-00239-7
Ames, B. N., Cathcart, R., Schwiers, E., and Hochstein, P. (1981). Uric Acid Provides an Antioxidant Defense in Humans against Oxidant- and Radical-Caused Aging and Cancer: a Hypothesis. Proc. Natl. Acad. Sci. U.S.A. 78 (11), 6858–6862. doi:10.1073/pnas.78.11.6858
Asgari, E., and Hilton, R. M. (2021). One Size Does Not Fit All: Understanding Individual Living Kidney Donor Risk. Pediatr. Nephrol. 36 (2), 259–269. doi:10.1007/s00467-019-04456-8
Batt, C., Phipps-Green, A. J., Black, M. A., Cadzow, M., Merriman, M. E., Topless, R., et al. (2014). Sugar-sweetened Beverage Consumption: a Risk Factor for Prevalent Gout withSLC2A9genotype-specific Effects on Serum Urate and Risk of Gout. Ann. Rheum. Dis. 73 (12), 2101–2106. doi:10.1136/annrheumdis-2013-203600
Beer, N. L., Tribble, N. D., McCulloch, L. J., Roos, C., Johnson, P. R. V., Orho-Melander, M., et al. (2009). The P446L Variant in GCKR Associated with Fasting Plasma Glucose and Triglyceride Levels Exerts its Effect through Increased Glucokinase Activity in Liver. Hum. Mol. Genet. 18 (21), 4081–4088. doi:10.1093/hmg/ddp357
Boocock, J., Leask, M., Okada, Y., Matsuo, H., Kawamura, Y., Shi, Y., et al. (2020). Genomic Dissection of 43 Serum Urate-Associated Loci Provides Multiple Insights into Molecular Mechanisms of Urate Control. Hum. Mol. Genet. 29 (6), 923–943. doi:10.1093/hmg/ddaa013
Chen, B.-D., Chen, X.-C., Pan, S., Yang, Y.-N., He, C.-H., Liu, F., et al. (2017). TT Genotype of Rs2941484 in the Human HNF4G Gene Is Associated with Hyperuricemia in Chinese Han Men. Oncotarget 8 (16), 26918–26926. doi:10.18632/oncotarget.15851
Chen, C.-J., Tseng, C.-C., Yen, J.-H., Chang, J.-G., Chou, W.-C., Chu, H.-W., et al. (2018). ABCG2 Contributes to the Development of Gout and Hyperuricemia in a Genome-wide Association Study. Sci. Rep. 8 (1), 3137. doi:10.1038/s41598-018-21425-7
Choe, J.-Y., Jung, H.-Y., Park, K.-Y., and Kim, S.-K. (2014). Enhanced P62 Expression through Impaired Proteasomal Degradation Is Involved in Caspase-1 Activation in Monosodium Urate Crystal-Induced Interleukin-1 Expression. Rheumatology 53 (6), 1043–1053. doi:10.1093/rheumatology/ket474
Choi, H. K., and Curhan, G. (2004). Beer, Liquor, and Wine Consumption and Serum Uric Acid Level: the Third National Health and Nutrition Examination Survey. Arthritis & Rheumatism 51 (6), 1023–1029. doi:10.1002/art.20821
Christensen, E. I., and Birn, H. (2002). Megalin and Cubilin: Multifunctional Endocytic Receptors. Nat. Rev. Mol. Cell Biol. 3 (4), 258–267. doi:10.1038/nrm778
Chung, S., and Kim, G.-H. (2021). Urate Transporters in the Kidney: What Clinicians Need to Know. Electrolyte Blood Press 19 (1), 1–9. doi:10.5049/ebp.2021.19.1.1
Dalbeth, N., Merriman, T. R., and Stamp, L. K. (2016). Gout. Lancet 388 (10055), 2039–2052. doi:10.1016/s0140-6736(16)00346-9
Dashti, H. S., Jones, S. E., Wood, A. R., Lane, J. M., van Hees, V. T., Wang, H., et al. (2019). Genome-wide Association Study Identifies Genetic Loci for Self-Reported Habitual Sleep Duration Supported by Accelerometer-Derived Estimates. Nat. Commun. 10 (1), 1100. doi:10.1038/s41467-019-08917-4
Dehghan, A., Köttgen, A., Yang, Q., Hwang, S.-J., Kao, W. L., Rivadeneira, F., et al. (2008). Association of Three Genetic Loci with Uric Acid Concentration and Risk of Gout: a Genome-wide Association Study. Lancet 372 (9654), 1953–1961. doi:10.1016/s0140-6736(08)61343-4
Diogo, D., Tian, C., Franklin, C. S., Alanne-Kinnunen, M., March, M., Spencer, C. C. A., et al. (2018). Phenome-wide Association Studies across Large Population Cohorts Support Drug Target Validation. Nat. Commun. 9 (1), 4285. doi:10.1038/s41467-018-06540-3
Dong, Z., Zhao, D., Yang, C., Zhou, J., Qian, Q., Ma, Y., et al. (2015). Common Variants in LRP2 and COMT Genes Affect the Susceptibility of Gout in a Chinese Population. PLoS One 10 (7), e0131302. doi:10.1371/journal.pone.0131302
Dong, Z., Zhou, J., Jiang, S., Li, Y., Zhao, D., Yang, C., et al. (2020). Epistatic Interaction between PKD2 and ABCG2 Influences the Pathogenesis of Hyperuricemia and Gout. Hereditas 157 (1), 2. doi:10.1186/s41065-020-0116-6
Dong, Z., Zhou, J., Jiang, S., Li, Y., Zhao, D., Yang, C., et al. (2017). Effects of Multiple Genetic Loci on the Pathogenesis from Serum Urate to Gout. Sci. Rep. 7, 43614. doi:10.1038/srep43614
Edenberg, H. J. (2007). The Genetics of Alcohol Metabolism: Role of Alcohol Dehydrogenase and Aldehyde Dehydrogenase Variants. Alcohol Res. Health 30 (1), 5–13.
Fernández-Torres, J., Martínez-Nava, G. A., Oliviero, F., López-Reyes, A. G., Martínez-Flores, K., Garrido-Rodríguez, D., et al. (2019). Common Gene Variants Interactions Related to Uric Acid Transport Are Associated with Knee Osteoarthritis Susceptibility. Connect. tissue Res. 60 (3), 219–229. doi:10.1080/03008207.2018.1483359
García-Nieto, V. M., Claverie-Martín, F., Moraleda-Mesa, T., Perdomo-Ramírez, A., Tejera-Carreño, P., Córdoba-Lanus, E., et al. (2022). La gota asociada a reducción de la excreción renal de ácido úrico. Esa tubulopatía que no tratamos los nefrólogos. Nefrología 42, 273–279. doi:10.1016/j.nefro.2021.03.013
Gbd, (2017). Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 328 Diseases and Injuries for 195 Countries, 1990-2016: a Systematic Analysis for the Global Burden of Disease Study 2016. Lancet 390 (10100), 1211–1259. doi:10.1016/s0140-6736(17)32154-2
González-Senac, N. M., Bailén, R., Torres, R. J., de Miguel, E., and Puig, J. G. (2014). Metabolic Syndrome in Primary Gout. Nucleosides, Nucleotides Nucleic Acids 33 (4-6), 185–191. doi:10.1080/15257770.2013.853785
Gosling, A. L., Boocock, J., Dalbeth, N., Harré Hindmarsh, J., Stamp, L. K., Stahl, E. A., et al. (2018). Mitochondrial Genetic Variation and Gout in Māori and Pacific People Living in Aotearoa New Zealand. Ann. Rheum. Dis. 77 (4), 571–578. doi:10.1136/annrheumdis-2017-212416
Granados, J. C., Richelle, A., Gutierrez, J. M., Zhang, P., Zhang, X., Bhatnagar, V., et al. (2021). Coordinate Regulation of Systemic and Kidney Tryptophan Metabolism by the Drug Transporters OAT1 and OAT3. J. Biol. Chem. 296, 100575. doi:10.1016/j.jbc.2021.100575
Hak, A. E., and Choi, H. K. (2008). Menopause, Postmenopausal Hormone Use and Serum Uric Acid Levels in US Women - the Third National Health and Nutrition Examination Survey. Arthritis Res. Ther. 10 (5), R116. doi:10.1186/ar2519
Higashino, T., Matsuo, H., Okada, Y., Nakashima, H., Shimizu, S., Sakiyama, M., et al. (2018). A Common Variant of MAF/c-MAF, Transcriptional Factor Gene in the Kidney, Is Associated with Gout Susceptibility. Hum. cell 31 (1), 10–13. doi:10.1007/s13577-017-0186-6
Hollis-Moffatt, J. E., Xu, X., Dalbeth, N., Merriman, M. E., Topless, R., Waddell, C., et al. (2009). Role of the Urate transporterSLC2A9gene in Susceptibility to Gout in New Zealand Māori, Pacific Island, and Caucasian Case-Control Sample Sets. Arthritis & Rheumatism 60 (11), 3485–3492. doi:10.1002/art.24938
Huang, Y., Cohen, J. C., and Hobbs, H. H. (2011). Expression and Characterization of a PNPLA3 Protein Isoform (I148M) Associated with Nonalcoholic Fatty Liver Disease. J. Biol. Chem. 286 (43), 37085–37093. doi:10.1074/jbc.M111.290114
Hughes, K., Flynn, T., de Zoysa, J., Dalbeth, N., and Merriman, T. R. (2014). Mendelian Randomization Analysis Associates Increased Serum Urate, Due to Genetic Variation in Uric Acid Transporters, with Improved Renal Function. Kidney Int. 85 (2), 344–351. doi:10.1038/ki.2013.353
Hutton, J., Fatima, T., Major, T. J., Topless, R., Stamp, L. K., Merriman, T. R., et al. (2018). Mediation Analysis to Understand Genetic Relationships between Habitual Coffee Intake and Gout. Arthritis Res. Ther. 20 (1), 135. doi:10.1186/s13075-018-1629-5
Imaki, J., Tsuchiya, K., Mishima, T., Onodera, H., Kim, J. I., Yoshida, K., et al. (2004). Developmental Contribution of C-Maf in the Kidney: Distribution and Developmental Study of C-Maf mRNA in Normal Mice Kidney and Histological Study of C-Maf Knockout Mice Kidney and Liver. Biochem. Biophysical Res. Commun. 320 (4), 1323–1327. doi:10.1016/j.bbrc.2004.05.222
Ji, A., Shaukat, A., Takei, R., Bixley, M., Cadzow, M., Topless, R. K., et al. (2021). Aotearoa New Zealand Māori and Pacific Population-Amplified Gout Risk Variants: CLNK Is a Separate Risk Gene at the SLC2A9 Locus. J. Rheumatol. 48 (11), 1736–1744. doi:10.3899/jrheum.201684
Jin, T.-b., Ren, Y., Shi, X., Jiri, M., He, N., Feng, T., et al. (2015). Genetic Variations in the CLNK Gene and ZNF518B Gene Are Associated with Gout in Case-Control Sample Sets. Rheumatol. Int. 35 (7), 1141–1147. doi:10.1007/s00296-015-3215-3
Joosten, L. A. B., Netea, M. G., Mylona, E., Koenders, M. I., Malireddi, R. K. S., Oosting, M., et al. (2010). Engagement of Fatty Acids with Toll-like Receptor 2 Drives Interleukin-1β Production via the ASC/caspase 1 Pathway in Monosodium Urate Monohydrate Crystal-Induced Gouty Arthritis. Arthritis & Rheumatism 62 (11), 3237–3248. doi:10.1002/art.27667
Jump, D. B., Tripathy, S., and Depner, C. M. (2013). Fatty Acid-Regulated Transcription Factors in the Liver. Annu. Rev. Nutr. 33, 249–269. doi:10.1146/annurev-nutr-071812-161139
Kamatani, Y., Matsuda, K., Okada, Y., Kubo, M., Hosono, N., Daigo, Y., et al. (2010). Genome-wide Association Study of Hematological and Biochemical Traits in a Japanese Population. Nat. Genet. 42 (3), 210–215. doi:10.1038/ng.531
Kanai, M., Akiyama, M., Takahashi, A., Matoba, N., Momozawa, Y., Ikeda, M., et al. (2018). Genetic Analysis of Quantitative Traits in the Japanese Population Links Cell Types to Complex Human Diseases. Nat. Genet. 50 (3), 390–400. doi:10.1038/s41588-018-0047-6
Kawaguchi, M., Nakayama, A., Aoyagi, Y., Nakamura, T., Shimizu, S., Kawamura, Y., et al. (2021). Both variants of A1CF and BAZ1B Genes Are Associated with Gout Susceptibility: a Replication Study and Meta-Analysis in a Japanese Population. Hum. Cell 34 (2), 293–299. doi:10.1007/s13577-021-00485-4
Kawaguchi, T., Shima, T., Mizuno, M., Mitsumoto, Y., Umemura, A., Kanbara, Y., et al. (2018). Risk Estimation Model for Nonalcoholic Fatty Liver Disease in the Japanese Using Multiple Genetic Markers. PLoS One 13 (1), e0185490. doi:10.1371/journal.pone.0185490
Ketharnathan, S., Leask, M., Boocock, J., Phipps-Green, A. J., Antony, J., O’Sullivan, J. M., et al. (2018). A Non-coding Genetic Variant Maximally Associated with Serum Urate Levels Is Functionally Linked to HNF4A-dependent PDZK1 Expression. Hum. Mol. Genet. 27 (22), 3964–3973. doi:10.1093/hmg/ddy295
Kolz, M., Johnson, T., Sanna, S., Teumer, A., Vitart, V., Perola, M., et al. (2009). Meta-analysis of 28,141 Individuals Identifies Common Variants within Five New Loci that Influence Uric Acid Concentrations. PLoS Genet. 5 (6), e1000504. doi:10.1371/journal.pgen.1000504
Köttgen, A., Albrecht, E., Teumer, A., Vitart, V., Krumsiek, J., Hundertmark, C., et al. (2013). Genome-wide Association Analyses Identify 18 New Loci Associated with Serum Urate Concentrations. Nat. Genet. 45 (2), 145–154. doi:10.1038/ng.2500
Köttgen, A., Albrecht, E., Teumer, A., Vitart, V., Krumsiek, J., Hundertmark, C., et al. (2013). Genome-wide Association Analyses Identify 18 New Loci Associated with Serum Urate Concentrations. Nat. Genet. 45 (2), 145–154. doi:10.1038/ng.2500
Kuo, C.-F., Grainge, M. J., Zhang, W., and Doherty, M. (2015). Global Epidemiology of Gout: Prevalence, Incidence and Risk Factors. Nat. Rev. Rheumatol. 11 (11), 649–662. doi:10.1038/nrrheum.2015.91
Lan, B., Chen, P., Jiri, M., He, N., Feng, T., Liu, K., et al. (2016). WDR1 and CLNK Gene Polymorphisms Correlate with Serum Glucose and High-Density Lipoprotein Levels in Tibetan Gout Patients. Rheumatol. Int. 36 (3), 405–412. doi:10.1007/s00296-015-3378-y
Lanaspa, M. A., Tapia, E., Soto, V., Sautin, Y., and Sánchez-Lozada, L. G. (2011). Uric Acid and Fructose: Potential Biological Mechanisms. Seminars Nephrol. 31 (5), 426–432. doi:10.1016/j.semnephrol.2011.08.006
Lane, J. M., Liang, J., Vlasac, I., Anderson, S. G., Bechtold, D. A., Bowden, J., et al. (2017). Genome-wide Association Analyses of Sleep Disturbance Traits Identify New Loci and Highlight Shared Genetics with Neuropsychiatric and Metabolic Traits. Nat. Genet. 49 (2), 274–281. doi:10.1038/ng.3749
Leask, M., Dowdle, A., Salvesen, H., Topless, R., Fadason, T., Wei, W., et al. (2018). Functional Urate-Associated Genetic Variants Influence Expression of lincRNAs LINC01229 and MAFTRR. Front. Genet. 9, 733. doi:10.3389/fgene.2018.00733
Leask, M. P., and Merriman, T. R. (2021). The Genetic Basis of Urate Control and Gout: Insights into Molecular Pathogenesis from Follow-Up Study of Genome-wide Association Study Loci. Best Pract. Res. Clin. Rheumatology 35 (4), 101721. doi:10.1016/j.berh.2021.101721
Leask, M. P., Sumpter, N. A., Lupi, A. S., Vazquez, A. I., Reynolds, R. J., Mount, D. B., et al. (2020). The Shared Genetic Basis of Hyperuricemia, Gout, and Kidney Function. Seminars Nephrol. 40 (6), 586–599. doi:10.1016/j.semnephrol.2020.12.002
Lee, M.-t. G., Hsu, T.-C., Chen, S.-C., Lee, Y.-C., Kuo, P.-H., Yang, J.-H., et al. (2019). Integrative Genome-wide Association Studies of eQTL and GWAS Data for Gout Disease Susceptibility. Sci. Rep. 9 (1), 4981. doi:10.1038/s41598-019-41434-4
Levine, A. J., and Puzio-Kuter, A. M. (2010). The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science 330 (6009), 1340–1344. doi:10.1126/science.1193494
Li, C., Li, Z., Liu, S., Wang, C., Han, L., Cui, L., et al. (2015). Genome-wide Association Analysis Identifies Three New Risk Loci for Gout Arthritis in Han Chinese. Nat. Commun. 6, 7041. doi:10.1038/ncomms8041
Li, R., Miao, L., Qin, L., Xiang, Y., Zhang, X., Peng, H., et al. (2015). A Meta-Analysis of the Associations between the Q141K and Q126X ABCG2 Gene Variants and Gout Risk. Int. J. Clin. Exp. Pathol. 8 (9), 9812–9823.
Liang, M., Woodard, L. E., Liang, A., Luo, J., Wilson, M. H., Mitch, W. E., et al. (2015). Protective Role of Insulin-like Growth Factor-1 Receptor in Endothelial Cells against Unilateral Ureteral Obstruction-Induced Renal Fibrosis. Am. J. Pathology 185 (5), 1234–1250. doi:10.1016/j.ajpath.2015.01.027
Lieber, C. S., Jones, D. P., Losowsky, M. S., and Davidson, C. S. (1962). Interrelation of Uric Acid and Ethanol Metabolism in Man*. J. Clin. Invest. 41 (10), 1863–1870. doi:10.1172/jci104643
Lin, Y.-H., Chang, H.-M., Chang, F.-P., Shen, C.-R., Liu, C.-L., Mao, W.-Y., et al. (2013). Protoporphyrin IX Accumulation Disrupts Mitochondrial Dynamics and Function in ABCG2-Deficient Hepatocytes. FEBS Lett. 587 (19), 3202–3209. doi:10.1016/j.febslet.2013.08.011
Liu, H. C., Jamshidi, N., Chen, Y., Eraly, S. A., Cho, S. Y., Bhatnagar, V., et al. (2016). An Organic Anion Transporter 1 (OAT1)-Centered Metabolic Network. J. Biol. Chem. 291 (37), 19474–19486. doi:10.1074/jbc.M116.745216
Liu, R., Han, C., Wu, D., Xia, X., Gu, J., Guan, H., et al. (2015). Prevalence of Hyperuricemia and Gout in Mainland China from 2000 to 2014: A Systematic Review and Meta-Analysis. BioMed Res. Int. 2015, 1–12. doi:10.1155/2015/762820
Luciani, A., Villella, V. R., Esposito, S., Brunetti-Pierri, N., Medina, D., Settembre, C., et al. (2010). Defective CFTR Induces Aggresome Formation and Lung Inflammation in Cystic Fibrosis through ROS-Mediated Autophagy Inhibition. Nat. Cell Biol. 12 (9), 863–875. doi:10.1038/ncb2090
Luo, Z., Saha, A. K., Xiang, X., and Ruderman, N. B. (2005). AMPK, the Metabolic Syndrome and Cancer. Trends Pharmacol. Sci. 26 (2), 69–76. doi:10.1016/j.tips.2004.12.011
Maaten, J. C. T., Voorburg, A., Heine, R. J., Wee, P. M. T., Donker, A. J. M., and Gans, R. O. B. (1997). Renal Handling of Urate and Sodium during Acute Physiological Hyperinsulinaemia in Healthy Subjects. Clin. Sci. 92 (1), 51–58. doi:10.1042/cs0920051
Macgregor, S., Lind, P. A., Bucholz, K. K., Hansell, N. K., Madden, P. A. F., Richter, M. M., et al. (2009). Associations of ADH and ALDH2 Gene Variation with Self Report Alcohol Reactions, Consumption and Dependence: an Integrated Analysis. Hum. Mol. Genet. 18 (3), 580–593. doi:10.1093/hmg/ddn372
Marrero, M. B., Banes-Berceli, A. K., Stern, D. M., and Eaton, D. C. (2006). Role of the JAK/STAT Signaling Pathway in Diabetic Nephropathy. Am. J. Physiology-Renal Physiology 290 (4), F762–F768. doi:10.1152/ajprenal.00181.2005
McKinney, C., Stamp, L. K., Dalbeth, N., Topless, R. K., Day, R. O., Kannangara, D. R., et al. (2015). Multiplicative Interaction of Functional Inflammasome Genetic Variants in Determining the Risk of Gout. Arthritis Res. Ther. 17, 288. doi:10.1186/s13075-015-0802-3
Mejías, E., Navas, J., Lluberes, R., and Martínez-Maldonado, M. (1989). Hyperuricemia, Gout, and Autosomal Dominant Polycystic Kidney Disease. Am. J. Med. Sci. 297 (3), 145–148. doi:10.1097/00000441-198903000-00002
Miner, J. N., Tan, P. K., Hyndman, D., Liu, S., Iverson, C., Nanavati, P., et al. (2016). Lesinurad, a Novel, Oral Compound for Gout, Acts to Decrease Serum Uric Acid through Inhibition of Urate Transporters in the Kidney. Arthritis Res. Ther. 18 (1), 214. doi:10.1186/s13075-016-1107-x
Nakamura, K., Sakurai, M., Miura, K., Morikawa, Y., Yoshita, K., Ishizaki, M., et al. (2012). Alcohol Intake and the Risk of Hyperuricaemia: A 6-year Prospective Study in Japanese Men. Nutr. Metabolism Cardiovasc. Dis. 22 (11), 989–996. doi:10.1016/j.numecd.2011.01.003
Nakatochi, M., Kanai, M., Nakayama, A., Hishida, A., Kawamura, Y., Ichihara, S., et al. (2019). Genome-wide Meta-Analysis Identifies Multiple Novel Loci Associated with Serum Uric Acid Levels in Japanese Individuals. Commun. Biol. 2, 115. doi:10.1038/s42003-019-0339-0
Nakatochi, M., Kanai, M., Nakayama, A., Hishida, A., Kawamura, Y., Ichihara, S., et al. (2019). Genome-wide Meta-Analysis Identifies Multiple Novel Loci Associated with Serum Uric Acid Levels in Japanese Individuals. Commun. Biol. 2, 115. doi:10.1038/s42003-019-0339-0
Nakayama, A., Matsuo, H., Shimizu, T., Ogata, H., Takada, Y., Nakashima, H., et al. (2013). A Common Missense Variant of Monocarboxylate Transporter 9 (MCT9/SLC16A9) Gene Is Associated with Renal Overload Gout, but Not with All Gout Susceptibility. Hum. Cell 26 (4), 133–136. doi:10.1007/s13577-013-0073-8
Nakayama, A., Matsuo, H., Shimizu, T., Takada, Y., Nakamura, T., Shimizu, S., et al. (2014). Common Variants of a Urate-Associated Gene LRP2 Are Not Associated with Gout Susceptibility. Rheumatol. Int. 34 (4), 473–476. doi:10.1007/s00296-013-2924-8
Nakayama, A., Nakaoka, H., Yamamoto, K., Sakiyama, M., Shaukat, A., Toyoda, Y., et al. (2017). GWAS of Clinically Defined Gout and Subtypes Identifies Multiple Susceptibility Loci that Include Urate Transporter Genes. Ann. Rheum. Dis. 76 (5), 869–877. doi:10.1136/annrheumdis-2016-209632
Namjou, B., Lingren, T., Lingren, T., Huang, Y., Parameswaran, S., Cobb, B. L., et al. (2019). GWAS and Enrichment Analyses of Non-alcoholic Fatty Liver Disease Identify New Trait-Associated Genes and Pathways across eMERGE Network. BMC Med. 17 (1), 135. doi:10.1186/s12916-019-1364-z
Narang, R. K., and Dalbeth, N. (2018). Management of Complex Gout in Clinical Practice: Update on Therapeutic Approaches. Best Pract. Res. Clin. Rheumatology 32 (6), 813–834. doi:10.1016/j.berh.2019.03.010
Neogi, T. (2010). Interleukin-1 Antagonism in Acute Gout: Is Targeting a Single Cytokine the Answer? Arthritis & Rheumatism 62 (10), 2845–2849. doi:10.1002/art.27635
Nieradko-Iwanicka, B. (2021). The Role of Alcohol Consumption in Pathogenesis of Gout. Crit. Rev. Food Sci. Nutr., 1–9. doi:10.1080/10408398.2021.1911928
Nishioka, K., Sumida, T., Iwatani, M., Kusumoto, A., Ishikura, Y., Hatanaka, H., et al. (2002). Influence of Moderate Drinking on Purine and Carbohydrate Metabolism. Alcohol Clin. Exp. Res. 26 (8 Suppl. l), 20s–25S. doi:10.1097/01.Alc.0000026829.60802.67
Okada, Y., Sim, X., Sim, X., Go, M. J., Wu, J.-Y., Gu, D., et al. (2012). Meta-analysis Identifies Multiple Loci Associated with Kidney Function-Related Traits in East Asian Populations. Nat. Genet. 44 (8), 904–909. doi:10.1038/ng.2352
Onuora, S. (2020). ABCG2 SNP Associated with Early-Onset Gout. Nat. Rev. Rheumatol. 16 (4), 186. doi:10.1038/s41584-020-0393-5
Padova, J., Onesti, G., Faludi, G., Bendersky, G., and Bendersky, G. (1964). THE EFFECT OF GLUCOSE LOADS ON RENAL URIC ACID EXCRETION IN DIABETIC PATIENTS. Metabolism 13, 507–512. doi:10.1016/0026-0495(64)90137-4
Park, J. s., Kim, Y., and Kang, J. (2021). Genome-wide Meta-Analysis Revealed Several Genetic Loci Associated with Serum Uric Acid Levels in Korean Population: an Analysis of Korea Biobank Data. J. Hum. Genet. 67, 231–237. doi:10.1038/s10038-021-00991-1
Perheentupa, J., and Raivio, K. (1967). Fructose-induced Hyperuricæmia. Lancet 290 (7515), 528–531. doi:10.1016/s0140-6736(67)90494-1
Prestin, K., Wolf, S., Feldtmann, R., Hussner, J., Geissler, I., Rimmbach, C., et al. (2014). Transcriptional Regulation of Urate Transportosome Member SLC2A9 by Nuclear Receptor HNF4α. Am. J. Physiology-Renal Physiology 307 (9), F1041–F1051. doi:10.1152/ajprenal.00640.2013
Puig, J. G., Miranda, M. E., Mateos, F. A., Picazo, M. L., Jiménez, M. L., Calvin, T. S., et al. (1993). Hereditary Nephropathy Associated with Hyperuricemia and Gout. Arch. Intern Med. 153 (3), 357–365. doi:10.1001/archinte.1993.00410030063009
Qing, Y.-F., Zhou, J.-G., Zhang, Q.-B., Wang, D.-S., Li, M., Yang, Q.-B., et al. (2013). Association of TLR4 Gene Rs2149356 Polymorphism with Primary Gouty Arthritis in a Case-Control Study. PLoS One 8 (5), e64845. doi:10.1371/journal.pone.0064845
Quinones Galvan, A., Natali, A., Baldi, S., Frascerra, S., Sanna, G., Ciociaro, D., et al. (1995). Effect of Insulin on Uric Acid Excretion in Humans. Am. J. Physiology-Endocrinology Metabolism 268, E1–E5. doi:10.1152/ajpendo.1995.268.1.E1
Rasheed, H., McKinney, C., Stamp, L. K., Dalbeth, N., Topless, R. K., Day, R., et al. (2016). The Toll-like Receptor 4 (TLR4) Variant Rs2149356 and Risk of Gout in European and Polynesian Sample Sets. PLoS One 11 (1), e0147939. doi:10.1371/journal.pone.0147939
Rasheed, H., Phipps-Green, A., Topless, R., Hollis-Moffatt, J. E., Hindmarsh, J., Franklin, C., et al. (2013). Association of the Lipoprotein Receptor-Related Protein 2 Gene with Gout and Non-additive Interaction with Alcohol Consumption. Arthritis Res. Ther. 15 (6), R177. doi:10.1186/ar4366
Rasheed, H., Stamp, L. K., Dalbeth, N., and Merriman, T. R. (2017). Interaction of the GCKR and A1CF Loci with Alcohol Consumption to Influence the Risk of Gout. Arthritis Res. Ther. 19 (1), 161. doi:10.1186/s13075-017-1369-y
Reginato, A. M., Mount, D. B., Yang, I., and Choi, H. K. (2012). The Genetics of Hyperuricaemia and Gout. Nat. Rev. Rheumatol. 8 (10), 610–621. doi:10.1038/nrrheum.2012.144
Rhyu, J., and Bhat, S. P. (2021). Skeletal Complications with GNAS Mutation: An Unusual Case with Osteoma Cutis, Gout, and Synovial Chondromatosis in a Patient with Pseudopseudohypoparathyroidism. AACE Clin. Case Rep. 7 (3), 180–183. doi:10.1016/j.aace.2020.11.036
Roberts, R. L., Wallace, M. C., Phipps-Green, A. J., Topless, R., Drake, J. M., Tan, P., et al. (2017). ABCG2 Loss-Of-Function Polymorphism Predicts Poor Response to Allopurinol in Patients with Gout. Pharmacogenomics J. 17 (2), 201–203. doi:10.1038/tpj.2015.101
Sakiyama, M., Matsuo, H., Nakaoka, H., Kawamura, Y., Kawaguchi, M., Higashino, T., et al. (2018). Common Variant of BCAS3 Is Associated with Gout Risk in Japanese Population: the First Replication Study after Gout GWAS in Han Chinese. BMC Med. Genet. 19 (1), 96. doi:10.1186/s12881-018-0583-z
Sandoval-Plata, G., Morgan, K., and Abhishek, A. (2021). Variants in Urate Transporters, ADH1B, GCKR and MEPE Genes Associate with Transition from Asymptomatic Hyperuricaemia to Gout: Results of the First Gout versus Asymptomatic Hyperuricaemia GWAS in Caucasians Using Data from the UK Biobank. Ann. Rheum. Dis. 80 (9), 1220–1226. doi:10.1136/annrheumdis-2020-219796
Schauer, C., Janko, C., Munoz, L. E., Zhao, Y., Kienhöfer, D., Frey, B., et al. (2014). Aggregated Neutrophil Extracellular Traps Limit Inflammation by Degrading Cytokines and Chemokines. Nat. Med. 20 (5), 511–517. doi:10.1038/nm.3547
Schmidt, M. I., Watson, R. L., Duncan, B. B., Metcalf, P., Brancati, F. L., Richey Sharrett, A., et al. (1996). Clustering of Dyslipidemia, Hyperuricemia, Diabetes, and Hypertension and its Association with Fasting Insulin and Central and Overall Obesity in a General Population. Metabolism 45 (6), 699–706. doi:10.1016/s0026-0495(96)90134-1
Shi, C.-S., Shenderov, K., Huang, N.-N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., et al. (2012). Activation of Autophagy by Inflammatory Signals Limits IL-1β Production by Targeting Ubiquitinated Inflammasomes for Destruction. Nat. Immunol. 13 (3), 255–263. doi:10.1038/ni.2215
Siniachenko, O. V., Diadyk, A. I., Nikolenko, Iu. I., and Khomenko, M. V. (1984). Circulating Immune Complexes in Gout. Vrach Delo (9), 61–63.
Smith, E., Hoy, D., Cross, M., Merriman, T. R., Vos, T., Buchbinder, R., et al. (2014). The Global Burden of Gout: Estimates from the Global Burden of Disease 2010 Study. Ann. Rheum. Dis. 73 (8), 1470–1476. doi:10.1136/annrheumdis-2013-204647
So, A., De Meulemeester, M., Pikhlak, A., Yücel, A. E., Richard, D., Murphy, V., et al. (2010). Canakinumab for the Treatment of Acute Flares in Difficult-To-Treat Gouty Arthritis: Results of a Multicenter, Phase II, Dose-Ranging Study. Arthritis & Rheumatism 62 (10), 3064–3076. doi:10.1002/art.27600
So, A., De Smedt, T., Revaz, S., and Tschopp, J. (2007). A Pilot Study of IL-1 Inhibition by Anakinra in Acute Gout. Arthritis Res. Ther. 9 (2), R28. doi:10.1186/ar2143
Spadaro, O., Goldberg, E. L., Camell, C. D., Youm, Y.-H., Kopchick, J. J., Nguyen, K. Y., et al. (2016). Growth Hormone Receptor Deficiency Protects against Age-Related NLRP3 Inflammasome Activation and Immune Senescence. Cell Rep. 14 (7), 1571–1580. doi:10.1016/j.celrep.2016.01.044
Sun, H., Tian, J., Xian, W., Xie, T., and Yang, X. (2015). miR-34a Inhibits Proliferation and Invasion of Bladder Cancer Cells by Targeting Orphan Nuclear Receptor HNF4G. Dis. markers 2015, 1–8. doi:10.1155/2015/879254
Tafaj, O., and Jüppner, H. (2017). Pseudohypoparathyroidism: One Gene, Several Syndromes. J. Endocrinol. Invest 40 (4), 347–356. doi:10.1007/s40618-016-0588-4
Tanner, C., Boocock, J., Stahl, E. A., Dobbyn, A., Mandal, A. K., Cadzow, M., et al. (2017). Population-Specific Resequencing Associates the ATP-Binding Cassette Subfamily C Member 4 Gene with Gout in New Zealand Māori and Pacific Men. Arthritis & Rheumatology 69 (7), 1461–1469. doi:10.1002/art.40110
Terkeltaub, R., Sundy, J. S., Schumacher, H. R., Murphy, F., Bookbinder, S., Biedermann, S., et al. (2009). The Interleukin 1 Inhibitor Rilonacept in Treatment of Chronic Gouty Arthritis: Results of a Placebo-Controlled, Monosequence Crossover, Non-randomised, Single-Blind Pilot Study. Ann. Rheumatic Dis. 68 (10), 1613–1617. doi:10.1136/ard.2009.108936
Tin, A., Marten, J., Halperin Kuhns, V. L., Li, Y., Wuttke, M., Kirsten, H., et al. (2019). Target Genes, Variants, Tissues and Transcriptional Pathways Influencing Human Serum Urate Levels. Nat. Genet. 51 (10), 1459–1474. doi:10.1038/s41588-019-0504-x
Tin, A., Marten, J., Halperin Kuhns, V. L., Li, Y., Wuttke, M., Kirsten, H., et al. (2019). Target Genes, Variants, Tissues and Transcriptional Pathways Influencing Human Serum Urate Levels. Nat. Genet. 51 (10), 1459–1474. doi:10.1038/s41588-019-0504-x
Tin, A., Li, Y., Brody, J. A., Nutile, T., Chu, A. Y., Huffman, J. E., et al. (2018). Large-scale Whole-Exome Sequencing Association Studies Identify Rare Functional Variants Influencing Serum Urate Levels. Nat. Commun. 9 (1), 4228. doi:10.1038/s41467-018-06620-4
Tin, A., Woodward, O. M., Kao, W. H. L., Liu, C.-T., Lu, X., Nalls, M. A., et al. (2011). Genome-wide Association Study for Serum Urate Concentrations and Gout Among African Americans Identifies Genomic Risk Loci and a Novel URAT1 Loss-Of-Function Allele. Hum. Mol. Genet. 20 (20), 4056–4068. doi:10.1093/hmg/ddr307
Tong, X., Zhao, F., Mancuso, A., Gruber, J. J., and Thompson, C. B. (2009). The Glucose-Responsive Transcription Factor ChREBP Contributes to Glucose-dependent Anabolic Synthesis and Cell Proliferation. Proc. Natl. Acad. Sci. U.S.A. 106 (51), 21660–21665. doi:10.1073/pnas.0911316106
Tsuchiya, M., Misaka, R., Nitta, K., and Tsuchiya, K. (2015). Transcriptional Factors, Mafs and Their Biological Roles. Wjd 6 (1), 175–183. doi:10.4239/wjd.v6.i1.175
Tu, H.-P., Chen, C.-J., Tovosia, S., Ko, A. M.-S., Lee, C.-H., Ou, T.-T., et al. (2010). Associations of a Non-synonymous Variant in SLC2A9 with Gouty Arthritis and Uric Acid Levels in Han Chinese Subjects and Solomon Islanders. Ann. Rheumatic Dis. 69 (5), 887–890. doi:10.1136/ard.2009.113357
Tu, H.-P., Ko, A. M.-S., Chiang, S.-L., Lee, S.-S., Lai, H.-M., Chung, C.-M., et al. (2014). Joint Effects of Alcohol Consumption and ABCG2 Q141K on Chronic Tophaceous Gout Risk. J. Rheumatol. 41 (4), 749–758. doi:10.3899/jrheum.130870
Tu, H.-P., Min-Shan Ko, A., Lee, S.-S., Lee, C.-P., Kuo, T.-M., Huang, C.-M., et al. (2018). Variants of ALPK1 with ABCG2, SLC2A9, and SLC22A12 Increased the Positive Predictive Value for Gout. J. Hum. Genet. 63 (1), 63–70. doi:10.1038/s10038-017-0368-9
Turan, S., and Bastepe, M. (2015). GNAS Spectrum of Disorders. Curr. Osteoporos. Rep. 13 (3), 146–158. doi:10.1007/s11914-015-0268-x
Urano, W., Taniguchi, A., Anzai, N., Inoue, E., Sekita, C., Endou, H., et al. (2010). Association between GLUT9 and Gout in Japanese Men. Ann. Rheumatic Dis. 69 (5), 932–933. doi:10.1136/ard.2009.111096
Urano, W., Taniguchi, A., Inoue, E., Sekita, C., Ichikawa, N., Koseki, Y., et al. (2013). Effect of Genetic Polymorphisms on Development of Gout. J. Rheumatol. 40 (8), 1374–1378. doi:10.3899/jrheum.121244
van der Gaag, M., van den Berg, R., van den Berg, H., Schaafsma, G., and Hendriks, H. (2000). Moderate Consumption of Beer, Red Wine and Spirits Has Counteracting Effects on Plasma Antioxidants in Middle-Aged Men. Eur. J. Clin. Nutr. 54 (7), 586–591. doi:10.1038/sj.ejcn.1601061
Wallace, M. C., Roberts, R. L., Nanavati, P., Miner, J. N., Dalbeth, N., Topless, R., et al. (2018). Association between ABCG2 Rs2231142 and Poor Response to Allopurinol: Replication and Meta-Analysis. Rheumatol. Oxf. 57 (4), 656–660. doi:10.1093/rheumatology/kex467
Wang, J., Liu, S., Wang, B., Miao, Z., Han, L., Chu, N., et al. (2012). Association between Gout and Polymorphisms in GCKR in Male Han Chinese. Hum. Genet. 131 (7), 1261–1265. doi:10.1007/s00439-012-1151-9
Wang, L., Ma, Q., Yao, H., He, L. J., Fang, B. B., Cai, W., et al. (2018). Association of GCKR Rs780094 Polymorphism with Circulating Lipid Levels in Type 2 Diabetes and Hyperuricemia in Uygur Chinese. Int. J. Clin. Exp. Pathol. 11 (9), 4684–4694.
Wang, X., Shaw, S., Amiri, F., Eaton, D. C., and Marrero, M. B. (2002). Inhibition of the JAK/STAT Signaling Pathway Prevents the High Glucose-Induced Increase in TGF-β and Fibronectin Synthesis in Mesangial Cells. Diabetes 51 (12), 3505–3509. doi:10.2337/diabetes.51.12.3505
Wen, C., Yee, S., Liang, X., Hoffmann, T., Kvale, M., Banda, Y., et al. (2015). Genome-wide Association Study Identifies ABCG2 (BCRP) as an Allopurinol Transporter and a Determinant of Drug Response. Clin. Pharmacol. Ther. 97 (5), 518–525. doi:10.1002/cpt.89
Wisely, G. B., Miller, A. B., Davis, R. G., Thornquest, A. D., Johnson, R., Spitzer, T., et al. (2002). Hepatocyte Nuclear Factor 4 Is a Transcription Factor that Constitutively Binds Fatty Acids. Structure 10 (9), 1225–1234. doi:10.1016/s0969-2126(02)00829-8
Wong, K., Briddon, S. J., Holliday, N. D., and Kerr, I. D. (2016). Plasma Membrane Dynamics and Tetrameric Organisation of ABCG2 Transporters in Mammalian Cells Revealed by Single Particle Imaging Techniques. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1863 (1), 19–29. doi:10.1016/j.bbamcr.2015.10.002
Wu, J. N., and Koretzky, G. A. (2004). The SLP-76 Family of Adapter Proteins. Seminars Immunol. 16 (6), 379–393. doi:10.1016/j.smim.2004.08.018
Yang, Q., Köttgen, A., Dehghan, A., Smith, A. V., Glazer, N. L., Chen, M.-H., et al. (2010). Multiple Genetic Loci Influence Serum Urate Levels and Their Relationship with Gout and Cardiovascular Disease Risk Factors. Circ. Cardiovasc Genet. 3 (6), 523–530. doi:10.1161/circgenetics.109.934455
Yang, Y., and Cvekl, A. (2007). Large Maf Transcription Factors: Cousins of AP-1 Proteins and Important Regulators of Cellular Differentiation. Ejbm 23 (1), 2–11. doi:10.23861/ejbm20072347
Yeung, B. H. Y., Law, A. Y. S., and Wong, C. K. C. (2012). Evolution and Roles of Stanniocalcin. Mol. Cell. Endocrinol. 349 (2), 272–280. doi:10.1016/j.mce.2011.11.007
Yi, X. L., Li, J., Meng, D. M., Liu, Y. J., Liu, Y. H., Ma, H. M., et al. (2018). An Intron Variant of SLC2A9 Increases the Risk for Type 2 Diabetes Mellitus Complicated with Hyperuricemia in Chinese Male Population. Iran. J. Public Health 47 (6), 844–851.
Yoon, J. C., Puigserver, P., Chen, G., Donovan, J., Wu, Z., Rhee, J., et al. (2001). Control of Hepatic Gluconeogenesis through the Transcriptional Coactivator PGC-1. Nature 413 (6852), 131–138. doi:10.1038/35093050
Yu, J., Riou, C., Davidson, D., Minhas, R., Robson, J. D., Julius, M., et al. (2001). Synergistic Regulation of Immunoreceptor Signaling by SLP-76-Related Adaptor Clnk and Serine/threonine Protein Kinase HPK-1. Mol. Cell Biol. 21 (18), 6102–6112. doi:10.1128/mcb.21.18.6102-6112.2001
Yu, K.-H., See, L.-C., Huang, Y.-C., Yang, C.-H., and Sun, J.-H. (2008). Dietary Factors Associated with Hyperuricemia in Adults. Seminars Arthritis Rheumatism 37 (4), 243–250. doi:10.1016/j.semarthrit.2007.04.007
Zhang, Y., Cai, W., Song, J., Miao, L., Zhang, B., Xu, Q., et al. (2014). Association between the PNPLA3 I148M Polymorphism and Non-alcoholic Fatty Liver Disease in the Uygur and Han Ethnic Groups of Northwestern China. PLoS One 9 (10), e108381. doi:10.1371/journal.pone.0108381
Zhao, J., Hu, Y., and Peng, J. (2021). Targeting Programmed Cell Death in Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): a Promising New Therapy. Cell Mol. Biol. Lett. 26 (1), 17. doi:10.1186/s11658-021-00254-z
Zhao, J., Jiang, P., Guo, S., Schrodi, S. J., and He, D. (2021). Apoptosis, Autophagy, NETosis, Necroptosis, and Pyroptosis Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid Arthritis. Front. Immunol. 12, 809806. doi:10.3389/fimmu.2021.809806
Zhou, W., Kanai, M., Wu, K-H. H., Humaira, R., Tsuo, K., Hirbo, J. B., et al. (2021). Global Biobank Meta-Analysis Initiative: Powering Genetic Discovery across Human Diseases. medRxiv. 2021.11.19.21266436. doi:10.1101/2021.11.19.21266436
Glossary
GWAS genome-wide association studies
GBMI Global Biobank Meta-analysis Initiative
URAT1 urate transporter-1
GLUT9 glucose transporter 9
OAT4 organic anion transporter 4
OAT10 organic anion transporter 4
ABCG2 TP-binding cassette superfamily G member 2
ABCC4 ATP Binding Cassette Subfamily C Member 4
ATP adenosine triphosphate
TLR Toll-like receptor
NLRP3 nod-like receptor pyrin domain 3
NF-kB nuclear factor-κB
IL interleukin
NSAIDs non-steroidal anti-inflammation drugs
SLC2A9 solute carrier family 2 member 9
SLC22A11 solute carrier family 22 member 11
SLC17A1 solute carrier family 17 member 1
SLC22A12 solute carrier family 22 member 12
MAF MAF BZIP transcription factor
SLC2A12 solute carrier family 2 member 12
SLC16A9 solute carrier family 16 member 9
GCKR glucokinase (hexokinase 4) regulator
PDZK1 PDZ domain-containing 1
HNF4A hepatocyte nuclear factor 4 alpha
SNP single-nucleotide polymorphism
C-MAF C-MAF BZIP transcription factor
cis-eQTL cis-expression quantitative trait loci
MAFTRR MAF transcriptional regulator RNA
HNF4G hepatocyte nuclear factor 4 gamma
PKD2 polycystin 2
ADH1B alcohol dehydrogenase 1B (Class I), beta polypeptide
MCT9 monocarboxylate transporter 9
FAM35A shieldin complex subunit 2
AMPD2 adenosine monophosphate deaminase 2
MLXIP MLX interacting protein
MLXIPL MLX interacting protein-like
PNPLA3 patatin-like phospholipase domain containing 3
MAFLD metabolism-related fatty liver disease
IGF1R insulin like growth factor 1 receptor
BMI body mass index
A1CF APOBEC1 complementation factor
ApoB apolipoprotein B
LRP2 lipoprotein receptor-related protein 2
STC1 stanniocalcin 1
CYP1A2 cytochrome P450 family 1 subfamily A member 2
HCRTR2 hypocretin receptor 2
CLNK cytokine-dependent hematopoietic cell linker
GNAS guanine nucleotide-binding protein a-stimulating polypeptide
SCL22A6 solute carrier family 22 member 6
BCAS3 breast cancer-amplified sequence 3
cAMP cyclic AMP
OAT1 organic anion transporter 1
MEPE matrix extracellular phosphoglycoprotein
PPM1K-DT protein phosphatase, Mg2+/Mn2+ dependent 1K- divergent transcript
ADH1B alcohol dehydrogenase 1B (Class I), beta polypeptide
Keywords: hyperuricemia, gout, genetic susceptibility loci, novel mechanism, inflammation introduction
Citation: Zhao J, Guo S, Schrodi SJ and He D (2022) Trends in the Contribution of Genetic Susceptibility Loci to Hyperuricemia and Gout and Associated Novel Mechanisms. Front. Cell Dev. Biol. 10:937855. doi: 10.3389/fcell.2022.937855
Received: 06 May 2022; Accepted: 31 May 2022;
Published: 23 June 2022.
Edited by:
Haitao Wang, National Cancer Institute, United StatesReviewed by:
Jing Guo, Stanford University, United StatesXin Yin, Penn State Milton S. Hershey Medical Center, United States
Yuying Huang, University of Texas MD Anderson Cancer Center, United States
Mingdian Tan, Stanford University, United States
Copyright © 2022 Zhao, Guo, Schrodi and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shicheng Guo, Shicheng.Guo@wisc.edu; Steven J. Schrodi, Schrodi@wisc.edu; Dongyi He, dongyihe@medmail.com.cn