 Jun Yang1†
Jun Yang1† Zejun Fang
Zejun Fang Ming Ye
Ming Ye Xi Xu
Xi Xu- 1Department of Orthopedic Surgery, Sanmen People’s Hospital of Zhejiang Province, Sanmenwan Branch of the First Affiliated Hospital, College of Medicine, Zhejiang University, Sanmen, China
- 2Central Laboratory, The Municipal Affiliated Hospital of Xuzhou Medical University, Xuzhou, China
- 3Central Laboratory, Sanmen People’s Hospital of Zhejiang Province, Sanmenwan Branch of the First Affiliated Hospital, College of Medicine, Zhejiang University, Sanmen, China
- 4Department of Pathophysiology, Zunyi Medical University, Zunyi, China
- 5Department of General Surgery, Sanmen People’s Hospital of Zhejiang Province, Sanmenwan Branch of the First Affiliated Hospital, College of Medicine, Zhejiang University, Sanmen, China
- 6Department of Pathology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Histone deacetylase 5 (HDAC5) is a class II HDAC. Aberrant expression of HDAC5 has been observed in multiple cancer types, and its functions in cell proliferation and invasion, the immune response, and maintenance of stemness have been widely studied. HDAC5 is considered as a reliable therapeutic target for anticancer drugs. In light of recent findings regarding the role of epigenetic reprogramming in tumorigenesis, in this review, we provide an overview of the expression, biological functions, regulatory mechanisms, and clinical significance of HDAC5 in cancer.
Introduction
Covalent modification of chromatin regulates gene transcription during cell differentiation; this includes histone acetylation and deacetylation, which modulate the binding of transcription factors to DNA. Modifications can occur at various sites, including the N-terminal amino acid residues of histones H3 and H4, and the N- and C-terminal amino acid residues of histones H2A, H2B, and H1. Histone acetylation of the ϵ-amino group of lysine, one of the earliest and most common posttranslational modifications of histones, is regulated by histone acetyltransferases and histone deacetylases (HDACs) (1–5).
The HDAC family has 18 members that can be divided into 4 classess based on their structure and function (6, 7). Class I includes HDAC1-3 and 8; class II includes HDAC4-7, 9, and 10; and class III includes sirtuin enzymes (SIRT 1-7). These 3 classes constitute the classical HDACs. Class IV comprises only HDAC11, which is structurally distinct from the other HDACs. Class II HDACs are further divided into class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6 and 10) based on their subcellular localization and expression pattern (8).
Class IIa HDACs have a high degree of homology to yeast hda1 (HDAC-1). In addition to the C-terminal catalytic core, all class IIa HDACs have a conserved N-terminal extension that can bind to transcription factors and the chaperone protein 14-3-3. For example, when phosphorylated by calcium/calmodulin dependent protein kinase (CaMK) or protein kinase D (PKD), class IIa HDACs bind to 14-3-3, inducing its disaggregation from the transcription factor myocyte specific enhancer factor 2 (MEF2) and bringing about its shuttling from the nucleus to the cytoplasm (9).
As a class IIa HDAC, HDAC5 not only catalyzes the deacetylation of nuclear histones, but also deacetylates or forms complexes with other proteins in various physiologic contexts including nerve regeneration and neuronal apoptosis (10, 11), glucose metabolism (12), and insulin resistance (13). Cancer is one of the leading causes of death worldwide, and the identification of further specific molecular markers of cancer will be useful for early diagnosis, therapeutic targeting, and treatment response monitoring. This review highlights the current state of knowledge of the roles and clinical significance of HDAC5 in tumorigenesis.
Structure and Distribution of HDAC5
Structure of HDAC5
HDAC5 was first identified in the mouse genome in 1999 (14). The gene encoding HDAC5 (also known as HD5 or NY-CO-9) in humans is located on chromosome 17q21 and spans 39,138 bp, comprising 26 exons. The protein consists of 1122 amino acids, has a molecular weight of 121.9 kDa, and has C-terminal deacetylase and N-terminal adapter domains. The former domain, which is also referred to as the HDAC domain, contains a nuclear export sequence(NES) and is highly conserved (80% homology) across class IIa HDACs; it is composed of 400-450 amino acids and shares 53% sequence similarity with yeast hda1 (15). The conserved 450-600 amino acid N-terminal adaptor domain has a nuclear localization sequence (NLS) and binds to transcription factors such as C-terminal-binding protein (CtBP), MEF2, and heterochromatin P1 (HP1) (3).
HDAC5 also has a flexible zinc-binding element outside the catalytic core. This element contains highly conserved cysteine and histidine residues that form a hydrophobic pocket near the substrate-binding channel. It may mediate substrate recognition and regulate enzymatic activity and interactions with other proteins (16). Moreover, HDAC5 also has a conserved histidine near the first zinc-binding site whose side chain turns outward and is far away from the catalytic active site (17). Thus, while HDAC5 by itself has no deacetylase activity, it can be induced by interaction with HDAC3 through the silencing mediator of retinoic acid and thyroid hormone receptor (SMRT)/nuclear receptor corepressor (NCoR) coinhibition complex (18). In other words, HDAC5 regulates gene expression by binding to transcription factors through its N terminal adapter domain and targeting the SMRT/NCoR-HDAC3 complex to a specific subcellular location through its C-terminal deacetylase domain.
Distribution of HDAC5
HDAC5 protein is expressed in lung, brain, myocardium, skeletal muscle, and placenta, and accumulating evidence indicates that it has variable expression and functions in different types of tumor: HDAC5 is overexpressed in breast cancer (19, 20), hepatocellular carcinoma (HCC) (21), lung cancer (22), pancreatic neuroendocrine cancer(pNET) (23) and colorectal cancer(CRC) (24). In contrast, although HDAC5 was shown to induce tissue invasion of gastric cancer cells (25), gene expression profiles of histone modifiers indicate that HDAC5 is downregulated in gastric cancer (26) (Table 1). These conflicting findings imply that HDAC5 exhibits dual functions in cancer development. Furthermore, HDAC5 mRNA and protein have been detected in the blood of patients with CRC (32, 33) and breast cancer (34), but not in that of healthy subjects or patients with nonrecurrent cancer, suggesting that circulating HDAC5 may serve as a potential biomarker for cancer diagnosis and prognosis.
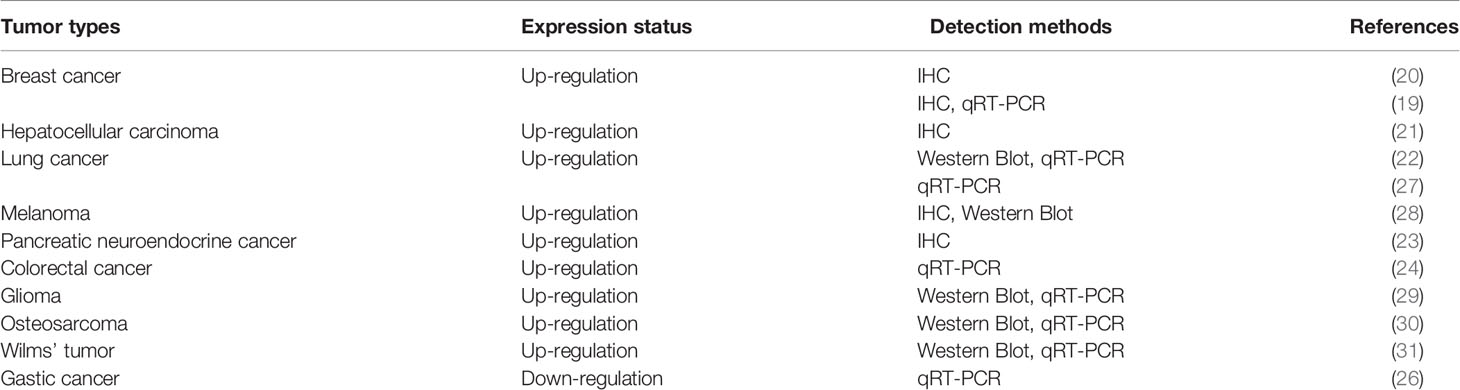
Table 1 The expression of HDAC5 in various tumors.
Regulation of HDAC5
Posttranslational Modification of HDAC5
Proteomics analysis combined with phosphomutant screening has revealed that there are at least 17 phosphorylation sites in the HDAC5 functional domains (35), highlighting that HDAC5 can be phosphorylated at multiple conserved residues by a variety of protein kinases. For example, PKD, CaMK II, and AMPK phosphorylate Ser259 and Ser498 on both sides of the HDAC5 NLS, which promotes the binding of 14-3-3 to HDAC5 and its shuttling from the nucleus to the cytoplasm (36–38). In myocyte-like cells, cAMP signaling prevents 14-3-3 binding to HDAC5 by bringing about Ser498 hypophosphorylation (39). Protein kinase C-related kinase 1/2 (PRK1/2) phosphorylate HDAC5 at Thr292 in the NLS, promoting its binding to 14-3-3 and preventing HDAC5 nuclear entry (40). Additionally, minibrain-related kinase (Mirk) phosphorylates HDAC5 at Ser279, preventing its translocation from the cytoplasm to the nucleus (41) (Figure 1).
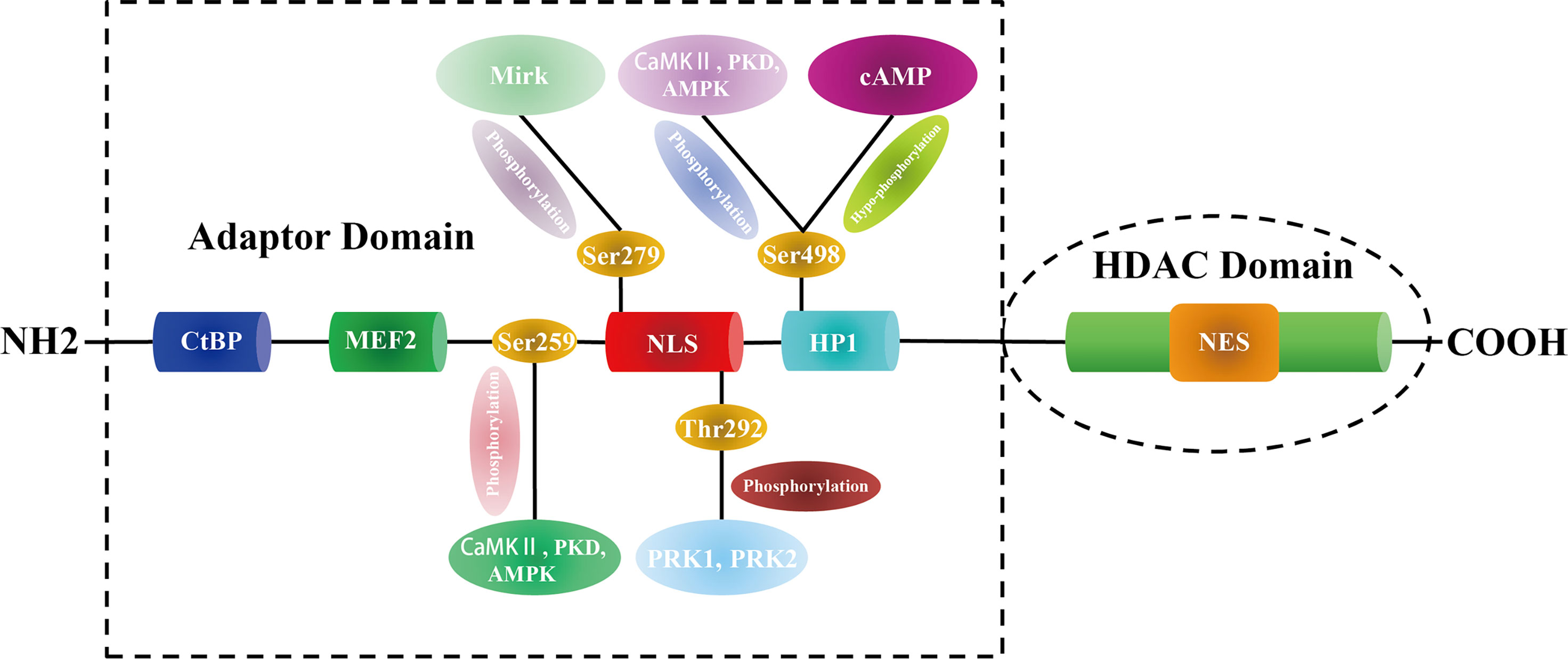
Figure 1 The structure and modification sites of HDAC5.
Non-Coding RNAs Modification to HDAC5
HDAC5 expression is regulated by miRNAs. miR-2861 was the first reported miRNA to directly regulate HDAC5, which it does by binding to the HDAC5 mRNA coding sequence (42). miR-9 suppresses HDAC5 activity (43) and inhibits the translation of HDAC5 transcript by binding to the 3′ untranslated region (44). Other miRNAs known to regulate HDAC5 are miR-124 (44, 45), miR-125a-5p (46), miR-589-5p (27), and miR-217 (47). Many miRNAs targeting HDAC5 have been identified using target prediction software (Supplementary Table 1), although most of these require experimental validation.
According to the competing endogenous RNA (ceRNA) hypothesis, long noncoding RNAs (lncRNAs) inhibit miRNA function by acting as endogenous miRNA sponges (48). The lncRNA SENEBLOC acts as a sponge regarding miR-3175, thereby upregulating HDAC5 (49). Additional studies investigating the role of lncRNAs in HDAC5 regulation are currently underway. The findings to date indicate that HDAC5 is epigenetically modified and regulated at the transcriptional level (Figure 2), providing new avenues for HDAC5-based cancer therapy.
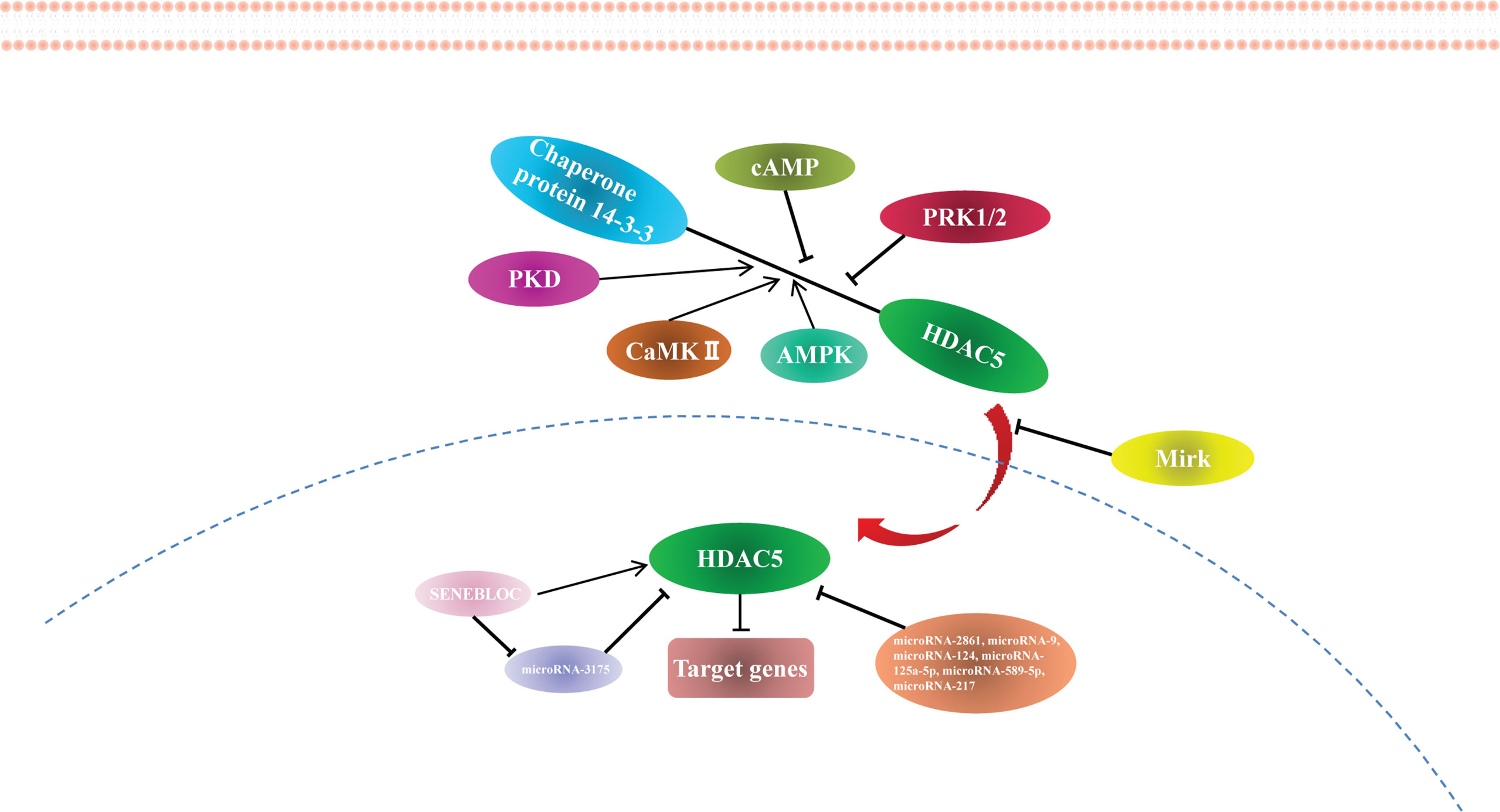
Figure 2 The regulation of HDAC5.
HDAC5 in Cancer
The functions of HDAC5 in tumorigenesis have been investigated in a variety of cancers. HDAC5 plays distinct roles in different cancer types. In this review, we summarize recent findings on the biological activities of HDAC5 in several common cancers and cancer-related processes (Table 2, Figure 3).
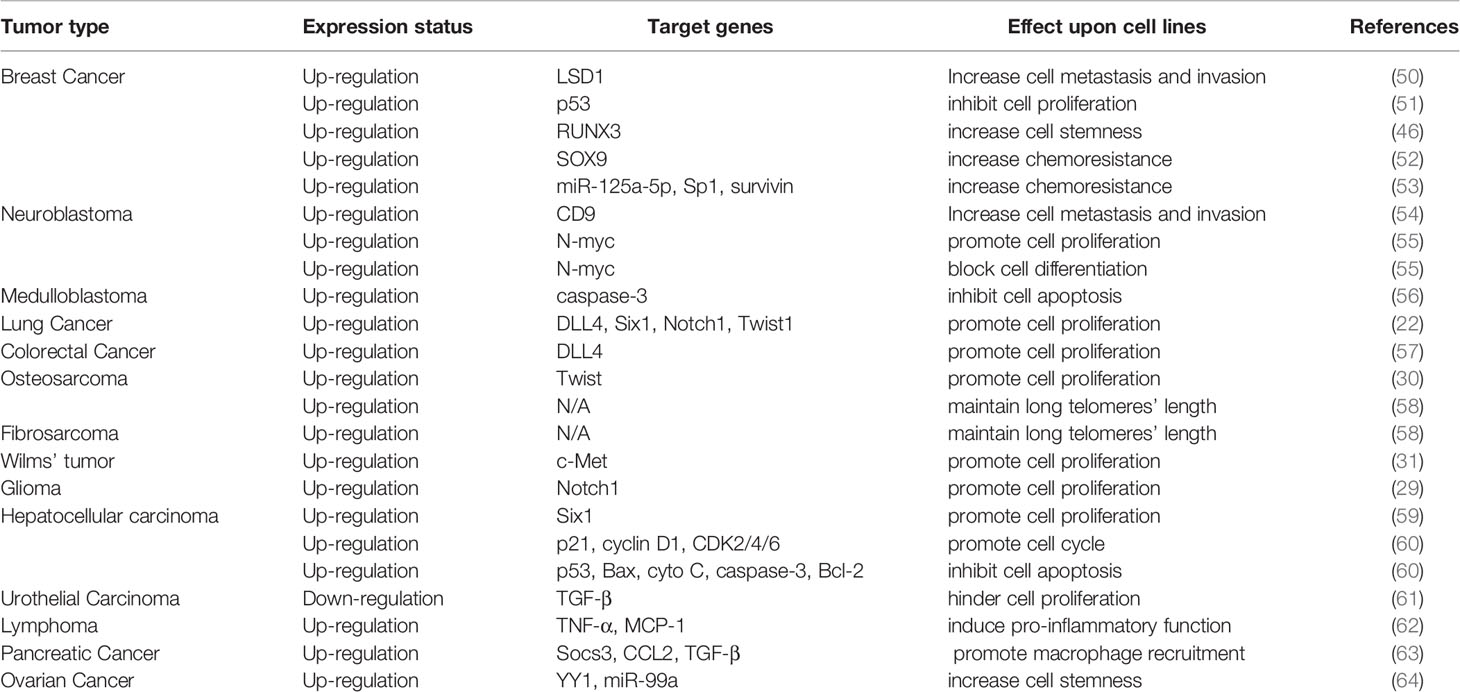
Table 2 Multiple cellular processes of HDAC5 in cancer management.
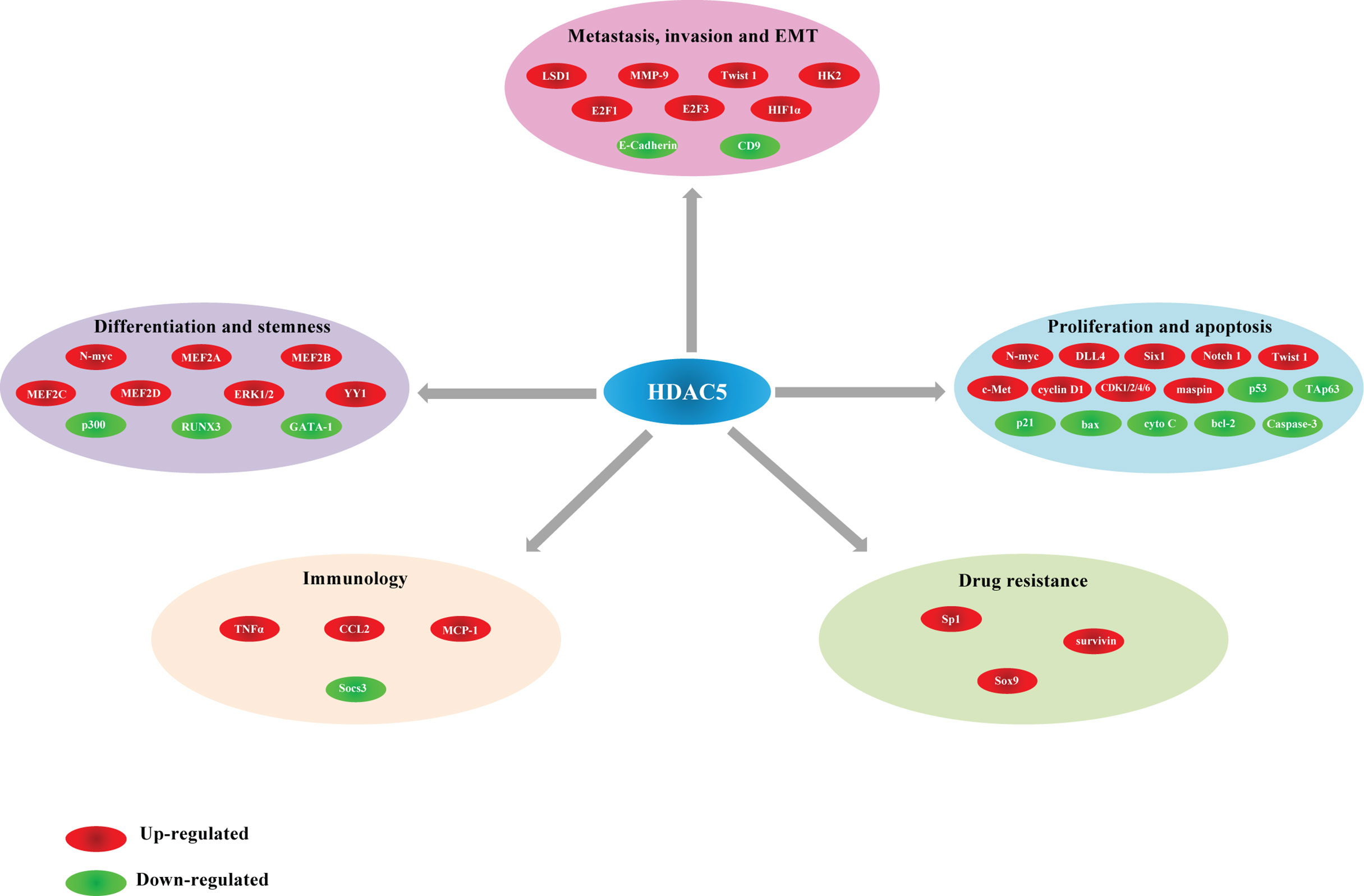
Figure 3 Different actions of HDAC5 and their corresponding target genes in cancer.
Role of HDAC5 in Cancer Metastasis and Invasion
PCR and immunohistochemical analyses have shown that HDAC5 was highly expressed in the cytoplasm of malignant epithelial cells, and HDAC5 expression was positively associated with distant metastasis and lymph node metastasis (65). HDAC5 expression was also found to be positively associated with intrahepatic metastasis and distant metastasis in HCC (21). In vitro and in vivo experiments have demonstrated that HDAC5 knockdown blocked metastasis of melanoma cells (28). Elevated HDAC5 expression is frequently observed in the luminal A and B subtypes of breast cancer, and HDAC5 silencing suppressed breast cancer cell motility and invasion (50). HDAC5 was also shown to promote cell invasion and metastasis in neuroblastoma (54), pancreatic cancer (66) and lung cancer (67).
In addition, HDAC5 enhanced the invasiveness of gastric cancer cell lines by stimulating protein kinase C (PKC)/matrix metalloproteinase 9 (MMP9) (25). Science MMP9 promotes tumor invasion and metastasis through extracellular matrix remodeling, regulation of cell adhesion, and degradation of vascular basement membrane and perivascular matrix during EMT (68, 69), the correlation between HDAC5 and EMT during tumorigenesis has drawn a great attention (Table 3).
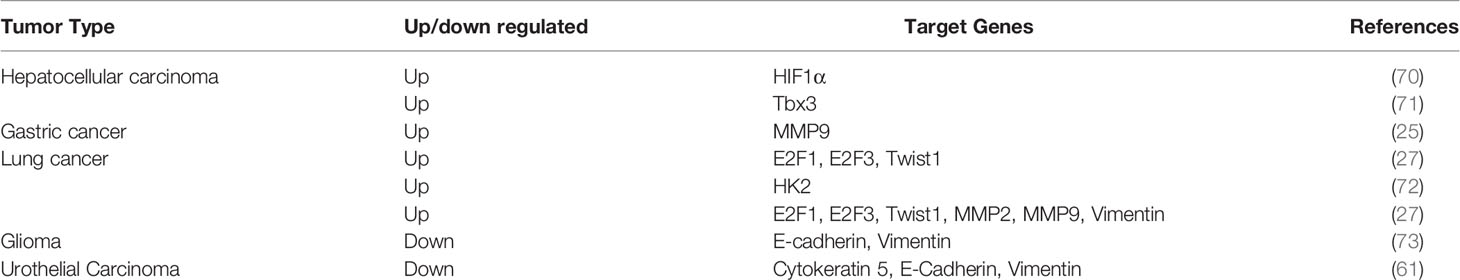
Table 3 The involvement of HDAC5 in epithelial-mesenchymal transition (EMT) processes.
In HCC, HDAC5 was shown to be involved in T-box 3 (Tbx3)-mediated EMT and metastasis, which was dependent on two HDAC5-interacting motifs (71). During HCC progression, application of the nonselective HDAC inhibitor (HDACi) panobinostat increased the expression of E-cadherin, an epithelial marker that is downregulated during EMT (74). HDAC5 was also found to promote doxorubicin-induced EMT in glioma cells, which could account for chemoresistance in glioma (73). On the other hand, it is worth noting that HDAC5 induced EMT but inhibited cell proliferation in urothelial carcinoma (UC) (61).
The basic helix-loop-helix transcription factor Twist1 can induce EMT by negatively regulating the transcription of E-cadherin (75). Twist1 is a downstream target of HDAC5 in osteosarcoma progression (30). In non-small cell lung cancer, HDAC5 activity was shown to induce the expression of EMT-related genes including the transcriptional regulators E2F1, E2F3, and Twist1 (27), providing evidence for its direct involvement in promoting EMT. As dysregulation of energy metabolism also contributes to EMT (76), it is possible that metabolic reprogramming is involved in HDAC5-mediated EMT. It was recently demonstrated that HDAC5 together with HDAC4 enhanced glycolysis by inducing the upregulation of hexokinase 2 (HK2), which was critical for EMT induced by hypoexpression of 5′ AMP-activated protein kinase (AMPK) in lung cancer (72).
Role of HDAC5 in Cancer Proliferation and Apoptosis
The contribution of HDAC5 to cancer cell proliferation has been investigated in many studies. For example, HDAC5 overexpression was shown to promote cell growth, while small interfering RNA (siRNA)-mediated silencing of HDAC5 caused cell cycle arrest at the G0 phase in medulloblastoma (56). In neuroblastoma, HDAC5 promoted cell proliferation but had little effect on cell death (55). Overexpression of HDAC5 increased the proliferation of lung cancer cells, possibly via upregulation of downstream target genes (22). HDAC5 also increased DLL4 expression in colorectal cancer (CRC) cells, thereby enhancing their proliferation (57). HDAC5 induced osteosarcoma cell proliferation in a Twist1-dependent manner, highlighting the varied functions of the HDAC5/Twist1 axis in tumor progression (30). HDAC5 also increased c-Met expression to promote Wilms’ tumor cell proliferation (31). Notch1 signaling plays an important role in cancer cell proliferation (77, 78); HDAC5 was shown to promote Notch1 expression in glioma cells, leading to increased glioma cell proliferation (29). In HCC, HDAC5 activated cell proliferation by inducing Six1 expression, providing the first evidence for the oncogenic role of HDAC5 in HCC development and progression (59). However, HDAC5 also inhibits tumor cell proliferation. For example, elevated levels of HDAC5 in UC cells suppressed cell proliferation, an effect that may involve transforming growth factor β (TGF-β) (61).
Electron microscopy and immunohistochemical analyses of HDAC5 subcellular localization revealed that HDAC5 was associated with heterochromatin in S and G1 phases, suggesting a role in DNA replication and cell cycle progression (79). For example, HDAC5 silencing shielded cell-cycle of prostate cancer cells from RB-mediated repression (80). Knockdown of HDAC5 also induced G1 cell cycle arrest, which hindered breast cancer proliferation (50). Overexrpression of HDAC5 increased S phase of non-small cell lung cancer (NSCLC), which demonstrated that HDAC5 promote NSCLC proliferation through inducing DNA replication (27).
Cancer occurrence and progression are caused not only by abnormal cell proliferation and differentiation, but also by dysregulation of apoptosis. In fact, inducing cell apoptosis has become an important therapeutic strategy in cancer (81), and clarifying the role of cell apoptosis in cancer can provide insight into the molecular basis of tumorigenesis as well as a basis for the development of effective therapies. Recent studies have demonstrated the involvement of HDAC5 in cancer cell apoptosis. HDAC5 knockdown in medulloblastoma cell lines increased caspase-3 activity, which is important for apoptosis induction (56); and upregulation of apoptosis-related proteins and morphologic changes associated with apoptosis were observed in HeLa and MCF-7 cells transfected with a siRNA targeting HDAC5 (79). Overexpression of HDAC5 in HCC cells was correlated with reduced expression of p21 and hyperactivation of cyclin D1 and cyclin-dependent kinase 2/4/6 (CDK2/4/6), indicating that HDAC5 promotes cell cycle progression and blocks apoptosis in HCC tumorigenesis (60). However, in another study, overexpression of HDAC5 in MCF-7 breast cancer cells inhibited proliferation and promoted apoptosis in a p53-independent manner (51). Thus, HDAC5 may have opposite functions depending on the cellular context and interaction partner.
HDAC5 and Immuno-Oncology
Malignant cells can escape immune surveillance and rapidly proliferate to form a tumor. HDACs have been implicated in the immune response (82–84), and the regulation of HDACs in cellular immunity plays an important role in tumorigenesis. HDAC5 interacts with the immune system—including immune cells and inflammatory cytokines—in cancer occurrence or progression. HDAC5 was shown to be involved in macrophage differentiation in lymphoma U937 cells (85), and HDAC5 depletion in U937 cells reduced the levels of tumor necrosis factor α (TNF-α) and monocyte chemoattractant protein 1 (MCP-1) via stimulation of NF-κB activity, suggesting a regulatory function for HDAC5 in the proinflammatory response of macrophages (62). In vitro and in vivo experiments have shown that the suppressor function of regulatory T cells (Tregs) was attenuated in HDAC5−/− mice. Meanwhile, HDAC5 silencing suppressed the switch from CD4+ T cells to Tregs under polarizing conditions and inhibited interferon γ (IFN-γ) production by CD8+ T cells (86). Another study demonstrated that HDAC5 recruited macrophages to the tumor microenvironment through the Suppressor of cytokine signaling 3 (Socs3)/C-C motif chemokine ligand 2 (CCL2) axis and promoted pancreatic cancer via TGF-β-dependent paracrine signaling (63). These findings indicate that targeting HDAC5 is a promising cancer immunotherapy strategy.
HDAC5 in Cancer Cell Differentiation and Stemness
Aberrant differentiation plays an important role in tumorigenesis. Epigenetic modifications including histone deacetylation are involved in cell differentiation (87). As such, clarifying the role and mechanisms of HDAC5 in cellular differentiation may provide a basis for new therapeutic strategies in cancer.
HDAC5 overexpression inhibited murine embryonic kidney(MEK) cell differentiation through transcriptional repression of GATA-1 and prevented the colocalization of the 2 proteins during erythroid differentiation (88). HDAC5 was also observed to block N-myc-mediated differentiation in neuroblastoma cells (55). MEF2 family proteins, which are recruited by HDAC5, include MEF2A, MEF2B, MEF2C, and MEF2D (89). Alternative splicing of MEF2 genes can yield protein isoforms with distinct functions (90). MEF2Cα2 engaged in a weaker interaction with HDAC5 than MEF2Cα1 in rhabdomyosarcoma (RMS) cell lines, which nonetheless enhanced the ability of the MEF2Cα2 isoform to promote RMS differentiation (91).
Cancer stem cells play a key role in tumorigenesis and can significantly influence the response to tumor therapy (92). HDAC5 increased the stemness of human breast cancer stem cells through inhibiting the binding of the Runt-related transcription factor 3(RUNX3)/p300 complex to the promoter of target genes (46). HDAC5 showed elevated expression in tumorspheres formed by H460 lung cancer cells. Application of the HDAC5 inhibitor LMK-235 reduced extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation in a dose-dependent manner, demonstrating that the HDAC5-ERK1/2 axis plays an important role in maintaining the stemness of lung cancer stem cells (93). Additionally, during ovarian cancer (OC) progression, HDAC5 interacted with YY1 to promote OC cell stemness by deacetylating the promoter of the microRNA miR-99a (64). These results imply that HDAC5 regulates cancer cell differentiation and stemness through interaction with different cofactors.
HDAC5 and Drug Resistance
Drug resistance is a major reason for cancer treatment failure. The main mechanisms of resistance are enhanced anti-apoptotic capacity, hyperactivation of cell proliferation, DNA replication, and cell cycle progression (94). Given its important role in cell proliferation, cell cycling, and apoptosis, HDAC5 may play a key role in the development of therapeutic resistance.
HDAC5 knockdown was shown to increase the sensitivity of MCF-7 and HeLa cells to doxorubicin and cisplatin by inducing heterochromatin decondensation (79). HDAC5 was illustrated to be involved in sorafenib resistance of HCC as well (95). Tamoxifen is a first-line treatment for breast cancer, HDAC5 deacethylated SOX9 and made it localized in the nucleus, which was responsible for tamoxifen resistant in breast cancer (52). HDAC5 expression was also elevated in estrogen-independent breast cancer cells and induced tamoxifen resistance via the miR-125a-5p/specificity protein 1 (Sp1)/survivin axis (53). Additionally, formononetin inhibited HDAC5 expression to attenuate doxorubicin-induced EMT, thereby promoting doxorubicin sensitivity in glioma cells (73).
Telomeres are small DNA-protein complexes located at the end of linear chromosomes in eukaryotic cells that maintain chromosome integrity and are involved in cell cycle regulation (96). During tumorigenesis, telomerase stimulates the re-synthesis of telomeric DNA, allowing cancer cells to proliferate continuously, which reduces the efficacy of chemotherapy (97). Telomeres are subject to various epigenetic modifications (98–100). HDAC5 preferentially localized at long telomeres and maintains their length, and HDAC5 silencing was found to increase the sensitivity of osteosarcoma and fibrosarcoma cells with long telomeres to chemotherapy (58).
HDAC5 and Oncogenetic Signaling Pathways
Studies have indicated that HDAC5 participated in cancer progression through various oncogenetic signaling pathways, such as TAp63/maspin (21), HIPK2/HIF1α (70), and p65/NF-κB pathways (95)(Figure 4).
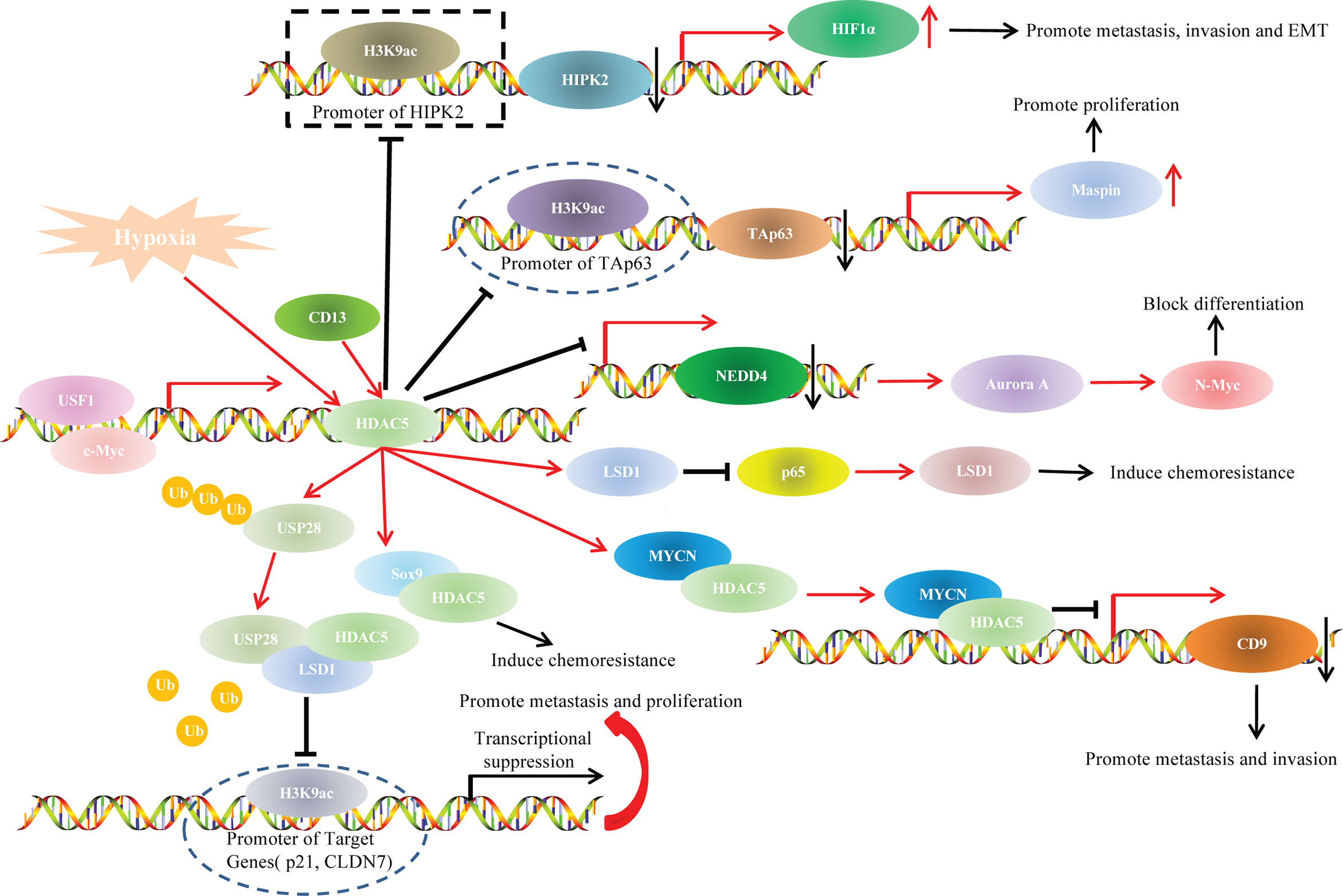
Figure 4 DAC5 and oncogenic signaling pathways.
HDAC5 and TAp63/maspin Signaling
P63 is a new member of the p53 gene family. Although its structure is highly homologous to p53, p63 encodes a special C-terminal sequence, which contains sterile α group and a transcriptional inhibition domain. The C-terminus can bind to lipid membranes and inhibit transcription (101). Up to 14 p63 homologs have been identified, which are produced by transcription from different promoters and selective cleavage. The three p63 homologs that are produced through alternative splicing of mRNA transcribed from the promoter upstream of intron 1 are named TAp63α, TAp63β and TAp63γ because they contain a transcription domain (TA) (102). Many studies have demonstrated that these homologs are important transcriptional activators of mammary serine protease inhibitor (Maspin) (103, 104). Maspin (SERPINB5), a member of the serine protease inhibitor superfamily, is not only a serine protease inhibitor gene, but also a new soluble protein that is found in the cytoplasm. It can inhibit angiogenesis, increase cell adhesion, induce apoptosis and inhibit growth and metastasis of cancer cells (105, 106).
In our previous study, we explored the role of HDAC5 in HCC and found that HDAC5 overexpression induced HCC proliferation and tumorigenesis in vitro. We also knocked down HDAC5 in Hep3B (p53-null) and HepG2 (p53-WT) cells and investigated the expression levels of p53 family members. The results indicated that HDAC5 downregulated p53, TAp63, and p73 at both the mRNA and protein levels. In addition, TAp63 gradually decreased with HDAC5 overexpression in immortalized liver cell line (THLE-3 cells). Chromatin immunoprecipitation (ChIP) showed that HDAC5 knockdown increased the acetylation at the TAp63 promoter (i.e., at Lys9 of histone H3 [H3K9ac]), which indicated the suppressive effect of HDAC5 on TAp63 transcription. Rescue experiments showed that HDAC5 promoted proliferation and tumorigenesis via the Tap63/Maspin axis. In summary, our findings demonstrated that HDAC5 knockdown increased acetylation at the TAp63 promoter, resulting in TAp63 hyperactivation and Maspin overexpression, which blocked HCC cell proliferation and tumorigenesis (21).
HDAC5 and HIPK2/HIF1α Signaling
Hypoxia plays an important role in tumorigenesis and metastasis. Hypoxia inducible factor 1 (HIF1) is a key transcription factor that can transmit the hypoxia signal and mediate the effect of hypoxia. HIF1α is a functional subunit of HIF-1. Under hypoxia, HIF1α activates downstream target genes through interacting with hypoxia-response elements, which thereby regulate proliferation, apoptosis, invasion, metastasis, angiogenesis, energy metabolism, and chemoradiotherapy resistance of cancer cells (107).
Epithelial-to-mesenchymal transition (EMT) is a process that occurs under normal physiologic conditions such as embryonic development and wound healing. During EMT, epithelial cells lose their polarity and acquire mesenchymal-like features along with the capacity to migrate and invade other tissues (108). An increasing number of studies have reported that the change in the oxygen level in cancer microenvironments and the HIF1α-induced hypoxia signal transduction pathway acted as a vital regulator of EMT, which played a key role in hypoxia-induced cancer invasion and metastasis (109, 110).
In our previous study, we illustrated that HDAC5 induced HCC migration, invasion and EMT only under hypoxia but not under normoxia. Although a dual luciferase reporter assay indicated increased transcription of HIF1α after ectopic HDAC5 expression under hypoxia, HDAC5 did not transactivate HIF1α directly even if under hypoxia. Further mechanistic analysis using ChIP assays unveiled that, under hypoxia, HDAC5 was recruited to the HIPK2 promoter; HDAC5 knockdown increased H3K9ac modification at this promoter, and the resultant upregulated HIPK2 bound to the HIF1α promoter to inhibit HIF1α expression. Our findings revealed that the HDAC5/HIPK2/HIF1α axis contributed to hypoxia-induced aggressiveness of HCC (70).
HDAC5 and LSD1 Signaling Pathways
Lysine-specific histone demethylase 1 (LSD1), also known as KDM1A or AOF2, is the first identified histone specific demethylase (111). LSD1 exhibits varying functions related to cancer progression in different cancer types, as the LSD1 protein is methylated in some cancer type and demethylated in others (112). HDAC5 was positively associated with LSD1 levels in breast cancer cells and tissue specimens. In vitro experiments unveiled that HDAC5 promoted invasion, metastasis and tumorigenic development via LSD1. Co-immunoprecipitation (Co-IP) and deletion mapping studies demonstrated that the NLS element was responsible for the HDAC5-LSD1 interaction. Protein ubiquitination assays indicated that HDAC5 stabilized LSD1 through blocking USP28 polyubiquitination. These results suggest an important role for the HDAC5/LSD1 axis in tumorigenesis (50).
In addition, HDAC5 also functioned as a downstream target gene of various upstream factors and participated in the regulatory pathways during cancer progression. For example, USF1 transactivated HDAC5 by physically binding to the HDAC5 promoter at −356 to −100 bp, thereby increasing the stability of LSD1 to induce chemoresistance of breast cancer (113).
HDAC5 and p65/NF-κB Signaling
NF-κB is a transcription factor involved in cell proliferation and transformation, apoptosis, immune responses and other important biological activities (114). NF-κB protein usually forms homodimer/heterodimer from p65 and p50, and is inactivated in cytoplasm due to the formation of trimer complex with inhibitor IKB (115). Therefore, the correlation between HDAC5 and p65/NF-κBsignaling during tumor progression has drawn researchers’ attention. Hu et al. illustrated that CD13 promoted sorafenib resistance in HCC via indirectly increasing p65 protein stability. Co-IP indicated a strong interaction between CD13 and HDAC5. Truncation assays demonstrated that the N-terminal region of HDAC5 protein (amino acids 1-684), and the N-terminal cytoplasmic domain of CD13 (amino acids 2-8) accounted for the HDAC5-CD13 interaction. Hence, CD13 interacted with HDAC5 to increase the stabilization of LSD1, which demethylated p65 (50, 116) and thereby stabilized it and increased p65/NF-κB signaling; this resulted in sorafenib resistance and HCC progression (95).
HDAC5 and Myc Family
The Myc gene family is one of the most widely studied family of nuclear oncogenes. This family includes three main members: c-myc, N-myc and L-myc. N-myc protein can be stabilized with phosphorylated at serine 62 (Ser62) or be degraded with phosphorylated at threonine 58 (Thr58) (117). HDAC5 knockdown reduced N-myc at protein levels but had little effect on N-myc mRNA levels, so HDAC5 upregulated N-myc at the posttranscriptional level. HDAC5 overexpression significantly upregulated total N-myc protein in cells with Ser62 mutant N-myc but not in cells with Thr58-mutant N-myc. This indicated that HDAC5 upregulates N-myc protein by increasing N-Myc protein stability. Microarray and ChIP assays revealed that HDAC5 knockdown led to the transactivation of NEDD4 (an E3 ubiquitin–protein ligase). This illustrated that HDAC5 stabilized N-myc protein through suppressing the transcription of NEDD4 (55). Interestingly, HDAC5 was also shown to interact with N-myc protooncogene protein (MYCN), and the complex colocalized to the CD9 promoter and attenuated CD9 expression in neuroblastoma cells, leading to tumor cell invasion and metastasis (54).
In addition, C-myc transactivited HDAC5 though directly binding to the −200/+20 bp region upstream of the transcription start site of HDAC5, which contained C-myc-specific binding sequence (CACGTG). In turn, HDAC5 promoted SOX9 nuclear localization via interaction with its HMGB domain (amino acids 1–181) in estrogen receptor-positive breast cancer cells. These results indicated that the C-myc/HDAC5/SOX9 axis is a potential target for estrogen receptor-positive breast cancer therapy (52).
Clinical Significance of HDAC5 in Cancer
HDAC5 and Cancer Diagnosis
Cancer cells release proteins into the peripheral blood during tumorigenesis. Thus, circulating proteins can serve as prognostic biomarkers for predicting disease recurrence and informing treatment decisions (118). Circulating HDAC5 is a potential marker for cancer diagnosis, given that it is detected at different levels in the peripheral blood of cancer patients as compared to healthy subjects or patients with nonrecurrent disease. One study found that the specificity of HDAC5 in distinguishing CRC patients from healthy subjects was 96.3%, indicating that it can be a diagnostic biomarker for CRC (32). Proteomic analysis with an antibody array identified proteins that were differentially expressed between breast cancer patients with and those without recurrence (34); moreover, HDAC5 was detected at a significantly higher level in the blood in patients with recurrent breast cancer than in those with recurrent triple-negative breast cancer.
Tumor antigens can provide insight into antitumor immune responses and the mechanisms of immune escape used by cancer cells, which can aid cancer diagnosis and the establishment of immunotherapy strategies. An antibody reactivity screen revealed HDAC5 as the only serum antigen that differed between CRC patients and healthy subjects (33). Thus, serum HDAC5 has clinical utility as a diagnostic biomarker for cancer.
HDAC5 and Cancer Therapy
HDAC5 and HDAC Inhibitors
Molecular targeted therapies have specific antitumor effects, which limits their toxicity. Some studies demonstrated that HDAC5 is a potential therapeutic target for HDAC inhibitors (HDACis) as anticancer drugs (17, 25, 46, 73, 74, 119–123)(Table 4). HDACis can be divided into 4 groups according to their chemical structure—namely, hydroxamic acids, benzamides, cyclic peptides, and carboxylic acids. The basic structural features of HDACis are a hydroxamic acid complexed with Zn2+, intermediate fatty chain linker, and lipophilic cap that engages in hydrophobic interaction with the binding pocket (127). HDACis could be divided into pan-HDACis and selective class IIA HDACis.
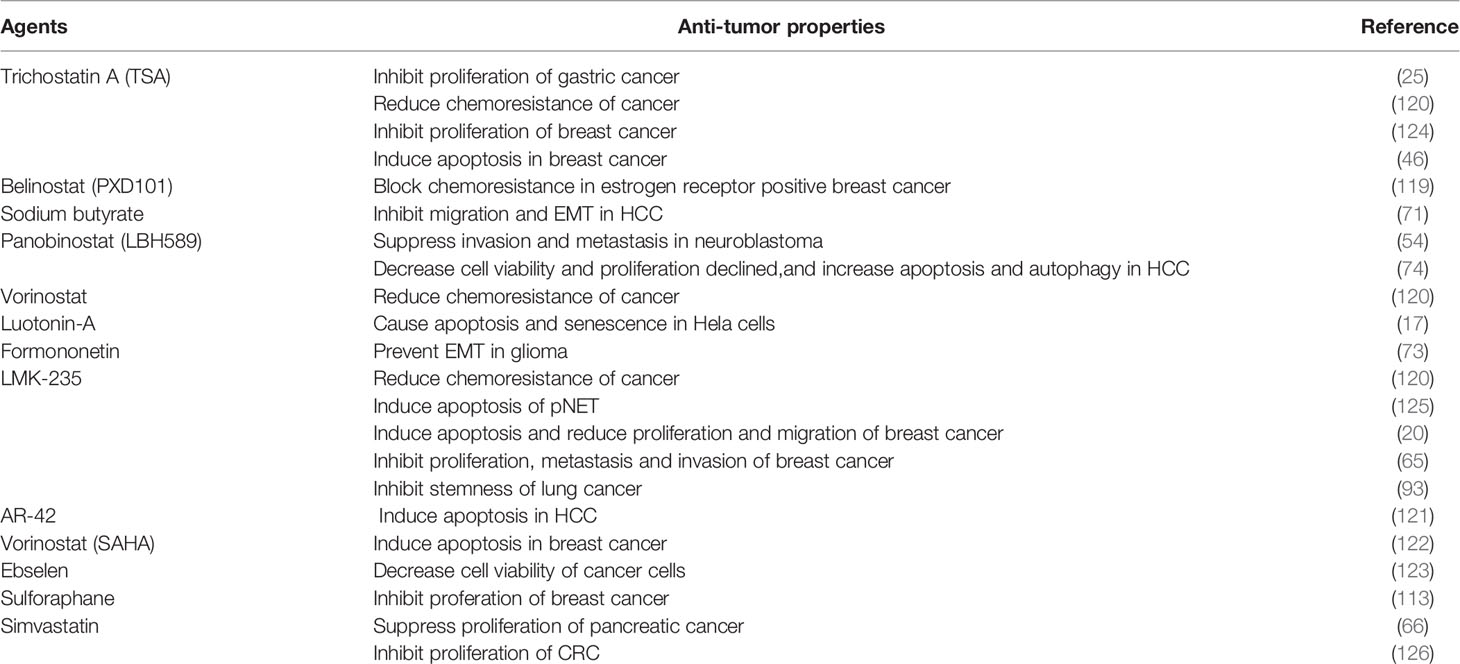
Table 4 Summary of HDAC5-targeted drugs in cancer therapy.
Trichostatin A (TSA)is the first identified pan-HDACis with an inhibitory activity depends on Zn2+-dependent HDAC enzymes. The crystal structure of TSA and HDAC analogues showed that the conserved deacetylase active center were made up by a tubular bag, a zinc binding region and two ASP his (aspartate histidine) charge relay networks. The long fatty chain of TSA bound to the inner part of the tubular bag, and the hydroxime group at the end of the fatty chain showed connection with the zinc binding region through carbonyl and hydroxyl groups, which thereby inhibiting the ability of HDACs including recognizing substrate, regulating enzymatic activity and interacting with other proteins (128). TSA was illustrated to inhibit HDAC5 to block malignant behavior of cancer cells, including chemoresistance (120), proliferation (124) and invasion (25). Other HDACis such as belinostat, givenostat, panobinostat and dacinostat have the same mechanism with TSA as well. Among them, belinostat(PXD101) and panobinostat(LBH589) were US FDA-approved for cancer therapy (129, 130). Belinostat has been revealed to attenuate chemoresistance in estrogen receptor positive breast cancer via targeting class IIA HDACs including HDAC5 (119). Besides, HDAC3 and HDAC5 had copy number gain during HCC progression, panobinostat might inhibit HDAC3 and HDAC5 to decrease proliferation and induce apoptosis and autophagy of HCC, and this anticancer effect could be augmented by combination therapy with panobinostat and sorafenib (74).
Vorinostat(SAHA) is one of HDACis approved by FDA for cancer therapy. The hydroxime group of SAHA, also bound to Zn2+ in the way similar with TSA, but the fatty chain of SAHA showed less connection to the tubular bag than that of TSA, which indicated that the inhibitory activity of SAHA is lower than TSA (131). Hence, combination therapy with SAHA and other therapeutic drugs has drawn researchers’ attention. Vasilatos et al. (122) explored the role of pargyline (an LSD1 inhibitor) and SAHA in breast cancer, the results demonstrated that both pargyline and SAHA suppressed cell proliferation and induced apoptosis through inhibiting HDAC5. Meanwhile, combination therapy with pargyline and SAHA led to superior anticancer effect. Interestingly, this anticancer effect was not found in non-TNBC counterparts or non-tumorigenic breast cells, which illustrated therapeutic efficacy of SAHA on breast cancer was subtype-dependent.
LMK-235, a selective HDACi showed preferable interactions with the catalytic zinc ion of class IIA HDACs, was illustrated to decrease phosphohistone H3 and Ki-67 levels in pancreatic neuroendocrine tumors (pNETs) while inducing pNET cell apoptosis and histone H3 acetylation, making it an effective targeted therapy for pNET that indirectly antagonizes the activity of HDAC4/5 (125). Similar findings were reported in breast (20, 65) and lung (93) cancers. However, LMK-235 induced HDAC5 expression in UC cells, indicating that HDAC5 is not a suitable therapeutic target in UC (132). Cao et al. (113) selected a series of HDACis including SAHA, TSA, LBH-589, PXD-101, MS-275, MC-1568, Romidepsin and Sulforaphane (SFN) to test their inhibitory effect towards HDAC5 expression on breast cancer cells. The results illustrated that SFN showed most significant inhibitory effect on HDAC5 expression at both mRNA level and protein level. Dual luciferase reporter assay indicated that SFN hindered the transcriptional activity of HDAC5 via affecting the region from −356 to −100 bp at HDAC5 promoter. In addition, USF1 could block the downregulation of HDAC5 mediated by SFN. These finding suggested SFN as a potential drug for breast cancer therapy.
HDAC5 and Metabolism Inhibitors
Hendrick et al. (133) demonstrated that knock down of HDAC5 induced cancer apoptosis through an iron-dependent reactive oxygen species (ROS) production. In addition, HDAC5-knock down cells adapted oxidative stress through glucose and glutamine metabolic reprogramming. Therefore, blocking glucose and glutamine metabolism in HDAC5-knock down cancer cells significantly induced cell death, which provided insight into a combination therapy with HDAC5 inhibitors and various inhibitors of metabolism as a new strategy for cancer treatment.
During tumorigenesis, cancer cells often undergone deregulated lipid metabolism (134). Statins, a class of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, can modulate the cell cycle, signal transduction pathways, and angiogenesis, and thus have therapeutic potential in cancer treatment (135–139). Simvastatin was shown to attenuate pancreatic cancer growth by inhibiting the oxysterol binding-related protein 5 (ORP5)/HDAC5 axis (66). Additionally, the combination of statins and other therapeutics was shown to prolong survival in cancer patients (140). For example, statins inhibited CRC cell proliferation by modulating the expression of enhancer of zeste homolog 2 (EZH2) and HDAC5, an effect that was enhanced in the presence of MC-1568, a class II HDACi. The underlying mechanism involved the upregulation of p27(KIP1) by statin via inhibition of EZH2; MC-1568 further increased p27(KIP1) expression by suppressing HDAC5, resulting in an antiproliferative effect in CRC (126).
Collectively, these findings highlight the clinical significance and utility of HDAC5 in targeted cancer therapy.
Conclusion
Accumulating evidence indicates that HDAC5 can serve as a biomarker for tumorigenesis in a variety of cancer types. HDAC5 expression is correlated with clinicopathologic features of cancer patients, highlighting its clinical value. Elucidating the targets and mechanisms of action of HDAC5 will enhance our understanding of the molecular basis of tumorigenesis and provide novel markers for early diagnosis, treatment response monitoring, and predicting disease prognosis, as well as an empirical basis for the development of effective cancer treatments.
Author Contributions
XX and MY contributed to the study conception and design. XX edited the manuscript language. JY and CG wrote the main manuscript text and prepared the figures and tables. QK, ZF, and XC provided advice regarding the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was mainly supported by the Natural Science Foundation of Zhejiang Province (LQ21H160009), the Medical and Health Research Project of Zhejiang Province (2019RC175), Science and Technology Plan Project of Taizhou (1902KY193), The Applied Basic Research Plan of Xuzhou (KH17043) and 2016 Joint Funds of Zunyi city and school (E-214). We thank Charlesworth for the English language editing of the article.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.661620/full#supplementary-material
Supplementary Table 1 | microRNAs predicted to regulate HDAC5.
References
1. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation Of Histones And Their Possible Role In the Regulation of RNA Synthesis. Proc Natl Acad Sci U S A (1964) 51:786–94. doi: 10.1073/pnas.51.5.786
2. Gray SG, Ekstrom TJ. The Human Histone Deacetylase Family. Exp Cell Res (2001) 262(2):75–83. doi: 10.1006/excr.2000.5080
3. Marks PA, Miller T, Richon VM. Histone Deacetylases. Curr Opin Pharmacol (2003) 3(4):344–51. doi: 10.1016/s1471-4892(03)00084-5
4. Marmorstein R, Roth SY. Histone Acetyltransferases: Function, Structure, and Catalysis. Curr Opin Genet Dev (2001) 11(2):155–61. doi: 10.1016/s0959-437x(00)00173-8
5. Roth SY, Denu JM, Allis CD. Histone Acetyltransferases. Annu Rev Biochem (2001) 70:81–120. doi: 10.1146/annurev.biochem.70.1.81
6. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone Deacetylases (HDACs): Characterization of the Classical HDAC Family. Biochem J (2003) 370(Pt 3):737–49. doi: 10.1042/BJ20021321
7. Gregoretti IV, Lee YM, Goodson HV. Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J Mol Biol (2004) 338(1):17–31. doi: 10.1016/j.jmb.2004.02.006
8. Haberland M, Montgomery RL, Olson EN. The Many Roles of Histone Deacetylases in Development and Physiology: Implications for Disease and Therapy. Nat Rev Genet (2009) 10(1):32–42. doi: 10.1038/nrg2485
9. Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of Skeletal Myogenesis by Association of the MEF2 Transcription Factor With Class II Histone Deacetylases. Mol Cell (2000) 6(2):233–44. doi: 10.1016/s1097-2765(00)00025-3
10. Pita-Thomas W, Mahar M, Joshi A, Gan D, Cavalli V. HDAC5 Promotes Optic Nerve Regeneration by Activating the mTOR Pathway. Exp Neurol (2019) 317:271–83. doi: 10.1016/j.expneurol.2019.03.011
11. Wei JY, Lu QN, Li WM, He W. Intracellular Translocation of Histone Deacetylase 5 Regulates Neuronal Cell Apoptosis. Brain Res (2015) 1604:15–24. doi: 10.1016/j.brainres.2015.01.043
12. Seo WD, Lee JH, Jia Y, Wu C, Lee SJ. Saponarin Activates AMPK in a Calcium-Dependent Manner and Suppresses Gluconeogenesis and Increases Glucose Uptake Via Phosphorylation of CRTC2 and HDAC5. Bioorg Medicinal Chem Lett (2015) 25(22):5237–42. doi: 10.1016/j.bmcl.2015.09.057
13. Kain V, Kapadia B, Viswakarma N, Seshadri S, Prajapati B, Jena PK, et al. Co-Activator Binding Protein PIMT Mediates TNF-alpha Induced Insulin Resistance in Skeletal Muscle Via the Transcriptional Down-Regulation of MEF2A and GLUT4. Sci Rep (2015) 5:15197. doi: 10.1038/srep15197
14. Verdel A, Khochbin S. Identification of a New Family of Higher Eukaryotic Histone Deacetylases. Coordinate Expression of Differentiation-Dependent Chromatin Modifiers. J Biol Chem (1999) 274(4):2440–5. doi: 10.1074/jbc.274.4.2440
15. Johnstone RW. Histone-Deacetylase Inhibitors: Novel Drugs for the Treatment of Cancer. Nat Rev Drug Discovery (2002) 1(4):287–99. doi: 10.1038/nrd772
16. Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, et al. Selective Class IIa Histone Deacetylase Inhibition Via A Nonchelating Zinc-Binding Group. Nat Chem Biol (2013) 9(5):319–25. doi: 10.1038/nchembio.1223
17. Venkatesh R, Ramaiah MJ, Gaikwad HK, Janardhan S, Bantu R, Nagarapu L, et al. Luotonin-a Based Quinazolinones Cause Apoptosis and Senescence Via HDAC Inhibition and Activation of Tumor Suppressor Proteins in HeLa Cells. Eur J Medicinal Chem (2015) 94:87–101. doi: 10.1016/j.ejmech.2015.02.057
18. Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, et al. Enzymatic Activity Associated With Class II HDACs Is Dependent on a Multiprotein Complex Containing HDAC3 and SMRT/N-Cor. Mol Cell (2002) 9(1):45–57. doi: 10.1016/s1097-2765(01)00429-4
19. Patani N, Jiang WG, Newbold RF, Mokbel K. Histone-Modifier Gene Expression Profiles are Associated With Pathological and Clinical Outcomes in Human Breast Cancer. Anticancer Res (2011) 31(12):4115–25.
20. Oltra SS, Cejalvo JM, Tormo E, Albanell M, Ferrer A, Nacher M, et al. HDAC5 Inhibitors as a Potential Treatment in Breast Cancer Affecting Very Young Women. Cancers (2020) 12(2). doi: 10.3390/cancers12020412
21. Gu H, Fang Z, Cai X, Song R, Lin M, Ye J, et al. Highly Expressed Histone Deacetylase 5 Promotes the Growth of Hepatocellular Carcinoma Cells by Inhibiting the TAp63-maspin Pathway. Am J Cancer Res (2018) 8(3):462–75.
22. Zhong L, Sun S, Yao S, Han X, Gu M, Shi J. Histone Deacetylase 5 Promotes the Proliferation and Invasion of Lung Cancer Cells. Oncol Rep (2018) 40(4):2224–32. doi: 10.3892/or.2018.6591
23. Klieser E, Urbas R, Stattner S, Primavesi F, Jager T, Dinnewitzer A, et al. Comprehensive Immunohistochemical Analysis of Histone Deacetylases in Pancreatic Neuroendocrine Tumors: HDAC5 as a Predictor of Poor Clinical Outcome. Hum Pathol (2017) 65:41–52. doi: 10.1016/j.humpath.2017.02.009
24. Stypula-Cyrus Y, Damania D, Kunte DP, Cruz MD, Subramanian H, Roy HK, et al. HDAC Up-Regulation in Early Colon Field Carcinogenesis is Involved in Cell Tumorigenicity Through Regulation of Chromatin Structure. PloS One (2013) 8(5):e64600. doi: 10.1371/journal.pone.0064600
25. Lee KH, Choi EY, Kim MK, Kim KO, Jang BI, Kim SW, et al. Inhibition of Histone Deacetylase Activity Down-Regulates Urokinase Plasminogen Activator and Matrix Metalloproteinase-9 Expression in Gastric Cancer. Mol Cell Biochem (2010) 343(1-2):163–71. doi: 10.1007/s11010-010-0510-x
26. Orenay-Boyacioglu S, Kasap E, Gerceker E, Yuceyar H, Demirci U, Bilgic F, et al. Expression Profiles of Histone Modification Genes in Gastric Cancer Progression. Mol Biol Rep (2018) 45(6):2275–82. doi: 10.1007/s11033-018-4389-z
27. Liu C, Lv D, Li M, Zhang X, Sun G, Bai Y, et al. Hypermethylation of miRNA-589 Promoter Leads to Upregulation of HDAC5 Which Promotes Malignancy in Non-Small Cell Lung Cancer. Int J Oncol (2017) 50(6):2079–90. doi: 10.3892/ijo.2017.3967
28. Liu J, Gu J, Feng Z, Yang Y, Zhu N, Lu W, et al. Both HDAC5 and HDAC6 are Required for the Proliferation and Metastasis of Melanoma Cells. J Trans Med (2016) 14:7. doi: 10.1186/s12967-015-0753-0
29. Liu Q, Zheng JM, Chen JK, Yan XL, Chen HM, Nong WX, et al. Histone Deacetylase 5 Promotes the Proliferation of Glioma Cells by Upregulation of Notch 1. Mol Med Rep (2014) 10(4):2045–50. doi: 10.3892/mmr.2014.2395
30. Chen J, Xia J, Yu YL, Wang SQ, Wei YB, Chen FY, et al. HDAC5 Promotes Osteosarcoma Progression by Upregulation of Twist 1 Expression. Tumour Biol J Int Soc Oncodevelop Biol Med (2014) 35(2):1383–7. doi: 10.1007/s13277-013-1189-x
31. Cao X, Liu DH, Zhou Y, Yan XM, Yuan LQ, Pan J, et al. Histone Deacetylase 5 Promotes Wilms’ Tumor Cell Proliferation Through the Upregulation of C-Met. Mol Med Rep (2016) 13(3):2745–50. doi: 10.3892/mmr.2016.4828
32. Chan CC, Fan CW, Kuo YB, Chen YH, Chang PY, Chen KT, et al. Multiple Serological Biomarkers for Colorectal Cancer Detection. Int J Cancer (2010) 126(7):1683–90. doi: 10.1002/ijc.24912
33. Scanlan MJ, Welt S, Gordon CM, Chen YT, Gure AO, Stockert E, et al. Cancer-Related Serological Recognition of Human Colon Cancer: Identification of Potential Diagnostic and Immunotherapeutic Targets. Cancer Res (2002) 62(14):4041–7.
34. Bera A, Russ E, Srinivasan M, Eidelman O, Eklund M, Hueman M, et al. Proteomic Analysis of Inflammatory Biomarkers Associated With Breast Cancer Recurrence. Military Med (2020) 185(Suppl 1):669–75. doi: 10.1093/milmed/usz254
35. Greco TM, Yu F, Guise AJ, Cristea IM. Nuclear Import of Histone Deacetylase 5 by Requisite Nuclear Localization Signal Phosphorylation. Mol Cell Proteomics MCP (2011) 10(2):M110 004317. doi: 10.1074/mcp.M110.004317
36. Haworth RS, Stathopoulou K, Candasamy AJ, Avkiran M. Neurohormonal Regulation of Cardiac Histone Deacetylase 5 Nuclear Localization by Phosphorylation-Dependent and Phosphorylation-Independent Mechanisms. Circ Res (2012) 110(12):1585–95. doi: 10.1161/CIRCRESAHA.111.263665
37. Mishra S, Gray CB, Miyamoto S, Bers DM, Brown JH. Location Matters: Clarifying the Concept of Nuclear and Cytosolic CaMKII Subtypes. Circ Res (2011) 109(12):1354–62. doi: 10.1161/CIRCRESAHA.111.248401
38. Fu X, Zhao JX, Liang J, Zhu MJ, Foretz M, Viollet B, et al. AMP-Activated Protein Kinase Mediates Myogenin Expression and Myogenesis Via Histone Deacetylase 5. Am J Physiol Cell Physiol (2013) 305(8):C887–95. doi: 10.1152/ajpcell.00124.2013
39. He T, Huang J, Chen L, Han G, Stanmore D, Krebs-Haupenthal J, et al. Cyclic AMP Represses Pathological MEF2 Activation by Myocyte-Specific Hypo-Phosphorylation of HDAC5. J Mol Cell Cardiol (2020) 145:88–98. doi: 10.1016/j.yjmcc.2020.05.018
40. Harrison BC, Huynh K, Lundgaard GL, Helmke SM, Perryman MB, McKinsey TA. Protein Kinase C-Related Kinase Targets Nuclear Localization Signals in a Subset of Class IIa Histone Deacetylases. FEBS Lett (2010) 584(6):1103–10. doi: 10.1016/j.febslet.2010.02.057
41. Deng X, Ewton DZ, Mercer SE, Friedman E. Mirk/dyrk1B Decreases the Nuclear Accumulation of Class II Histone Deacetylases During Skeletal Muscle Differentiation. J Biol Chem (2005) 280(6):4894–905. doi: 10.1074/jbc.M411894200
42. Li H, Xie H, Liu W, Hu R, Huang B, Tan YF, et al. A Novel microRNA Targeting HDAC5 Regulates Osteoblast Differentiation in Mice and Contributes to Primary Osteoporosis in Humans. J Clin Invest (2009) 119(12):3666–77. doi: 10.1172/JCI39832
43. Roccaro AM, Sacco A, Jia X, Azab AK, Maiso P, Ngo HT, et al. microRNA-dependent Modulation of Histone Acetylation in Waldenstrom Macroglobulinemia. Blood (2010) 116(9):1506–14. doi: 10.1182/blood-2010-01-265686
44. Gu X, Fu C, Lin L, Liu S, Su X, Li A, et al. miR-124 and miR-9 Mediated Downregulation of HDAC5 Promotes Neurite Development Through Activating MEF2C-GPM6A Pathway. J Cell Physiol (2018) 233(1):673–87. doi: 10.1002/jcp.25927
45. Dong N, Xu B, Shi H, Tang X. Baicalein Inhibits Amadori-Glycated Albumin-Induced MCP-1 Expression in Retinal Ganglion Cells Via a MicroRNA-124-Dependent Mechanism. Invest Ophthalmol Visual Sci (2015) 56(10):5844–53. doi: 10.1167/iovs.15-17444
46. Hsieh TH, Hsu CY, Tsai CF, Long CY, Wu CH, Wu DC, et al. HDAC Inhibitors Target HDAC5, Upregulate microRNA-125a-5p, and Induce Apoptosis in Breast Cancer Cells. Mol Ther J Am Soc Gene Ther (2015) 23(4):656–66. doi: 10.1038/mt.2014.247
47. Shi L, Tian Z, Fu Q, Li H, Zhang L, Tian L, et al. Mir-217-Regulated MEF2D-HDAC5/ND6 Signaling Pathway Participates in the Oxidative Stress and Inflammatory Response After Cerebral Ischemia. Brain Res (2020) 1739:146835. doi: 10.1016/j.brainres.2020.146835
48. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell (2011) 146(3):353–8. doi: 10.1016/j.cell.2011.07.014
49. Xu CL, Sang B, Liu GZ, Li JM, Zhang XD, Liu LX, et al. SENEBLOC, a Long non-Coding RNA Suppresses Senescence Via p53-Dependent and Independent Mechanisms. Nucleic Acids Res (2020) 48(6):3089–102. doi: 10.1093/nar/gkaa063
50. Cao C, Vasilatos SN, Bhargava R, Fine JL, Oesterreich S, Davidson NE, et al. Functional Interaction of Histone Deacetylase 5 (HDAC5) and Lysine-Specific Demethylase 1 (LSD1) Promotes Breast Cancer Progression. Oncogene (2017) 36(1):133–45. doi: 10.1038/onc.2016.186
51. Huang Y, Tan M, Gosink M, Wang KK, Sun Y. Histone Deacetylase 5 is Not a p53 Target Gene, But its Overexpression Inhibits Tumor Cell Growth and Induces Apoptosis. Cancer Res (2002) 62(10):2913–22.
52. Xue Y, Lian W, Zhi J, Yang W, Li Q, Guo X, et al. HDAC5-Mediated Deacetylation and Nuclear Localisation of SOX9 is Critical for Tamoxifen Resistance in Breast Cancer. Br J Cancer (2019) 121(12):1039–49. doi: 10.1038/s41416-019-0625-0
53. Huang WT, Tsai YH, Chen SH, Kuo CW, Kuo YL, Lee KT, et al. HDAC2 and HDAC5 Up-Regulations Modulate Survivin and Mir-125a-5p Expressions and Promote Hormone Therapy Resistance in Estrogen Receptor Positive Breast Cancer Cells. Front Pharmacol (2017) 8:902. doi: 10.3389/fphar.2017.00902
54. Fabian J, Opitz D, Althoff K, Lodrini M, Hero B, Volland R, et al. MYCN and HDAC5 Transcriptionally Repress CD9 to Trigger Invasion and Metastasis in Neuroblastoma. Oncotarget (2016) 7(41):66344–59. doi: 10.18632/oncotarget.11662
55. Sun Y, Liu PY, Scarlett CJ, Malyukova A, Liu B, Marshall GM, et al. Histone Deacetylase 5 Blocks Neuroblastoma Cell Differentiation by Interacting With N-Myc. Oncogene (2014) 33(23):2987–94. doi: 10.1038/onc.2013.253
56. Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, et al. HDAC5 and HDAC9 in Medulloblastoma: Novel Markers for Risk Stratification and Role in Tumor Cell Growth. Clin Cancer Res an Off J Am Assoc Cancer Res (2010) 16(12):3240–52. doi: 10.1158/1078-0432.CCR-10-0395
57. He P, Liang J, Shao T, Guo Y, Hou Y, Li Y. HDAC5 Promotes Colorectal Cancer Cell Proliferation by Up-Regulating DLL4 Expression. Int J Clin Exp Med (2015) 8(4):6510–6.
58. Novo CL, Polese C, Matheus N, Decottignies A, Londono-Vallejo A, Castronovo V, et al. A New Role for Histone Deacetylase 5 in the Maintenance of Long Telomeres. FASEB J Off Publ Fed Am Societies Exp Biol (2013) 27(9):3632–42. doi: 10.1096/fj.12-224204
59. Feng GW, Dong LD, Shang WJ, Pang XL, Li JF, Liu L, et al. HDAC5 Promotes Cell Proliferation in Human Hepatocellular Carcinoma by Up-Regulating Six1 Expression. Eur Rev Med Pharmacol Sci (2014) 18(6):811–6.
60. Fan J, Lou B, Chen W, Zhang J, Lin S, Lv FF, et al. Down-Regulation of HDAC5 Inhibits Growth of Human Hepatocellular Carcinoma by Induction of Apoptosis and Cell Cycle Arrest. Tumour Biol J Int Soc Oncodevelop Biol Med (2014) 35(11):11523–32. doi: 10.1007/s13277-014-2358-2
61. Jaguva Vasudevan AA, Hoffmann MJ, Beck MLC, Poschmann G, Petzsch P, Wiek C, et al. Hdac5 Expression in Urothelial Carcinoma Cell Lines Inhibits Long-Term Proliferation But Can Promote Epithelial-to-Mesenchymal Transition. Int J Mol Sci (2019) 20(9):2135. doi: 10.3390/ijms20092135
62. Poralla L, Stroh T, Erben U, Sittig M, Liebig S, Siegmund B, et al. Histone Deacetylase 5 Regulates the Inflammatory Response of Macrophages. J Cell Mol Med (2015) 19(9):2162–71. doi: 10.1111/jcmm.12595
63. Hou P, Kapoor A, Zhang Q, Li J, Wu CJ, Li J, et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discovery (2020) 10(7):1058–77. doi: 10.1158/2159-8290.CD-19-0597
64. Qian S, Wang W, Li M. Transcriptional Factor Yin Yang 1 Facilitates the Stemness of Ovarian Cancer Via Suppressing miR-99a Activity Through Enhancing its Deacetylation Level. Biomed Pharmacother = Biomed Pharmacother (2020) 126:110085. doi: 10.1016/j.biopha.2020.110085
65. Li A, Liu Z, Li M, Zhou S, Xu Y, Xiao Y, et al. HDAC5, a Potential Therapeutic Target and Prognostic Biomarker, Promotes Proliferation, Invasion and Migration in Human Breast Cancer. Oncotarget (2016) 7(25):37966–78. doi: 10.18632/oncotarget.9274
66. Ishikawa S, Nagai Y, Masuda T, Koga Y, Nakamura T, Imamura Y, et al. The Role of Oxysterol Binding Protein-Related Protein 5 in Pancreatic Cancer. Cancer Sci (2010) 101(4):898–905. doi: 10.1111/j.1349-7006.2009.01475.x
67. Gong S, Ying L, Fan Y, Sun Z. Fentanyl Inhibits Lung Cancer Viability and Invasion Via Upregulation of miR-331-3p and Repression of HDAC5. OncoTargets Ther (2020) 13:13131–41. doi: 10.2147/OTT.S281095
68. Huang H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors (2018) 18(10):3249. doi: 10.3390/s18103249
69. Scheau C, Badarau IA, Costache R, Caruntu C, Mihai GL, Didilescu AC, et al. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal Cell Pathol (2019) 2019:9423907. doi: 10.1155/2019/9423907
70. Ye M, Fang Z, Gu H, Song R, Ye J, Li H, et al. Histone Deacetylase 5 Promotes the Migration and Invasion of Hepatocellular Carcinoma Via Increasing the Transcription of Hypoxia-Inducible factor-1alpha Under Hypoxia Condition. Tumour Biol J Int Soc Oncodevelop Biol Med (2017) 39(6):1010428317705034. doi: 10.1177/1010428317705034
71. Dong L, Dong Q, Chen Y, Li Y, Zhang B, Zhou F, et al. Novel HDAC5-interacting Motifs of Tbx3 are Essential for the Suppression of E-cadherin Expression and for the Promotion of Metastasis in Hepatocellular Carcinoma. Signal Transduct Targe Ther (2018) 3:22. doi: 10.1038/s41392-018-0025-6
72. Feng S, Zhang L, Liu X, Li G, Zhang B, Wang Z, et al. Low Levels of AMPK Promote Epithelial-Mesenchymal Transition in Lung Cancer Primarily Through HDAC4- and HDAC5-mediated Metabolic Reprogramming. J Cell Mol Med (2020). 24(14):7789–7801. doi: 10.1111/jcmm.15410
73. Liu Q, Sun Y, Zheng JM, Yan XL, Chen HM, Chen JK, et al. Formononetin Sensitizes Glioma Cells to Doxorubicin Through Preventing EMT Via Inhibition of Histone Deacetylase 5. Int J Clin Exp Pathol (2015) 8(6):6434–41.
74. Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, et al. Combination Therapy for Hepatocellular Carcinoma: Additive Preclinical Efficacy of the HDAC Inhibitor Panobinostat With Sorafenib. J Hepatolo (2012) 56(6):1343–50. doi: 10.1016/j.jhep.2012.01.009
75. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell (2004) 117(7):927–39. doi: 10.1016/j.cell.2004.06.006
76. Lai X, Li Q, Wu F, Lin J, Chen J, Zheng H, et al. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front Cell Dev Biol (2020) 8:760. doi: 10.3389/fcell.2020.00760
77. Feng L, Wang K, Tang P, Chen S, Liu T, Lei J, et al. Deubiquitinase USP18 Promotes the Progression of Pancreatic Cancer Via Enhancing the Notch1-c-Myc Axis. Aging (2020) 12(19):19273–92. doi: 10.18632/aging.103760
78. Vinson KE, George DC, Fender AW, Bertrand FE, Sigounas G. The Notch Pathway in Colorectal Cancer. Int J Cancer (2016) 138(8):1835–42. doi: 10.1002/ijc.29800
79. Peixoto P, Castronovo V, Matheus N, Polese C, Peulen O, Gonzalez A, et al. HDAC5 is Required for Maintenance of Pericentric Heterochromatin, and Controls Cell-Cycle Progression and Survival of Human Cancer Cells. Cell Death Differentiation (2012) 19(7):1239–52. doi: 10.1038/cdd.2012.3
80. Zhou Y, Jin X, Ma J, Ding D, Huang Z, Sheng H, et al. HDAC5 Loss Impairs RB Repression of Pro-Oncogenic Genes and Confers Cdk4/6 Inhibitor Resistance in Cancer. Cancer Res (2021) 81(6):1486–99. doi: 10.1158/0008-5472.CAN-20-2828
81. Murray D, Mirzayans R. Cellular Responses to Platinum-Based Anticancer Drugs and UVC: Role of p53 and Implications for Cancer Therapy. Int J Mol Sci (2020) 21(16):5766. doi: 10.3390/ijms21165766
82. Dequiedt F, Kasler H, Fischle W, Kiermer V, Weinstein M, Herndier BG, et al. HDAC7, a Thymus-Specific Class II Histone Deacetylase, Regulates Nur77 Transcription and TCR-mediated Apoptosis. Immunity (2003) 18(5):687–98. doi: 10.1016/s1074-7613(03)00109-2
83. Barneda-Zahonero B, Roman-Gonzalez L, Collazo O, Rafati H, Islam AB, Bussmann LH, et al. HDAC7 is a Repressor of Myeloid Genes Whose Downregulation is Required for Transdifferentiation of Pre-B Cells Into Macrophages. PloS Genet (2013) 9(5):e1003503. doi: 10.1371/journal.pgen.1003503
84. de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW. Inhibition of HDAC9 Increases T Regulatory Cell Function and Prevents Colitis in Mice. Gastroenterology (2010) 138(2):583–94. doi: 10.1053/j.gastro.2009.10.037
85. Baek YS, Haas S, Hackstein H, Bein G, Hernandez-Santana M, Lehrach H, et al. Identification of Novel Transcriptional Regulators Involved in Macrophage Differentiation and Activation in U937 Cells. BMC Immunol (2009) 10:18. doi: 10.1186/1471-2172-10-18
86. Xiao H, Jiao J, Wang L, O’Brien S, Newick K, Wang LC, et al. HDAC5 Controls the Functions of Foxp3(+) T-Regulatory and CD8(+) T Cells. Int J Cancer (2016) 138(10):2477–86. doi: 10.1002/ijc.29979
87. Zhao M, Tao Y, Peng GH. The Role of Histone Acetyltransferases and Histone Deacetylases in Photoreceptor Differentiation and Degeneration. Int J Med Sci (2020) 17(10):1307–14. doi: 10.7150/ijms.43140
88. Watamoto K, Towatari M, Ozawa Y, Miyata Y, Okamoto M, Abe A, et al. Altered Interaction of HDAC5 With GATA-1 During MEL Cell Differentiation. Oncogene (2003) 22(57):9176–84. doi: 10.1038/sj.onc.1206902
89. Black BL, Olson EN. Transcriptional Control of Muscle Development by Myocyte Enhancer Factor-2 (MEF2) Proteins. Annu Rev Cell Dev Biol (1998) 14:167–96. doi: 10.1146/annurev.cellbio.14.1.167
90. Potthoff MJ, Olson EN. MEF2: A Central Regulator of Diverse Developmental Programs. Development (2007) 134(23):4131–40. doi: 10.1242/dev.008367
91. Zhang M, Zhu B, Davie J. Alternative Splicing of MEF2C pre-mRNA Controls its Activity in Normal Myogenesis and Promotes Tumorigenicity in Rhabdomyosarcoma Cells. J Biol Chem (2015) 290(1):310–24. doi: 10.1074/jbc.M114.606277
92. Vinogradov S, Wei X. Cancer Stem Cells and Drug Resistance: The Potential of Nanomedicine. Nanomedicine (2012) 7(4):597–615. doi: 10.2217/nnm.12.22
93. Zhao M, Li L, Zhou J, Cui X, Tian Q, Jin Y, et al. Mir-2861 Behaves as a Biomarker of Lung Cancer Stem Cells and Regulates the HDAC5-ERK System Genes. Cell Reprogramming (2018) 20(2):99–106. doi: 10.1089/cell.2017.0045
94. Longley DB, Johnston PG. Molecular Mechanisms of Drug Resistance. J Pathol (2005) 205(2):275–92. doi: 10.1002/path.1706
95. Hu B, Xu Y, Li YC, Huang JF, Cheng JW, Guo W, et al. CD13 Promotes Hepatocellular Carcinogenesis and Sorafenib Resistance by Activating HDAC5-LSD1-NF-kappaB Oncogenic Signaling. Clin Trans Med (2020) 10(8):e233. doi: 10.1002/ctm2.233
96. Gabet AS, Mortreux F, Charneau P, Riou P, Duc-Dodon M, Wu Y, et al. Inactivation of hTERT Transcription by Tax. Oncogene (2003) 22(24):3734–41. doi: 10.1038/sj.onc.1206468
97. Shammas MA, Koley H, Beer DG, Li C, Goyal RK, Munshi NC. Growth Arrest, Apoptosis, and Telomere Shortening of Barrett’s-Associated Adenocarcinoma Cells by a Telomerase Inhibitor. Gastroenterology (2004) 126(5):1337–46. doi: 10.1053/j.gastro.2004.01.026
98. Garcia-Cao M, O’Sullivan R, Peters AH, Jenuwein T, Blasco MA. Epigenetic Regulation of Telomere Length in Mammalian Cells by the Suv39h1 and Suv39h2 Histone Methyltransferases. Nat Genet (2004) 36(1):94–9. doi: 10.1038/ng1278
99. Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, et al. Role of the RB1 Family in Stabilizing Histone Methylation at Constitutive Heterochromatin. Nat Cell Biol (2005) 7(4):420–8. doi: 10.1038/ncb1235
100. Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, et al. DNA Methyltransferases Control Telomere Length and Telomere Recombination in Mammalian Cells. Nat Cell Biol (2006) 8(4):416–24. doi: 10.1038/ncb1386
101. Sauer M, Bretz AC, Beinoraviciute-Kellner R, Beitzinger M, Burek C, Rosenwald A, et al. C-Terminal Diversity Within the p53 Family Accounts for Differences in DNA Binding and Transcriptional Activity. Nucleic Acids Res (2008) 36(6):1900–12. doi: 10.1093/nar/gkn044
102. Mangiulli M, Valletti A, Caratozzolo MF, Tullo A, Sbisa E, Pesole G, et al. Identification and Functional Characterization of Two New Transcriptional Variants of the Human p63 Gene. Nucleic Acids Res (2009) 37(18):6092–104. doi: 10.1093/nar/gkp674
103. Kim S, Han J, Kim J, Park C. Maspin Expression is Transactivated by p63 and is Critical for the Modulation of Lung Cancer Progression. Cancer Res (2004) 64(19):6900–5. doi: 10.1158/0008-5472.CAN-04-1657
104. Ito R, Nakayama H, Yoshida K, Oda N, Yasui W. Loss of Maspin Expression is Associated With Development and Progression of Gastric Carcinoma With p53 Abnormality. Oncol Rep (2004) 12(5):985–90. doi: 10.3892/or.12.5.985
105. Wang N, Chang LL. Maspin Suppresses Cell Invasion and Migration in Gastric Cancer Through Inhibiting EMT and Angiogenesis Via ITGB1/FAK Pathway. Hum Cell (2020) 33(3):663–75. doi: 10.1007/s13577-020-00345-7
106. Sun Q, Zhang K, Li H, Chen W, Liu L, Huang G, et al. The Overexpression of Maspin Increases the Sensitivity of Lung Adenocarcinoma Drug-Resistant Cells to Docetaxel In Vitro and In Vivo. Ann Trans Med (2020) 8(22):1522. doi: 10.21037/atm-20-7053
107. Akanji MA, Rotimi D, Adeyemi OS. Hypoxia-Inducible Factors as an Alternative Source of Treatment Strategy for Cancer. Oxid Med Cell Longevity (2019) 2019:8547846. doi: 10.1155/2019/8547846
108. Jonckheere S, Adams J, De Groote D, Campbell K, Berx G, Goossens S. Epithelial-Mesenchymal Transition (EMT) as a Therapeutic Target. Cells Tissues Organs (2021) 1–26. doi: 10.1159/000512218
109. Chatterjee R, Ghosh B, Mandal M, Nawn D, Banerjee S, Pal M, et al. Pathophysiological Relationship Between Hypoxia Associated Oxidative Stress, Epithelial-mesenchymal Transition, Stemness Acquisition and Alteration of Shh/ Gli-1 Axis During Oral Sub-Mucous Fibrosis and Oral Squamous Cell Carcinoma. Eur J Cell Biol (2020) 100(1):151146. doi: 10.1016/j.ejcb.2020.151146
110. Wang N, Muhetaer G, Zhang X, Yang B, Wang C, Zhang Y, et al. Sanguisorba Officinalis L. Suppresses Triple-Negative Breast Cancer Metastasis by Inhibiting Late-Phase Autophagy Via Hif-1alpha/Caveolin-1 Signaling. Front Pharmacol (2020) 11:591400. doi: 10.3389/fphar.2020.591400
111. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell (2004) 119(7):941–53. doi: 10.1016/j.cell.2004.12.012
112. Wang GG, Allis CD, Chi P. Chromatin Remodeling and Cancer, Part I: Covalent Histone Modifications. Trends Mol Med (2007) 13(9):363–72. doi: 10.1016/j.molmed.2007.07.003
113. Cao C, Wu H, Vasilatos SN, Chandran U, Qin Y, Wan Y, et al. HDAC5-LSD1 Axis Regulates Antineoplastic Effect of Natural HDAC Inhibitor Sulforaphane in Human Breast Cancer Cells. Int J Cancer (2018) 143(6):1388–401. doi: 10.1002/ijc.31419
114. Ramadass V, Vaiyapuri T, Tergaonkar V. Small Molecule NF-kappaB Pathway Inhibitors in Clinic. Int J Mol Sci (2020) 21(14):5164. doi: 10.3390/ijms21145164
115. Giridharan S, Srinivasan M. Mechanisms of NF-kappaB p65 and Strategies for Therapeutic Manipulation. J Inflammation Res (2018) 11:407–19. doi: 10.2147/JIR.S140188
116. Kim D, Nam HJ, Lee W, Yim HY, Ahn JY, Park SW, et al. PKCalpha-LSD1-NF-kappaB-Signaling Cascade Is Crucial for Epigenetic Control of the Inflammatory Response. Mol Cell (2018) 69(3):398–411 e6. doi: 10.1016/j.molcel.2018.01.002
117. Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell (2009) 15(1):67–78. doi: 10.1016/j.ccr.2008.12.005
118. Chen H, Werner S, Tao S, Zornig I, Brenner H. Blood Autoantibodies Against Tumor-Associated Antigens as Biomarkers in Early Detection of Colorectal Cancer. Cancer Lett (2014) 346(2):178–87. doi: 10.1016/j.canlet.2014.01.007
119. Chi F, Liu J, Brady SW, Cosgrove PA, Nath A, McQuerry JA, et al. A `One-Two Punch’ Therapy Strategy to Target Chemoresistance in Estrogen Receptor Positive Breast Cancer. Trans Oncol (2021) 14(1):100946. doi: 10.1016/j.tranon.2020.100946
120. Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, Kassack MU, et al. Histone Deacetylase (HDAC) Inhibitors With A Novel Connecting Unit Linker Region Reveal a Selectivity Profile for HDAC4 and HDAC5 With Improved Activity Against Chemoresistant Cancer Cells. J Medicinal Chem (2013) 56(2):427–36. doi: 10.1021/jm301254q
121. Zhang M, Pan Y, Dorfman RG, Chen Z, Liu F, Zhou Q, et al. AR-42 Induces Apoptosis in Human Hepatocellular Carcinoma Cells Via HDAC5 Inhibition. Oncotarget (2016) 7(16):22285–94. doi: 10.18632/oncotarget.8077
122. Vasilatos SN, Katz TA, Oesterreich S, Wan Y, Davidson NE, Huang Y. Crosstalk Between Lysine-Specific Demethylase 1 (LSD1) and Histone Deacetylases Mediates Antineoplastic Efficacy of HDAC Inhibitors in Human Breast Cancer Cells. Carcinogenesis (2013) 34(6):1196–207. doi: 10.1093/carcin/bgt033
123. Wang Y, Wallach J, Duane S, Wang Y, Wu J, Wang J, et al. Developing Selective Histone Deacetylases (HDACs) Inhibitors Through Ebselen and Analogs. Drug Design Dev Ther (2017) 11:1369–82. doi: 10.2147/DDDT.S124977
124. Clarke CJ, Shamseddine AA, Jacob JJ, Khalife G, Burns TA, Hannun YA. ATRA Transcriptionally Induces nSMase2 Through CBP/p300-Mediated Histone Acetylation. J Lipid Res (2016) 57(5):868–81. doi: 10.1194/jlr.M067447
125. Wanek J, Gaisberger M, Beyreis M, Mayr C, Helm K, Primavesi F, et al. Pharmacological Inhibition of Class IIa HDACs by LMK-235 in Pancreatic Neuroendocrine Tumor Cells. Int J Mol Sci (2018) 19(10):3128. doi: 10.3390/ijms19103128
126. Ishikawa S, Hayashi H, Kinoshita K, Abe M, Kuroki H, Tokunaga R, et al. Statins Inhibit Tumor Progression Via An Enhancer of Zeste Homolog 2-Mediated Epigenetic Alteration in Colorectal Cancer. Int J Cancer (2014) 135(11):2528–36. doi: 10.1002/ijc.28672
127. Marks PA, Xu WS. Histone Deacetylase Inhibitors: Potential in Cancer Therapy. J Cell Biochem (2009) 107(4):600–8. doi: 10.1002/jcb.22185
128. Woo SH, Frechette S, Abou Khalil E, Bouchain G, Vaisburg A, Bernstein N, et al. Structurally Simple Trichostatin A-Like Straight Chain Hydroxamates As Potent Histone Deacetylase Inhibitors. J Medicinal Chem (2002) 45(13):2877–85. doi: 10.1021/jm020154k
129. Sawas A, Radeski D, O’Connor OA. Belinostat in Patients With Refractory or Relapsed Peripheral T-cell Lymphoma: A Perspective Review. Ther Adv Hematol (2015) 6(4):202–8. doi: 10.1177/2040620715592567
130. McDermott J, Jimeno A. Belinostat for the Treatment of Peripheral T-cell Lymphomas. Drugs Today (2014) 50(5):337–45. doi: 10.1358/dot.2014.50.5.2138703
131. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, et al. Structures of a Histone Deacetylase Homologue Bound to the TSA and SAHA Inhibitors. Nature (1999) 401(6749):188–93. doi: 10.1038/43710
132. Kaletsch A, Pinkerneil M, Hoffmann MJ, Jaguva Vasudevan AA, Wang C, Hansen FK, et al. Effects of Novel HDAC Inhibitors on Urothelial Carcinoma Cells. Clin Epigenet (2018) 10(1):100. doi: 10.1186/s13148-018-0531-y
133. Hendrick E, Peixoto P, Blomme A, Polese C, Matheus N, Cimino J, et al. Metabolic Inhibitors Accentuate the Anti-Tumoral Effect of HDAC5 Inhibition. Oncogene (2017) 36(34):4859–74. doi: 10.1038/onc.2017.103
134. Matsushita Y, Nakagawa H, Koike K. Lipid Metabolism in Oncology: Why It Matters, How to Research, and How to Treat. Cancers (2021) 13(3):474. doi: 10.3390/cancers13030474
135. Thibault A, Samid D, Tompkins AC, Figg WD, Cooper MR, Hohl RJ, et al. Phase I Study of Lovastatin, an Inhibitor of the Mevalonate Pathway, in Patients With Cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (1996) 2(3):483–91.
136. Larner J, Jane J, Laws E, Packer R, Myers C, Shaffrey M. A Phase I-II Trial of Lovastatin for Anaplastic Astrocytoma and Glioblastoma Multiforme. Am J Clin Oncol (1998) 21(6):579–83. doi: 10.1097/00000421-199812000-00010
137. Kawata S, Yamasaki E, Nagase T, Inui Y, Ito N, Matsuda Y, et al. Effect of Pravastatin on Survival in Patients With Advanced Hepatocellular Carcinoma. A Randomized Controlled Trial. Br J Cancer (2001) 84(7):886–91. doi: 10.1054/bjoc.2000.1716
138. Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR. Lovastatin Augments Apoptosis Induced by Chemotherapeutic Agents in Colon Cancer Cells. Clin Cancer Res an Off J Am Assoc Cancer Res (1999) 5(8):2223–9.
139. Mace AG, Gantt GA, Skacel M, Pai R, Hammel JP, Kalady MF. Statin Therapy is Associated With Improved Pathologic Response to Neoadjuvant Chemoradiation in Rectal Cancer. Dis Colon Rectum (2013) 56(11):1217–27. doi: 10.1097/DCR.0b013e3182a4b236
140. Lee J, Jung KH, Park YS, Ahn JB, Shin SJ, Im SA, et al. Simvastatin Plus Irinotecan, 5-Fluorouracil, and Leucovorin (FOLFIRI) As First-Line Chemotherapy in Metastatic Colorectal Patients: A Multicenter Phase II Study. Cancer Chemother Pharmacol (2009) 64(4):657–63. doi: 10.1007/s00280-008-0913-5
Keywords: HDAC5, cancer, biological function, clinical application, biomarker
Citation: Yang J, Gong C, Ke Q, Fang Z, Chen X, Ye M and Xu X (2021) Insights Into the Function and Clinical Application of HDAC5 in Cancer Management. Front. Oncol. 11:661620. doi: 10.3389/fonc.2021.661620
Received: 31 January 2021; Accepted: 18 May 2021;
Published: 10 June 2021.
Edited by:
Lorenzo Gerratana, University of Udine, ItalyReviewed by:
Jian Zhu, Shandong University, ChinaViviana Moresi, National Research Council (CNR), Italy
Copyright © 2021 Yang, Gong, Ke, Fang, Chen, Ye and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Xu, zdxuxi@zju.edu.cn; Ming Ye, yemingsm@163.com
†These authors have contributed equally to this work