Inflammasomes and their roles in arthritic disease pathogenesis
 Gabsik Yang
Gabsik Yang Han Chang Kang
Han Chang Kang Yong-Yeon Cho
Yong-Yeon Cho Hye Suk Lee
Hye Suk Lee Joo Young Lee
Joo Young Lee- 1Department of Pharmacology, College of Korean Medicine, Woosuk University, Jeonju, South Korea
- 2College of Pharmacy, The Catholic University of Korea, Seoul, South Korea
The inflammasome is a molecular platform that is created in the cytosolic compartment to mediate the host immunological response to cellular injury and infection. Caspase-1 may be activated by the inflammasome, which leads to the generation of the inflammatory cytokines interleukin-1β (IL-1β) and IL-18 and the beginning of pyroptosis, which is a type of proinflammatory cell death. Scientists have identified a number of different inflammasomes in the last 2 decades. The NLRP3 inflammasome has been studied the most, and its activity may be triggered by a broad range of different inducers. However, activation of the NLRP3 inflammasome in a manner that is not properly controlled is also a factor in the etiology of many human illnesses. Accumulating evidence indicates that the NLRP3 inflammasome plays a significant role in the innate and adaptive immune systems and the development of various arthritic illnesses, such as rheumatoid arthritis, ankylosing spondylitis, and gout. The present review provides a concise summary of the biological properties of the NLRP3 inflammasome and presents the fundamental processes behind its activation and control. We discuss the role of the inflammasome in the pathogenesis of arthritic diseases, such as rheumatoid arthritis, ankylosing spondylitis, and gout, and the potential of newly developed therapies that specifically target the inflammasome or its products for the treatment of inflammatory diseases, with a particular emphasis on treatment and clinical application.
Introduction
Autoimmune disorders, such as rheumatoid arthritis (RA), gout, systemic lupus erythematosus (SLE), ankylosing spondylitis (AS), juvenile idiopathic spinal arthritis, and Sjögren’s syndrome (SS), are characterized by the loss of immunological tolerance to autoantigens and persistent autoresponsive immune responses, which culminate in the overproduction of autoantibodies and organ damage (Zhernakova et al., 2013). However, the precise process of autoimmune disease initiation is not clear, which makes the treatment of these conditions challenging.
Inflammation is a congenital immune response that is primarily caused by macrophages (myeloid immune cells) and initiated by pattern recognition receptors (PRRs) recognition of pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) induced by infection or intrinsic stress (Janeway and Medzhitov 2002). PRRs include receptors that are present on the cell membrane, such as Toll-like receptors (TLRs), that are present in the cytoplasm, such as nucleotide-binding domain-like receptors (NLRs) and inflammasomes (Schroder et al., 2010). Inflammasomes are protein complexes comprised of a receptor protein, an adaptor, and procaspase-1. These are “canonical” inflammasomes, with NLRs and absent in melanoma 2 (AIM2) as a receptor protein and apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) as an adaptor protein. Formation of inflammasomes is induced upon a variety of stimuli, including PAMPs derived from infection and DAMPs derived from host damage, which culminate in the maturation of pro-inflammatory cytokines, such as interleukin (IL)-1β and IL-18, that activate host immunity. A “non-canonical” inflammasome pathway leads to activation of caspase-4 and -5 in humans and caspase-11 in mice in response to intracellular LPS. The NLR family has at least 22 members, including NLRP1, NLRP3, and NLRC4, with a central nucleotide binding and oligomerization (NACHT) domain, caspase recruitment (CARD) domain, and leucine-rich repeats (LRRs) or a pyrin (PYD) domain. The NLRP1 inflammatory complex is a protein complex consisting of NLRP1, ASC, and pro-caspase-1 (Lamkanfi and Dixit 2014), was first identified in the NLR family. The pro-inflammatory cytokines pro-IL-1β and pro-IL-18 are cleaved to release the active cytokines IL-1β and IL-18, respectively (Broz and Dixit 2016). The NLRP3 inflammasome is predominantly expressed in peripheral blood leukocytes, and an NLRP3 inflammatory complex coupled with ASC and pro-caspase-1 rapidly forms in response to inflammatory stimuli. Formation of the NLRP3 inflammatory complex activates caspase-1, which leads to maturation of the pro-inflammatory cytokines IL-1β and IL-18 (Seok et al., 2020). IL-1β plays a multifunctional role in the immune response to induce cytokine production, enhance T-cell activation and antigen recognition, guide innate immune cells to the site of infection (Dinarello 2011; van de Veerdonk and Netea 2013), and induce activation of the NF-κB signaling cascade to induce cytokine production, which results in the transcriptional activation of genes encoding chemokines and various pro-inflammatory mediators (Kanneganti et al., 2007). The NLRP3 inflammasome is linked with the sensitivity, disease severity, and treatment outcomes of autoimmune disorders (Seok et al., 2021). This review examines the function of the NLRP3 inflammasome in the development of autoimmune disorders, such as RA, AS, and gout, based on previous studies and highlights current research on the involvement of inflammasomes in inflammatory and rheumatic autoimmune disorders.
The pathology of rheumatic autoimmune disorders
Rheumatoid arthritis (RA) is an autoimmune disease that is characterized by chronic inflammation and joint pain. RA involves the inflammation of active joints, cartilage destruction, bone erosion, and calcification around the joints, which result in joint dysfunction. RA is one of the most common inflammatory autoimmune disorders worldwide (George et al., 2020), and 1%–2% of the world’s population suffers from RA. Its incidence is increasing 8.2% annually (Crowson et al., 2011; Safiri et al., 2019). Autoimmunity is the first step in RA pathogenesis, and high serum concentrations of autoantibodies, such as anticitrullineated peptide antibodies (ACPAs), are hallmarks of RA, although some patients are seronegative for RA (Firestein 2003; McInnes and Schett 2011). The exact pathogenesis of RA is not clear, but genetics, smoking, obesity, infection, periodontal disease, and the gut microbiota are likely involved (Muruve et al., 2008).
Ankylosing spondylitis (AS) is a chronic inflammatory disease that primarily causes inflammation and new bone formation in the axial skeleton and peripheral joints (Dougados and Baeten 2011). AS is a type of spondyloarthritis (SpA), and the pathological feature of SpA is the occurrence of ankylosing in the axial joint (Maksymowych et al., 2012). Maksymowych divided the symptoms of AS into inflammation, destruction, recovery, and extra-articular features (Maksymowych 2010): 1) inflammatory features of peripheral synovitis and adhesions; 2) destructive characteristics, such as loss of cartilage and bone; 3) restorative repair characteristics of new bone formation; and 4) inflammatory bowel disease (IBD), acute anterior encephalitis (AAU), and extra-articular features, such as psoriasis. Inflammation, which is the main initial symptom of AS, gradually progresses to bone abnormality (bone loss), joint stiffness, and joint fusion, which result in ankylosing of the spine (Quaden et al., 2016). Whether there is a correlation between bone formation and inflammation, which is one of the main features of AS, remains controversial (Maksymowych et al., 2012). However, the mechanism and pathogenesis of AS are not clear despite active research. AS is a multifactorial disease that is affected by genetic factors, such as HLA-B27, environmental factors, such as intestinal bacteria, and immunological factors (Tsui et al., 2014). Epidemiological studies showed that AS primarily occurred in men their twenties and forties (Zhu et al., 2019), with a worldwide prevalence of 0.1%–1.4% (Akkoc 2008).
Gout is the most common form of inflammatory arthritis (Stamp and Farquhar 2021), and it is caused by the accumulation of monosodium urate (MSU) crystals in joints, tendons, and other tissues due to elevated serum urate levels (Dehlin et al., 2020). According to recent data from the National Institute of Health and Nutrition (NIHN), the prevalence of gout is generally higher in men (5.2%) than women (2.7%) (Chen-Xu et al., 2019), and the risk of developing gout increases with age (Dehlin et al., 2020). A tophus is a granuloma that histologically surrounds urate crystals and tissue debris (Schweyer et al., 2000). Acute gout attacks are caused by the body’s immune response to tophi (Lee, 2011).
Rheumatoid arthritis and the inflammasome
Genetic factors play a pivotal role in RA pathogenesis, and several genes contribute to disease induction (Crowson et al., 2011; George et al., 2020). Most of the genetic mutations related to RA development are related to the immune response, and mutations in human leukocyte antigen (HLA) and the inflammatory complex are major genetic risk factors (Crowson et al., 2011). Variations in the NLRP1 gene increase NLRP1 expression, which is a risk factor for the development of inflammatory diseases, including RA, type 1 intrinsic diabetes, vitiligo, and associated autoimmune diseases (Jin et al., 2007; McInnes and Schett 2011; Sui et al., 2012; Levandowski et al., 2013). Mutations in the NLRP3 gene play an important role in several diseases, including gouty arthritis and type I diabetes (Pontillo et al., 2010; Zhang et al., 2018), and the expression of NLRP3 inflammatory complex-related genes (NLRP3, ASC, and CASP1) in peripheral blood mononuclear cells (PBMCs) may play an important role in RA pathogenesis and disease activity (Cheng et al., 2021). Overactivation of the NLRP3 inflammatory complex leads to excessive inflammation and unnecessary host tissue damage, which contribute to the severity of RA (He et al., 2016). The activity and expression of NLRP3 are upregulated in RA patients, which further suggests that the NLRP3 inflammatory complex may play an essential role in RA pathogenesis (Mathews et al., 2014; Choulaki et al., 2015; Ruscitti et al., 2015; Kim H et al., 2017; Addobbati et al., 2018). The expression of NLRC4 was markedly higher in monocytes derived from Brazilian RA patients (Addobbati et al., 2018).
Cartilage decomposition in RA occurs due to an excessive immune response (Komatsu and Takayanagi, 2022). Pro-inflammatory cytokines, such as IL-1, tumor necrosis factor (TNF), IL-6, and IL-17, induce cartilage-degrading enzymes, such as matrix metalloproteinases (MMPs) and aggrecanases (e.g., ADAM metallopeptidase with thrombospondin type 1 motif [ADAMTS]-4 and ADAMTS-5). These factors induce the production of synovial fluid and inhibit extracellular matrix production by chondrocytes in synovial fibroblasts (Pap and Korb-Pap, 2015; Araki and Mimura, 2017). This inflammatory reaction primarily occurs via the formation of an inflammatory complex called an inflammasome, which results in the formation and secretion of inflammasome-specific cytokines, such as IL-1β and IL-18, in macrophages and the inflammatory necrosis of macrophages called pyroptosis (Figure 1) (Lamkanfi and Dixit, 2014; Broz and Dixit, 2016). Pro-inflammatory cytokines, such as TNF, IL-6, and IL-1, which are abundant in the synovial membrane and synovial fluid of patients with RA, promote RANKL expression by synovial fibroblasts, and TNF and M-CSF induce osteoclast production (Takayanagi, 2007; Okamoto et al., 2017; Shim et al., 2018). IL-1β, IL-6, IL-18, and TNF promote Th17 cell differentiation, reduce the synthesis of cartilage components (Alippe and Mbalaviele 2019), and inhibit osteoblast generation via several mechanisms (Shim et al., 2018).
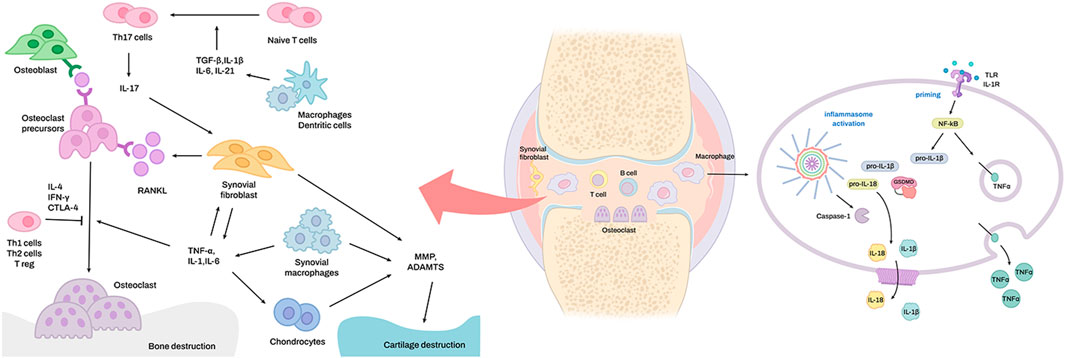
FIGURE 1. The signaling pathways for inflammasome activation in rheumatoid arthritis. Overexpression of TNF-α in the synovial macrophages of rheumatoid arthritis joints leads to activation of the classical NF-κB pathway, which leads to activation of the transcription factor RelA. RelA constructively regulates the expression of NLRP3. In conclusion, a higher level of NLRP3 expression results in more robust activation of the NLRP3 inflammasome.
After encountering APCs, antigens, and other microenvironmental variables, a native CD4 T cell’s trip to becoming an antigen-specific Th17 cell may be separated into three terrains. During the first terrain, TCR–pMHC interaction strength, costimulation strength, and other “non-cytokine"-induced variables work with APC-generated cytokines (e.g., IL-6) to trigger activation of STAT proteins (e.g., STAT3, STAT1) and “pioneer” TFs such as BATF and IRF4. STAT proteins and pioneer transcription factors (TFs) begin lineage-specific development by inducing STAT-responsive and IRF4/BATF-responsive genes, including RORt. STAT3-induced RORt works with “pioneer” TFs to epigenetically modify important Th17-related genes to make them transcriptionally permissive. In its final passage through the third terrain, an orchestration of complex signaling events modulated by RORt along with lineage-associated TFs (e.g., Runx1, AhR, and c-Maf) determines the stability of the Th17 developmental program by integrating pro-inflammatory and anti-inflammatory environmental cues. Interplay of varied elements across three terrains determines Th17 cell plasticity (Bhaumik and Basu 2017). Autoimmunity to a number of self-proteins has been linked to the onset and development of RA. Some of the antigens described come from joints, like type II collagen and human cartilage-derived glycoprotein HCgp39 (Tsark et al., 2002). Other antigens are stress-associated proteins, like grp78/BiP, which is an intracellular chaperone involved in endoplasmic reticulum stress and angiogenesis in proliferating RA synovial tissue (Bläss et al., 2001; Yoo et al., 2012). Endoplasmic reticulum stress can be caused by many things, such as proinflammatory cytokines, cell death, proteins in the endoplasmic reticulum that do not fold correctly, and reactive oxygen species (Hasnain et al., 2012). Because of the flow of Ca2+ inside the cell, the peptidyl arginine deiminase enzymes are turned on, which speeds up the deamination of arginine to citrullination (Anzilotti et al., 2010). The function of NLRP3 in mediating DC-dependent Th17 cell responses has also been seen in Bordetella pertussis and Candida albicans infection models (Dunne et al., 2010; van de Veerdonk et al., 2011). NLRP3 hyperactivation, on the other hand, generates increased IL-1 synthesis from APCs, resulting in enhanced Th17 cell differentiation and Th17 cell-dominant immunopathology (Meng et al., 2009). In autoimmune, infectious, and inflammatory scenarios, activation of the NLPR3-mediated inflammasome by DCs promotes Th17 cell development through IL-1 and/or IL-18 (Huang et al., 2012). In addition, when the inflammasome is turned on in murine macrophages, citrullination is caused (Mishra et al., 2019).
Tofacitinib (TOF) is a Janus kinase (JAK) inhibitor that ameliorated joint inflammation and damage in a collagen-induced arthritis (CIA) model by suppressing γδT17 cell activation mediated via inhibition of the NLRP3 inflammasome (Yang et al., 2021), which shows the relationship between NLRP3 and Th17 cells. Osteoblasts and osteoclasts precisely control bone tissue, which are controlled by growth factors and hormones (Siddiqui and Partridge 2016). Macrophages, monocytes, neutrophils, and Th17 cells in bone tissue are also involved in homeostasis regulation via signal exchange with these cells (Sato et al., 2006; Chen et al., 2020). Increased inflammasome activity in these cells may change the functions of osteoblasts and osteoclasts to change bone mass and bone quality. During osteolysis by osteoclast activity, factors derived from the bone matrix act as DAMPs to further promote the inflammatory response and bone loss. Conversely, inhibition of bone resorption reduces the inflammasome activity of bone constituent cells (Alippe et al., 2017; Madel et al., 2019). Therefore, inflammasome activation is a cellular action that promotes bone resorption but may also be the result of inflammatory bone loss. These findings show that inflammasome activation plays a positive feedback role in inflammatory bone loss. NLRP3 inflammasome activity increases bone resorption activity in osteoclast lineage cells and promotes IL-1β and IL-18 production. The increase in pathogen-associated molecular patterns (PAMPs) and DAMPs involved in osteolysis promotes polynuclearization and IL-1β and IL-18 production and increases inflammasome activity in osteoclasts and osteoclast progenitors (Kim H. W et al., 2017). IL-1β induces proliferation and multinucleation of osteoclasts derived from hematopoietic lineage cells in the presence of macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor kappa B ligand (RANKL) (Cao et al., 2016). However, it also induces the migration of osteoclast precursors, which results in quantitative changes in M-CSF, osteoprotegerin (OPG), and C-X3-C motif ligand 1 (CX3CL1) involved in hyperdifferentiation (Roper et al., 2020).
DAMPs are classified as endogenous stimuli produced by dying cells or extracellular matrix (ECM) (Frevert et al., 2018). Internal cellular components such as heat shock proteins and the extracellular matrix (ECM) are common sources of DAMPs (e.g., biglycan, decorin). They are released into the extracellular milieu upon tissue damage, where they may either exacerbate inflammation or promote tissue regeneration through activation of pattern recognition receptors (PRRs) (Figure 2) (Frevert et al., 2018). The biological activity of these DAMPs is dependent on the activation of PRRs including Toll-like receptors (TLRs), NOD-like receptors (NLRs), and Receptor for Advanced Glycosylation End products (RAGEs) (Barreto et al., 2015). Barreto et al. also demonstrated that soluble biglycan is commonly detected in knee synovial fluid of patients with advanced knee OA or rheumatoid arthritis (RA) (Barreto et al., 2015).
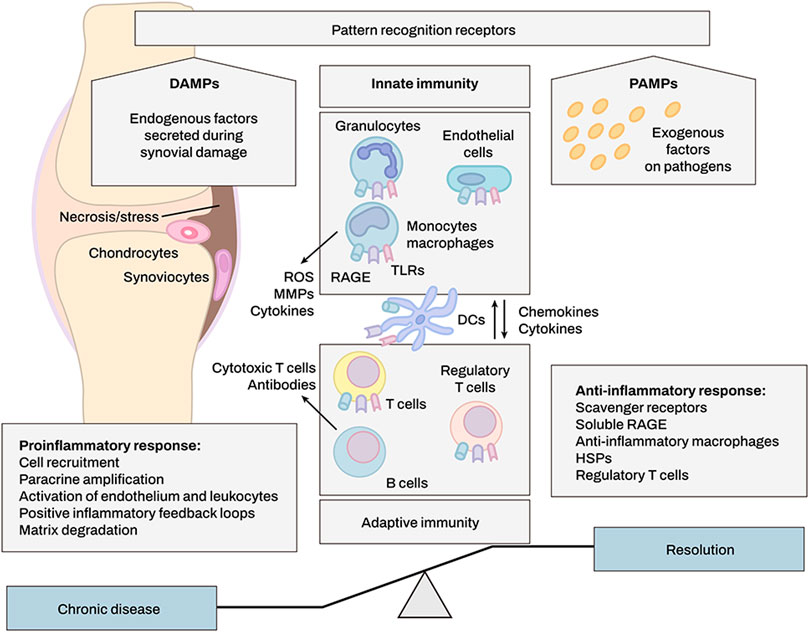
FIGURE 2. DAMPs produced upon synovial tissue damage regulate immune responses. DAMP molecules activate immune cells after cell injury, infection, or inflammation. Innate immune system may enhance and alleviate inflammation by eliminating or modulating adaptive immune responses, e.g., through regulatory T cells. Positive feedback loops involving DAMPs increase leukocyte recruitment and cause persistent rheumatoid arthritis.
In vivo investigations have shown that the NLRP3 inflammasome promotes bone resorption under estrogen deprivation and prolonged parathyroid hormone exposure, and that NLRP3 deletion lowers bone loss in numerous high bone turnover scenarios (Alippe et al., 2017). In osteoclasts exposed to high glucose concentrations in the rat model of diabetic osteoporosis, NLRP3 inflammasome and associated proteins are upregulated, bone density is decreased, and osteoclast markers are upregulated (Zhang et al., 2021). MSC exosomes may suppress the overactive NLRP3 inflammasome (Zhang et al., 2021). The NF-κB pathway initiates NLRP3 inflammasome activation. NF-κB regulates inflammation downstream of TLRs and the immune response’s traditional inflammatory pathways. NF-κB pathway affects formation and maturation of osteoblasts and osteoclasts, according to many studies (Novack 2011). High glucose upregulates NLRP3 inflammasome expression, which is controlled by ROS/MAPKs/NF-κB. NF-κB inhibitors diminish NLRP3 expression and bone resorption (An et al., 2019). The NLRP3 inflammasome and the conventional inflammatory pathway mutually conflict and affect osteoclast development, confirming NF-κB’s function in initiating the NLRP3 inflammasome (Jiang et al., 2021). IL-1β suppresses osteogenic differentiation by dampening the BMP/Smad pathway and other osteogenic markers such RUNX2, OCN, and ALP. IL-1β promotes activated osteoclasts via a RANKL-RANK independent pathway after binding with IL-1R on T cells or B cells and inducing the production of RANKL on osteoblasts (Jiang et al., 2021).
Ankylosing spondylitis and the inflammasome
The initial stage of AS is inflammation, and controlling inflammation may be the key to finding ways to prevent other major symptoms of AS. is Inflammasome activation is a hallmark of an inflammatory response mediated primarily by macrophages (Yi 2018). Although few studies investigated the relationship between AS and the NLRP3 inflammasome, there is accumulating evidence of the constituents of the NLRP3 inflammasome or related expression factors in AS pathology. Levels of caspase-1 in inflammasomes were higher in AS than other types of arthritis (Kim et al., 2022). High NLRP3 and ASC gene expression and a correlation between AS and the NLRP3 inflammasome were reported (Kim et al., 2018). However, another study observed intestinal overexpression of NLRs and AIM2 in patients with AS and suggested a link between the microbiome and inflammasome activation (Guggino et al., 2021). Anakinra, which inhibits IL-1, is effective in treating AS (Tan et al., 2004; Yi 2018). MicroRNA (miR)-21 expression in response to JAK2/STAT3 signaling activation by TNF-α exposure caused osteogenesis in AS (Zou et al., 2020), which suggests a correlation between the NLRP3 inflammasome and AS (Zhu et al., 2021). There is an association between various inflammatory cytokines and AS related to the NLRP3 inflammasome (Zeng et al., 2011; Yan et al., 2014; Maxwell et al., 2015). However, the relationship between AS and the NLRP3 inflammasome has been revealed indirectly, and further research is needed to determine the direct correlation between AS and the NLRP3 inflammasome.
Gout and the inflammasome
The NLRP3 inflammasome plays a pivotal role in the development of acute gout because the MSU crystal is an agonist of the NLRP3 inflammasome (Martinon et al., 2006). The activated NLRP3 inflammasome degrades pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, respectively (Figure 3) (Dinarello 1998; Sutterwala et al., 2006). Increased IL-1β production is a critical pathogenic hallmark of gouty inflammation and attack.
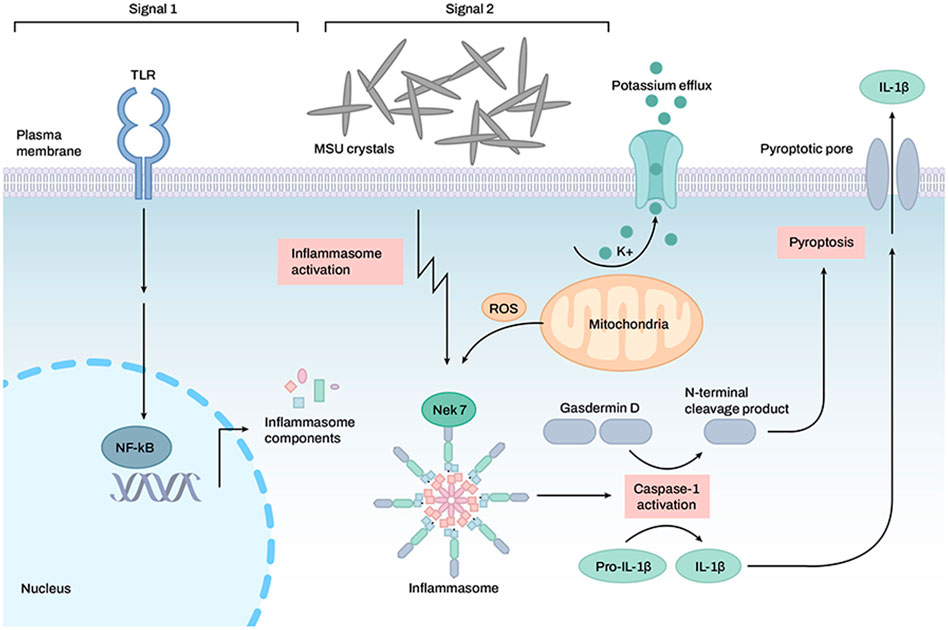
FIGURE 3. Activation of the inflammasome by monosodium uric acid crystals in the induction of gouty arthritis symptoms. TLR receptors are activated, and inflammasome formation is mediated by monosodium uric acid crystals. Once the inflammasome is activated, caspase-1 is also activated, which allows for the cleavage of GSDMD, pro-IL-1β, and pro-IL-18. Active IL-1 and IL-18 may be exported to the extracellular space via a transmembrane channel formed by the N-terminal GSDMD.
Uric acid is released from damaged cells and acts as a DAMP to activate the NLRP3 inflammasome (Shi et al., 2003; Martinon et al., 2006). Once uric acid crystals are phagocytosed by macrophages, phagosomes are ruptured by crystals, and phagosomal contents, such as cathepsins, proteases, and Ca2+, are released into the cytosol (Hornung et al., 2008). Uric acid crystal-induced activation of the NLRP3 inflammasome is accompanied by mitochondrial ROS generation and K+ efflux (So and Martinon 2017). Uric acid crystals induce the binding of TXNIP with NLRP3, which may occur via the production of ROS, and initiate NLRP3 inflammasome assembly (Kim et al., 2019).
The critical roles of the NLRP3 inflammasome and IL-1β in gout pathology have been revealed, and a blocking strategy for these pathways has emerged as a promising new therapeutic for gout attack. NLRP3 inflammasome inhibitors and IL-1 inhibitors, such as anakinra (an IL-1R antagonist), rilonacept (IL-1 TRAP), and canakinumab (monoclonal anti-IL-1β antibody), have been tested in clinical trials and showed efficacy in the treatment of acute and chronic gout patients.
Therapeutic approaches
In the acute phase response, NSAIDs (naproxen, ibuprofen, coxibs) are used to decrease pain by reducing inflammation. NSAIDs exert their pharmacological action by inhibiting cyclooxygenase (COX), particularly inflammation-induced COX-2. However, the potential of injury must be addressed, since suppression of prostaglandins may result in major adverse effects, such as bleeding, gastrointestinal ulcers, renal failure, heart failure, rashes, disorientation, confusion, seizures, and more. Using COX-2-selective NSAIDs (celecoxib, rofecoxib, valdecoxib) may help prevent some of the negative effects (Crofford 2013). It has been known that rheumatoid arthritis may be effectively treated by inhibiting cytokines. Anakinra is a recombinant human IL-1 receptor antagonist that inhibits IL-1α and IL-1β competitively (Schwaid and Spencer 2021). Although the Food and Drug Administration (FDA) has licensed Anakinra for the treatment of rheumatoid arthritis in certain individuals (Mertens and Singh 2009), it is only moderately effective and inferior to TNF-α inhibitors, showing its limited application (Ramírez and Cañete 2018). Additionally, canakinumab, a human monoclonal IL-1β antibody, and rilonacept, a decoy receptor of IL-1α and IL-1β, have been targeted in rheumatoid arthritis (Swanson et al., 2019).
Many compounds inhibit the NLRP3 inflammasome (Figure 4) and show potential therapeutic efficacy for rheumatic diseases (Table 1). MCC950 inhibits AIM2, NLRC4, and NLRP1 activation in mouse macrophages, human monocyte–derived macrophages, human PBMCs, and canonical and noncanonical NLRP3 activation (Coll et al., 2015). MCC950 treatment effectively attenuated the inflammatory symptoms of NLRP3-related diseases, including experimental autoimmune meningitis and cryopyrin-associated periodic syndrome (CAPS) (Coll et al., 2015). OLT1177 is composed of β-sulfonyl nitrile that inhibits canonical and noncanonical NLRP3 activation (Marchetti et al., 2018a). OLT1177 lowers caspase-1 activity and IL-1 production in monocytes from patients with CAPS, limits adenosine triphosphatase (ATPase) activity, and binds directly to the NLRP3 protein. It also reduces LPS-induced systemic inflammation in rats (Marchetti et al., 2018b). The NLRP3 inflammasome inhibitors BAY 11-7082, BOT-4-one, parthenolide, INF 39, NSC 170724, and OLT1177 have median inhibitory concentration (IC50) values between 1 and 10 μM. However, this effect does not lead to a direct and specific binding action to NLRP3. The off-target effects cause BAY 11-7082, BOT-4-one, parthenolide, and INF 39 to exhibit a variety of behaviors (Jiang et al., 2017). Because the biological activity of NSC 170724 and OLT1177 in an NLRP3-dependent model could not be assessed, the real mechanism of inhibition is not clear. Oridonin creates a covalent link with NLRP3 while simultaneously inhibiting the activity of NF-κB and mitogen-activated protein kinase (MAPK) (He et al., 2018). Although CY-09 was designed as a derivative of a cystic fibrosis transmembrane conductance regulator (CFTR) inhibitor, a subsequent study showed that CY-09 preferentially bound to the NACHT ATPase site of NLRP3. IL-1β has an IC50 value of 5 μM (Jiang et al., 2017). β-Carotene inhibits macrophage activation by ATP, MSU crystals, and nigericin. β-Carotene reduced inflammation in a mouse model of gouty arthritis caused by MSU crystals (Yang et al., 2020a) and reduced IL-1β release in human synovial cells from patients with gout, which suggest an antigout action (Yang et al., 2020b). So far, three firms have entered the clinic with NLRP3 inhibitors (Schwaid and Spencer 2021). Olatec has published phase II results in acute gout for their chemical dapansutrile. This is the most clinically progressed NLRP3 inhibitor at the present (Jansen et al., 2019). Dapansutrile reduced joint discomfort by more than 50% after 3 days of therapy in their trial. Dapansutrile was given in doses of 100, 300, 1,000, or 2000 mg per day. Notably, there was no improvement in effectiveness beyond the 300 mg/day level, and treatment responses were identical in all groups after 3 days. Pain relief ranged from 56 to 68% on day 3 in individuals treated with 300 mg/day, which is comparable to pain relief found with NSAIDs or prednisolone (Klück et al., 2020).
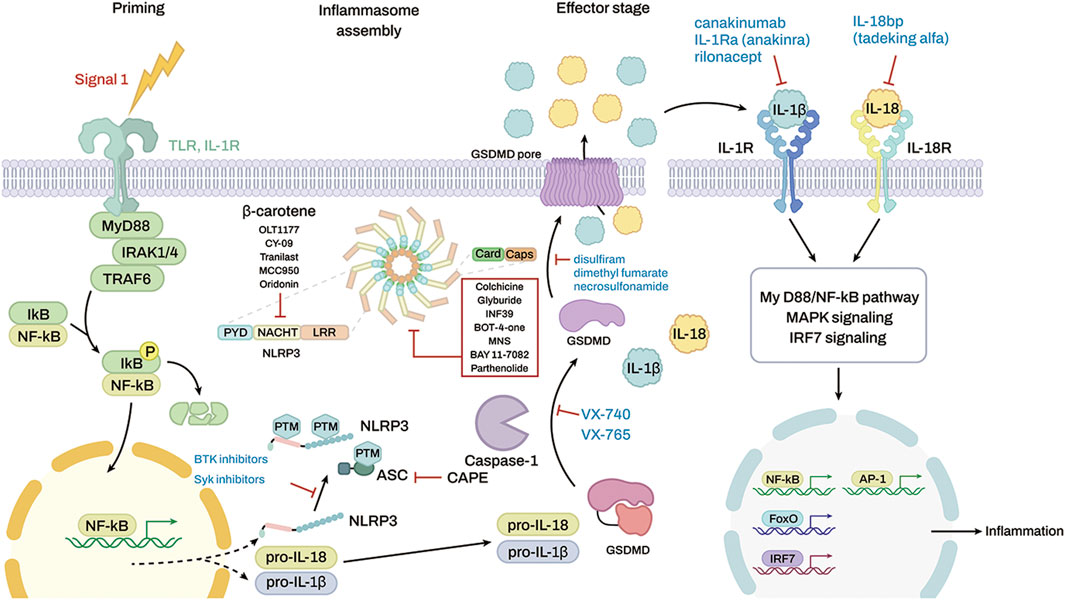
FIGURE 4. Targeting inflammasomes by inhibitors and the molecular targets of some anti-inflammatory drugs. The red box shows a list of inhibitors that stop the inflammasome function, but the molecular targets are unknown.
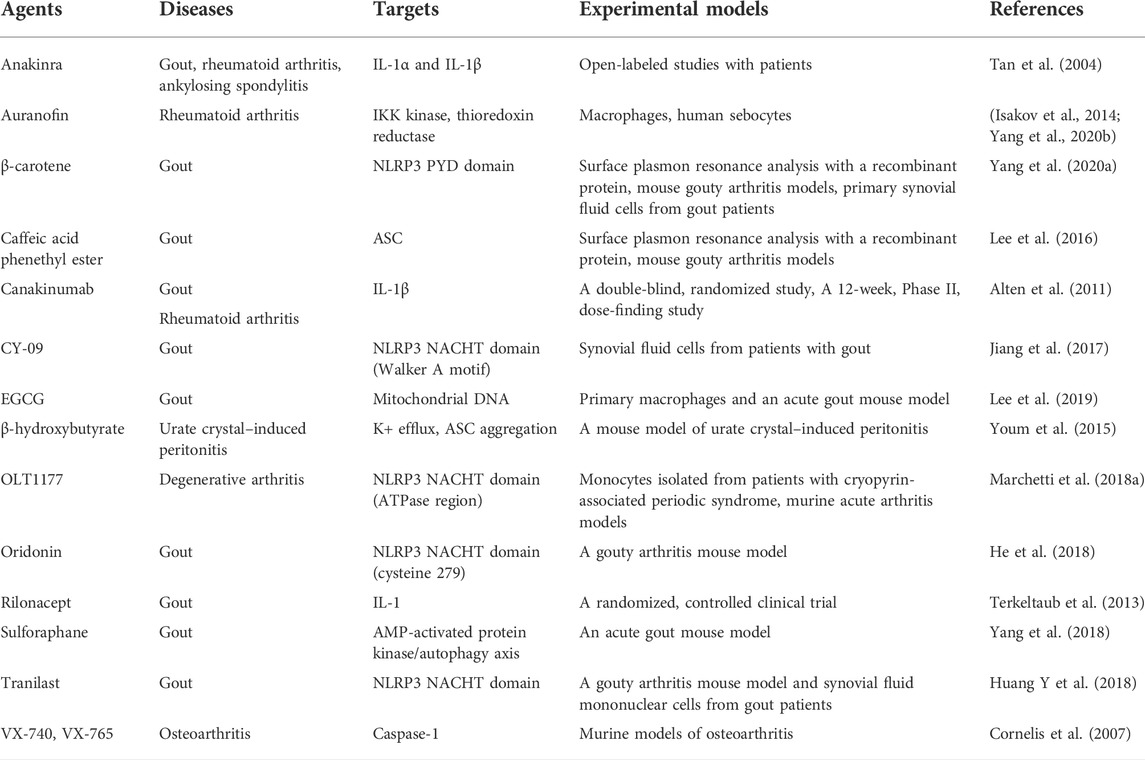
TABLE 1. Inhibitors of the NLRP3 inflammasome in rheumatic diseases.
Small endogenous non-coding RNAs called microRNAs (miRNAs) play a role in the post-transcriptional control of gene expression. They have a length of around 22 nucleotides. A growing body of research has shown that miRNAs are crucial in the development of certain autoimmune illnesses, such as RA, systemic lupus erythematosus (Xie and Xu 2018; Senousy et al., 2019), Sjögren’s syndrome (Jang et al., 2019), and systemic sclerosis (Iwamoto et al., 2016). Several microRNAs (miRNAs) post-transcriptionally regulate NLRP3 expression in RA. Reduced levels of miR-20a (Jin et al., 2021) and miR-21 (Dong et al., 2014; Guggino et al., 2018) aggravate RA by triggering the NLRP3 inflammasome pathway and boosting STAT3 expression, while lowering STAT5 expression, both of which are linked with an imbalance of Th17/Treg cells (Huang Q et al., 2018). MiR-223 is a negative regulator of NLRP3, and miR-223–3p targets the 3′-UTR of NLRP3 to reduce its expression in fibroblast-like synoviocytes (Bauernfeind et al., 2012; Wu et al., 2020). MiR-30a reduces the expression of NLRP3 in synovial macrophages via direct binding to the 3ʹ UTR, and AAV-miR-30a injection inhibited NLRP3 inflammasome activation in mice. A20 is a de-ubiquitinase and an E3 ligase that is induced by inflammatory cytokines, and it acts as a negative regulator of NF-κB signaling in the TLR and TNF-α pathways. Single nucleotide polymorphisms (SNPs) of A20 are associated with high susceptibility to autoimmune diseases and rheumatoid arthritis. Lack of A20 in myeloid cells initiated a spontaneous polyarthritis in mice and enhanced the expression levels of NLRP3 and pro-IL-1β in macrophages (Vande Walle et al., 2014). Deficiency of NLRP3 or caspase-1/-11 in A20 knockout mice protected against the inflammatory symptoms of rheumatoid arthritis, which showed that A20 negatively regulated NLRP3 inflammasome activity in rheumatoid arthritis (Vande Walle et al., 2014). These results suggest that intracellular signaling targets regulate NLRP3 inflammasome activity and directly target inflammasome components. The recent research suggests that miRNAs are promising candidates for better understanding the pathogenetic processes of RA and designing more effective treatment strategies.
Although there is currently no clinically accepted treatment that targets or modulates NLRP3 inflammasome activation, these investigational compounds may regulate its anti-inflammatory action in arthritic disease via several signaling pathways.
Conclusion and perspective
The role of the NLRP3 inflammasome in autoinflammatory disorders has been established. Patients with arthritis benefit enormously from the introduction of inflammasome-targeted biologics. Research on the activation of diverse inflammasomes has expanded, and the results have allowed for the development of highly targeted and effective inhibitors. Human cell models, rather than animal models, are required for the successful translation of newly identified inflammasome inhibitors into (pre)clinical trials. Differences between human and murine inflammasome biology alter the treatment effectiveness results drawn from murine illness models. The development of appropriate cellular models harboring patient mutations and the direct use of patient-derived cells may help identify therapeutically useful medicines more quickly and reliably.
Author contributions
GY wrote the first draft of the manuscript. HK, Y-YC, and HL helped writing, reviewing, and editing of the manuscript. JL finalized writing the manuscript.
Funding
This study was supported by grants from the National Research Foundation of Korea (NRF) (NRF-2019R1A2C2085739 and NRF-2020R1A4A2002894) funded by the Korean government (Ministry of Science, ICT and Future Planning).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Addobbati, C., da Cruz, H. L. A., Adelino, J. E., Melo Tavares Ramos, A. L., Fragoso, T. S., Domingues, A., et al. (2018). Polymorphisms and expression of inflammasome genes are associated with the development and severity of rheumatoid arthritis in Brazilian patients. Inflamm. Res. 67 (3), 255–264.
Akkoc, N. (2008). Are spondyloarthropathies as common as rheumatoid arthritis worldwide? A review. Curr. Rheumatol. Rep. 10 (5), 371–378.
Alippe, Y., and Mbalaviele, G. (2019). Omnipresence of inflammasome activities in inflammatory bone diseases. Semin. Immunopathol. 41 (5), 607–618. doi:10.1007/s00281-019-00753-4
Alippe, Y., Wang, C., Ricci, B., Xiao, J., Qu, C., Zou, W., et al. (2017). Bone matrix components activate the NLRP3 inflammasome and promote osteoclast differentiation. Sci. Rep. 7 (1), 6630. doi:10.1038/s41598-017-07014-0
Alten, R., Gomez-Reino, J., Durez, P., Beaulieu, A., Sebba, A., Krammer, G., et al. (2011). Efficacy and safety of the human anti-IL-1β monoclonal antibody canakinumab in rheumatoid arthritis: results of a 12-week, phase II, dose-finding study. BMC Musculoskelet. Disord. 12, 153. doi:10.1186/1471-2474-12-153
An, Y., Zhang, H., Wang, C., Jiao, F., Xu, H., Wang, X., et al. (2019). Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. Faseb J. 33 (11), 12515–12527. doi:10.1096/fj.201802805RR
Anzilotti, C., Pratesi, F., Tommasi, C., and Migliorini, P. (2010). Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun. Rev. 9 (3), 158–160. doi:10.1016/j.autrev.2009.06.002
Araki, Y., and Mimura, T. (2017). Matrix metalloproteinase gene activation resulting from disordred epigenetic mechanisms in rheumatoid arthritis. Int. J. Mol. Sci. 18 (5), E905. doi:10.3390/ijms18050905
Barreto, G., Soininen, A., Ylinen, P., Sandelin, J., Konttinen, Y. T., Nordström, D. C., et al. (2015). Soluble biglycan: A potential mediator of cartilage degradation in osteoarthritis. Arthritis Res. Ther. 17, 379. doi:10.1186/s13075-015-0902-0
Bauernfeind, F., Rieger, A., Schildberg, F. A., Knolle, P. A., Schmid-Burgk, J. L., and Hornung, V. (2012). NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 189 (8), 4175–4181. doi:10.4049/jimmunol.1201516
Bhaumik, S., and Basu, R. (2017). Cellular and molecular dynamics of Th17 differentiation and its developmental plasticity in the intestinal immune response. Front. Immunol. 8, 254. doi:10.3389/fimmu.2017.00254
Bläss, S., Union, A., Raymackers, J., Schumann, F., Ungethüm, U., Müller-Steinbach, S., et al. (2001). The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. 44 (4), 761–771. doi:10.1002/1529-0131(200104)44:4<761:AID-ANR132>3.0.CO;2-S
Broz, P., and Dixit, V. M. (2016). Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16 (7), 407–420. doi:10.1038/nri.2016.58
Cao, Y., Jansen, I. D., Sprangers, S., Stap, J., Leenen, P. J., Everts, V., et al. (2016). IL-1β differently stimulates proliferation and multinucleation of distinct mouse bone marrow osteoclast precursor subsets. J. Leukoc. Biol. 100 (3), 513–523. doi:10.1189/jlb.1A1215-543R
Chen, K., Jiao, Y., Liu, L., Huang, M., He, C., He, W., et al. (2020). Communications between bone marrow macrophages and bone cells in bone Remodeling. Front. Cell Dev. Biol. 8, 598263. doi:10.3389/fcell.2020.598263
Chen-Xu, M., Yokose, C., Rai, S. K., Pillinger, M. H., and Choi, H. K. (2019). Contemporary prevalence of gout and Hyperuricemia in the United States and decadal Trends: The national health and Nutrition Examination Survey, 2007-2016. Arthritis Rheumatol. 71 (6), 991–999. doi:10.1002/art.40807
Cheng, L., Liang, X., Qian, L., Luo, C., and Li, D. (2021). NLRP3 gene polymorphisms and expression in rheumatoid arthritis. Exp. Ther. Med. 22 (4), 1110. doi:10.3892/etm.2021.10544
Choulaki, C., Papadaki, G., Repa, A., Kampouraki, E., Kambas, K., Ritis, K., et al. (2015). Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res. Ther. 17 (1), 257. doi:10.1186/s13075-015-0775-2
Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Muñoz-Planillo, R., Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21 (3), 248–255. doi:10.1038/nm.3806
Cornelis, S., Kersse, K., Festjens, N., Lamkanfi, M., and Vandenabeele, P. (2007). Inflammatory caspases: Targets for novel therapies. Curr. Pharm. Des. 13 (4), 367–385. doi:10.2174/138161207780163006
Crofford, L. J. (2013). Use of NSAIDs in treating patients with arthritis. Arthritis Res. Ther. 15 (3), S2. doi:10.1186/ar4174
Crowson, C. S., Matteson, E. L., Myasoedova, E., Michet, C. J., Ernste, F. C., Warrington, K. J., et al. (2011). The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 63 (3), 633–639. doi:10.1002/art.30155
Dehlin, M., Jacobsson, L., and Roddy, E. (2020). Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 16 (7), 380–390. doi:10.1038/s41584-020-0441-1
Dinarello, C. A. (2011). A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur. J. Immunol. 41 (5), 1203–1217. doi:10.1002/eji.201141550
Dinarello, C. A. (1998). Interleukin-1 beta, interleukin-18, and the interleukin-1 beta converting enzyme. Ann. N. Y. Acad. Sci. 856, 1–11. doi:10.1111/j.1749-6632.1998.tb08307.x
Dong, L., Wang, X., Tan, J., Li, H., Qian, W., Chen, J., et al. (2014). Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J. Cell. Mol. Med. 18 (11), 2213–2224. doi:10.1111/jcmm.12353
Dougados, M., and Baeten, D. (2011). Spondyloarthritis. Lancet 377 (9783), 2127–2137. doi:10.1016/S0140-6736(11)60071-8
Dunne, A., Ross, P. J., Pospisilova, E., Masin, J., Meaney, A., Sutton, C. E., et al. (2010). Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J. Immunol. 185 (3), 1711–1719. doi:10.4049/jimmunol.1000105
Firestein, G. S. (2003). Evolving concepts of rheumatoid arthritis. Nature 423 (6937), 356–361. doi:10.1038/nature01661
Frevert, C. W., Felgenhauer, J., Wygrecka, M., Nastase, M. V., and Schaefer, L. (2018). Danger-associated molecular patterns derived from the extracellular matrix provide Temporal control of innate immunity. J. Histochem. Cytochem. 66 (4), 213–227. doi:10.1369/0022155417740880
George, G., Shyni, G. L., and Raghu, K. G. (2020). Current and novel therapeutic targets in the treatment of rheumatoid arthritis. Inflammopharmacology 28 (6), 1457–1476. doi:10.1007/s10787-020-00757-9
Guggino, G., Mauro, D., Rizzo, A., Alessandro, R., Raimondo, S., Bergot, A. S., et al. (2021). Inflammasome activation in ankylosing spondylitis is associated with gut dysbiosis. Arthritis Rheumatol. 73 (7), 1189–1199. doi:10.1002/art.41644
Guggino, G., Orlando, V., Saieva, L., Ruscitti, P., Cipriani, P., La Manna, M. P., et al. (2018). Downregulation of miRNA17-92 cluster marks Vγ9Vδ2 T cells from patients with rheumatoid arthritis. Arthritis Res. Ther. 20 (1), 236. doi:10.1186/s13075-018-1740-7
Hasnain, S. Z., Lourie, R., Das, I., Chen, A. C., and McGuckin, M. A. (2012). The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 90 (3), 260–270. doi:10.1038/icb.2011.112
He, H., Jiang, H., Chen, Y., Ye, J., Wang, A., Wang, C., et al. (2018). Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 9 (1), 2550. doi:10.1038/s41467-018-04947-6
He, Y., Hara, H., and Núñez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends biochem. Sci. 41 (12), 1012–1021. doi:10.1016/j.tibs.2016.09.002
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9 (8), 847–856. doi:10.1038/ni.1631
Huang, G., Wang, Y., and Chi, H. (2012). Regulation of TH17 cell differentiation by innate immune signals. Cell. Mol. Immunol. 9 (4), 287–295. doi:10.1038/cmi.2012.10
Huang, Q., Chen, S. S., Li, J., Tao, S. S., Wang, M., Leng, R. X., et al. (2018). miR-210 expression in PBMCs from patients with systemic lupus erythematosus and rheumatoid arthritis. Ir. J. Med. Sci. 187 (1), 243–249. doi:10.1007/s11845-017-1634-8
Huang, Y., Jiang, H., Chen, Y., Wang, X., Yang, Y., Tao, J., et al. (2018). Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 10 (4), e8689. doi:10.15252/emmm.201708689
Isakov, E., Weisman-Shomer, P., and Benhar, M. (2014). Suppression of the pro-inflammatory NLRP3/interleukin-1β pathway in macrophages by the thioredoxin reductase inhibitor auranofin. Biochim. Biophys. Acta 1840 (10), 3153–3161. doi:10.1016/j.bbagen.2014.07.012
Iwamoto, N., Vettori, S., Maurer, B., Brock, M., Pachera, E., Jüngel, A., et al. (2016). Downregulation of miR-193b in systemic sclerosis regulates the proliferative vasculopathy by urokinase-type plasminogen activator expression. Ann. Rheum. Dis. 75 (1), 303–310. doi:10.1136/annrheumdis-2014-205326
Janeway, C. A., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi:10.1146/annurev.immunol.20.083001.084359
Jang, S. I., Tandon, M., Teos, L., Zheng, C., Warner, B. M., and Alevizos, I. (2019). Dual function of miR-1248 links interferon induction and calcium signaling defects in Sjögren's syndrome. EBioMedicine 48, 526–538. doi:10.1016/j.ebiom.2019.09.010
Jansen, T., Klück, V., Janssen, M., Comarniceanu, A., Efdé, M., Scribner, C., et al. (2019). P160 the first phase 2A proof-of-concept study of a selective NLRP3 inflammasome inhibitor, dapansutrile™ (OLT1177™), in acute gout. Ann. Rheumatic Dis. 78 (1), A70–A71.
Jiang, H., He, H., Chen, Y., Huang, W., Cheng, J., Ye, J., et al. (2017). Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 214 (11), 3219–3238. doi:10.1084/jem.20171419
Jiang, N., An, J., Yang, K., Liu, J., Guan, C., Ma, C., et al. (2021). NLRP3 inflammasome: A new target for prevention and control of osteoporosis? Front. Endocrinol. 12, 752546. doi:10.3389/fendo.2021.752546
Jin, S., Sun, S., Ling, H., Ma, J., Zhang, X., Xie, Z., et al. (2021). Protectin DX restores Treg/T(h)17 cell balance in rheumatoid arthritis by inhibiting NLRP3 inflammasome via miR-20a. Cell Death Dis. 12 (3), 280. doi:10.1038/s41419-021-03562-6
Jin, Y., Mailloux, C. M., Gowan, K., Riccardi, S. L., LaBerge, G., Bennett, D. C., et al. (2007). NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 356 (12), 1216–1225. doi:10.1056/NEJMoa061592
Kanneganti, T. D., Lamkanfi, M., and Núñez, G. (2007). Intracellular NOD-like receptors in host defense and disease. Immunity 27 (4), 549–559. doi:10.1016/j.immuni.2007.10.002
Kim, H., Walsh, M. C., Takegahara, N., Middleton, S. A., Shin, H. I., Kim, J., et al. (2017). The purinergic receptor P2X5 regulates inflammasome activity and hyper-multinucleation of murine osteoclasts. Sci. Rep. 7 (1), 196. doi:10.1038/s41598-017-00139-2
Kim, H. W., Kwon, Y. J., Park, B. W., Song, J. J., Park, Y. B., and Park, M. C. (2017). Differential expressions of NOD-like receptors and their associations with inflammatory responses in rheumatoid arthritis. Clin. Exp. Rheumatol. 35 (4), 630–637.
Kim, S. H., Lee, J. H., Jeong, H. J., Kim, J. M., Baek, W. K., Kim, T. H., et al. (2022). Clinical significance of elevated serum caspase-1 levels in patients with ankylosing spondylitis. Ann. Lab. Med. 42 (2), 293–295. doi:10.3343/alm.2022.42.2.293
Kim, S. K., Cho, Y. J., and Choe, J. Y. (2018). NLRP3 inflammasomes and NLRP3 inflammasome-derived proinflammatory cytokines in peripheral blood mononuclear cells of patients with ankylosing spondylitis. Clin. Chim. Acta. 486, 269–274. doi:10.1016/j.cca.2018.08.022
Kim, S. K., Choe, J. Y., and Park, K. Y. (2019). TXNIP-mediated nuclear factor-κB signaling pathway and intracellular shifting of TXNIP in uric acid-induced NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 511 (4), 725–731. doi:10.1016/j.bbrc.2019.02.141
Klück, V., Jansen, T., Janssen, M., Comarniceanu, A., Efdé, M., Tengesdal, I. W., et al. (2020). Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: An open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2 (5), e270–e280. doi:10.1016/s2665-9913(20)30065-5
Komatsu, N., and Takayanagi, H. (2022). Mechanisms of joint destruction in rheumatoid arthritis - immune cell-fibroblast-bone interactions. Nat. Rev. Rheumatol. 18 (7), 415–429. doi:10.1038/s41584-022-00793-5
Lamkanfi, M., and Dixit, V. M. (2014). Mechanisms and functions of inflammasomes. Cell 157 (5), 1013–1022. doi:10.1016/j.cell.2014.04.007
Lee, H. E., Yang, G., Kim, N. D., Jeong, S., Jung, Y., Choi, J. Y., et al. (2016). Targeting ASC in NLRP3 inflammasome by caffeic acid phenethyl ester: A novel strategy to treat acute gout. Sci. Rep. 6, 38622. doi:10.1038/srep38622
Lee, H. E., Yang, G., Park, Y. B., Kang, H. C., Cho, Y. Y., Lee, H. S., et al. (2019). Epigallocatechin-3-Gallate prevents acute gout by suppressing NLRP3 inflammasome activation and mitochondrial DNA synthesis. Molecules 24 (11), E2138. doi:10.3390/molecules24112138
Lee, M. S. (2011). Role of innate immunity in diabetes and metabolism: recent progress in the study of inflammasomes. Immune Netw. 11 (2), 95–99. doi:10.4110/in.2011.11.2.95
Levandowski, C. B., Mailloux, C. M., Ferrara, T. M., Gowan, K., Ben, S., Jin, Y., et al. (2013). NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1β processing via the NLRP1 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 110 (8), 2952–2956. doi:10.1073/pnas.1222808110
Madel, M. B., Ibáñez, L., Wakkach, A., de Vries, T. J., Teti, A., Apparailly, F., et al. (2019). Immune function and diversity of osteoclasts in Normal and pathological conditions. Front. Immunol. 10, 1408. doi:10.3389/fimmu.2019.01408
Maksymowych, W. P. (2010). Disease modification in ankylosing spondylitis. Nat. Rev. Rheumatol. 6 (2), 75–81. doi:10.1038/nrrheum.2009.258
Maksymowych, W. P., Elewaut, D., and Schett, G. (2012). Motion for debate: The development of ankylosis in ankylosing spondylitis is largely dependent on inflammation. Arthritis Rheum. 64 (6), 1713–1719. doi:10.1002/art.34442
Marchetti, C., Swartzwelter, B., Gamboni, F., Neff, C. P., Richter, K., Azam, T., et al. (2018a). OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. U. S. A. 115 (7), E1530–E1539. doi:10.1073/pnas.1716095115
Marchetti, C., Swartzwelter, B., Koenders, M. I., Azam, T., Tengesdal, I. W., Powers, N., et al. (2018b). NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res. Ther. 20 (1), 169. doi:10.1186/s13075-018-1664-2
Martinon, F., Pétrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440 (7081), 237–241. doi:10.1038/nature04516
Mathews, R. J., Robinson, J. I., Battellino, M., Wong, C., Taylor, J. C., Eyre, S., et al. (2014). Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann. Rheum. Dis. 73 (6), 1202–1210. doi:10.1136/annrheumdis-2013-203276
Maxwell, L. J., Zochling, J., Boonen, A., Singh, J. A., Veras, M. M., Tanjong Ghogomu, E., et al. (2015). TNF-alpha inhibitors for ankylosing spondylitis. Cochrane Database Syst. Rev. 4, Cd005468. doi:10.1002/14651858.CD005468.pub2
McInnes, I. B., and Schett, G. (2011). The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 365 (23), 2205–2219. doi:10.1056/NEJMra1004965
Meng, G., Zhang, F., Fuss, I., Kitani, A., and Strober, W. (2009). A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity 30 (6), 860–874. doi:10.1016/j.immuni.2009.04.012
Mertens, M., and Singh, J. A. (2009). Anakinra for rheumatoid arthritis: A systematic review. J. Rheumatol. 36 (6), 1118–1125. doi:10.3899/jrheum.090074
Mishra, N., Schwerdtner, L., Sams, K., Mondal, S., Ahmad, F., Schmidt, R. E., et al. (2019). Cutting Edge: Protein arginine deiminase 2 and 4 regulate NLRP3 inflammasome-dependent IL-1β maturation and ASC speck formation in macrophages. J. Immunol. 203 (4), 795–800. doi:10.4049/jimmunol.1800720
Muruve, D. A., Pétrilli, V., Zaiss, A. K., White, L. R., Clark, S. A., Ross, P. J., et al. (2008). The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452 (7183), 103–107. doi:10.1038/nature06664
Novack, D. V. (2011). Role of NF-κB in the skeleton. Cell Res. 21 (1), 169–182. doi:10.1038/cr.2010.159
Okamoto, K., Nakashima, T., Shinohara, M., Negishi-Koga, T., Komatsu, N., Terashima, A., et al. (2017). Osteoimmunology: The Conceptual Framework Unifying the immune and skeletal systems. Physiol. Rev. 97 (4), 1295–1349. doi:10.1152/physrev.00036.2016
Pap, T., and Korb-Pap, A. (2015). Cartilage damage in osteoarthritis and rheumatoid arthritis--two unequal siblings. Nat. Rev. Rheumatol. 11 (10), 606–615. doi:10.1038/nrrheum.2015.95
Pontillo, A., Brandao, L., Guimaraes, R., Segat, L., Araujo, J., and Crovella, S. (2010). Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity 43 (8), 583–589. doi:10.3109/08916930903540432
Quaden, D. H., De Winter, L. M., and Somers, V. (2016). Detection of novel diagnostic antibodies in ankylosing spondylitis: An overview. Autoimmun. Rev. 15 (8), 820–832. doi:10.1016/j.autrev.2016.06.001
Ramírez, J., and Cañete, J. D. (2018). Anakinra for the treatment of rheumatoid arthritis: A safety evaluation. Expert Opin. Drug Saf. 17 (7), 727–732. doi:10.1080/14740338.2018.1486819
Roper, P. M., Shao, C., and Veis, D. J. (2020). Multitasking by the OC lineage during bone infection: Bone resorption, immune modulation, and microbial Niche. Cells 9 (10), E2157. doi:10.3390/cells9102157
Ruscitti, P., Cipriani, P., Di Benedetto, P., Liakouli, V., Berardicurti, O., Carubbi, F., et al. (2015). Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1β via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: A possible implication for therapeutic decision in these patients. Clin. Exp. Immunol. 182 (1), 35–44. doi:10.1111/cei.12667
Safiri, S., Kolahi, A. A., Hoy, D., Smith, E., Bettampadi, D., Mansournia, M. A., et al. (2019). Global, regional and national burden of rheumatoid arthritis 1990-2017: A systematic analysis of the Global Burden of disease study 2017. Ann. Rheum. Dis. 78 (11), 1463–1471. doi:10.1136/annrheumdis-2019-215920
Sato, K., Suematsu, A., Okamoto, K., Yamaguchi, A., Morishita, Y., Kadono, Y., et al. (2006). Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 203 (12), 2673–2682. doi:10.1084/jem.20061775
Schroder, K., Zhou, R., and Tschopp, J. (2010). The NLRP3 inflammasome: A sensor for metabolic danger? Science 327 (5963), 296–300. doi:10.1126/science.1184003
Schwaid, A. G., and Spencer, K. B. (2021). Strategies for targeting the NLRP3 inflammasome in the clinical and Preclinical space. J. Med. Chem. 64 (1), 101–122. doi:10.1021/acs.jmedchem.0c01307
Schweyer, S., Hemmerlein, B., Radzun, H. J., and Fayyazi, A. (2000). Continuous recruitment, co-expression of tumour necrosis factor-alpha and matrix metalloproteinases, and apoptosis of macrophages in gout tophi. Virchows Arch. 437 (5), 534–539. doi:10.1007/s004280000282
Senousy, M. A., Helmy, H. S., Fathy, N., Shaker, O. G., and Ayeldeen, G. M. (2019). Association of MTMR3 rs12537 at miR-181a binding site with rheumatoid arthritis and systemic lupus erythematosus risk in Egyptian patients. Sci. Rep. 9 (1), 12299. doi:10.1038/s41598-019-48770-5
Seok, J. K., Kang, H. C., Cho, Y. Y., Lee, H. S., and Lee, J. Y. (2020). Regulation of the NLRP3 inflammasome by post-Translational Modifications and small molecules. Front. Immunol. 11, 618231. doi:10.3389/fimmu.2020.618231
Seok, J. K., Kang, H. C., Cho, Y. Y., Lee, H. S., and Lee, J. Y. (2021). Therapeutic regulation of the NLRP3 inflammasome in chronic inflammatory diseases. Arch. Pharm. Res. 44 (1), 16–35. doi:10.1007/s12272-021-01307-9
Shi, Y., Evans, J. E., and Rock, K. L. (2003). Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425 (6957), 516–521. doi:10.1038/nature01991
Shim, J. H., Stavre, Z., and Gravallese, E. M. (2018). Bone loss in rheumatoid arthritis: Basic mechanisms and clinical implications. Calcif. Tissue Int. 102 (5), 533–546. doi:10.1007/s00223-017-0373-1
Siddiqui, J. A., and Partridge, N. C. (2016). Physiological bone Remodeling: Systemic regulation and growth factor involvement. Physiol. (Bethesda) 31 (3), 233–245. doi:10.1152/physiol.00061.2014
So, A. K., and Martinon, F. (2017). Inflammation in gout: Mechanisms and therapeutic targets. Nat. Rev. Rheumatol. 13 (11), 639–647. doi:10.1038/nrrheum.2017.155
Stamp, L. K., and Farquhar, H. (2021). Treatment advances in gout. Best. Pract. Res. Clin. Rheumatol. 35 (4), 101719. doi:10.1016/j.berh.2021.101719
Sui, J., Li, H., Fang, Y., Liu, Y., Li, M., Zhong, B., et al. (2012). NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han Chinese. Arthritis Rheum. 64 (3), 647–654. doi:10.1002/art.33370
Sutterwala, F. S., Ogura, Y., Szczepanik, M., Lara-Tejero, M., Lichtenberger, G. S., Grant, E. P., et al. (2006). Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24 (3), 317–327. doi:10.1016/j.immuni.2006.02.004
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19 (8), 477–489. doi:10.1038/s41577-019-0165-0
Takayanagi, H. (2007). Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 7 (4), 292–304. doi:10.1038/nri2062
Tan, A. L., Marzo-Ortega, H., O'Connor, P., Fraser, A., Emery, P., and McGonagle, D. (2004). Efficacy of anakinra in active ankylosing spondylitis: A clinical and magnetic resonance imaging study. Ann. Rheum. Dis. 63 (9), 1041–1045. doi:10.1136/ard.2004.020800
Terkeltaub, R. A., Schumacher, H. R., Carter, J. D., Baraf, H. S., Evans, R. R., Wang, J., et al. (2013). Rilonacept in the treatment of acute gouty arthritis: A randomized, controlled clinical trial using indomethacin as the active comparator. Arthritis Res. Ther. 15 (1), R25. doi:10.1186/ar4159
Tsark, E. C., Wang, W., Teng, Y. C., Arkfeld, D., Dodge, G. R., and Kovats, S. (2002). Differential MHC class II-mediated presentation of rheumatoid arthritis autoantigens by human dendritic cells and macrophages. J. Immunol. 169 (11), 6625–6633. doi:10.4049/jimmunol.169.11.6625
Tsui, F. W., Tsui, H. W., Akram, A., Haroon, N., and Inman, R. D. (2014). The genetic basis of ankylosing spondylitis: New insights into disease pathogenesis. Appl. Clin. Genet. 7, 105–115. doi:10.2147/TACG.S37325
van de Veerdonk, F. L., Joosten, L. A., Shaw, P. J., Smeekens, S. P., Malireddi, R. K., van der Meer, J. W., et al. (2011). The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur. J. Immunol. 41 (8), 2260–2268. doi:10.1002/eji.201041226
van de Veerdonk, F. L., and Netea, M. G. (2013). New insights in the Immunobiology of IL-1 family members. Front. Immunol. 4, 167. doi:10.3389/fimmu.2013.00167
Vande Walle, L., Van Opdenbosch, N., Jacques, P., Fossoul, A., Verheugen, E., Vogel, P., et al. (2014). Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512 (7512), 69–73. doi:10.1038/nature13322
Wu, Z. M., Luo, J., Shi, X. D., Zhang, S. X., Zhu, X. B., and Guo, J. (2020). Icariin alleviates rheumatoid arthritis via regulating miR-223-3p/NLRP3 signalling axis. Autoimmunity 53 (8), 450–458. doi:10.1080/08916934.2020.1836488
Xie, L., and Xu, J. (2018). Role of MiR-98 and its Underlying mechanisms in systemic lupus erythematosus. J. Rheumatol. 45 (10), 1397–1405. doi:10.3899/jrheum.171290
Yan, J. W., Wang, Y. J., Peng, W. J., Tao, J. H., Wan, Y. N., Li, B. Z., et al. (2014). Therapeutic potential of interleukin-17 in inflammation and autoimmune diseases. Expert Opin. Ther. Targets 18 (1), 29–41. doi:10.1517/14728222.2013.843669
Yang, G., Lee, H. E., Moon, S. J., Ko, K. M., Koh, J. H., Seok, J. K., et al. (2020a). Direct binding to NLRP3 pyrin domain as a novel strategy to prevent NLRP3-driven inflammation and gouty arthritis. Arthritis Rheumatol. 72 (7), 1192–1202. doi:10.1002/art.41245
Yang, G., Lee, S. J., Kang, H. C., Cho, Y. Y., Lee, H. S., Zouboulis, C. C., et al. (2020b). Repurposing auranofin, an anti-rheumatic Gold compound, to treat Acne Vulgaris by targeting the NLRP3 inflammasome. Biomol. Ther. 28 (5), 437–442. doi:10.4062/biomolther.2020.004
Yang, G., Yeon, S. H., Lee, H. E., Kang, H. C., Cho, Y. Y., Lee, H. S., et al. (2018). Suppression of NLRP3 inflammasome by oral treatment with sulforaphane alleviates acute gouty inflammation. Rheumatol. Oxf. 57 (4), 727–736. doi:10.1093/rheumatology/kex499
Yang, X., Zhan, N., Jin, Y., Ling, H., Xiao, C., Xie, Z., et al. (2021). Tofacitinib restores the balance of γδTreg/γδT17 cells in rheumatoid arthritis by inhibiting the NLRP3 inflammasome. Theranostics 11 (3), 1446–1457. doi:10.7150/thno.47860
Yi, Y. S. (2018). Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J. Physiol. Pharmacol. 22 (1), 1–15. doi:10.4196/kjpp.2018.22.1.1
Yoo, S. A., You, S., Yoon, H. J., Kim, D. H., Kim, H. S., Lee, K., et al. (2012). A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. J. Exp. Med. 209 (4), 871–886. doi:10.1084/jem.20111783
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21 (3), 263–269. doi:10.1038/nm.3804
Zeng, L., Lindstrom, M. J., and Smith, J. A. (2011). Ankylosing spondylitis macrophage production of higher levels of interleukin-23 in response to lipopolysaccharide without induction of a significant unfolded protein response. Arthritis Rheum. 63 (12), 3807–3817. doi:10.1002/art.30593
Zhang, L., Wang, Q., Su, H., and Cheng, J. (2021). Exosomes from adipose derived mesenchymal stem cells alleviate diabetic osteoporosis in rats through suppressing NLRP3 inflammasome activation in osteoclasts. J. Biosci. Bioeng. 131 (6), 671–678. doi:10.1016/j.jbiosc.2021.02.007
Zhang, Q. B., Qing, Y. F., He, Y. L., Xie, W. G., and Zhou, J. G. (2018). Association of NLRP3 polymorphisms with susceptibility to primary gouty arthritis in a Chinese Han population. Clin. Rheumatol. 37 (1), 235–244. doi:10.1007/s10067-017-3900-6
Zhernakova, A., Withoff, S., and Wijmenga, C. (2013). Clinical implications of shared genetics and pathogenesis in autoimmune diseases. Nat. Rev. Endocrinol. 9 (11), 646–659. doi:10.1038/nrendo.2013.161
Zhu, H., Jian, Z., Zhong, Y., Ye, Y., Zhang, Y., Hu, X., et al. (2021). Janus kinase inhibition Ameliorates Ischemic Stroke injury and Neuroinflammation through reducing NLRP3 inflammasome activation via JAK2/STAT3 pathway inhibition. Front. Immunol. 12, 714943. doi:10.3389/fimmu.2021.714943
Zhu, W., He, X., Cheng, K., Zhang, L., Chen, D., Wang, X., et al. (2019). Ankylosing spondylitis: Etiology, pathogenesis, and treatments. Bone Res. 7, 22. doi:10.1038/s41413-019-0057-8
Zou, Y. C., Yan, L. M., Gao, Y. P., Wang, Z. Y., and Liu, G. (2020). miR-21 may act as a potential mediator between inflammation and abnormal bone formation in ankylosing spondylitis based on TNF-α concentration-dependent manner through the JAK2/STAT3 pathway. Dose. Response. 18 (1), 1559325819901239. doi:10.1177/1559325819901239
Keywords: inflammation, pharmacological inhibitors, therapeutic targets, immunity, pattern recognition receptors
Citation: Yang G, Kang HC, Cho Y-Y, Lee HS and Lee JY (2022) Inflammasomes and their roles in arthritic disease pathogenesis. Front. Mol. Biosci. 9:1027917. doi: 10.3389/fmolb.2022.1027917
Received: 25 August 2022; Accepted: 17 October 2022;
Published: 28 October 2022.
Edited by:
Jinhui Tao, University of Science and Technology of China, ChinaReviewed by:
Jaime Díaz-Zúñiga, University of Chile, ChileWeiming Ouyang, United States Food and Drug Administration, United States
Copyright © 2022 Yang, Kang, Cho, Lee and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joo Young Lee, joolee@catholic.ac.kr