 Xinjie Xu1,2,3†
Xinjie Xu1,2,3† Shengkang Huang1,2†
Shengkang Huang1,2† Xinyi Xiao1,2†
Xinyi Xiao1,2† Qihang Sun1,2
Qihang Sun1,2 Xiaoqian Liang1,2
Xiaoqian Liang1,2 Sifei Chen1,2
Sifei Chen1,2 Zijing Zhao1,2Zhaochang Huo1,2
Zijing Zhao1,2Zhaochang Huo1,2 Sanfang Tu1*
Sanfang Tu1* Yuhua Li1*
Yuhua Li1*- 1Department of Hematology, Zhujiang Hospital, Southern Medical University, Guangzhou, China
- 2The Second School of Clinical Medicine, Southern Medical University, Guangzhou, China
- 3State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Chimeric antigen receptor (CAR) T-cell therapy exhibits desirable and robust efficacy in patients with acute lymphoblastic leukemia (ALL). Stimulated by the revolutionized progress in the use of FDA-approved CD19 CAR T cells, novel agents with CAR designs and targets are being produced in pursuit of superior performance. However, on the path from bench to bedside, new challenges emerge. Accessibility is considered the initial barrier to the transformation of this patient-specific product into a commercially available product. To ensure infusion safety, profound comprehension of adverse events and proactive intervention are required. Additionally, resistance and relapse are the most critical and intractable issues in CAR T-cell therapy for ALL, thus precluding its further development. Understanding the limitations through up-to-date insights and characterizing multiple strategies will be critical to leverage CAR T-cell therapy flexibly for use in clinical situations. Herein, we provide an overview of the application of CAR T-cell therapy in ALL, emphasizing the main challenges and potential clinical strategies in an effort to promote a standardized set of treatment paradigms for ALL.
Introduction
Chimeric antigen receptor (CAR) T-cell therapy has achieved impressive outcomes in the treatment of hematological malignancies, especially providing a potentially curative option for patients with relapsed/refractory B-cell acute lymphoblastic leukemia (R/R B-ALL). Before the emergence of CAR T-cell therapy, the prognosis of patients with R/R ALL was poor, with a 5-year survival rate of 21% in children (1) and 10% in adults (2). However, CAR T-cell therapy has improved the situation remarkably, with a high complete remission (CR) rate of 57% to 93% (Table 1). In 2017, the Food and Drug Administration (FDA) approved the first CAR T-cell therapy drug, tisagenlecleucel, for the treatment of R/R B-ALL patients under 25 years old (15). Afterwards, a remarkable growth spurt of clinical application was observed, and the developments of CAR T-cell therapy in R/R B-ALL are quite rapid and promising.
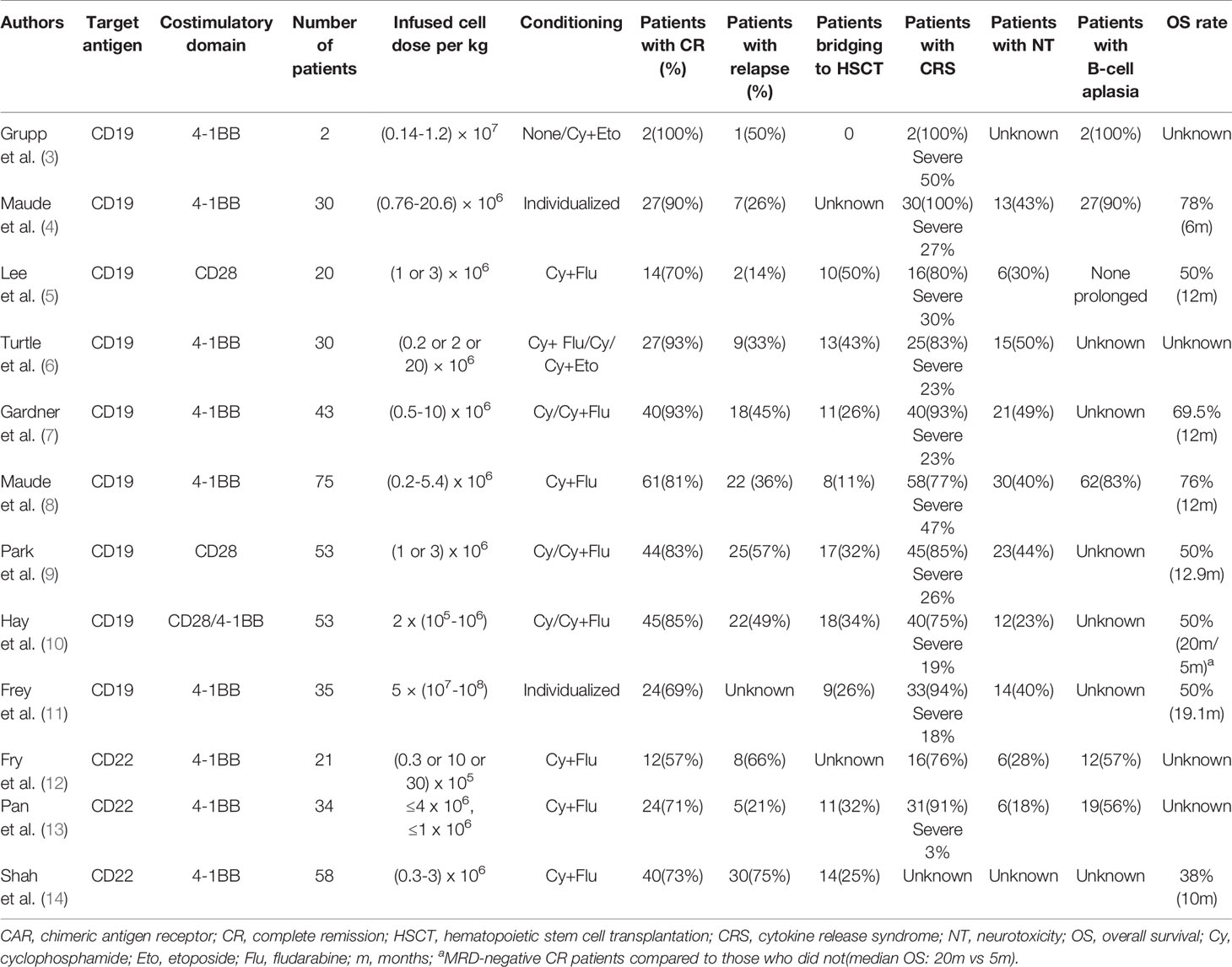
Table 1 Selected published clinical trials of CAR T-cell therapy.
CAR T-cells are genetically modified T-cells that recognize specific antigens in a human leukocyte antigen (HLA)-independent method. The basic structure is composed of an extracellular domain containing single-chain variable fragments (scFvs) for antigen recognition, a transmembrane domain mainly for CAR stability support, and an endodomain including a costimulatory signaling domain (commonly CD28 or 4-1BB) and an intracellular T-cell receptor signaling domain (typically CD3ζ) for T-cell activation. Based on different intracellular domains and other modifications, CAR T-cell products can be divided into four generations. Traditional CAR T-cell administration involves patient screening, enrollment and apheresis for T-cell collection, CAR T-cell generation, pretreatment and infusion. Efficacy and safety assessments and long-term follow-up are required (Figure 1).
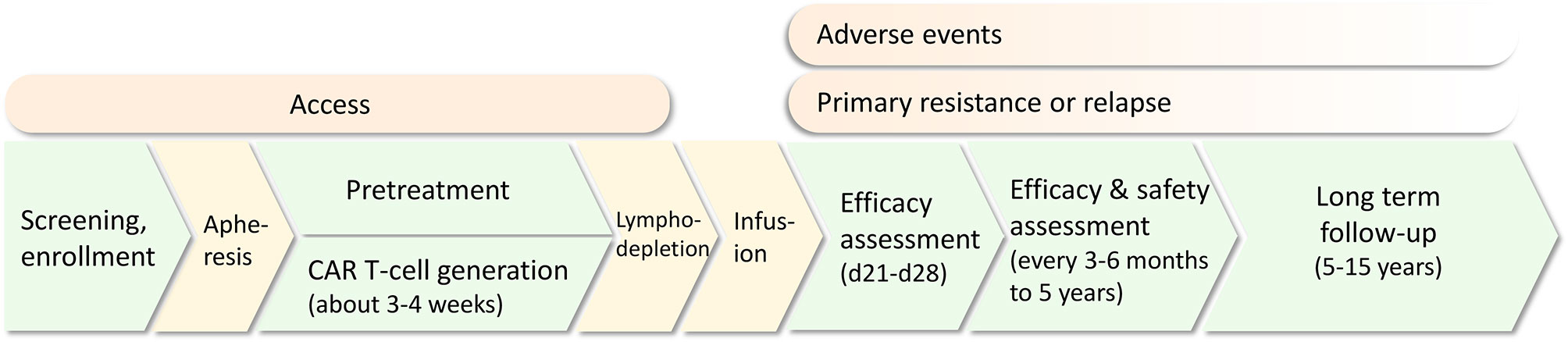
Figure 1 The common treatment protocol of CAR T-cell therapy for relapsed/refractory acute lymphoblastic leukemia (R/R ALL). Challenges including access, adverse events, primary resistance and relapse are present in different treatment procedures.
Although the procedure has been gradually standardized, there are still some limitations in several stages that impede the expanding application of CAR T-cell therapy in the clinic. Access to therapy cannot be guaranteed for every patient; adverse events highlight safety issues; and more importantly, disease relapse is an intractable issue restricting the development of CAR T-cell therapy. Correspondingly, multiple preventive and therapeutic strategies are being proposed to solve these challenges, from developing the CAR structure or optimizing clinical administration protocols. In this review, we summarize the application of CAR T-cell therapy in ALL and its development, emphasizing the main challenges and corresponding strategies. CAR engineering strategies in basic research are beyond the scope of this review; rather, we focus our attention on clinical strategies in an effort to facilitate the clinical practice of these attractive therapies.
The Application of CAR T-Cell Therapy in B-All
Target antigen selection is the key for accurate killing. Ideal targets ought to be highly expressed in all tumor cells, with no expression in normal tissues, and do not downregulate expression under immune pressure. For ALL, at present, there are several relatively widely accepted targets in clinical applications with extraordinary outcomes; furthermore, many emerging alternative targets are being proposed and are under active research (Table 2).
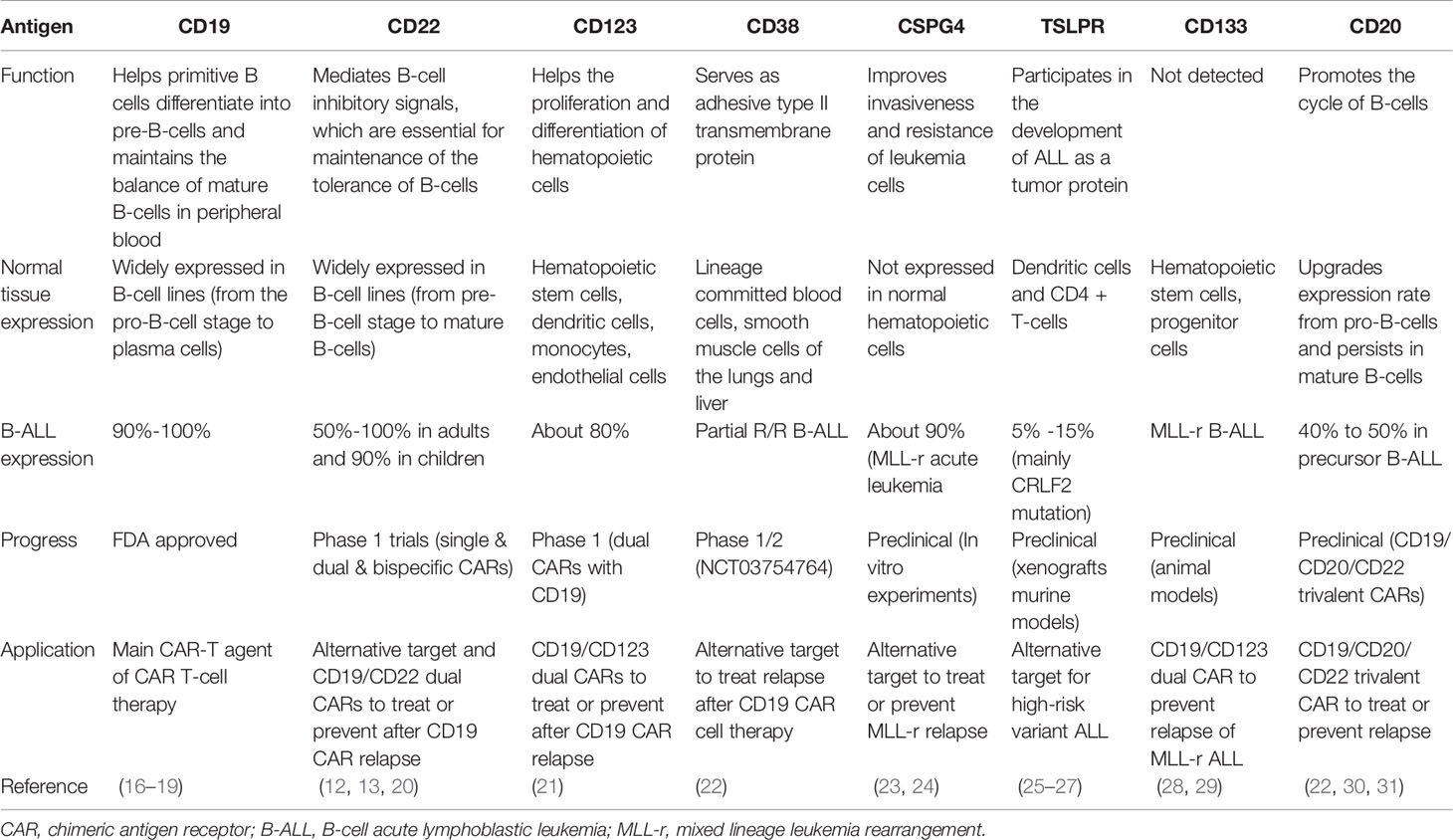
Table 2 Target antigens of CAR T-cell therapy.
CD19
CD19 is currently the most widely used and highly developed option for the treatment of B-ALL and is applied in almost 70% of CAR T-cell therapies (32, 33). CD19 is universally expressed in B-cell lines and plays an important role in the maturation of B-cells. Since the first two patients treated with tisagenlecleucel (CTL019) were reported in 2013 (3), further studies have developed rapidly. A global, multicenter, phase II clinical trial, ELIANA (NCT02435849), became a crucial study for the clinical application of CAR T-cell therapy (8), in which 75 ALL patients from 25 research centers had an overall response rate of 81% with minimal residual disease (MRD) negativity after the infusion of CTL019. The rates of event-free survival (EFS) and overall survival (OS) for 6 months were 73% and 60%, respectively, and for one year, 63% and 46%, respectively, which favorably achieved the FDA’s audit standards for approval. These data also manifested significant superiority in contrast with data of other FDA-approved agents, such as clofarabine and blinatumomab (34, 35). Simultaneously, different constructs of CD19 CAR T-cells have been tested with various outcomes (5–7, 9). A meta-analysis (36) included 35 clinical trials with different costimulatory domains, scFv clones, or T-cell origins and reported that the pooled CR was 80% (95% CI, 75.5–84.8; I²=56.96%). However, most completed trials are early phase single-arm trials. Further comparison studies are warranted to confirm the efficacy and determine associated influential factors.
CD22
CD22 is a common alternative target in clinical trials. CD22 is also restricted to the B-cell lineage, expressed in 50% to 100% of adult ALL patients and approximately 90% of pediatric patients (37, 38). A common expression threshold of eligibility for anti-CD22-directed therapy trials is at least 20% to 30% of tumor cells (39). Pan et al. (13) reported a 70.5% (24/34) total CR rate induced by CD22 CAR T-cells on day 30. Among these patients, 91% (31/34) previously exhibited CD19 CAR T-cell therapy failure. Fry et al. (12) published initial data in 2018 that CR induced by their CD22 CAR T-cells was 57% (11/21). Recently, the study was updated to include 55 B-ALL patients (51 after prior CD19-targeted therapy) infused with CD22 CAR T-cells and adopted an improved manufacturing method. The CR rate was 70%, and the median OS was 13.4 months (95% CI, 7.7–20.3 months), validating CD22 CAR T-cell therapy as an effective salvage regimen for patients who fail CD19-targeted therapies (14). However, there are still some limitations of CD19 and CD22 CAR T-cells, which will be discussed below.
CD123
CD123 is widely expressed in the hematological system without lineage restriction, both in normal cells such as hematopoietic stem cells, dendritic cells, monocytes, endothelial cells, and malignant cells such as acute myeloid leukemia and ALL (21). Ruella et al. (40) proved the robust potency of CD123 CAR T-cells on primitive B-ALL cells and CD19-negative B-ALL cells in vitro and in a murine model. Because CD123 is expressed in most CD19-negative relapsed or inherent CD19-resistant subpopulations, CD123 CAR T-cell therapy is anticipated to be an ideal prevention or remedy for post-CD19 CAR relapse. However, CD123 is also expressed on normal hematopoietic stem cells, and irreversible myeloablative impacts of CD123 CAR T-cells were reported by previous studies (41, 42). On-target off-tumor toxicity should be considered carefully when translating this therapy into clinical practice.
CD38
CD38, an adhesive type II transmembrane protein (22), is expressed in monocytes and smooth muscle cells in the liver and lung and activates T-cells in normal tissues. It could also be detected in R/R B-ALL (43, 44) and some attempts have been made to apply anti-CD38 CAR T-cells in a phase 1/2 clinical trial (NCT03754764). Guo et al. (45) reported a preliminary case of CD38 CAR T-cells in an R/R B-ALL patient after bispecific CD19/CD22 CAR T-cell failure. CD38 CAR T-cells reduced the tumor burden in bone marrow and blood but caused uncontrollable cytokine release syndrome (CRS). Obvious off-tumor effects have been found due to CD38 expression in normal cells, especially in CAR T-cells, resulting in fratricide and short-term survival. Locking CD38 with antibodies or proteins may be capable of avoiding autolysis, ensuring the continuous proliferation and long-term potency of CD38 CAR T-cells in future clinical applications (46). Ongoing efforts to confirm persistence and safety issues of CD38 CAR T-cell therapy in the field of leukemia are underway.
BAFF-R
B-cell activating factor receptor (BAFF-R, also known as TNFRSF13C), as the main receptor for BAFF, is responsible for B-cell maturation, survival and activation of the T-cell–mediated immune response. BAFF-R is universally expressed in mature B-cells of healthy people but abnormally expressed in precursor cells of patients with B-ALL (47–49). Turazzi et al. (50) constructed an efficient BAFF-R CAR (INVsh.BAFFR. CAR), which can proliferate and secrete cytokines to lyse ALL cell lines. Qin et al. (51) also verified the efficiency of BAFF-R CAR T-cells on CD19-negative ALL cells in blinatumomab relapse patient-derived xenografts in vivo. It has been suggested that BAFF-R is preserved in relapsed tumor cells since the BAFF/BAFF-R signaling pathway is essential for the survival of ALL cells and may result in a low rate of downregulated expression (52). Targeting BAFF-R or combining it with CD19 CAR is a potential direction to treat or reduce the risk of CD19-negative relapses.
CSPG4
Chondroitin sulfate proteoglycan 4 (CSPG4, also known as neuron-glial antigen-2, NG2) is a type of single transmembrane protein that is expressed on a variety of tumors, including melanoma, breast cancer, malignant gliomas and leukemia (53–55). Regarding hematological tumors, CSPG4 is mainly expressed on KMT2A rearrangement (56–59), or the more commonly called mixed lineage leukemia rearrangement (MLL-r) phenotype. MLL-r is characterized by chromosome 11 translocation and is generally insensitive to common chemotherapy regimens with a poor prognosis (60). MLL-r ALL patients undergoing CD19 CAR T-cell therapy also have high risks of relapse due to lineage switching (6, 61). Compared with lineage-restricted targets, such as CD19 and CD22, CSPG4 can be detected in both MLL-r ALL and AML cells, and its expression is not affected by lineage switching. Harrer et al. (62) provided basic evidence for the application of CSPG4 CAR T-cells in MLL B-ALL. The constructed MLL-r leukemia cell line KOPN8 can activate cocultured CSPG4 CAR T-cells, secreting cytokines and eradicating targeted cells. More studies are warranted to further validate their therapeutic potential.
TSLPR
Thymic stromal lymphopoietin receptor (TSLPR) is a heterodimeric complex formed by the TSLPR subunit and CD127 subunit. The former is encoded by the cytokine receptor-like factor 2 (CRLF2) gene (63). The overexpression of TSLPR in 5% to 15% of ALL patients is mainly due to CRLF2 translocations or changes in promoter regions (64–66). CRLF2 rearrangement could also cause Philadelphia chromosome-like (Ph-like) ALL, a high-risk phenotype with resistance to traditional chemotherapy and poor outcomes (64, 66). Qin et al. (25) constructed an effective anti-TSLPR CAR T-cell design to eliminate TSLPR-overexpressing ALL cells in animal models, with comparable antileukemia efficacy of CD19 CARs. It has been confirmed that abnormalities in the TSLPR signaling pathway are closely associated with cell canceration (67, 68). TSLPR is of great importance in the occurrence of leukemia cells and is less likely to downregulate expression for relapse. Therefore, TSLPR CAR T-cells are expected to become a novel therapeutic scheme for high-risk leukemia.
Challenges
Access
The premise of benefitting from CAR T-cell therapy is based on successful reception of effective products. Currently, the main access is through FDA-approved drugs or enrollment in clinical trials. However, several obstacles restrict patients receiving treatment, including cost issues, limitations of inclusion criteria and unexpected status of the gap period between leukapheresis and infusion (Figure 2).
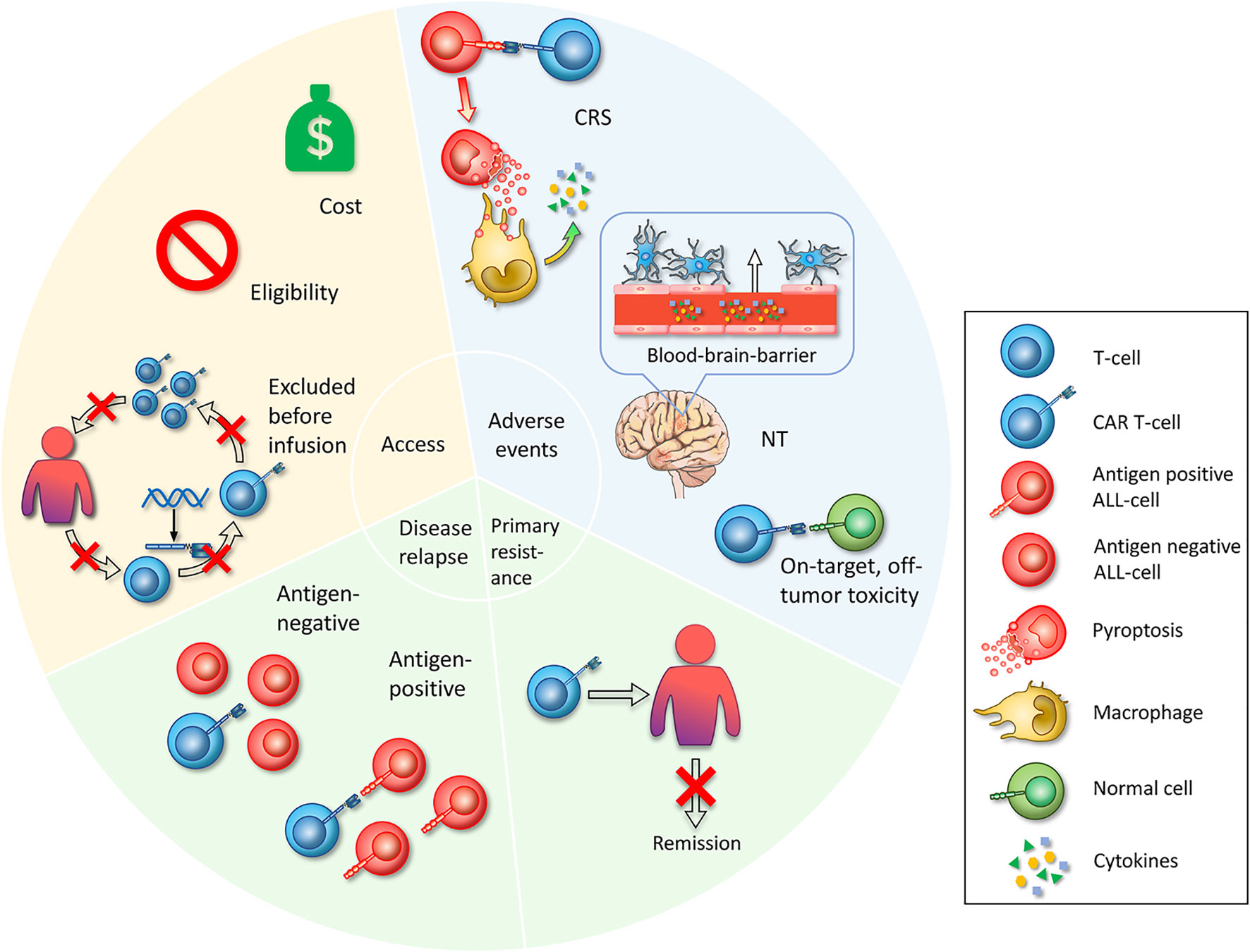
Figure 2 Challenges in CAR T-cell therapy. First, access is whether the patient can start CAR T-cell therapy. Limited access includes the cost of the manufacture of CAR T-cell therapy itself and other attendant expenses during the treatment is unaffordable for common patients. Eligibility, referring to specific admission or exclusion criteria set by clinical trials, impeding broad enrollment. Excluded before infusion refers to patients who have been enrolled and have experienced leukapheresis, who exhibit adverse disease progression during the preparation of CAR T-cells, or who fail to produce CAR T-cell products. The second challenge is serious adverse events that occur during the treatment with CAR T-cells. The mechanisms of CRS are that CAR T-cells specifically recognize ALL cells and lead to widespread pyroptosis, with the pyroptosis-released factors then activating macrophages to produce inflammatory cytokines. Neurotoxicity is a toxic encephalopathy state because of the disruption of the BBB endothelium. On-target, off-tumor toxicities mean that CAR T-cells recognize and attack normal cells. Third, primary resistance refers to the inability to induce remission after CAR T-cell infusion. Fourth, relapse includes antigen-positive relapse with the presence of target antigen and antigen-negative relapse with antigen loss or dim expression.
Cost
CAR T-cells, as tailored and gene-editing drugs, are undoubtedly costly. Tisagenlecleucel, priced at $475,000 in the USA for a single infusion, is one of the most expensive agents for cancer treatment (69, 70). The current manufacture of CAR T-cells involves obtaining peripheral blood mononuclear cells from apheresis using anti-CD3/CD28 beads for activation, retroviral or lentiviral transduction and expansion of modified cells in bags. Expenditures mainly come from CAR T-cell generation, logistics transportation and clinical medication. In particular, GMP-graded viral vectors for CAR gene transduction place extremely high demands on professional technicians and equipment (71). Costs with medical staff and supportive care services for complication management and hospitalization should be considered simultaneously. Time cost is another aspect. The manufacturing process usually takes 12 to 17 days for the autologous cell product, during which patients are at risk of disease progression.
Eligibility
In addition to FDA-approved CAR T-cells, patients could also be enrolled in clinical trials. However, eligibility criteria are also strict, varying with different characters of each product. Expert consensus of CAR T-cell therapy management guidelines proposed by a multidisciplinary and interprofessional team recommended that the minimum eligibility criteria of new CAR T-cell trials should be based upon the FDA approved indications and former experience of pivotal studies (72). It has been suggested that graft-versus-host disease (GVHD), uncontrollable infections, recent donor-lymphocyte infusion (DLI) treatment, and the sites of active disease should be evaluated carefully, especially for central nervous system (CNS) pathology, due to potential neurotoxicity associated with CAR T-cell therapy (72). In addition, protocol-specific criteria included age, weight, and prior treatment status, such as the history of HSCT, the history of CD19-targeted therapy, absolute lymphocyte counts (ALCs), and liver and kidney function based on their research purposes (7, 73).
Excluded Before Infusion
Before infusion, patients can still be excluded from clinical trials due to infection, disease progression or production failure (8, 9). After apheresis, patients usually receive lymphodepletion, making them vulnerable to infection. ALL progresses rapidly, and as a rescue plan, enrolled patients are generally in poor condition. Therefore, they are likely to experience disease deterioration or infections during the manufacturing period. On the other hand, there are chances for manufacturing failure (8, 9). Poor T-cell quality and insufficient collection are the main barriers. T-cells collected from the peripheral blood of ALL patients are directly affected by their age, differences in antigen exposure, and chemotherapy drugs (74–76). Chemotherapy could induce T-cell deletion and/or dysfunctional metabolism through mitochondrial damage (77). Other factors include manufacturing samples that mostly contain terminal effector T-cells (TEff) and effector memory cells (TEM) with poor proliferation capability (78), and culture systems contain suppressive components, such as myeloid-derived monocytes, inhibiting the expansion of T-cells (79). The risk of production failure caused by manufacturing technology and transportation is decreasing gradually, benefiting from the emergence of substantially successful experiences and the proposal of production guidelines (80, 81).
Adverse Events
Cytokine Release Syndrome
CRS is the most common adverse event following CAR T-cell therapy, with a prevalence of 75% to 100% in patients (Table 1), which refers to a systemic inflammatory response mediated by the release of excessive cytokines, including IL-6, IL-1, IL-8, IFN-γ, GM-CSF, macrophage inflammatory protein-1B (MIP-1B), and monocyte chemoattractant protein-1 (MCP-1) (82, 83). The clinical manifestations can range from fever to severe complications, such as hypotension, hypoxia, capillary leakage, and multiple organ failure (84). Many parameters are associated with the incidence of CRS, including the infusion dose of CARs, tumor burden, chemotherapy regimen, and CAR construct (85–87).
IL-6, the hallmark cytokine of CRS, was greatly elevated in the serum of patients with CRS post CAR T-cell infusion. In addition, the confirmed efficacy of IL-6 and IL-6 receptor (IL-6R) antagonists for treatment underscores that IL-6 signaling instigates the amplification cytokine cascade that contributes to the pathophysiology of CRS (5, 6). IL-6 activates Janus kinases (JAKs) and the signal transducer and activator of transcription 3 (STAT3) pathway through classic cis signaling or trans signaling. The former induces pleiotropic effects on immune cells through membrane-bound IL-6R (mIL-6R), while trans signaling is activated by the soluble form of IL-6R (sIL-6R) in many cell types, including endothelial cells. This results in the production and secretion of large amounts of cytokines by downstream cells (88).
For the generation of IL-6, monocytes and macrophages are considered the main sources. Norelli et al. (82) found that depletion of circulating monocytes from mice before CD19 CAR T-cells could prevent CRS and enable the complete suppression of CRS incidence and CRS-related mortality. At the single-cell level, monocytes specifically expressed high levels of IL6 and IL-1β. On the other hand, Giavridis et al. (89) showed that macrophages are the main source of IL-6 based on RNA sequence analyses and cell enumeration data. They further hypothesized that macrophages secrete this key factor via engineered CD40 ligand (CD40L)-CD40 interaction between macrophages and CAR T-cells. A recent study proposed that CAR T-cell–induced pyroptosis of targeT-cells rather than apoptosis is a critical reason for CRS (90). CD19 CAR T-cells specifically recognize ALL cells and release a mass of perforin/granzyme B, thereby activating caspase 3 and further lysing highly expressed gasdermin E on ALL cells, which leads to pore-forming activity and widespread pyroptosis of ALL cells. Furthermore, pyroptotic cells release large amounts of damage-associated molecular pattern molecules (DAMPs), which activate caspase 1 for gasdermin D cleavage in macrophages, resulting in the release of proinflammatory cytokines, such as IL-6 and IL-1β and the subsequent occurrence of CRS (Figure 2). Ordinary tumor-specific T-cells kill B leukemic cells, leading to apoptosis, which will not activate macrophages. Another study also confirmed that CD19 CAR T-cells can cause pyroptosis of target cells through granzyme A and gasdermin B (91).
Neurotoxicity
Neurotoxicity (NT) is defined as a toxic encephalopathy state following CAR T-cell infusion, accompanied by confusion, unconsciousness, delirium, tremor, aphasia, seizures, and cerebral edema (92), which is another prominent toxicity occurring in 17.6% to 50% of patients (Table 1). NT is associated with CRS, with clinical data showing that 90% of NT is concurrent with or after CRS (4, 93).
The occurrence of NT after CD19 CAR T-cell therapy is not fully understood. The mechanism might be associated with endothelial activation and blood brain barrier (BBB) disruption (94). Some of the CRS-released cytokines, such as IFN-γ, IL-6, IL-8, and MCP-1, can activate endothelial cells (95, 96). Biomarkers of endothelial activation, such as angiopoietin-2 (ANG2), high ratios of ANG2/ANG-1, and elevated von Willebrand factor (vWF), were higher in patients with NT. Endothelial dysfunction contributes to disruption of the BBB, and the BBB was not able to shield cerebrospinal fluid (CSF) from high levels of serum cytokines, which induced stress in brain vascular pericytes and secretion of endothelium-activating cytokines and eventually resulted in severe NT (Figure 2) (93, 94, 97). On the other hand, a recent study revealed CD19 expression in human brain mural cells using single-cell RNA sequencing analysis and confirmed perivascular staining at the protein level. Mural cells surround the endothelium and maintain the integrity of the BBB. This finding indicated that CD19 CAR T-cells might directly attack brain mural cells and cause increased leakiness of the BBB (98). Less severe NT incidence post CD22 CAR T-cell infusion than CD19 CAR T-cells may also be associated with this finding (14).
On-Target, Off-Tumor Toxicities
The targets recognized by CAR T-cells are the most common tumor-associated antigens (TAAs), whereas the target antigens exist on some normal cells, which makes normal tissues inevitably attacked by CAR T-cell–specific recognition, thereby leading to on-target, off-tumor toxicities (Figure 2). CD19 or CD22 CAR T-cells target B-ALL cells and simultaneously damage healthy B-cells. More than 50% of patients receiving CD19 or CD22 CARs (Table 1) developed B-cell aplasia, plasma cell deficiency, or even severe hypogammaglobulinemia so that they were susceptible to infection due to low immunity. Myeloablation induced by CD123 CAR T-cells and fratricide killing of CD38 CAR T-cells are more thorny off-tumor toxicities than B-cell aplasia that impede their development.
Other Adverse Events
Other adverse events deserve attention. CAR T-cell–associated hemophagocytic lymphohistiocytosis (HLH)/macrophage activation syndrome (MAS) is attributed to a group of severe immunological disorders characterized by hyperactivation of macrophages and lymphocytes, proinflammatory cytokine production, lymphohistiocytic tissue infiltration, and immune-mediated multiorgan failure. Patients experience a wide range of clinical syndromes, including high fevers, hepatosplenomegaly, liver/renal dysfunction, coagulopathy and cytopenia. Several clinical trials have indicated that CD19 CAR T-cell therapy could induce HLH/MAS (99), and a recent CD22 CAR T-cell clinical trial also reported a 38% incidence of HLH/MAS-like toxicities at an average time of 14 days after infusion (14). Moreover, owing to the robust antitumor efficacy of CAR T-cells, massive malignant cells are dissolved and cause tumor lysis syndrome (TLS). Lytic ALL cells rapidly release large amounts of intracellular substances into the blood, which surpass the capability of liver metabolism and renal excretion, resulting in metabolite accumulation and a series of electrolyte disorders, such as hyperuricemia, hyperkalemia, hyperphosphatemia, hypocalcemia, and metabolic acidosis, which in turn even lead to life-threatening arrhythmias or acute renal failure (100).
Primary Resistance
Primary resistance refers to the inability to induce remission after CAR T-cell infusion (Figure 2). It is closely related to the intrinsic T-cell functional state at the starting material (SM) stage, which is largely a consequence of their differentiation status strongly correlating with the antitumor activity of adoptively transferred T-cells (101–104). Studies have shown that at the SM stage, compared with patients who achieved partial remission (PR) or no remission (NR), patients with CR have a higher ratio of CD45RO-CD27+CD8+ T-cells and stem cell memory T-cells (TSCM) (105). Numerous studies have proven that generating CAR T-cells with a less differentiated phenotype, such as central memory T-cells (TCM) and TSCM, has greater in vivo efficacy than TEff (6, 106–108). TSCM is a subset of memory T-cells with superior self-renewal capacity and can also differentiate into TCM and TEM in vitro (109). This subtype can even facilitate CAR T-cell homing to secondary lymphoid organs and continuous proliferation (110, 111). Other phenotypes might also be associated with primary resistance. At the SM stage, LAGhigh/TNF-αlow CD8+ T-cells herald a high risk for primary resistance (101). Patients with CR had a lower rate of CAR T-cells with a CD8+ PD-1+ phenotype than those with PR and NR. CD8+PD-1+ CTL019 coexpresses LAG-3+ and TIM-3+, indicating poorer prognosis of patients (105).
Primary resistance may also be associated with the inherent biological characteristics of tumors. Nathan et al. (112) found that death receptor ligands in tumors, such as Fas ligand (FasL) and TNF-related apoptosis inducing ligand (TRAIL), are of critical importance for CAR T-cell cytotoxic killing. They used a CRISPR-based genome-wide loss-of-function screen in an ALL cell line under immune pressure from CD19 CAR T-cells and found that impaired death receptor signaling in ALL may lead to significantly reduced CAR T-cell cytotoxic activity and primary resistance to CAR T-cell therapy, which in turn mediate CAR T-cell dysfunction. Moreover, tumor burden serving as an indicator for primary resistance remains controversial. Some studies demonstrated that upon overexposure to target cells, CAR T-cells are more rapidly cleared and inactivated (5, 9), whereas Finney et al. found that a high antigen burden could promote the expansion of CAR T-cells (101). Moreover, some characteristics of patients, including extramedullary diseases other than CNS, increased levels of Tregs, and high-risk cytogenetic/molecular abnormalities such as E2A/PBX1, often lead to a low remission rate (113). Further studies are needed to unveil the underlying correlation.
Relapse
Although the CR rate of ALL in CD19 or CD22 CAR T-cell treatment is as high as 57% to 93%, the relapse rate reaches 14% to 66% (Table 1), which becomes one of the most significant issues limiting CAR T-cell application. Relapse includes two patterns: antigen-positive relapse and antigen-negative relapse.
Antigen-Positive Relapse
Antigen-positive relapse (Figure 2) is usually associated with short persistence and low potency of CAR T-cells (4). Components of CAR constructs, such as costimulatory domains and scFv, can influence the potency and persistence of CAR T-cells. It was found in clinical studies that 4-1BB–based CAR T-cells have greater persistence than CD28-based CAR T-cells (5, 8, 114). The CD28 costimulatory domain initiated intensive cytokine release but short persistence through rapid and robust signaling of the STAT3/PI3K/AKT pathway, which can result in more differentiated memory T-cells and a reduction in mitochondrial biogenesis (115, 116). In contrast, 4-1BB–based CARs have longer persistence through tumor necrosis factor receptor-associated factors (TRAFs) and the NF-κB pathway, leading to an increase in fatty acid oxidation and TCM differentiation. 4-1BB can also ameliorate the exhaustion of CAR T-cells induced by clustering of CAR scFvs and tonic CAR CD3ζ phosphorylation (117). Immunogenicity derived from murine scFv may trigger the host immune response and limit the persistence of CAR T-cells, while replacement with humanized scFv can reduce immunogenicity (118). In addition, in a study, 12 of 14 ALL patients achieved CR treated with low-affinity scFv CAR T-cells, which showed better proliferation than higher-affinity scFv (119).
The choice of T-cell subset at the SM stage will also influence antitumor efficacy and persistence (101, 105). CD8+ CAR T-cells exhibited higher lytic activity than CD4+ CAR T-cells, while the production of abundant IL-2 by CD4+ CAR T-cells might augment the proliferation and efficacy of CD8+ CAR-T-cells. A defined 1:1 CD4/CD8 ratio is the most widely used, and the optimal CD4/CD8 ratio is under intensive investigation (120). In addition, age-related T-cell changes reflected by gene expression patterns and secretory profiles might influence CAR T-cell persistence (121). A study showed that CAR T-cells derived from young donors had greater expansion ability with more memory-like phenotypes but inferior cytotoxicity than those derived from geriatric donors (122). Moreover, c-Jun overexpression (123) and the immune-suppressive bone marrow microenvironment (124) are likely to be parameters related to antigen-positive relapse in CAR T-cell therapy.
Antigen-Negative Relapse
Antigen modulation or loss on the surface of tumor cells, which makes them incapable of being recognized by CAR T-cells, is another pattern of relapse (Figure 2). The mechanism by which 10% to 20% (125) of patients develop antigen-negative relapse following CD19 CAR T-cell treatment is complicated, including gene mutation, selective splicing, lineage switching, immune selection, trogocytosis and antigen masking. Sotillo et al. (126) found that tumor cells in relapsed patients can induce epitope loss through frameshift mutations and selective splicing. Although CD19 of this subtype can also transmit signals, it can no longer be recognized by FMC63 on CD19 CAR T-cells due to the lack of exon 2 expression. SRSF3 is one of the splicing factors whose function is to retain exon 2. Insufficient expression of SRSF3 in relapsed ALL cells may be a major reason for skipping the exon 2 isoform of CD19 (126). Lineage switching is another mechanism related to antigen loss. Some patients develop myeloid leukemia relapse, with an altered antigen expression profile. Generally, ALL patients with KMT2A (MLL) rearrangement develop myeloid conversion under CD19 immune pressure (6, 61). BCR-ABL1-positive and TCF3-ZNF384 fusion-positive B-ALL patients have also been found to undergo myeloid transformation after CD19-targeted immunotherapy (127, 128).
Immune selection means that under the pressure of CD19 CAR T-cells, inherently resistant CD19-negative leukemia cells are retained and evolve into a dominant clonal community. Fischer et al. (129) revealed that the total deletion (ex2-isoform) and partial deletion of exon 2 (ex2part-isoform) of CD19 in B cells, which cannot be recognized by CD19 CAR T-cells, existed in CD19+ B-ALL patients and healthy people before treatment. Grupp et al. (3) performed a flow cytometry test on a CD19-negative relapse case after CAR T-cell therapy and validated that CD19-negative clones pre-existed. Furthermore, in B-ALL xenograft studies, Hamieh et al. (130) reported trogocytosis phenomena in which CD19 antigen is transferred from ALL cells to CAR T-cells, thus resulting in antigen escape of ALL cells as well as fratricide killing of CAR T-cells. Intriguingly, there is a clinical case of mask CD19 epitope relapse (131), which means that the CAR gene was accidentally introduced into B leukemia cells, masking the CD19 epitope from detection of flow cytometry and recognition of CD19 CAR T-cells.
Regarding CD22 CAR T-cell therapy, there are also cases of CD22-negative or CD22-dim relapse. Because the potency of CD22 CAR T-cells is greatly affected by cell surface antigen density, ALL cells below a certain threshold can escape. In the trial of Fry et al. (12), seven of eight relapsed patients had a decline in CD22 site density with no CD22 gene mutation or mRNA change observed, indicating that the downregulation mechanism may be related to posttranscriptional regulation.
Clinical Strategies
For Access
To overcome the limitations of tailored products, numerous optimization strategies for manufacturing techniques have been proposed. Using an automation system (CliniMACS Prodigy System) (132), nonviral transposon-based systems (133–135), a type II CRISPR/Cas9 system (136) and transferring CAR coding sequences in vivo (137, 138) could reduce the manufacturing time and cost as well as manifest the high potency and proliferation capacity of CAR T-cells. A new platform, “FasT”, using electroporation to transduce the CAR gene and shortening manufacturing time to 24 h, generated less differentiated phenotypes with superior expansion capacity in a first-in-human clinical trial (139). Moreover, recent advances have paid intensive attention to allogeneic, off-the-shelf CAR T-cells.
Allogeneic, Off-the-Shelf CAR T-Cell
To solve the problems of autologous T-cells with poor quantity and quality, allogeneic healthy donor-derived T-cells have become an attractive alternative. In Zhang’s study (140), six R/R B-ALL patients who received donor-derived CAR T-cells stayed alive with complete donor chimerism and remained in MRD-negative remission with a median follow-up of 243.5 days. Tu et al. (141) enrolled three R/R ALL patients in the study, and all reached CR after administering pooled donor-derived fourth generation CAR T-cells targeting CD19 and CD123. A meta-analysis showed that the pooled CR rate of different allogeneic CD19 CAR T-cell trials was 55% (95% CI, 30.6–79.0%), the pooled CRS rate was 53.9% (95% CI, 10.7–94.2%), and the pooled NT rate was 3.1% (95% CI, 0.0–23.0) (36).
Recent advances in allogeneic CAR T-cells have focused on off-the-shelf products, also called universal CAR (UCAR). The third-party products are based on a detachable, universal, programmable CAR system and gene editing technology (142), making standardized batch preparation possible (Figure 3A). In a process superior to case-by-case manufacture, recipients with rapidly progressive diseases could have access to UCAR T-cells in time at a lower cost, which would make the UCAR more competitive in the future market. Ongoing UCAR19 clinical trials, PALL (NCT02808442) and CALM (NCT02746952) (143), have reported that 14 (66.7%) of the 21 R/R B-ALL patients who received UCART19 infusion achieved CR/CRi. The phase 1 trial of UCAR22 is also underway (NCT04150497). Hu et al. reported the latest clinical results of a CRISPR/Cas9-engineered universal CD19/CD22-targeting CAR T-cell product (CTA101) in B-ALL patients. A total of 5/6 (83.3%) patients achieved MRD-CR with manageable adverse events (144).
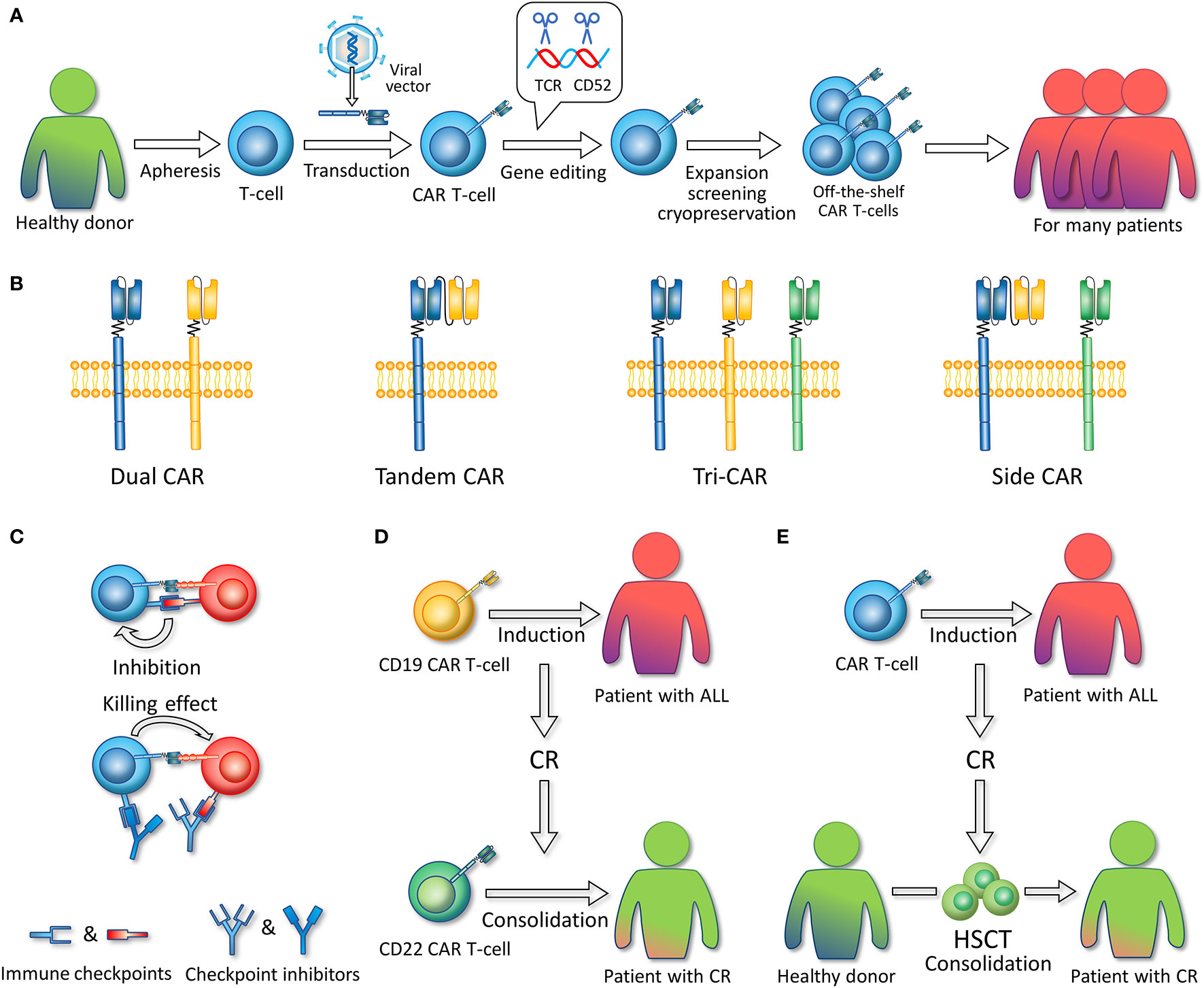
Figure 3 (A) Allogeneic, off-the-shelf CAR T-cells. The figure illustrates the general preparation process of allogeneic, off-the-shelf CAR T-cells. T-cells are extracted from healthy donors. Viral vectors are used to introduce CAR-encoding genes into T-cells. Gene editing technology is used to remove the gene fragments encoding TCR and CD52. CAR T-cells are expanded, screened, and cryopreserved. These CAR T-cell products can serve as timely treatment for many patients. (B) Multi-antigen targeted CAR. Dual CAR refers to two different mono-CARs in one T-cell. Tandem CAR refers to a CAR structure that contains two single-chain variable fragments. Tri-CAR T-cell coexpresses three different mono-CARs on a single T-cell. Side CAR T-cell expresses a Tandem CAR and a mono-CAR. (C) In combination with immune checkpoint inhibitors. Immune checkpoint inhibitors such as PD-1/PD-L1 inhibitors can specifically block the binding of CAR T-cell or ALL-expressed immune checkpoint molecules to the corresponding receptors on CAR T-cells, thereby enabling CAR T-cell activation and the killing of tumor cells. (D) Sequential infusion. CD19 CAR T-cells are first infused to induce CR, and after patients achieve CR, CD22 CAR T-cells are infused as consolidation. (E) Bridging to HSCT. CAR T-cells are infused to induce CR, and after patients achieve CR, hematopoietic stem cell transplantation (HSCT) is conducted as consolidation.
Nonetheless, the application of allogeneic CAR T-cells met additional challenges, including GVHD and graft rejection mediated by the presence of class I HLA (HLA I) and HLA II overexpression (145). Gene editing technology (142) and protein expression blockers (PEBLs) (146) are expected to be used for T-cell receptor (TCR) modulation to prevent GVHD and to date, severe GVHDs have rarely been reported in clinical trials (147, 148). In terms of graft rejection, the sources of third-party T-cells could be selected seriously from donors with high compatibility of HLA (i.e., from previous allo-HSCT donors). Simultaneously, chemotherapy regimens such as Cy/Flu for lymphodepletion and serum treatments such as the anti-CD52 monoclonal antibody alemtuzumab are advocated to suppress immune cells, which can result in long-term lymphodepletion and thereby reduce allo-rejection (149), while UCAR can be preserved for the knock-off of the CD52 gene. However, the optimal intensity and duration of immunosuppression remain to be determined and are required careful evaluation in clinical trials.
For Adverse Events
For Cytokine Release Syndrome
Recent consensus on grading and management for CRS was proposed (72, 150). Finding potential biomarkers to predict CRS is also of importance. Studies suggest that IFN-γ, IL-6, and sIL2Rα are strongly correlated with severe CRS (33). The cytokine combination of sgp130, MCP1 and eosinophil chemokines in predicting severe CRS has a sensitivity of 86% and a specificity of 97% (33). For prevention, CRS can be controlled more effectively with a fractionated dosing scheme of CAR T-cells. In a trial by Frey et al. (11), patients in the high-dose fractionated group received infusion with 10% of the dose on day 1 (D1), 30% on D2, and 60% on D3. D2 and D3 were withheld for early signs of CRS, such as fever. Compared with the high-dose single infusion and low-dose group, the high-dose fractionated group had a superior CR rate (50% vs. 33% vs. 90%, P=0.004) and lower CRS incidence (50% vs. 22% vs. 5%, P=0.017).
With regard to management, the IL-6 receptor blocker tocilizumab, which has been approved by the FDA for the treatment of CRS (151), is the first-line medication for CRS without affecting the efficacy of CAR T-cells (152). Other drugs, such as siltuximab (an IL-6 blocker) (153), anakinra (an IL-1 receptor blocker) (154), and dasatinib (a tyrosine kinase inhibitor) (155), have been applied in clinical trials with significant effectiveness. For the administration of corticosteroids, their impact on CAR T-cell activity remains controversial (156, 157). When corticosteroids are used, the dose should be controlled and individualized according to the patient’s response.
For Neurotoxicity
Recent grading and management guidelines for NT were developed (72, 150). For clinical parameters, Ang-2 and vWF can estimate the occurrence of NT. Low platelet counts (PLT<60) are significantly associated with severe NT (158). MCP-1 proved to be the best predictor of severe NT within 36 h after CAR T-cell administration, with high specificity and sensitivity (94). With regard to the treatment of NT, tocilizumab has limited efficacy due to its difficulty in penetrating the BBB and even increasing the CSF IL-6 level because of the peripheral blockade of IL-6R (94, 159). For this reason, some agencies regard corticosteroids, such as dexamethasone, as the first-line therapy for NT (160), but the thresholds for administration and dosing schemes vary and have not been prospectively compared. On the other hand, CAR engineering strategies to overcome CRS and NT have been proposed, including “on/off” switch systems, suicide gene systems and direct antagonism approaches (161, 162).
For On-Target, Off-Tumor Toxicities
B-cell aplasia driven by CD19 or CD22 CAR T-cell therapy is tolerated and treatable. The administration of antibiotics and gamma globulin is capable of preventing infections and improving immunity. Furthermore, repeating vaccine series are a promising direction for patients who have achieved B-cell recovery, and this remains an active area in upcoming research (163). Myeloablation caused by CD123 CAR T- cells is risky and requires high caution, and treatment must be terminated if necessary. Generally, selecting safer TAAs and optimizing CAR constructs, such as improving specificity through targeting multiple antigens, logic gating and conditional expression systems, are the ultimate strategies to avoid on-target, off-tumor toxicities (161, 162, 164).
For Other Adverse Events
At present, diagnostic criteria for CAR T-cell–related HLH have been proposed (99). Allopurinol can be used for prevention before cell infusion, and IL-6 inhibitors and glucocorticoids can serve as effective medications (163). Anakinra and corticosteroids were used in a trial, and all treated participants had alleviation of HLH/MAS-like toxicities without any apparent negative impact on response or CAR T-cell expansion. In addition, etoposide and cytarabine may have therapeutic effects on HLH (165–167). For TLS, in addition to applying high-dose corticosteroids or corresponding cytokine inhibitors such as tocilizumab for treatment, reducing the infusion dose or administering different types of CAR T-cell populations can alleviate the clinical symptoms (100).
For Primary Resistance and Relapse
Engineering strategies to improve the persistence and antitumor activity of CAR T-cells have been reviewed recently (161, 162), which is not the focus of our article.
Multiantigen-Targeted CAR T-Cell
Multiantigen-targeted CAR T-cells are a promising direction to overcome single antigen relapse issues since the “OR” Boolean logic gate is utilized to activate T-cells in the presence of either validated antigen. It mainly refers to dual CAR T-cells, bispecific CAR and trivalent CAR T-cells (Figure 3B). Dual CAR T-cells are designed to contain two different CARs on one T cell. CD19/CD22 and CD19/CD123 dual CAR T-cells for ALL have been successfully constructed and show greater potency than single CAR T-cells or a pool of both CAR T-cells in mice (40, 130, 168). Amrolia et al. (169) infused CD19/CD22 dual CAR T-cells to treat seven R/R ALL children, with a 100% molecular remission rate and a favorable safety profile. No more than grade 3 CRS or grade 2 neurotoxicity was reported.
Bispecific CAR refers to a CAR protein containing two scFv domains, which is also called Tandem CAR (TanCAR). The CD19/CD20 bispecific CAR T-cell constructed by Martyniszyn et al. was effective on a mixed CD19+CD20+/CD20-negative phenotype from the blood and bone marrow of transplanted mice, while anti-CD20 CAR T-cells left CD20-negative leukemic cells behind without curing the disease (170). In the trial of Dai et al. (171), six out of six patients with R/R B-ALL achieved MRD-negative CR after infusion of CD19/CD22 bispecific CAR T-cells, with one patient with mild CRS and no patient with NT. Since CD133 is expressed in MLL-r B-ALL cells, preclinical studies have also proposed the application of CD19/CD133 bispecific CAR T-cells (29), with the findings showing the synergistic effect of CD19 and CD133 and that T-cells simultaneously targeting CD19 and CD133 possess greater recognition efficacy than single-targeted T-cells in vitro and mouse models.
There are two patterns of trivalent CAR T-cells: TriCAR expressing CD19 CAR, CD20 CAR, and CD22 CAR on a single T-cell and side CAR expressing CD20/CD19 TanCAR combined with CD19 CAR (31). Fousek et al. (172) constructed CD19/CD20/CD22 TriCAR T-cells that manifested strong killing activity on both ALL cells and CD19-negative relapsed cells in vitro and in murine models, while CD19 CAR T-cells were ineffective.
Nonetheless, there are CD19-negative/CD22-low relapse cases after both CD19/CD22 dual and bispecific CAR T-cell–induced CR (169, 171), in which the mechanism remains to be elucidated. In addition, poor T-cell persistence is also a relatively common situation and contributes to a proportion of relapses. It is worth noting that membrane binding location, linker length between the heavy and light chains and other spatial considerations are all important for the efficacy and safety of CAR T-cells, which is more complex in the incorporation of multiple scFvs (51, 173, 174). More basic studies are required to optimize the structure of multitargeting CARs in pursuit of better clinical outcomes. On the other hand, the utilization of the “AND” “NOT” logic-gated system in multi-CAR could serve as a promising strategy for avoiding attacks on normal tissue and alleviating on-target, off-tumor toxicities (175).
In Combination With Immune Checkpoint Inhibitors
Immune checkpoints (PD-1, CTLA-4, TIM-3, etc.) serve as a brake on immune cell overactivity and prevent autoimmune reactivity, whereas tumor cells can escape human immune surveillance by upregulating the expression of immune checkpoints, thereby leading to tumor recurrence (176–178). On this basis, immune checkpoint inhibitors can be used to regulate the intrinsic T-cell functional state and promote the efficacy of CAR T-cells. Among them, inhibitors (e.g., nivolumab, pembrolizumab or atezolizumab) targeting the programmed cell death 1 receptor (PD-1) and its ligand PD-L1 have attracted attention (Figure 3C), and clinical trials in combination with CAR T-cell therapy are underway. In the study by Maude et al. (179), four children with relapsed B-ALL after CD19 CAR T-cell treatment presented poor CAR T-cell persistence. Subsequently, they received the PD-1 inhibitor pembrolizumab (PEM), and increased persistence of CAR T-cells in circulation was detected by flow cytometry. Li et al. (180) reported 13 pediatric patients with relapsed B-ALL who received CD19 CAR T-cells and PEM in combination. Enhanced CAR T-cell function was observed in three of the patients, and two partial and two complete responses were observed in four patients who previously had relapse or no response for CAR T-cells after the administration of PEM. Remarkably, significant CAR T-cell proliferation was detected in one patient within days of pembrolizumab administration. However, thus far, few clinical trials have evaluated the efficacy of CAR T-cells and PD-1 blockers in combination, and the rationality of this combination needs to be further clarified.
Sequential Infusion
Sequential infusion refers to the process of infusing CD19 CAR T-cells as induction to CR, followed by infusion of CD22 CAR T-cells for consolidation (Figure 3D). Compared with dual CAR T-cells and bispecific CAR T-cells, this two-cycle therapy is more convenient and cost-effective because it does not require the generation of complex CAR T-cell products (181). Wang et al. (182) conducted a pilot study on 51 ALL patients to evaluate the effectiveness and safety of the sequential infusion of two monospecific CAR T-cells, CD19 and CD22. Drawing on the findings that the MRD-negative rate was 96.0%, the median progression-free survival (PFS) was 13.6 months, and the median OS was 31 months with no relapse of antigen loss observed. In addition, Pan et al. (183) performed sequential infusion on 20 pediatric patients with R/R B-ALL, and the median interval between the two cycles of infusion was 1.65 months. During the 20-month follow-up period, 17 patients remained in CR, and three patients relapsed, resulting in a leukemia-free survival (LFS) rate of 79.5% at 12 months and OS rates of 92.3% at 18 months. In addition, regarding adverse events, 4 of 20 patients developed no CRS; mild or moderate (grade 1–2) CRS was observed in 15 of 20 patients, and grade 1 NT occurred in 3 of 20 patients, indicating that sequential infusion of two CAR T-cell products is a safe strategy. The data from the above clinical trials show that sequential infusion of CD19 and CD22 CAR T-cells is effective and safe and may be a feasible strategy to prevent relapse of single-targeted CAR T-cell therapy. However, the limited CAR T-cell persistence and the interval lymphodepleting chemotherapy that probably eradicates previous CAR T-cells should be considered, and longer follow-up is needed (181).
Consolidative HSCT
HSCT is considered to be the only potentially curative therapy for R/R B-ALL (184, 185). Serving it as a consolidative therapy after CAR T-cell–induced CR may improve the long-term outcome (Figure 3E). However, whether HSCT after CAR T-cell therapy benefits patients remains controversial. Park et al. (9) reported that 17 of 53 patients received allo-HSCT after 19–28z CAR T-cell therapy, and their OS or LFS was not significantly different from those who did not undergo HSCT. In contrast, there is accumulating evidence supporting the benefit of consolidative HSCT (186–188). A multicenter retrospective study indicated that haploidentical HSCT after CD19 CAR T-cell therapy could benefit patients, with great improvement in LFS and OS compared with the nontransplant group (65.6% versus 32.8% P < 0.001; 77.0% versus 36.4%, P < 0.001) and no increased risk of treatment-related toxicity compared with previous values. It also identified pretransplant MRD negativity as an important independent predictor of high LFS and OS (186). Gu et al. (187) reported similar conclusions after analyzing 56 adults with R/R Ph+ ALL who received allo-HSCT post-CD19 CAR T-cell therapy. Moreover, they further compared the 2-year treatment-related mortality between the transplant and nontransplant groups and detected no significant difference (14.3%, CI 7.6–21% vs. 9.8%, CI 3.2–16.4%, p =0.804), confirming the safety of this therapy. Bridging CD22 CAR T-cells to HSCT also seems to be feasible; 11 of 24 CR patients received transplantation after CD22 CAR T-cell therapy, with a one-year LFS rate of 71.6% (95% CI, 44.2–99.0) and a 1-year relapse rate of 9.1% (95% CI, 0–26.2), while four of the seven CR patients without further treatment had a relapse at 1.7 to 6 months (13). More randomized controlled studies with longer follow-ups are warranted to verify the efficacy and safety of these strategies.
Discussion
In this article, we reviewed the whole process of CAR T-cell administration in ALL patients, with a focus on its clinical application, existing challenges and clinical coping strategies. Regardless of the verified effectiveness, CAR T-cell therapy still faces certain challenges. Resistance and relapse are the main barriers restricting the development of CAR T-cell therapy. The underlying mechanism has not been unveiled completely, especially for antigen-negative relapse. More studies are warranted to clarify them to promote the proposal of potential strategies. On the one hand, CAR engineering strategies are a critical part of tackling antigen-positive relapse issues by enhancing the potency and persistence of T-cells so far. On the other hand, clinically, multiantigen-targeted CAR T-cells, combinatorial therapy and sequential therapy are in the spotlight. Multiantigen-targeted CAR T-cells require the search for more rational target combinations and optimization of multi-CAR constructs to augment the specific killing effects without impairing the efficacy. In terms of combinatorial therapy, combining checkpoint inhibitors, antitumor vaccines or other novel agents may exert synergic effects, but further clinical trials are required to confirm their efficacy and safety. In sequential therapy, whether to bridge HSCT is a hotspot. An increasing number of studies have recently demonstrated its benefits, especially for patients reaching MRD-negative CR after CAR T-cell therapy. Further confirmation of the specific indications, such as age, high-risk mutation, disease risk stratification, CAR T-cell persistence in vivo, B-cell aplasia duration and donor type of transplantation, is necessary.
With regard to safety issues, CRS and NT deserve intensive attention, and their mechanisms are not completely understood. A growing number of in vitro and animal experiments have been launched to better characterize these systemic cytokine toxicities. It has been confirmed that the cytokine storm is the result of multicell crossover, while the interactions between CAR T-cells, targeted cells, other immune cells, and various cytokines are worthy of further study. Even though consensus on grading and management is introduced, there still exists controversy in individualized medication. Novel drugs and optimization schemes are needed. Additionally, off-the-shelf CAR T-cells may be a promising direction to mitigate the obstacle of access and promote the commercialization process, while manufacturing techniques and the efficacy of the products need further improvements. Generally, the challenges brought by CAR T-cell therapy in turn promote its development. In the future, more innovative and clinical controlled studies are needed to optimize and confirm this promising therapy.
Author Contributions
XXu, ST and YL designed the study. XXu, SH, XXiao, and QS drafted the manuscript. QS, XXiao, XL, and SH prepared the tables and figures. SC, ZZ, and ZH participated in data collection. All authors participated in the revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by grants from the Clinical Research Startup Program of Southern Medical University by High-level University Construction Funding of Guangdong Provincial Department of Education (no. LC2016ZD027), the Guangdong Science and Technology Department (no. 2017A020215183), the Major Program for Health Medical Collaborative Innovation of Guangzhou (no. 201704020216), research funds from Natural Science Foundation of Guangdong Province, China (no. 2018B030311042) and Frontier Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (no. 2018GZR110105014).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
XXu, SH, XXiao, QS, XL, SC, ZZ, and ZH are the Small Small bird (SSb) group members and acknowledge the strong support from SSb.
References
1. Nguyen K, Devidas M, Cheng SC, La M, Raetz EA, Carroll WL, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a children’s oncology group study. Leukemia (2008) 22(12):2142–50. doi: 10.1038/leu.2008.251
2. Oriol A, Vives S, Hernández-Rivas JM, Tormo M, Heras I, Rivas C, et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the pethema study group. Haematologica (2010) 95(4):589–96. doi: 10.3324/haematol.2009.014274
3. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
4. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371(16):1507–17. doi: 10.1056/NEJMoa1407222
5. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3
6. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126(6):2123–38. doi: 10.1172/JCI85309
7. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood (2017) 129(25):3322–31. doi: 10.1182/blood-2017-02-769208
8. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
9. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med (2018) 378(5):449–59. doi: 10.1056/NEJMoa1709919
10. Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood (2019) 133(15):1652–63. doi: 10.1182/blood-2018-11-883710
11. Frey NV, Shaw PA, Hexner EO, Pequignot E, Gill S, Luger SM, et al. Optimizing chimeric antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol (2020) 38(5):415–22. doi: 10.1200/JCO.19.01892
12. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med (2018) 24(1):20–8. doi: 10.1038/nm.4441
13. Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia (2019) 33(12):2854–66. doi: 10.1038/s41375-019-0488-7
14. Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a Phase I Anti-CD22 CAR T-cell trial. J Clin Oncol (2020) 38(17):1938–50. doi: 10.1200/JCO.19.03279
15. US Food and Drug Administration. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome (2017). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome (Accessed June 3, 2020).
16. Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, et al. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol (2008) 9(1):63–72 doi: doi:10.1038/ni1547
17. Tedder TF, Zhou LJ, Engel P. The CD19/CD21 signal transduction complex of B lymphocytes. Immunol Today (1994) 15(9):437–42. doi: 10.1016/0167-5699(94)90274-7
18. Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol (2012) 1(1):36. doi: 10.1186/2162-3619-1-36
19. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet (2020) 395(10230):1146–62. doi: 10.1016/S0140-6736(19)33018-1
20. Nitschke L. CD22 and Siglec-G: B-cell inhibitory receptors with distinct functions. Immunol Rev (2009) 230(1):128–43. doi: 10.1111/j.1600-065X.2009.00801.x
21. Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res (2014) 2(1):4. doi: 10.1186/2050-7771-2-4
22. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev (2008) 88(3):841–86. doi: 10.1152/physrev.00035.2007
23. Lopez-Millan B, Sanchéz-Martínez D, Roca-Ho H, Gutiérrez-Agüera F, Molina O, Diaz de la Guardia R, et al. NG2 antigen is a therapeutic target for MLL-rearranged B-cell acute lymphoblastic leukemia. Leukemia (2019) 33(7):1557–69. doi: 10.1038/s41375-018-0353-0
24. Menendez P, Bueno C. Expression of NG2 antigen in MLL-rearranged acute leukemias: how complex does it get? Leuk Res (2011) 35(8):989–90. doi: 10.1016/j.leukres.2011.03.015
25. Qin H, Cho M, Haso W, Zhang L, Tasian SK, Oo HZ, et al. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood (2015) 126(5):629–39. doi: 10.1182/blood-2014-11-612903
26. Lo Kuan E, Ziegler SF. Thymic stromal lymphopoietin and cancer. J Immunol (2014) 193(9):4283–8. doi: 10.4049/jimmunol.1400864
27. Ziegler SF, Liu YJ. Thymic stromal lymphopoietin in normal and pathogenic T cell development and function. Nat Immunol (2006) 7(7):709–14. doi: 10.1038/ni1360
28. Wuchter C, Ratei R, Spahn G, Schoch C, Harbott J, Schnittger S, et al. Impact of CD133 (AC133) and CD90 expression analysis for acute leukemia immunophenotyping. Haematologica (2001) 86(2):154–61. doi: 10.3324/%25x
29. Li D, Hu Y, Jin Z, Zhai Y, Tan Y, Sun Y, et al. TanCAR T cells targeting CD19 and CD133 efficiently eliminate MLL leukemic cells. Leukemia (2018) 32(9):2012–6. doi: 10.1038/s41375-018-0212-z
30. Thomas DA, O’Brien S, Kantarjian HM. Monoclonal antibody therapy with rituximab for acute lymphoblastic leukemia. Hematol Oncol Clin North Am (2009) 23(5):949–71, v. doi: 10.1016/j.hoc.2009.07.005
31. Fousek K, Watanabe J, George A, An X, Samaha H, Navai SA, et al. Targeting primary pre-B cell acute lymphoblastic leukemia and CD19-negative relapses using trivalent CAR T cells. Blood (2017) 130(Suppl. 1):4614. doi: 10.1182/blood.V130.Suppl_1.4614.4614
32. Yu JX, Upadhaya S, Tatake R, Barkalow F, Hubbard-Lucey VM. Cancer cell therapies: the clinical trial landscape. Nat Rev Drug Discov (2020) 19(9):583–4. doi: 10.1038/d41573-020-00099-9
33. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov (2016) 6(6):664–79. doi: 10.1158/2159-8290.CD-16-0040
34. von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/Phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol (2016) 34(36):4381–9. doi: 10.1200/JCO.2016.67.3301
35. Jeha S, Gaynon PS, Razzouk BI, Franklin J, Kadota R, Shen V, et al. Phase II study of clofarabine in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. J Clin Oncol (2006) 24(12):1917–23. doi: 10.1200/JCO.2005.03.8554
36. Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematol (2020) 7(11):e816–e26. doi: 10.1016/S2352-3026(20)30277-5
37. Iwamoto S, Deguchi T, Ohta H, Kiyokawa N, Tsurusawa M, Yamada T, et al. Flow cytometric analysis of de novo acute lymphoblastic leukemia in childhood: report from the japanese pediatric leukemia/lymphoma study group. Int J Hematol (2011) 94(2):185–92. doi: 10.1007/s12185-011-0900-1
38. Kantarjian H, Thomas D, Jorgensen J, Kebriaei P, Jabbour E, Rytting M, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer (2013) 119(15):2728–36. doi: 10.1002/cncr.28136
39. FitzGerald DJ, Wayne AS, Kreitman RJ, Pastan I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res (2011) 71(20):6300–9. doi: 10.1158/0008-5472.CAN-11-1374
40. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest (2016) 126(10):3814–26. doi: 10.1172/JCI87366
41. Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood (2013) 122(18):3138–48. doi: 10.1182/blood-2012-12-474056
42. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood (2014) 123(15):2343–54. doi: 10.1182/blood-2013-09-529537
43. Blatt K, Menzl I, Eisenwort G, Cerny-Reiterer S, Herrmann H, Herndlhofer S, et al. Phenotyping and target expression profiling of CD34(+)/CD38(-) and CD34(+)/CD38(+) stem- and progenitor cells in acute lymphoblastic leukemia. Neoplasia (2018) 20(6):632–42. doi: 10.1016/j.neo.2018.04.004
44. Naik J, Themeli M, de Jong-Korlaar R, Ruiter RWJ, Poddighe PJ, Yuan H, et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica (2019) 104(3):e100–e3. doi: 10.3324/haematol.2018.192757
45. Guo Y, Feng K, Tong C, Jia H, Liu Y, Wang Y, et al. Efficiency and side effects of anti-CD38 CAR T cells in an adult patient with relapsed B-ALL after failure of bi-specific CD19/CD22 CAR T cell treatment. Cell Mol Immunol (2020) 17(4):430–2. doi: 10.1038/s41423-019-0355-5
46. Gao Z, Tong C, Wang Y, Chen D, Wu Z, Han W. Blocking CD38-driven fratricide among T cells enables effective antitumor activity by CD38-specific chimeric antigen receptor T cells. J Genet Genomics (2019) 46(8):367–77. doi: 10.1016/j.jgg.2019.06.007
47. Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, et al. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science (2001) 293(5537):2108–11. doi: 10.1126/science.1061965
48. Ng LG, Sutherland AP, Newton R, Qian F, Cachero TG, Scott ML, et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J Immunol (2004) 173(2):807–17. doi: 10.4049/jimmunol.173.2.807
49. Parameswaran R, Müschen M, Kim YM, Groffen J, Heisterkamp N. A functional receptor for B-cell-activating factor is expressed on human acute lymphoblastic leukemias. Cancer Res (2010) 70(11):4346–56. doi: 10.1158/0008-5472.CAN-10-0300
50. Turazzi N, Fazio G, Rossi V, Rolink A, Cazzaniga G, Biondi A, et al. Engineered T cells towards TNFRSF13C (BAFFR): a novel strategy to efficiently target B-cell acute lymphoblastic leukaemia. Br J Haematol (2018) 182(6):939–43. doi: 10.1111/bjh.14899
51. Qin H, Dong Z, Wang X, Cheng WA, Wen F, Xue W, et al. CAR T cells targeting BAFF-R can overcome CD19 antigen loss in B cell malignancies. Sci Transl Med (2019) 11:511. doi: 10.1126/scitranslmed.aaw9414
52. Fazio G, Turazzi N, Cazzaniga V, Kreuzaler M, Maglia O, Magnani CF, et al. TNFRSF13C (BAFFR) positive blasts persist after early treatment and at relapse in childhood B-cell precursor acute lymphoblastic leukaemia. Br J Haematol (2018) 182(3):434–6. doi: 10.1111/bjh.14794
53. Campoli MR, Chang CC, Kageshita T, Wang X, McCarthy JB, Ferrone S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): a melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit Rev Immunol (2004) 24(4):267–96. doi: 10.1615/CritRevImmunol.v24.i4.40
54. Yadavilli S, Hwang EI, Packer RJ, Nazarian J. The role of NG2 proteoglycan in glioma. Transl Oncol (2016) 9(1):57–63. doi: 10.1016/j.tranon.2015.12.005
55. Wang X, Osada T, Wang Y, Yu L, Sakakura K, Katayama A, et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst (2010) 102(19):1496–512. doi: 10.1093/jnci/djq343
56. Behm FG, Smith FO, Raimondi SC, Pui CH, Bernstein ID. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood (1996) 87(3):1134–9. doi: 10.1182/blood.V87.3.1134.bloodjournal8731134
57. Schwartz S, Rieder H, Schläger B, Burmeister T, Fischer L, Thiel E. Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia (2003) 17(8):1589–95. doi: 10.1038/sj.leu.2402989
58. Hilden JM, Dinndorf PA, Meerbaum SO, Sather H, Villaluna D, Heerema NA, et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the children’s oncology group. Blood (2006) 108(2):441–51. doi: 10.1182/blood-2005-07-3011
59. Wuchter C, Harbott J, Schoch C, Schnittger S, Borkhardt A, Karawajew L, et al. Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia (2000) 14(7):1232–8. doi: 10.1038/sj.leu.2401840
60. Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol (2012) 7:283–301. doi: 10.1146/annurev-pathol-011811-132434
61. Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood (2016) 127(20):2406–10. doi: 10.1182/blood-2015-08-665547
62. Harrer DC, Schuler G, Dörrie J, Schaft N. CSPG4-specific CAR T cells for high-risk childhood B cell precursor leukemia. Int J Mol Sci (2019) 20(11):2764. doi: 10.3390/ijms20112764
63. Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med (2011) 208(5):901–8. doi: 10.1084/jem.20110580
64. Loh ML, Zhang J, Harvey RC, Roberts K, Payne-Turner D, Kang H, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the children’s oncology group TARGET project. Blood (2013) 121(3):485–8. doi: 10.1182/blood-2012-04-422691
65. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and down syndrome-associated acute lymphoblastic leukemia. Nat Genet (2009) 41(11):1243–6. doi: 10.1038/ng.469
66. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med (2014) 371(11):1005–15. doi: 10.1056/NEJMoa1403088
67. van Bodegom D, Zhong J, Kopp N, Dutta C, Kim MS, Bird L, et al. Differences in signaling through the B-cell leukemia oncoprotein CRLF2 in response to TSLP and through mutant JAK2. Blood (2012) 120(14):2853–63. doi: 10.1182/blood-2012-02-413252
68. Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood (2012) 120(4):833–42. doi: 10.1182/blood-2011-12-389932
69. Whittington MD, McQueen RB, Campbell JD. Valuing chimeric antigen receptor T-cell therapy: current evidence, uncertainties, and payment implications. J Clin Oncol (2020) 38(4):359–66. doi: 10.1200/JCO.19.01558
70. Inserro A. CMS approves extra payments for CAR T, increases other payments in final rule AJMC Newsroom (2018). Available at: https://www.ajmc.com/newsroom/cms-approves-extra-payments-for-car-t-increases-other-payments-in-final-rule (Accessed June 3, 2020).
71. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev (2017) 4:92–101. doi: 10.1016/j.omtm.2016.12.006
72. Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat Rev Clin Oncol (2019) 16(1):45–63. doi: 10.1038/s41571-018-0075-2
73. Mejstríková E, Hrusak O, Borowitz MJ, Whitlock JA, Brethon B, Trippett TM, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J (2017) 7(12):659. doi: 10.1038/s41408-017-0023-x
74. Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood (1997) 89(10):3700–7. doi: 10.1182/blood.V89.10.3700
75. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med (2002) 8(4):379–85. doi: 10.1038/nm0402-379
76. Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell (2014) 159(4):814–28. doi: 10.1016/j.cell.2014.10.026
77. Das R, Storm J, Barrett D. Abstract 1631: T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit chimeric antigen receptor potential. Cancer Res (2018) 78(Suppl. 13):1631. doi: 10.1158/1538-7445.AM2018-1631
78. Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med (2016) 8(320):320ra3. doi: 10.1126/scitranslmed.aad5222
79. Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy (2016) 18(7):893–901. doi: 10.1016/j.jcyt.2016.04.003
80. Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol (2018) 53:164–81. doi: 10.1016/j.copbio.2018.01.025
81. Perica K, Curran KJ, Brentjens RJ, Giralt SA. Building a CAR garage: preparing for the delivery of commercial CAR T cell products at memorial sloan kettering cancer center. Biol Blood Marrow Transpl (2018) 24(6):1135–41. doi: 10.1016/j.bbmt.2018.02.018
82. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med (2018) 24(6):739–48. doi: 10.1038/s41591-018-0036-4
83. Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res (2018) 6:4. doi: 10.1186/s40364-018-0116-0
84. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124(2):188–95. doi: 10.1182/blood-2014-05-552729
85. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood (2017) 130(21):2295–306. doi: 10.1182/blood-2017-06-793141
86. Jin Z, Xiang R, Qing K, Li X, Zhang Y, Wang L, et al. The severe cytokine release syndrome in phase I trials of CD19-CAR-T cell therapy: a systematic review. Ann Hematol (2018) 97(8):1327–35. doi: 10.1007/s00277-018-3368-8
87. Rafei H, Kantarjian HM, Jabbour EJ. Recent advances in the treatment of acute lymphoblastic leukemia. Leuk Lymphoma (2019) 60(11):2606–21. doi: 10.1080/10428194.2019.1605071
88. Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol (2018) 18(12):773–89. doi: 10.1038/s41577-018-0066-7
89. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med (2018) 24(6):731–8. doi: 10.1038/s41591-018-0041-7
90. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
91. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
92. Rice J, Nagle S, Randall J, Hinson HE. Chimeric antigen receptor T cell-related neurotoxicity: mechanisms, clinical presentation, and approach to treatment. Curr Treat Options Neurol (2019) 21(8):40. doi: 10.1007/s11940-019-0580-3
93. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov (2018) 8(8):958–71. doi: 10.1158/2159-8290.CD-17-1319
94. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov (2017) 7(12):1404–19. doi: 10.1158/2159-8290.CD-17-0698
95. Gragnano F, Sperlongano S, Golia E, Natale F, Bianchi R, Crisci M, et al. The role of von willebrand factor in vascular inflammation: from pathogenesis to targeted therapy. Mediators Inflamm (2017) 2017:5620314. doi: 10.1155/2017/5620314
96. Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med (2006) 12(2):235–9. doi: 10.1038/nm1351
97. Gust J, Taraseviciute A, Turtle CJ. Neurotoxicity associated with CD19-targeted CAR-T cell therapies. CNS Drugs (2018) 32(12):1091–101. doi: 10.1007/s40263-018-0582-9
98. Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell (2020) 183(1):126–42.e17. doi: 10.1016/j.cell.2020.08.022
99. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol (2018) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
100. Howard SC, Trifilio S, Gregory TK, Baxter N, McBride A. Tumor lysis syndrome in the era of novel and targeted agents in patients with hematologic malignancies: a systematic review. Ann Hematol (2016) 95(4):563–73. doi: 10.1007/s00277-015-2585-7
101. Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest (2019) 129(5):2123–32. doi: 10.1172/JCI125423
102. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest (2005) 115(6):1616–26. doi: 10.1172/JCI24480
103. Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci USA (2009) 106(41):17469–74. doi: 10.1073/pnas.0907448106
104. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. doi: 10.1038/nm.2446
105. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
106. Singh H, Figliola MJ, Dawson MJ, Olivares S, Zhang L, Yang G, et al. Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PloS One (2013) 8(5):e64138. doi: 10.1371/journal.pone.0064138
107. Priesner C, Aleksandrova K, Esser R, Mockel-Tenbrinck N, Leise J, Drechsel K, et al. Automated enrichment, transduction, and expansion of clinical-scale CD62L(+) T cells for manufacturing of gene therapy medicinal products. Hum Gene Ther (2016) 27(10):860–9. doi: 10.1089/hum.2016.091
108. Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood (2016) 127(24):2980–90. doi: 10.1182/blood-2015-12-686725
109. McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol (2019) 97(7):664–74. doi: 10.1111/imcb.12254
110. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood (2014) 123(24):3750–9. doi: 10.1182/blood-2014-01-552174
111. Ali R, Babad J, Follenzi A, Gebe JA, Brehm MA, Nepom GT, et al. Genetically modified human CD4(+) T cells can be evaluated in vivo without lethal graft-versus-host disease. Immunology (2016) 148(4):339–51. doi: 10.1111/imm.12613
112. Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov (2020) 10(4):552–67. doi: 10.1158/2159-8290.CD-19-0813
113. An F, Wang H, Liu Z, Wu F, Zhang J, Tao Q, et al. Influence of patient characteristics on chimeric antigen receptor T cell therapy in B-cell acute lymphoblastic leukemia. Nat Commun (2020) 11(1):5928. doi: 10.1038/s41467-020-19774-x
114. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med (2014) 6(224):224ra25. doi: 10.1126/scitranslmed.3008226
115. Zheng W, O’Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia (2018) 32(5):1157–67. doi: 10.1038/s41375-017-0008-6
116. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T Cells. Immunity (2016) 44(2):380–90. doi: 10.1016/j.immuni.2016.01.021
117. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
118. Cao J, Wang G, Cheng H, Wei C, Qi K, Sang W, et al. Potent anti-leukemia activities of humanized CD19-targeted chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am J Hematol (2018) 93(7):851–8. doi: 10.1002/ajh.25108
119. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med (2019) 25(9):1408–14. doi: 10.1038/s41591-019-0549-5
120. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi: 10.1038/leu.2015.247
121. Guha P, Cunetta M, Somasundar P, Espat NJ, Junghans RP, Katz SC. Frontline Science: Functionally impaired geriatric CAR-T cells rescued by increased α5β1 integrin expression. J Leukoc Biol (2017) 102(2):201–8. doi: 10.1189/jlb.5HI0716-322RR
122. Kotani H, Li G, Yao J, Mesa TE, Chen J, Boucher JC, et al. Aged CAR T cells exhibit enhanced cytotoxicity and effector function but shorter persistence and less memory-like phenotypes. Blood (2018) 132(Suppl. 1):2047. doi: 10.1182/blood-2018-99-115351
123. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature (2019) 576(7786):293–300. doi: 10.1038/s41586-019-1805-z
124. Nie Y, Lu W, Chen D, Tu H, Guo Z, Zhou X, et al. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark Res (2020) 8:18. doi: 10.1186/s40364-020-00197-1
125. Xu X, Sun Q, Liang X, Chen Z, Zhang X, Zhou X, et al. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front Immunol (2019) 10:2664. doi: 10.3389/fimmu.2019.02664
126. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov (2015) 5(12):1282–95. doi: 10.1158/2159-8290.CD-15-1020
127. Nagel I, Bartels M, Duell J, Oberg HH, Ussat S, Bruckmueller H, et al. Hematopoietic stem cell involvement in BCR-ABL1-positive ALL as a potential mechanism of resistance to blinatumomab therapy. Blood (2017) 130(18):2027–31. doi: 10.1182/blood-2017-05-782888
128. Oberley MJ, Gaynon PS, Bhojwani D, Pulsipher MA, Gardner RA, Hiemenz MC, et al. Myeloid lineage switch following chimeric antigen receptor T-cell therapy in a patient with TCF3-ZNF384 fusion-positive B-lymphoblastic leukemia. Pediatr Blood Cancer (2018) 65(9):e27265. doi: 10.1002/pbc.27265
129. Fischer J, Paret C, El Malki K, Alt F, Wingerter A, Neu MA, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother (2017) 40(5):187–95. doi: 10.1097/CJI.0000000000000169
130. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature (2019) 568(7750):112–6. doi: 10.1038/s41586-019-1054-1
131. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med (2018) 24(10):1499–503. doi: 10.1038/s41591-018-0201-9
132. Mock U, Nickolay L, Philip B, Cheung GW, Zhan H, Johnston ICD, et al. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy (2016) 18(8):1002–11. doi: 10.1016/j.jcyt.2016.05.009
133. Vargas JE, Chicaybam L, Stein RT, Tanuri A, Delgado-Cañedo A, Bonamino MH. Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J Transl Med (2016) 14(1):288. doi: 10.1186/s12967-016-1047-x
134. Ghassemi S, Nunez-Cruz S, O’Connor RS, Fraietta JA, Patel PR, Scholler J, et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol Res (2018) 6(9):1100–9. doi: 10.1158/2326-6066.CIR-17-0405
135. Chicaybam L, Abdo L, Bonamino MH. Generation of CAR+ T lymphocytes using the sleeping beauty transposon system. Methods Mol Biol (2020) 2086:131–7. doi: 10.1007/978-1-0716-0146-4_9
136. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543(7643):113–7. doi: 10.1038/nature21405
137. Agarwal S, Weidner T, Thalheimer FB, Buchholz CJ. In vivo generated human CAR T cells eradicate tumor cells. Oncoimmunology (2019) 8(12):e1671761. doi: 10.1080/2162402X.2019.1671761
138. Frank AM, Buchholz CJ. Surface-engineered lentiviral vectors for selective gene transfer into subtypes of lymphocytes. Mol Ther Methods Clin Dev (2019) 12:19–31. doi: 10.1016/j.omtm.2018.10.006
139. Yang J, He J, Zhang X, Wang Z, Zhang Y, Cai S, et al. A feasibility and safety study of a new CD19-directed fast CAR-T therapy for refractory and relapsed B cell acute lymphoblastic leukemia. Blood (2019) 134(Suppl. 1):825. doi: 10.1182/blood-2019-121751
140. Zhang R, He Y, Yang D, Wang Y, Feng S, Wang J, et al. Donor-derived second generation of CD19 CAR-T cell therapy for relapsed B-cell acute lymphoblastic leukemia after allogenic stem cell transplantation. Blood (2018) 132(Suppl. 1):2179. doi: 10.1182/blood-2018-99-114950
141. Tu S, Deng L, Huang R, Zhou X, Yang J, Zhou W, et al. A novel chimeric antigen receptor T cells therapy strategy that dual targeting CD19 and CD123 to treat relapsed acute lymphoblastic leukemia after allogeneic hematopoietic stem cell transplantation. Blood (2018) 132(Suppl. 1):4015. doi: 10.1182/blood-2018-99-118526
142. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol (2018) 11(1):132. doi: 10.1186/s13045-018-0677-2
143. Benjamin R, Graham C, Yallop D, Jozwik A, Ciocarlie O, Jain N, et al. Preliminary data on safety, cellular kinetics and anti-leukemic activity of UCART19, an allogeneic anti-CD19 CAR T-cell product, in a pool of adult and pediatric patients with high-risk CD19+ relapsed/refractory B-cell acute lymphoblastic leukemia. Blood (2018) 132(Suppl. 1):896. doi: 10.1182/blood-2018-99-111356
144. Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. The safety and efficacy of a CRISPR/Cas9-engineered universal CAR-T Cell Product (CTA101) in patients with relapsed/refractory b-cell acute lymphoblastic leukemia. Blood (2020) 136(Suppl. 1):52. doi: 10.1182/blood-2020-142262
145. Ruella M, Kenderian SS. Next-generation chimeric antigen receptor T-cell therapy: Going off the shelf. BioDrugs (2017) 31(6):473–81. doi: 10.1007/s40259-017-0247-0
146. Kamiya T, Wong D, Png YT, Campana D. A novel method to generate T-cell receptor-deficient chimeric antigen receptor T cells. Blood Adv (2018) 2(5):517–28. doi: 10.1182/bloodadvances.2017012823
147. Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol (2016) 34(10):1112–21. doi: 10.1200/JCO.2015.64.5929
148. Chen Y, Cheng Y, Suo P, Yan C, Wang Y, Chen Y, et al. Donor-derived CD19-targeted T cell infusion induces minimal residual disease-negative remission in relapsed B-cell acute lymphoblastic leukaemia with no response to donor lymphocyte infusions after haploidentical haematopoietic stem cell transplantation. Br J Haematol (2017) 179(4):598–605. doi: 10.1111/bjh.14923
149. Qasim W. Allogeneic CAR T cell therapies for leukemia. Am J Hematol (2019) 94(S1):S50–s4. doi: 10.1002/ajh.25399
150. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transpl (2019) 25(4):625–38. doi: 10.1016/j.bbmt.2018.12.758
151. Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, et al. FDA approval summary: Tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist (2018) 23(8):943–7. doi: 10.1634/theoncologist.2018-0028
152. Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood (2019) 134(24):2149–58. doi: 10.1182/blood.2019001463
153. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Locke FL, Lin Y, et al. Toxicity management after chimeric antigen receptor T cell therapy: one size does not fit ‘ALL’. Nat Rev Clin Oncol (2018) 15(4):218. doi: 10.1038/nrclinonc.2018.20
154. Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood (2015) 125(26):4103–13. doi: 10.1182/blood-2015-02-628354
155. Brattås MK, Reikvam H, Tvedt THA, Bruserud Ø. Dasatinib as an investigational drug for the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia in adults. Expert Opin Investig Drugs (2019) 28(5):411–20. doi: 10.1080/13543784.2019.1597052
156. Chen H, Wang F, Zhang P, Zhang Y, Chen Y, Fan X, et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front Med (2019) 13(5):610–7. doi: 10.1007/s11684-019-0714-8
157. Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J (2020) 10(2):15. doi: 10.1038/s41408-020-0280-y
158. Park JH, Santomasso B, Riviere I, Senechal B, Wang X, Purdon T, et al. Baseline and early post-treatment clinical and laboratory factors associated with severe neurotoxicity following 19-28z CAR T cells in adult patients with relapsed B-ALL. J Clin Oncol (2017) 35(Suppl. 15):7024. doi: 10.1200/JCO.2017.35.15_suppl.7024
159. Chen F, Teachey DT, Pequignot E, Frey N, Porter D, Maude SL, et al. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J Immunol Methods (2016) 434:1–8. doi: 10.1016/j.jim.2016.03.005
160. Gutierrez C, Brown ART, Herr MM, Kadri SS, Hill B, Rajendram P, et al. The chimeric antigen receptor-intensive care unit (CAR-ICU) initiative: Surveying intensive care unit practices in the management of CAR T-cell associated toxicities. J Crit Care (2020) 58:58–64. doi: 10.1016/j.jcrc.2020.04.008
161. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
162. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell (2020) 38(4):473–88. doi: 10.1016/j.ccell.2020.07.005
163. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127(26):3321–30. doi: 10.1182/blood-2016-04-703751
164. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell (2016) 164(4):770–9. doi: 10.1016/j.cell.2016.01.011
165. Rajasekaran S, Kruse K, Kovey K, Davis AT, Hassan NE, Ndika AN, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children*. Pediatr Crit Care Med (2014) 15(5):401–8. doi: 10.1097/PCC.0000000000000078
166. Wohlfarth P, Agis H, Gualdoni GA, Weber J, Staudinger T, Schellongowski P, et al. Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically III adult patients with hemophagocytic lymphohistiocytosis. J Intensive Care Med (2019) 34(9):723–31. doi: 10.1177/0885066617711386
167. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48(2):124–31. doi: 10.1002/pbc.21039
168. Qin H, Ramakrishna S, Nguyen S, Fountaine TJ, Ponduri A, Stetler-Stevenson M, et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol Ther Oncolytics (2018) 11:127–37. doi: 10.1016/j.omto.2018.10.006
169. Amrolia PJ, Wynn R, Hough RE, Vora A, Bonney D, Veys P, et al. Phase I study of AUTO3, a bicistronic chimeric antigen receptor (CAR) T-cell therapy targeting CD19 and CD22, in pediatric patients with relapsed/refractory B-cell acute lymphoblastic leukemia (r/r B-ALL): Amelia Study. Blood (2019) 134(Suppl. 1):2620. doi: 10.1182/blood-2019-123424
170. Martyniszyn A, Krahl AC, André MC, Hombach AA, Abken H. CD20-CD19 bispecific CAR T cells for the treatment of B-cell malignancies. Hum Gene Ther (2017) 28(12):1147–57. doi: 10.1089/hum.2017.126
171. Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol (2020) 13(1):30. doi: 10.1186/s13045-020-00856-8
172. Fousek K, Watanabe J, Joseph SK, George A, An X, Byrd TT, et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia (2020) 35(1):75–89. doi: 10.1038/s41375-020-0792-2
173. Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood (2020) 136(14):1632–44. doi: 10.1182/blood.2020005278
174. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res (2016) 4(6):498–508. doi: 10.1158/2326-6066.CIR-15-0231
175. Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol (2019) 12(1):128. doi: 10.1186/s13045-019-0813-7
176. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
177. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
178. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
179. Maude SL, Hucks GE, Seif AE, Talekar MK, Teachey DT, Baniewicz D, et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J Clin Oncol (2017) 35(Suppl. 15):103. doi: 10.1200/JCO.2017.35.15_suppl.103
180. Li AM, Hucks GE, Dinofia AM, Seif AE, Teachey DT, Baniewicz D, et al. Checkpoint inhibitors augment CD19-directed Chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood (2018) 132(Suppl. 1):556. doi: 10.1182/blood-2018-99-112572
181. Shah NN. The one-two punch (of CAR T cells). Blood (2020) 135(5):303–4. doi: 10.1182/blood.2019004272
182. Wang N, Hu X, Cao W, Li C, Xiao Y, Cao Y, et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood (2020) 135(1):17–27. doi: 10.1182/blood.2019000017
183. Pan J, Zuo S, Deng B, Xu X, Li C, Zheng Q, et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood (2020) 135(5):387–91. doi: 10.1182/blood.2019003293
184. Jabbour E, Short NJ, Jorgensen JL, Yilmaz M, Ravandi F, Wang SA, et al. Differential impact of minimal residual disease negativity according to the salvage status in patients with relapsed/refractory B-cell acute lymphoblastic leukemia. Cancer (2017) 123(2):294–302. doi: 10.1002/cncr.30264
185. Stein A, Palmer J, Tsai NC, Al Malki MM, Aldoss I, Ali H, et al. Phase I trial of total marrow and lymphoid irradiation transplantation conditioning in patients with relapsed/refractory acute leukemia. Biol Blood Marrow Transpl (2017) 23(4):618–24. doi: 10.1016/j.bbmt.2017.01.067
186. Zhao H, Wei J, Wei G, Luo Y, Shi J, Cui Q, et al. Pre-transplant MRD negativity predicts favorable outcomes of CAR-T therapy followed by haploidentical HSCT for relapsed/refractory acute lymphoblastic leukemia: a multi-center retrospective study. J Hematol Oncol (2020) 13(1):42. doi: 10.1186/s13045-020-00873-7
187. Gu B, Shi BY, Zhang X, Zhou SY, Chu JH, Wu XJ, et al. Allogeneic haematopoietic stem cell transplantation improves outcome of adults with relapsed/refractory Philadelphia chromosome-positive acute lymphoblastic leukemia entering remission following CD19 chimeric antigen receptor T cells. Bone Marrow Transpl (2020) 56(1):91–100. doi: 10.1038/s41409-020-0982-6
Keywords: CAR T-cell, application, challenges, access, adverse events, resistance, relapse, clinical strategies
Citation: Xu X, Huang S, Xiao X, Sun Q, Liang X, Chen S, Zhao Z, Huo Z, Tu S and Li Y (2021) Challenges and Clinical Strategies of CAR T-Cell Therapy for Acute Lymphoblastic Leukemia: Overview and Developments. Front. Immunol. 11:569117. doi: 10.3389/fimmu.2020.569117
Received: 03 June 2020; Accepted: 22 December 2020;
Published: 10 February 2021.
Edited by:
Jose A. Garcia-Sanz, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Francisco Martin, Andalusian Autonomous Government of Genomics and Oncological Research (GENYO), SpainYi Luo, Zhejiang University, China
Copyright © 2021 Xu, Huang, Xiao, Sun, Liang, Chen, Zhao, Huo, Tu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanfang Tu, doctortutu@163.com; Yuhua Li, liyuhua2011gz@163.com
†These authors have contributed equally to this work