Mechanoregulation of Vascular Endothelial Growth Factor Receptor 2 in Angiogenesis
 Bronte Miller
Bronte Miller Mary Kathryn Sewell-Loftin
Mary Kathryn Sewell-Loftin- 1Department of Biomedical Engineering, University of Alabama at Birmingham, Birmingham, AL, United States
- 2O'Neal Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, AL, United States
The endothelial cells that compose the vascular system in the body display a wide range of mechanotransductive behaviors and responses to biomechanical stimuli, which act in concert to control overall blood vessel structure and function. Such mechanosensitive activities allow blood vessels to constrict, dilate, grow, or remodel as needed during development as well as normal physiological functions, and the same processes can be dysregulated in various disease states. Mechanotransduction represents cellular responses to mechanical forces, translating such factors into chemical or electrical signals which alter the activation of various cell signaling pathways. Understanding how biomechanical forces drive vascular growth in healthy and diseased tissues could create new therapeutic strategies that would either enhance or halt these processes to assist with treatments of different diseases. In the cardiovascular system, new blood vessel formation from preexisting vasculature, in a process known as angiogenesis, is driven by vascular endothelial growth factor (VEGF) binding to VEGF receptor 2 (VEGFR-2) which promotes blood vessel development. However, physical forces such as shear stress, matrix stiffness, and interstitial flow are also major drivers and effectors of angiogenesis, and new research suggests that mechanical forces may regulate VEGFR-2 phosphorylation. In fact, VEGFR-2 activation has been linked to known mechanobiological agents including ERK/MAPK, c-Src, Rho/ROCK, and YAP/TAZ. In vascular disease states, endothelial cells can be subjected to altered mechanical stimuli which affect the pathways that control angiogenesis. Both normalizing and arresting angiogenesis associated with tumor growth have been strategies for anti-cancer treatments. In the field of regenerative medicine, harnessing biomechanical regulation of angiogenesis could enhance vascularization strategies for treating a variety of cardiovascular diseases, including ischemia or permit development of novel tissue engineering scaffolds. This review will focus on the impact of VEGFR-2 mechanosignaling in endothelial cells (ECs) and its interaction with other mechanotransductive pathways, as well as presenting a discussion on the relationship between VEGFR-2 activation and biomechanical forces in the extracellular matrix (ECM) that can help treat diseases with dysfunctional vascular growth.
Introduction
Angiogenesis, or the development of blood vessels from preexisting vasculature, is a pillar of normal development and growth; however, various disease states are associated with dysregulation of blood vessel growth. For example, tumors express irregular and tortuous vasculature, while ischemia is the lack of functional vessels, creating hypoxic environments and ultimately leading to tissue death. Much research has focused on either inhibiting or promoting angiogenic processes to treat diseases, but these therapeutic approaches have shown mixed results. Perhaps some of these failures are due in part to the focus on biochemical regulation of angiogenesis while ignoring mechanical cues that could also be controlling blood vessel development. For this review, we will use biochemical signaling to refer to the binding of vascular endothelial growth factor (VEGF) to VEGF receptor 2 (VEGFR-2), while biomechanical signaling will refer to VEGFR-2 activation or downstream signaling induced by non-ligand binding cues.
Mechanotransduction is the process by which mechanical signals from the extracellular matrix (ECM), nearby cells, or surrounding fluids interact with a cell and are subsequently processed into a biochemical signaling cascade of various proteins to regulate the cellular response to the mechanical cue. In the vasculature, the endothelial cells (ECs) that comprise the vessel walls are in a constant state of mechanical stimulation, through a variety of forces (Figure 1). This includes not only shear stress from blood flow within the vessels, but also interstitial flow over the vessels from the surrounding tissues (1–3). There are compressive strains due to matrix composition or stiffness, as well as regulation of the vascular tone by vascular smooth muscle cells (VSMCs) or pericytes (4, 5). Also, the geometry of blood vessels induces curvature stresses in the endothelial cells (1, 3, 5). Finally, in larger vessels of the arterial system, a pulsatile fluid pressure wave is experienced with every heartbeat (3). Together, these forces may alter endothelial cell behaviors in underappreciated manners that could affect the growth and development of new vasculature. During development, these forces guide cell activity, but many disease states possess an altered biomechanical milieu that dysregulates the vasculature. For example, in the tumor microenvironment (TME), matrix remodeling initiated by stromal and tumor cells increases matrix stiffness, usually through increased collagen deposition and organization. This increases both compressive forces on the cells and vessels, forcing higher levels of interstitial flow as fluids are “squeezed” from the vessels and generating irregular shear stresses. These types of changes have been associated with the regulation of tumor-associated angiogenesis, as will be discussed below. Additionally, arterial stiffening, which is common with increased age, is due to changes in elastin and collagen content of the arterial wall. These changes alter forces induced by blood flow and could significantly impact the endothelial cell signaling processes. Understanding how mechanotransduction by endothelial cells regulates angiogenesis will provide critical knowledge in how to promote or inhibit blood vessel growth in the dysregulated biomechanical environments of various disease states.
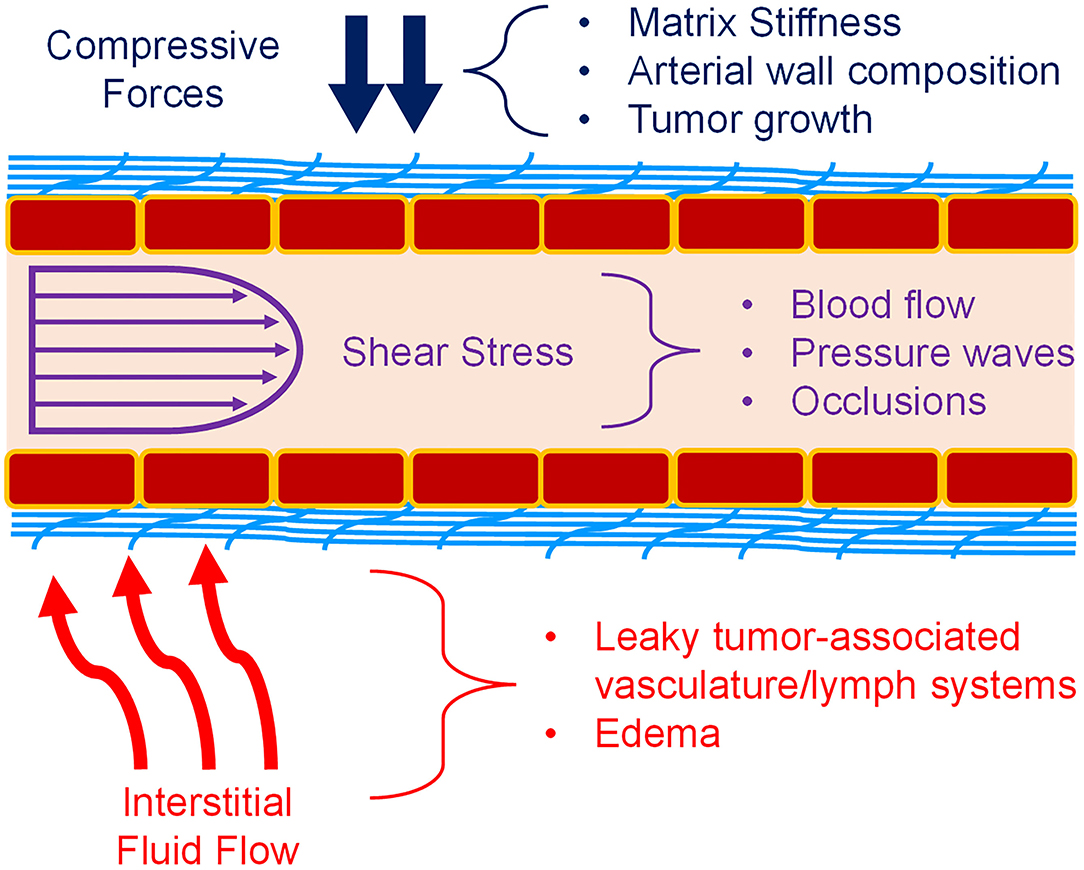
Figure 1. Mechanical forces in blood vessels. Vascular ECs (red boxes) experience a variety of forces. Compressive forces (blue) can be caused by matrix (light blue) stiffness, tumor expansion, and surrounding cells. Shear stress (purple) caused by blood flow is also a major component of the vascular environment. Pulsatile flow and atherosclerosis also affect fluid flow and the resulting shear stress. Interstitial fluid flow and pressure (red) is also present during angiogenesis, and especially in the TME. Permeable vasculature is commonly present in disease states such as tumors and edema. Figure was derived based on the following references: (1–13).
The ligand VEGF and one of its receptors VEGFR-2 are the major regulators of angiogenesis; however, VEGFR-2 has been known to be activated independent of its ligand (14, 15). Several studies have demonstrated that mechanical forces can regulate VEGFR-2 expression and activation (15–21). For example, ECs exposed to shear stress showed formation of a VEGFR-2 and VE-cadherin-β-catenin complex that acted as a mechanotransducer and allowed the cells to activate downstream pathways such as p38 and Akt (22). Since VEGFR-2 can promote angiogenesis, changes in the physical environment could inhibit or promote blood vessel development. This paper will focus on how VEGFR-2 on ECs has been shown to be mechanically-activated, which downstream mechanotransductive factors are regulated by VEGFR-2 signaling, and how knowledge and future studies of this phenomenon could help develop treatments for various disease states.
VEGFR-2
Considered one of the key components of endothelial proliferation and vascular growth, VEGFR-2, also known as fetal liver kinase 1 (Flk-1) or kinase insert domain receptor (KDR), is mostly found within vascular ECs, though it is also weakly expressed in other cells such as osteoblasts, hematopoietic cells, and megakaryocytes (23–26). Although this receptor tyrosine kinase (RTK) is primarily known for its angiogenic signaling effects, it is also involved in embryonic development, differentiation, and EC migration (27–29). Regulation of these processes is controlled by additional signaling pathways and kinase activity including Rho/ROCK, ERK, and Src (30, 31). In fact, many Rho-GTPases and mechanotransductive transcription factors are downstream of VEGF binding to VEGFR-2 (Figure 2). There are three domains of VEGFR-2: extracellular, transmembrane, and intracellular. Ligands bind to the extracellular region, which is rich in immunoglobulin domains, and the transmembrane domain stabilizes receptor dimerization (39, 40). The intracellular region contains a kinase domain and several tyrosine residues, which permit VEGFR-2 to act as an RTK and activate a variety of signaling cascades within ECs (32). Some of the prominent phosphorylation locations are Y1054/Y1059 which are necessary for angiogenic activation (41). The Y951 residue is more specific, causing Src activation and VE-cadherin phosphorylation; these effects lead to increased vascular permeability and mitogenesis (42, 43). Another well-studied phosphorylation site is Y1175, which interacts with Shb and activates PLCγ/MAPK, promoting migration and proliferation (44–46). Furthermore, Y1214 is a prominent site commonly associated with activation of p38, cdc42, Akt, and ERK pathways (33, 47). There are seven members of the VEGF ligand family: placenta growth factor (PIGF), four mammalian VEGFs (VEGF-A, VEGF-B, VEGF-C, VEGF-D), viral VEGF-E, and VEGF-F from snake venom (23). Each of these members of the VEGF ligand family has splice variant isoforms of varying molecular weights, with VEGF-A isoforms being the most prominent in directing blood vessel growth (23, 34, 48). VEGFR-2 dimerizes when activated, and though it can bind to other VEGF receptor family members, VEGFR-1 and VEGFR-3, to control migration and lymphangiogenesis, a VEGFR-2 homodimer is the primary regulator of blood vessel development (32, 39, 49–51). When inactive, VEGFR-2 colocalizes with caveolin-1, which resides in caveolae of the plasma membrane and is involved in cell signaling roles such as negatively regulating VEGFR-2 (52). However, once activated VEGFR-2 dissociates from caveolin-1, and VEGFR-2 is subsequently phosphorylated, endocytosed, and degraded (35, 52, 53). This phosphorylation and internalization of VEGFR-2 then leads to the activation of various signaling pathways, causing different reactions such as increased vascular permeability due to VE-cadherin endocytosis (54).
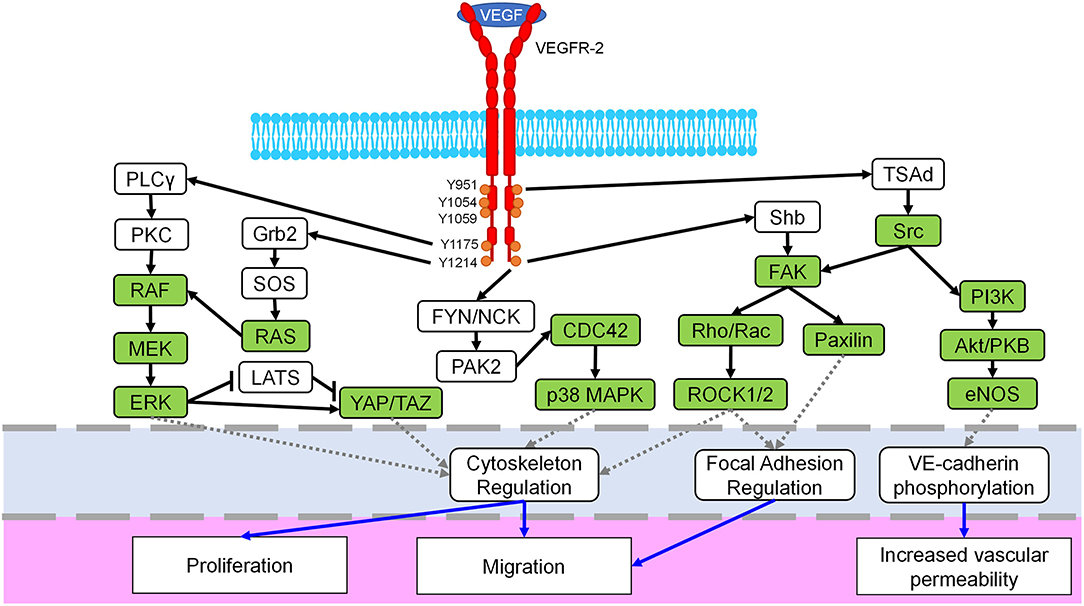
Figure 2. Mechanosignaling in the VEGFR-2 pathway. The VEGFR-2 receptor (red) dimerizes when bound by the VEGF ligand, spans the cell membrane (blue) and possesses multiple phosphorylation sites in the intracellular domain (orange). Downstream signaling initiated by VEGFR-2 includes several known mechanotransductive proteins and enzymes (green) including the MEK/ERK, Rho/ROCK, Src, and YAP/TAZ pathways. Crosstalk between related pathways, i.e., ERK/p38, are not shown for the sake of clarity. The primary target of each pathway is identified with a dashed gray line (light blue region) and includes cytoskeletal regulation, focal adhesion regulation, and VE-cadherin signaling. Finally, resulting cellular phenotypes for each mechanosignaling pathway are indicated with a solid blue arrow in the lowest region of the figure. Figure was derived based on the following references: (23, 24, 32–38).
Two other members of the VEGF receptor family are VEGFR-1 and VEGFR-3, which also bind members from the VEGF family (34). Neuropilin-1 (NRP-1) and neuropilin-2 (NRP-2) act as co-receptors to members of the VEGF-receptor family and have been shown to play roles in vascular growth in developmental angiogenesis as well as tumor-associated angiogenesis (23, 55, 56). Similar to VEGFR-2, VEGFR-1 also regulates angiogenesis, specifically by inhibiting pro-angiogenic signals due to binding kinetics with the VEGF ligand. The extracellular domain of VEGFR-1 has higher affinity for VEGF than VEGFR-2, and evidence suggests this binding is responsible for angiogenic inhibition; however, VEGFR-2 has a higher level of tyrosine kinase activity, resulting in the more prominent role in angiogenesis (23, 32, 34). Alternative splicing can form a soluble VEGFR-1 (sVEGFR-1) that contains no transmembrane or intracellular domain, and this variant has been shown to block proliferation of vascular ECs (57). On the other hand, VEGFR-3 is commonly associated with the development of the lymphatic system, or lymphangiogenesis, though it has a role in regulating other angiogenic properties such as VEGFR-2 expression (58–60). Similar to VEGFR-2, VEGFR-3 can be mechanically-activated by sheer stress, and it complexes with VEGFR-2 and VE-cadherin on the cellular membrane (61). Further discussion of the other VEGF receptors can be found in the following reviews: Melincovici et al. as well as Koch and Claesson-Welsh (23, 32).
Angiogenesis
Angiogenesis is the growth of new blood vessels from preexisting vasculature, as opposed to vasculogenesis, which is the development of vessels de novo (62). Both angiogenesis and vasculogenesis have been shown to play roles in progression of diseases including cancer, ischemia, and arterial stiffening (63, 64). The process of angiogenesis can be classified as either intussusceptive, also known as splitting, or sprouting angiogenesis. Splitting angiogenesis is less studied and occurs when one vessel develops into two parallel vessels through the growth and fusion of tissue pillars in the middle of the original capillary or vessel (63, 65). Sprouting angiogenesis is more widely understood and involves nascent vessels entering a previously un-vascularized tissue region, which is common in embryonic development and certain disease states including the TME (66–68). Sprouting angiogenesis begins with a stimulus, such as hypoxia which affects oxygen sensors including hypoxia inducible factors (HIF), causing cancer or stromal cells to produce VEGF that diffuses to a preexisting vessel before binding VEGFR-2 (69, 70). The binding of VEGF to VEGFR-2 causes a breakdown of the basement membrane, and these ECs undergo a phenotypic shift into tip cells, with increased cell-matrix interactions, matrix remodeling, and high migratory potential. When a tip cell is formed, the VEGFR-2 receptors on filopodia follow the VEGF gradient, or other pro-angiogenic stimulus, to lead the growth of the sprouting blood vessel. As these cells migrate toward the angiogenic signal, stalk cells behind them proliferate and form the new vascular structure (63, 66, 69, 71). This basic understanding of angiogenesis is incomplete, however, because other biomechanical forces can also induce and direct vasculature development. For example, the highly invasive breast cancer cell line MDA-MB-231 migrates against flow, and increased matrix stiffness can decrease the EC vascular sprouting (72–75). Moreover, recent work has shown that angiogenesis can be driven through biomechanical strains induced in a matrix by cancer-associated fibroblasts (CAFs) or through mechanical manipulation with a magnetic bead system, in a manner that is not dependent on secreted VEGF (76, 77). Physical, and not solely chemical, signals in the ECM can result in altered angiogenic patterns, and such changes in physical or biomechanical signals are commonly seen in many disease states. As the role of biochemical regulation of the VEGFR-2 pathway is still only partially understood, the objective of this review is to describe key themes in VEGFR-2 mechanoactivation and mechanotransduction and how these are present in various pathological conditions.
Angiogenesis in Disease
Dysregulation of angiogenesis occurs in various pathologies, and often this is caused or amplified by changes in the ECM. One common disease state is arterial stiffening which becomes more prevalent with age; this stiffening is also observed in several of the diseases discussed below, including peripheral arterial disease and ischemia (64). Late-passage or senescent ECs are more spread out and have decreased proliferative and angiogenic behaviors, with an increased chance to become apoptotic (78, 79). Understanding why this behavioral shift is associated with age could help to treat or even prevent further cardiovascular complications (79). In vascular walls, elastin and collagen help provide flexibility and rigidity, respectively; however, as an individual ages, concentration of elastin decreases and collagen increases (6, 7). As a result, the ECs of the blood vessel walls experience increased matrix stiffness and a higher arterial pulse pressure (80, 81). Furthermore, stiffer blood vessels can have increased permeability and become more likely to develop plaque accumulation, which can lead to further complications (82). Transcription factors YAP/TAZ, which reside downstream of VEGF/VEGFR-2, are upregulated on stiffer matrices and in disturbed flow, and in vivo inhibition of YAP/TAZ caused decreased atherosclerosis and EC inflammation (83, 84). Since many cells are known to respond to environmental mechanical cues, the natural stiffening of arteries could be a contributing factor of further cardiovascular diseases that are more common with age. Understanding how angiogenesis is affected by these physical changes would help develop new treatments that would target an overall cause of these illnesses instead of effects of arterial stiffening.
Another major disease state that involves angiogenesis is cancer progression, which has elucidated how the mechanical environment can affect blood vessel growth. When tumors are small, they can rely on simple diffusion for delivery of nutrients including oxygen; however, as the tumor increases in size, vasculature is required to provide cells with the increased nutrients for the increased cell numbers (2, 85). The TME is significantly different than normal, healthy tissue in both chemical and mechanical factors. First, there is an increase in proangiogenic factors such as VEGF within tumors (2, 75). Regarding mechanical components, tumor matrices are stiffer due to higher collagen production from stromal cells, and these collagen levels have been linked to Rho/ROCK and FAK regulation (8–11). New blood vessels grown during tumor progression are often not fully inter-connected with each other or the existing vasculature and can feature poor cell-cell contacts, leading to increased permeability or leakiness (12, 13). Furthermore, the interstitial fluid pressure is higher within tumors due to vessel compression leading to leaky, tortuous structures (12, 13). The combination of chemical and mechanical changes in the TME causes changes in angiogenic activity; understanding how such changes affect tumor growth and metastasis will help predict and treat this disease.
Unlike in the TME where angiogenesis is upregulated, some diseases are caused by or related to inhibited blood vessel growth, function, or survival. For example, peripheral arterial disease (PAD) causes hypoxic conditions in extremities often as a result of arterial blockage limiting blood flow. Late stages of this disease can lead to critical limb ischemia and eventually the need for limb amputation. Those that have PAD are at a higher risk of stroke and myocardial infarction, and many suffer from claudication, or pain from insufficient blood supply (86, 87). Mechanical changes such as disrupted fluid shear stress and stiffer arteries can result from plaque build-up and vessel narrowing present in many PAD cases (88–90). Also, patients suffering from PAD have been shown to have higher blood viscosity, especially when they experience regular claudication (91). Patients with PAD also show elevated levels of VEGF and lower levels of VEGFR-2 (92). Most current treatments focus on improving general cardiovascular health, with more drastic procedures such as stents being used if the condition worsens. Research is being done to determine if pro-angiogenic factors such as VEGF and fibroblast growth factor (FGF) could promote revascularization; however, these trials have shown limited success, suggesting that the angiogenic mechanisms of PAD are not fully understood (86, 93, 94). Since mechanical changes are present in the PAD disease state, how those changes affect the growth and deterioration of vasculature should be examined to produce more effective treatments aimed at revascularizing affected areas.
Related to PAD, ischemia occurs when tissue does not receive sufficient nutrients and oxygen due to insufficient or inefficient vasculature, creating a hypoxic environment and eventually leading to tissue death (95). Ischemia is classified based on the affected tissues. For example, myocardial ischemia is where cardiac tissue does not receive sufficient oxygen which can lead to multiple adverse events including arrythmias or myocardial infarctions (96). Myocardial infarctions cause heart tissue damage and scarring, with a higher amount of collagen deposited by cardiac fibroblasts during post-infarction events (97). This results in a stiffer matrix which is shown to express upregulated VEGF levels (98). The increased contractility of the myocardium, due to increased stiffness in the scar region, prevents revascularization of the affected area and severely impacts overall cardiac performance (99). Limb ischemia occurs when blood flow is limited to extremities such as the hands and legs. Both types of ischemia can be caused by atherosclerosis, or a build-up of plaque in the arteries, and limb ischemia can also be a complication of other diseases such as diabetes or PAD (96, 100, 101). Studies have shown upregulation of HIF, VEGF, and VEGFR-2 in limb ischemia, especially in acute limb ischemia as opposed to chronic (102, 103). Since limb ischemia is so similar to PAD, many of the mechanical changes present in PAD are also in ischemia, such as increased arterial stiffening and increased blood pressure (104). Studies have explored whether pro-angiogenic factors could promote vascularization in ischemic tissues, although so far these strategies have demonstrated mixed success, with single factors appearing to be less effective than combined treatments (93, 105, 106). A broader understanding of the relationship between angiogenic cytokines, VEGFR-2 mechanoactivation and mechanosignaling, and vessel reperfusion could benefit the development of this potential treatment strategy.
Angiogenesis in Development
The cardiovascular system of the developing fetus is the first functional system generated, occurring even before circulation is established. Since an early embryo does not have any blood vessels, vasculogenesis must initially occur before angiogenesis later in development (62). VEGF and VEGFR-2 are both necessary in vasculogenesis and angiogenesis, with higher VEGF expression levels present during the former and lower expression during the latter (107, 108). Expression of VEGFR-2 is also upregulated early in development, especially after FGF is secreted (109). Knockdowns of VEGFR-2 and VEGF are embryonically lethal, demonstrating their significant roles in development (27, 110, 111). During development, cells naturally produce mechanical forces which can influence many factors including differentiation, migration, and angiogenesis as the tissues of the fetus form. These rapid changes in organization cause cells to experience stretch, pressure, and shear stress, which cause mechanoactivation of various proteins, such as cadherins, caveolins, Rho, and cdc42, and aid in development in normal physiological conditions (4). However, deletion of vegfr-2 in a genetically-modified mouse model is embryonically lethal by stage E10.5 and showed no vasculature, suggesting that VEGFR-2 is required for vasculogenesis in development (27). Studying how these dynamic forces, and what additional cell types or matrix changes cause them, alter VEGFR-2 signaling could provide information about congenital vascular diseases and strategies for controlling blood vessel growth.
Angiogenesis in Tissue Engineering
While there has been a significant rise in interest for tissue engineered scaffolds for replacement of disease tissues or organs in recent years, generation of large-scale systems has been limited by size restrictions due to limited nutrient diffusion. Currently tissue scaffolds with cells can be no more than approximately 200 μm away from a capillary, which represents the limit of oxygen diffusion in these tissue mimics (112, 113). Although many tissue engineering strategies have attempted to incorporate or promote vascularization during scaffold generation, there have been mixed results for creating functional and mature blood vessels, often because of the vascularization of the new tissue can take weeks (114, 115). Many studies have used pro-angiogenic factors, such as VEGF and FGF, to promote blood vessel growth. However, the concentrations and gradients of these factors is complex in native tissues, and imprecise use can limit vessel maturity, function, permeability, and structure (113, 115, 116). In addition to incorporating vascular growth factors, several groups have attempted to use biomechanical stimulation to prime these engineered scaffolds to generate more physiologically-relevant tissue functionality (117, 118). Understanding the unique ways in which biomechanical forces regulate blood vessel growth and vascular function through mechanosignaling related to the VEGFR-2 pathway could provide alternative strategies for developing larger tissues or even organs with a fully functional and interconnected vascular network.
Stromal Cells
Other cells in the perivascular matrix can also act as a source of biomechanical signals to the ECs of the vasculature, potentially impacting the VEGFR-2 signaling axis. While some of these stromal cells, including vascular smooth muscle cells (VSMCs), weakly express VEGFR-2 and can play a role in ischemic conditions, the VEGFR-2 expression levels in ECs are significantly higher (23, 119–121). This includes VSMCs and pericytes in normal vasculature and stromal cells including cancer-associated fibroblasts (CAFs) in the tumor microenvironment. VSMCs comprise a concentric layer around the vascular lumen formed by endothelial cells in the tunica media and are most typically found on larger arterial vessels and veins; the overall function of these cells is to facilitate vasodilation or vasoconstriction based on physiological cues from the nervous system and driven by somatic need (122). These cells typically express alpha-smooth muscle actin and smooth muscle myosin heavy chain and can play a significant role in cardiovascular diseases including hypertension, ischemia, and atherosclerosis (123). Through regulation of vascular tone, VSMCs can directly lead to increased compressive forces on ECs in blood vessels as well as increases in shear stresses in the vessel lumen. Additionally, VSMCs demonstrate VEGFR-2 expression when exposed to hypoxic conditions (124). An additional example of mechanosensors present on both ECs and VSMCs is Piezo1, which is a non-selective cation channel that controls cellular response to shear stresses (125, 126). Pericytes, on the other hand, act as support cells in smaller vascular networks, specifically in capillary beds (122). There are multiple markers for pericytes including alpha smooth muscle actin, NG2 proteoglycan, platelet derived growth factor receptor beta, N-cadherin, and CD106 (127–129). Pericytes have been shown to directly interact with ECs in growing vascular networks, relying on heterotopic Notch signaling as well as heterotypic cadherin bonds to control sprouting angiogenesis and cell migration (130–132). These studies examined pericyte function and behavior in a variety of in vitro and ex vivo model systems in both cardiovascular development and disease as well as cancer progression. Moreover, expression of VEGFR-1 on pericytes acts as a regulator of tip cell formation in mice retinas and inhibiting this signal causes overgrowth of the vascular field (133). A full discussion of the mechanobiological roles of pericytes is beyond the scope of this review but can be found in a recent publication by Dessales et al. (5). In the context of the tumor microenvironment, CAFs are a mechanically-active stromal cell that promotes enhanced angiogenesis through growth factor signaling, matrix remodeling, and contractile behaviors (20, 76, 77, 134–138). More specifically for angiogenesis, CAFs have been shown to increase expression of growth factors including VEGF-A and stromal cell-derived factor 1 (SDF-1) after exposure to either cyclic strains or compressive forces, leading to increased angiogenesis in in vitro model systems (134, 137). The activation of VEGFR-2 and dissociation from VE-cadherin is regulated by a Src-dependent process that can be induced by tensile strains on pulmonary artery ECs (20). Overall, the regulation of angiogenesis by stromal cells, including VSMCs, pericytes, and CAFs, is a highly complex process based on interacting biomechanical and biochemical cues. Careful consideration of such factors should be considered when investigating therapeutic strategies that target inhibiting or promotion vascular growth through the VEGFR-2 signaling axis.
Models to Study VEGFR-2 Signaling
As it is highly difficult to control independent biomechanical forces in in vivo models, most of what we know about VEGFR-2 mechanotransduction comes from in vitro systems. For example, pulsatile or laminar flow can reduce inflammation and stabilize the vascular wall, while a multidirectional disturbed flow can cause inflammation and lead to atherosclerosis (61, 139–144). One study examined how treating HUVECs with VEGFR-2 inhibitor ZM323881 in non-uniform shear stress conditions caused decreased expression of adhesion molecules such as VE-cadherin; however, static conditions did not show this change (145). Inflammation and atherosclerosis are involved in various cardiovascular diseases, and identifying whether VEGFR-2 signaling is involved in this mechanotransductive pathway is an essential step in harnessing the receptor to treat disease states. Though in vitro experiments allow for easier control of mechanical and chemical stimulants, researchers still attempt to incorporate animal models to ensure that all aspects of the in vivo system are taken into account (21, 146). As the knockout of VEGFR-2 is embryonically lethal in murine models, alternative transgenic systems including inducible knockout or Cre-Lox targeted knockout must be used to determine the role of VEGFR-2 in disease progression (147–149). Some studies instead focus on heterozygous VEGFR-2 mouse models and show altered patterns of endothelial migration during embryogenesis and decreases in angiogenesis in tumor models (149, 150). One study analyzed the effects that a Y949F VEGFR-2 mutation had on ECs in mouse aortas. Cell polarity and alignment was disrupted due to this mutation, which mimicked the in vitro experiment using HUVECs and VEGFR-2 inhibitors SU1498 and ZM323881 (21). In mouse models utilizing a VE-cadherin Cre-based knockdown of VEGFR-2 in endothelial cells, dramatic decreases in angiogenesis were seen in retina samples, which correlated with dysregulation of VEGFR-2 patterning in cell membranes (151). Additional animal models studying VEGFR-2 signaling in myocardial ischemia involve both mouse and porcine samples and implicate TAZ interactions with VEGFR-2 (152). While such systems provide some useful information, it can result in mosaic expression of VEGFR-2 due to inconsistent Cre expression which, in combination with incomplete control over biomechanical stimuli, generates confounding data and unclear mechanisms of action. In fact, this extremely high level of physiological complexity with limited understanding of mechanobiological factors that affect VEGFR-2 may be partially responsible for the limited efficacy of therapeutic strategies that target this pathway. To address this, a variety of in vitro systems, including polyacrylamide hydrogels for substrate stiffness studies and microfluidic flow systems for shear stress studies, have been useful in elucidating VEGFR-2 responses to specific and highly controllable biomechanical stimuli (77, 153–160). Many such systems have been described through this review article and have generated considerable knowledge of VEGFR-2 signaling pathways. The next generation of model systems to study VEGFR-2 mechanoregulation should work to incorporate the in vivo complexity with the highly controllable biomechanical parameters of in vitro models. There is substantial clinical evidence for mutations in or alterations of VEGFR-2 signaling in not only cancer, PAD, and ischemia, but also neurological disorders such as Alzheimer's Disease, endometrial disorders, retinal degeneration, and bleeding disorders due to arteriovenous malformations (160–163). While these studies do not directly demonstrate mechanobiological regulation of VEGFR-2, there is substantial and increasing evidence that biomechanical forces are involved in all of these pathologies (3, 164–166). Therefore, a more fundamental understanding of VEGFR-2 mechanosignaling provides context and elucidation for a wide variety of disease states that may offer potential therapeutic avenues.
Signaling
The role of biomechanical forces as regulators of cell and tissue behaviors is appreciated across many fields including cancer biology, stem cell differentiation, cardiovascular regulation, tissue engineering, and many other areas (1, 167–170). For example, a stiff bone-like matrix can cause mesenchymal stem cells (MSCs) to present traits similar to osteoblasts instead of like neural or muscle tissue (170). Studies like this revealed that the physical environment can affect cellular pathways and represent seminal foundations of the field of cellular mechanics. In recent years, there have been more descriptions of many proteins as mechanotransducers such as integrins, cadherins, and the components of the cytoskeleton (4). The biochemical signaling that results from cellular interpretations or responses to biomechanical cues is commonly referred to as mechanotransduction; the term mechanobiology includes the wider understanding of the communication of biomechanical features between a cell and its environment. In addition to regulating angiogenesis, VEGFR-2 can also be considered a mechanoreceptor, which is activated in ECs by forces such as shear stress and cyclic stretch, and this mechanoactivation causes activation of various downstream mechanotransductive signaling pathways (15, 20, 21). Through these mechanical signals, VEGFR-2 can promote angiogenesis without its VEGF ligand (76, 77). This section of the review will describe how VEGFR-2 acts as a mechanoreceptor and closely interacts with other well-characterized mechanotransductive factors, demonstrating that altered physical forces caused by disease states could affect angiogenesis. A summary of specific pathways, inhibitors, and the resulting effects of vascular growth can be found in Table 1.

Table 1. Effects of inhibitors on EC mechanobiological pathways.
Receptor-Ligand Interactions
Several different isoforms of VEGF-A are produced by alternative splicing, with some of the most well-studied being VEGF121, VEGF165, VEGF189, and VEGF206; this growth factor can be secreted by many cell types including macrophages, tumor cells, and fibroblasts (23, 171). The lowest molecular weight isoform, VEGF121, is the most soluble and easily able to diffuse throughout the ECM. However, other heavier isoforms such as VEGF189 and VEGF206 remain bound to the ECM, with matrix-bound VEGF165, VEGF189, and VEGF206 all able to promote EC proliferation (172, 173). Studies have shown that the manner of ligand presentation to a receptor, either as a soluble cue or bound to the matrix, affects cell pathways and activity (174). For example, bone morphogenic protein (BMP)-2 can be soluble or matrix-bound, and cells grown on a stiff film showed slower internalization of bound BMP-2 compared to soluble (175). Binding a ligand to a matrix alters the interaction with a receptor, which in turn changes cell response, so the different presentations of VEGF could cause variable mechanoactivation of VEGFR-2. The most expressed isoform of VEGF-A is VEGF165, which can be found either in soluble form or bound to the ECM via proteoglycans (172, 173). Chen et al. studied this phenomenon by comparing VEGFR-2 phosphorylation and signaling caused by soluble (Vs) or matrix-bound (Vb) VEGF165 (16). To make Vb, VEGF was incorporated into a collagen gel. When analyzing VEGFR-2 phosphorylation as a whole, the receptor showed more sustained phosphorylation when activated by Vb than Vs. Upon studying individual VEGFR-2 tyrosine residues, researchers identified Y1214 to be the cause of this extended activation; it maintained phosphorylation for 15 min only when presented with Vb, compared to 5 min by Vs. The Y1214 phosphorylation site is associated with the ERK1/2 pathway, which is a well-known mechanosensitive pathway. This association reinforces the conclusion that VEGFR-2 phosphorylation, especially at Y1214, and activation are affected by mechanical forces within the environment (33). Further study of mechanoactivation of VEGFR-2 and the subsequent signaling cascades would provide a deeper understanding of VEGFR-2 mechanoregulation that could potentially identify novel targets for anti-angiogenic therapies in cancer treatments.
ERK/MAPK Pathway
A well-characterized mechanosignaling pathway is the mitogen activated protein kinase, or MAPK, pathway which involves numerous kinases including the extracellular-signal-regulated kinase (ERK) (176, 177). This pathway has been implicated in several diseases including multiple types of cancers and cardiovascular diseases (178, 179). After receiving a signal from a membrane bound receptor, the MAPK pathway activates when RAS, a GTPase, activates a phosphorylation cascade involving three kinases RAF, MEK, and ERK (180). The MAPK pathway further regulates various transcription factors, including but not limited to HSP27, p38, and c-Myc, which control cell activities such as proliferation, differentiation, and migration (177, 181). ERK is a known mechanotransductive factor, translating biomechanical or biophysical signals external to the cell into a biochemical signal that controls cell response (15, 182, 183). For example, stretching or tensile strain can cause ERK activation, which leads to Madin Darby canine kidney epithelial cell (MDCK) contraction, propagation of the ERK signal in neighboring cells, and collective cell migration (184). Furthermore, ERK works with Piezo1, another known mechanoreceptor, to cause proliferation when MDCKs are stretched and apoptosis when cells are heavily confluent (185).
Several studies have demonstrated a link between VEGFR-2 and the ERK pathway, where VEGFR-2 acts upstream of ERK phosphorylation (186–188). Activation of the ERK pathway is one of the major targets of VEGFR-2 activation, and this link between ERK and VEGFR-2 is necessary for adipose mesenchymal stem cell (AMSC) to EC differentiation (186, 187). One study demonstrated that shear stress upon an EC is sufficient for VEGFR-2 phosphorylation and subsequent ERK upregulation, without the biochemical stimulation of the VEGF ligand (17). Specifically, phosphorylation of VEGFR-2 at Y1175 and Y1214 are linked to ERK activation (16, 33, 45, 189). In a study by LaValley et al., matrix stiffness was shown to be connected to both VEGFR-2 and ERK activity (19). Plating human umbilical vein endothelial cells (HUVECs) on 1 or 10 kPa collagen-coated gels showed that stiffer matrices increased the levels of phosphorylated VEGFR-2 and ERK in sub-confluent cultures, consequently increasing proliferation (175). Combining VEGF with the 10 kPa gel resulted in the highest cell growth, and the addition of the ERK inhibitor PD98059 decreased this growth. Increased matrix stiffness also promotes VEGFR-2 internalization, which then causes ERK phosphorylation and cell proliferation (19). Another study linked VEGFR-2 and ERK with mechanoactivation using pulsatile flow. In osteocytes, pulsatile fluid flow can prevent cells from undergoing apoptosis; this flow also causes an increase in ERK phosphorylation. However, when treated with the VEGFR-2 inhibitor SU5416, the osteocytes displayed increased apoptosis and decreased ERK phosphorylation (18). The link between ERK mechanotransduction and VEGFR-2 activation indicates that the receptor plays a previously underappreciated role in cellular responses to various mechanical factors. While the MAPK pathway is ubiquitous in numerous cell types, including both healthy and diseased tissues, the specificity of VEGFR-2 as a mechanoreceptor during angiogenesis could provide a unique target for promotion or inhibition of angiogenic vessel growth during disease progression.
c-SRC Pathway
Another mechanotransductive protein downstream of VEGFR-2 signaling is Src, known as c-Src in humans, which is a cytoplasmic tyrosine kinase associated with the cell membrane or endosomal membranes. The Src protein contains Src homology (SH) domains and a kinase domain (190, 191). The role of Src has been described in many cell processes including adhesion, motility, proliferation, and differentiation, spanning across embryological development, healthy tissue homeostasis, and several disease pathways (192–198). Furthermore, Src activity is known to be at least partially regulated by mechanical cues such as shear stress and matrix adhesion, and it resides upstream of other effectors such as the ERK pathway (199, 200). In addition, Src serves as a key regulator of transforming growth factor beta (TGFβ) signaling in contractility responses of valvular interstitial cells during heart valve disease (197). These studies demonstrate the role of Src as a key intersection in numerous mechanotransductive pathways.
Moreover, Src has been shown to phosphorylate tyrosine residues on VEGFR-2, ultimately generating a positive feedback loop with further activation of both VEGFR-2 and Src (201). In one study, ECs were exposed to laminar flow in a cone and plate viscometer, which caused VEGFR-2 phosphorylation even when cells were treated with a VEGF inhibitor. However, exposure to Src inhibitor PP2 counteracted this effect and caused a decrease in phosphorylation levels of VEGFR-2 (15). This suggests that mechanical forces via shear stress cause Src to activate VEGFR-2, which could further support VEGFR-2 as a mechanoreceptor. In another study, aortic ECs from mice were studied to elucidate how VEGFR-2 and Src regulate cell alignment and polarity with respect to shear flow. Mice with a VEGFR-2 mutation Y949F, which is Y951 in humans, presented altered alignment and polarity, while mice with an inducible Src knockout in endothelial cells, only had impaired polarity (21). This reaffirms that Y951 of VEGFR-2 is linked to Src activity and also suggests that VEGFR-2 and Src interact to respond to the physical environment to control cellular polarity. Cell polarity is a necessary aspect of cell migration, development, and tissue organization; impairment of cellular polarity regulation leads to disease states including cystic kidney disease and birth defects such as neural tube defects (202–204). The role of Src as a mechanotransductive factor in the regulation of activation levels of VEGFR-2 suggest this mechanosignaling axis plays a role in vascular growth and could be targeted in novel therapeutic strategies.
Rho/ROCK Pathway
Another well-studied protein that has been linked to both mechanoactivation of cells and VEGFR-2 is Rho. This is a GTPase that causes downstream activation of Rho-associated coiled-coil containing protein kinase (ROCK), which is a critical regulator of cytoskeletal components involved in cell migration and is typically referred to as the Rho/ROCK pathway (10, 205–210). Rho can be activated by a variety of receptors and cytokines, and ROCK is activated when phosphorylated by Rho, causing ROCK to undergo a conformation change that increases its kinase activity (205, 211). This pathway is involved in cytoskeleton regulation, fibronectin matrix control, differentiation, and apoptosis (212–215). Several studies have identified a connection between physical forces and Rho activation (10, 214, 216–218). When rat embryonic fibroblasts were subjected to mechanical strain, Rho activity, as measured by Rho being bound to GTP instead of GDP, increased compared to non-strain controls (219). A different study observed the effect of strain on capillary ECs in tumor and control environments, and the addition of strain upregulated Rho activity in the control environment but not in the tumor. Without strain, both Rho and ROCK were upregulated in tumor ECs compared to control, and since there was no significant difference between Rho activity in the strained control conditions and the tumor ECs, mechanical factors in the TME may be causing activation of the Rho/ROCK pathway (220). In another study, ECs exposed to shear stress showed a downregulation of Rho activity due to integrin binding to the ECM, which allowed the ECs to reorganize their cytoskeletal elements and align with the flow (221). Together with Src and integrins, a substantial body of work has been completed on the Rho/ROCK pathway to describe mechanotransduction in a wide variety of cells in both physiological and pathological conditions (222). Mechanical forces can work with other receptors and enzymes to increase or decrease Rho/ROCK activity in order to guide the cell cycle, differentiation, and migration.
In addition, VEGFR-2 signaling has been shown to lead to Rho/ROCK activation which then affects downstream enzymes such as focal adhesion kinase (FAK) and transcription factors such as signal transducer and activator of transcription (STAT) (30, 223, 224). One study focused on Rho inhibitor p190RhoGAP, which is naturally synthesized in cells, to see how decreases in Rho signaling altered VEGFR-2 expression and if VEGFR-2 can be mechanically-activated (146). The pseudoenzyme p190RhoGAP contains pseudo GTPase domains and can act as a Rho regulator (223). Knockdown of 190RhoGAP in ECs using siRNA caused an increase of VEGFR-2 mRNA levels compared to the control. Additionally, VEGFR-2 mRNA was shown to be expressed at higher levels on 4,000 Pa gels compared to a 150 Pa gel. When treated with p190RhoGAP siRNA, the 150 Pa sample VEGFR-2 mRNA levels increased (146). This Rho inhibitor controlled VEGFR-2 levels according to the substrate stiffness used for cell culture. HUVECs grown on a stiffer gel of 10 kPa showed more VEGFR-2 endocytosis, a measure of activation, compared to HUVECs grown on a more compliant matrix of 1 kPa (19). Cells grown on a 10 kPa gel and treated with a ROCK inhibitor Y-27632 showed more VEGFR-2 on the cell membrane, suggesting that there was less activation and uptake of the receptor. The amount of VEGFR-2 present was similar to HUVECs grown on the 1 kPa gel without Y-27632. This experiment shows that the Rho/ROCK pathway is involved in regulation of VEGFR-2 endocytosis and subsequent activation in response to matrix stiffness. Understanding this complex relationship will help researchers to identify targets that could help control angiogenic activity.
Hippo Pathway
The Hippo pathway involves a kinase cascade leading to downstream transcription factors YAP and TAZ, which are involved in organ development as well as cell contractility, migration, and proliferation (225). This pathway has thus been connected to both regenerative medicine, because of its role in development and tissue growth, and cancer, because of its influence on proliferation and cell survival (225, 226). When phosphorylated due to Hippo pathway activation, YAP and TAZ become inactive; however, VEGF promotes the opposite effect and induces YAP/TAZ activity and translocation to the nucleus (151, 227, 228). These transcription factors are key mediators of angiogenesis, and mice with an endothelial knockout of these proteins resulted in major vascular dysregulation in development (151). YAP, TAZ, and VEGFR-2 have also been linked with other angiogenic factors such as BMPs, which have shown changes in expression after ischemic conditions (152). Mechanical cues influence YAP and TAZ activity, though these effects have been shown to be at least partially independent of the Hippo pathway (225). Mammary epithelial cells (MECs) grown on stiffer matrices of 15–40 kPa show higher nuclear localization of these factors than cells grown on compliant matrices of 0.7–1 kPa, resulting in increased proliferation and decreased apoptosis (229). However, cells with a YAP/TAZ knockdown acted similarly to those grown on the compliant matrices. Since YAP/TAZ are known mechanotransducers, they likely play a role with VEGFR-2 in angiogenic response to mechanical forces. One study demonstrated that HUVECs grown on a 1 kPa matrix caused downregulation of YAP activity and upregulation of VEGFR-2 transcription levels (83). When VEGF was added, VEGFR-2 levels increased on the 1 kPa gels. YAP activation by lysophosphatidic acid causes increased delta-like ligand 4 (DLL4) expression, which is a Notch1 ligand (230). However, in HUVECs, DLL4 showed the highest expression on 1 kPa gels when YAP is least active, and addition of YAP inhibitor verteporfin also caused increased DLL4 expression in HUVECs plated on a stiff 25 kPa gel (83). Even though previous work has shown that YAP activity can promote DLL4 expression, different combinations of matrix stiffnesses and soluble VEGF leads to varied results, exemplifying how complicated mechanobiology can be. Biomechanical and biochemical signaling involves complex pathway crosstalk which needs further study in order to determine how YAP/TAZ, VEGFRs, and other angiogenic factors regulate blood vessel growth.
Crosstalk With Other Mechanoignaling Pathways
The VEGFR-2 pathway interacts with several known important mechanotransduction pathways, as previously discussed (21, 61, 139–146). However, the crosstalk of mechanobiological agents is not exclusive to downstream signaling targets in this context. Considered as primary mechanosensors on ECs, signaling through both integrins and cell-cell adhesion molecules can lead to increases in expression of mechanotransductive factors including ERK, Src, and PI3K/Akt, among others (231–235). The interaction of adherens junctions and VEGFR-2 is required to drive responses to shear stress in ECs (22). Moreover, it has been shown that VEGF stimulation also promotes enhanced growth factor secretion in ECs including TGF-β1 and connective tissue growth factor (CTGF); inhibition of this process led to decreased basement membrane thicknesses in mouse retains (236). E-cadherin stimulation can cause increase in epidermal growth factor signaling which in turns activates the PI3K pathway (232, 237). It has also been shown that growth factor signaling can in and of itself be regulated through mechanical strain, in that activation of TGF-β1 from its latent form is increased when cells are treated with tensile forces (238, 239). Due to these confounding effects, precise model systems are needed to elucidate independent roles and reactions of VEGFR-2 in mechanosignaling.
Anti-VEGF Treatments, Side Effects, and Efficacy
Current clinically available anti-VEGF therapeutics are either derived from antibodies or are small molecule inhibitors of the tyrosine kinase receptors, including VEGFR-2 that VEGF interacts with to promote angiogenesis. The first such therapies were available in the early 2000s, with bevacizumab approved in 2004 for colorectal cancer which was soon expanded to other types of cancer treatment including lung, glioblastoma multiforme, ovarian, renal, and metastatic breast cancer (240, 241). Other drugs approved by the USA-FDA include ranibizumab, sunitinib and sorafenib, which also have been expanded from anti-angiogenic therapies in cancers to treat a variety of cardiovascular diseases including PAD and ischemia (242). For anti-cancer treatments, the efficacy of anti-VEGF-based therapies is substantially less in clinical applications compared to preclinical in vitro and in vivo models; for example, disease free progression of breast cancer is not significantly increased when bevacizumab is delivered with other first line chemotherapeutics (240, 241, 243, 244). Based on several studies with these limited benefits, the FDA revoked approval for bevacizumab for breast cancer treatments (245). For cardiovascular diseases, such as PAD, anti-VEGF treatment strategies show little clinical improvements, despite promising preclinical work in limb ischemia models (242). Finally, in ocular vascular degenerative disorders, several anti-VEGF therapies have been approved for clinical use but show limited improvements in many clinical trials (246). While some benefits have been observed in several studies for the various diseases and conditions discussed here, the side effects associated with anti-VEGF or anti-VEGFR-2 therapies can be severe and include heightened risk of arterial thromboembolic events, hypothyroidism, wound healing complications, GI perforations, neutropenia and hematological effects and increased risks for other cardiac adverse events (245, 247–249). If future treatments combine anti-VEGFR-2 and anti-mechanotransductive elements, additional side effects could be expected if the latter pathways are ubiquitous in healthy and diseased tissues. An example of this is fasudil, a selective ROCK inhibitor that has been used to treat pulmonary hypertension, ALS, subarachnoid hemorrhages, dementia, stroke, and atherosclerosis (250–258). While long term side effects of fasudil treatment are not known, it is expected that the inhibitor will affect the cardiovascular system and potentially lead to adverse cardiac events or hypertension; one study suggested that side effects in a clinical trial were mild-moderate and ranged from skin rashes to bleeding disorders (252). On the other hand, vascular normalization therapies, where increases in VEGF signaling would promote blood vessel growth or enhanced function, demonstrate their own challenges most typically related to delivery kinetics, administration routes, and ability to evaluate clinical outcomes (259–261). To address these challenges, some groups have begun investigating gene therapy as a technique to create a more sustained and controllable deliver of VEGF; an example of this is recent data from a clinical trial where VEGF-D delivered by adeno-associated viruses decreased angina associated symptoms in a majority of patients, with similar levels of major cardiac adverse events compared to control groups (262).
Discussion
Studies have observed complicated interactions between the biomechanical and biochemical environment which can prompt blood vessels to undergo angiogenesis or degrade functioning of existing vessels. LaValley et al. showed that individual effects of VEGF addition and a stiffer gel on cell proliferation was significantly less than when the two factors were combined (19). Understanding only the independent biomechanical or biochemical aspects of cellular signaling does not provide a full view of developmental processes or disease progression; both biochemical and biomechanical features must be studied to acquire a complete view of cellular regulation and potential novel treatment strategies.
All the diseases discussed in this paper cause both biochemical and physical changes in the matrix surrounding the cells and blood vessels, which alters normal function. For example, the cardiovascular diseases mentioned—PAD, ischemia, arterial stiffening—result in some form of matrix stiffening. In addition, the TME contains higher levels of collagen, while arterial stiffening, which is common in PAD and ischemia, is a result of less elastin and more collagen (6, 8). The vascular ECs in these diseases are surrounded by a stiffer matrix, and studies have shown less angiogenic sprouting, increased VEGFR-2 activation, and more cell growth in stiffer gels (19, 75). Additionally, the abnormal vasculature in tumors and the atherosclerosis in cardiovascular diseases both can cause abnormal shear stress within the vessels (2, 90). Patients that experience claudication often exercise less which causes lower fluid shear stress, and regular intervals of high shear stress is known to promote healthy vasculature (263). Flow regulates various cell activities such as the phosphorylation of proteins like VEGFR-2 and ERK and the orientation and polarity of the cell body (15, 18, 21). Because of mechanical changes present in disease states and in a developing embryo, further understanding of the relationship between cytokines, physical forces, and mechanotransducers will help researchers to determine how to either inhibit or promote angiogenesis to treat diseases or develop improved vascularization strategies for tissue engineering.
Future Directions
Regarding VEGFR-2 and angiogenesis, further investigation is required to understand how different types of physical forces such as pressure, matrix stiffness, tensile strains, and fluid shear stress affect VEGFR-2 activation and internalization. Furthermore, investigations of different VEGFR-2 tyrosine residues and how changes in biomechanical factors cause and sustain phosphorylation would help to clarify the interactions VEGFR-2 has with other enzymes and transcription factors. Identifying the exact phosphorylation site triggered by specific biomechanical stimuli and the resulting signaling cascades would help researchers develop ways to promote or inhibit relevant signals. As mentioned previously, additional research could be directed toward the complex relationship and interaction between chemical and mechanical cues. Since the combination of the two can produce the greatest effect and act synergistically, understanding these processes would open more directions of study that could ultimately lead to better control of angiogenesis. Specifically, a rise in sophisticated microfluidic systems designed to investigate individual and independent biomechanical and biochemical signals will be crucial to fully elucidate these complex interactions (72, 77, 154, 264). Finally, there are several members of the VEGF ligand family, each with various isoforms. The most common, VEGF-A, has some isoforms that are mainly soluble and other which are mainly matrix-bound, and Chen et al. discovered a difference caused by soluble vs. matrix-bound VEGF (16). This type of prolonged and site-specific activation represents an understudied phenomenon in VEGFR-2 regulation. The relationship between the VEGF and VEGFR families is complex due to the various members and isoforms, but each plays a role that needs to be more fully understood.
Conclusion
Mechanical forces, along with various cytokines, regulate the formation and maintenance of the vasculature. Thus, angiogenesis is a process that is central not only to development but also in many diseases. Additionally, the field of regenerative medicine should focus not only on chemical stimuli but also mechanical stimuli in order to most effectively develop new vasculature in scaffold structures. Investigations of how VEGFR-2, along with other connected pathways such as ERK/MAPK, Src, Rho/ROCK, and YAP/TAZ are mechanically controlled have demonstrated that mechanics are an essential focus in understanding how to evolve therapeutic angiogenesis or in preventing angiogenesis in the TME. Further study of VEGFR-2, its related pathways, and the interaction of biochemical and biomechanical signaling that control vascular growth will aid researchers in developing new treatments that more efficiently treat the underlying cause of various disease states.
Author Contributions
BM wrote the first draft of the manuscript. BM and MS-L contributed to manuscript revision, read, and approved the submitted version. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institutes of Health (CA230202 MS-L), O'Neal Comprehensive Cancer Center (IMPACT Award MS-L).
Conflict of Interest
MS-L receives compensation for consulting services for CerFlux, Incorporated.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yu H, Mouw JK, Weaver VM. Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol. (2011) 21:47–56. doi: 10.1016/j.tcb.2010.08.015
2. Zanotelli MR, Reinhart-King CA. Mechanical forces in tumor angiogenesis. Adv Exp Med Biol. (2018) 1092:91–112. doi: 10.1007/978-3-319-95294-9_6
3. Roman BL, Pekkan K. Mechanotransduction in embryonic vascular development. Biomech Model Mechanobiol. (2012) 11:1149–68. doi: 10.1007/s10237-012-0412-9
4. Ayad NME, Kaushik S, Weaver VM. Tissue mechanics, an important regulator of development and disease. Philos Trans R Soc Lond B Biol Sci. (2019) 374:20180215. doi: 10.1098/rstb.2018.0215
5. Dessalles CA, Babataheri A, Barakat AI. Pericyte mechanics and mechanobiology. J Cell Sci. (2021) 134:jcs240226. doi: 10.1242/jcs.240226
6. Schlatmann TJ, Becker AE. Histologic changes in the normal aging aorta: implications for dissecting aortic aneurysm. Am J Cardiol. (1977) 39:13–20. doi: 10.1016/S0002-9149(77)80004-0
7. Avolio A, Jones D, Tafazzoli-Shadpour M. Quantification of alterations in structure and function of elastin in the arterial media. Hypertension. (1998) 32:170–5. doi: 10.1161/01.HYP.32.1.170
8. Kauppila S, Stenback F, Risteli J, Jukkola A, Risteli L. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J Pathol. (1998) 186:262–8. doi: 10.1002/(SICI)1096-9896(1998110)186:3<262::AID-PATH191>3.0.CO;2-3
9. Huijbers IJ, Iravani M, Popov S, Robertson D, Al-Sarraj S, Jones C, et al. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PLoS ONE. (2010) 5:e9808. doi: 10.1371/journal.pone.0009808
10. Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. (2003) 163:583–95. doi: 10.1083/jcb.200305010
11. Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. (2009) 28:4326–43. doi: 10.1038/onc.2009.299
12. Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. (2004) 427:695. doi: 10.1038/427695a
13. Stylianopoulos T, Martin JD, Snuderl M, Mpekris F, Jain SR, Jain RK. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Res. (2013) 73:3833–41. doi: 10.1158/0008-5472.CAN-12-4521
14. Warren CM, Ziyad S, Briot A, Der A, Iruela-Arispe ML. A ligand-independent VEGFR2 signaling pathway limits angiogenic responses in diabetes. Sci Signal. (2014) 7:ra1. doi: 10.1126/scisignal.2004235
15. Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res. (2003) 93:354–63. doi: 10.1161/01.RES.0000089257.94002.96
16. Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol. (2010) 188:595–609. doi: 10.1083/jcb.200906044
17. Chen KD, Li YS, Kim M, Li S, Yuan S, Chien S, et al. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J Biol Chem. (1999) 274:18393–400. doi: 10.1074/jbc.274.26.18393
18. de Castro LF, Maycas M, Bravo B, Esbrit P, Gortazar A. VEGF receptor 2 (VEGFR2) activation is essential for osteocyte survival induced by mechanotransduction. J Cell Physiol. (2015) 230:278–85. doi: 10.1002/jcp.24734
19. LaValley DJ, Zanotelli MR, Bordeleau F, Wang W, Schwager SC, Reinhart-King CA. Matrix stiffness enhances VEGFR-2 internalization, signaling, and proliferation in endothelial cells. Converg Sci Phys Oncol. (2017) 3:044001. doi: 10.1088/2057-1739/aa9263
20. Tian Y, Gawlak G, O'Donnell JJ III, Birukova AA, Birukov KG. Activation of vascular endothelial growth factor (VEGF) receptor 2 mediates endothelial permeability caused by cyclic stretch. J Biol Chem. (2016) 291:10032–45. doi: 10.1074/jbc.M115.690487
21. Vion AC, Perovic T, Petit C, Hollfinger I, Bartels-Klein E, Frampton E, et al. Endothelial cell orientation and polarity are controlled by shear stress and VEGF through distinct signaling pathways. Front Physiol. (2020) 11:623769. doi: 10.3389/fphys.2020.623769
22. Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, et al. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci USA. (2002) 99:9462–7. doi: 10.1073/pnas.142224299
23. Melincovici CS, Bosca AB, Susman S, Marginean M, Mihu C, Istrate M, et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. (2018) 59:455–67.
24. Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. (2013) 273:114–27. doi: 10.1111/joim.12019
25. Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. (2006) 39:469–78. doi: 10.5483/BMBRep.2006.39.5.469
26. Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE. (2001) 2001:re21. doi: 10.1126/stke.2001.112.re21
27. Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. (1995) 376:62–6. doi: 10.1038/376062a0
28. Dikov MM, Ohm JE, Ray N, Tchekneva EE, Burlison J, Moghanaki D, et al. Differential roles of vascular endothelial growth factor receptors 1 and 2 in dendritic cell differentiation. J Immunol. (2005) 174:215–22. doi: 10.4049/jimmunol.174.1.215
29. Genet G, Boye K, Mathivet T, Ola R, Zhang F, Dubrac A, et al. Endophilin-A2 dependent VEGFR2 endocytosis promotes sprouting angiogenesis. Nat Commun. (2019) 10:2350. doi: 10.1038/s41467-019-10359-x
30. Sulpice E, Ding S, Muscatelli-Groux B, Berge M, Han ZC, Plouet J, et al. Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells. Biol Cell. (2009) 101:525–39. doi: 10.1042/BC20080221
31. Meyer RD, Sacks DB, Rahimi N. IQGAP1-dependent signaling pathway regulates endothelial cell proliferation and angiogenesis. PLoS ONE. (2008) 3:e3848. doi: 10.1371/journal.pone.0003848
32. Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. (2012) 2:a006502. doi: 10.1101/cshperspect.a006502
33. Testini C, Smith RO, Jin Y, Martinsson P, Sun Y, Hedlund M, et al. Myc-dependent endothelial proliferation is controlled by phosphotyrosine 1212 in VEGF receptor-2. EMBO Rep. (2019) 20:e47845. doi: 10.15252/embr.201947845
34. Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci. (2005) 109:227–41. doi: 10.1042/CS20040370
35. Simons M. An inside view: VEGF receptor trafficking and signaling. Physiology. (2012) 27:213–22. doi: 10.1152/physiol.00016.2012
36. Martino F, Perestrelo AR, Vinarsky V, Pagliari S, Forte G. Cellular mechanotransduction: from tension to function. Front Physiol. (2018) 9:824. doi: 10.3389/fphys.2018.00824
37. Nilsson M, Heymach JV. Vascular endothelial growth factor (VEGF) pathway. J Thorac Oncol. (2006) 1:768–70. doi: 10.1097/01243894-200610000-00003
38. Azad T, Ghahremani M, Yang X. The role of YAP and TAZ in angiogenesis and vascular mimicry. Cells. (2019) 8:823–35. doi: 10.3390/cells8050407
39. Ruch C, Skiniotis G, Steinmetz MO, Walz T, Ballmer-Hofer K. Structure of a VEGF-VEGF receptor complex determined by electron microscopy. Nat Struct Mol Biol. (2007) 14:249–50. doi: 10.1038/nsmb1202
40. Sarabipour S, Ballmer-Hofer K, Hristova K. VEGFR-2 conformational switch in response to ligand binding. eLife. (2016) 5:e13876. doi: 10.7554/eLife.13876
41. Kendall RL, Rutledge RZ, Mao X, Tebben AJ, Hungate RW, Thomas KA. Vascular endothelial growth factor receptor KDR tyrosine kinase activity is increased by autophosphorylation of two activation loop tyrosine residues. J Biol Chem. (1999) 274:6453–60. doi: 10.1074/jbc.274.10.6453
42. Matsumoto T, Bohman S, Dixelius J, Berge T, Dimberg A, Magnusson P, et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. (2005) 24:2342–53. doi: 10.1038/sj.emboj.7600709
43. Li X, Padhan N, Sjostrom EO, Roche FP, Testini C, Honkura N, et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat Commun. (2016) 7:11017. doi: 10.1038/ncomms11017
44. Holmqvist K, Cross MJ, Rolny C, Hagerkvist R, Rahimi N, Matsumoto T, et al. The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J Biol Chem. (2004) 279:22267–75. doi: 10.1074/jbc.M312729200
45. Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. (2001) 20:2768–78. doi: 10.1093/emboj/20.11.2768
46. Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci USA. (2005) 102:1076–81. doi: 10.1073/pnas.0404984102
47. Lamalice L, Houle F, Jourdan G, Huot J. Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene. (2004) 23:434–45. doi: 10.1038/sj.onc.1207034
48. Bates DO, Harper SJ. Regulation of vascular permeability by vascular endothelial growth factors. Vasc Pharmacol. (2002) 39:225–37. doi: 10.1016/S1537-1891(03)00011-9
49. Dixelius J, Makinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, et al. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J Biol Chem. (2003) 278:40973–9. doi: 10.1074/jbc.M304499200
50. Cudmore MJ, Hewett PW, Ahmad S, Wang KQ, Cai M, Al-Ani B, et al. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat Commun. (2012) 3:972. doi: 10.1038/ncomms1977
51. Mac Gabhann F, Popel AS. Dimerization of VEGF receptors and implications for signal transduction: a computational study. Biophys Chem. (2007) 128:125–39. doi: 10.1016/j.bpc.2007.03.010
52. Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. (2003) 14:334–47. doi: 10.1091/mbc.e02-07-0379
53. Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, et al. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. (2006) 7:1270–82. doi: 10.1111/j.1600-0854.2006.00462.x
54. Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. (2006) 8:1223–34. doi: 10.1038/ncb1486
55. Gelfand MV, Hagan N, Tata A, Oh WJ, Lacoste B, Kang KT, et al. Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. eLife. (2014) 3:e03720. doi: 10.7554/eLife.03720
56. Luo X, He JY, Xu J, Hu SY, Mo BH, Shu QX, et al. Vascular NRP2 triggers PNET angiogenesis by activating the SSH1-cofilin axis. Cell Biosci. (2020) 10:113. doi: 10.1186/s13578-020-00472-6
57. Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci USA. (1993) 90:10705–9. doi: 10.1073/pnas.90.22.10705
58. Makinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. (2001) 7:199–205. doi: 10.1038/84651
59. Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, et al. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. (2001) 20:1223–31. doi: 10.1093/emboj/20.6.1223
60. Heinolainen K, Karaman S, D'Amico G, Tammela T, Sormunen R, Eklund L, et al. VEGFR3 modulates vascular permeability by controlling VEGF/VEGFR2 signaling. Circ Res. (2017) 120:1414–25. doi: 10.1161/CIRCRESAHA.116.310477
61. Coon BG, Baeyens N, Han J, Budatha M, Ross TD, Fang JS, et al. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J Cell Biol. (2015) 208:975–86. doi: 10.1083/jcb.201408103
62. Swift MR, Weinstein BM. Arterial-venous specification during development. Circ Res. (2009) 104:576–88. doi: 10.1161/CIRCRESAHA.108.188805
63. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. (2011) 146:873–87. doi: 10.1016/j.cell.2011.08.039
64. Jia G, Aroor AR, Jia C, Sowers JR. Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1802–9. doi: 10.1016/j.bbadis.2018.08.008
65. Makanya AN, Hlushchuk R, Djonov VG. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis. (2009) 12:113–23. doi: 10.1007/s10456-009-9129-5
66. Adair TH, Montani JP. Angiogenesis. San Rafael, CA: Morgan & Claypool Life Sciences (2010). doi: 10.4199/C00017ED1V01Y201009ISP010
67. Nucera S, Biziato D, De Palma M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int J Dev Biol. (2011) 55:495–503. doi: 10.1387/ijdb.103227sn
68. Fang S, Salven P. Stem cells in tumor angiogenesis. J Mol Cell Cardiol. (2011) 50:290–5. doi: 10.1016/j.yjmcc.2010.10.024
69. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. (2011) 473:298–307. doi: 10.1038/nature10144
70. Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, et al. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. (2002) 16:2684–98. doi: 10.1101/gad.242002
71. Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. (2007) 8:464–78. doi: 10.1038/nrm2183
72. Polacheck WJ, Charest JL, Kamm RD. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc Natl Acad Sci USA. (2011) 108:11115–20. doi: 10.1073/pnas.1103581108
73. Wu Y, Al-Ameen MA, Ghosh G. Integrated effects of matrix mechanics and vascular endothelial growth factor (VEGF) on capillary sprouting. Ann Biomed Eng. (2014) 42:1024–36. doi: 10.1007/s10439-014-0987-7
74. Sieminski AL, Hebbel RP, Gooch KJ. The relative magnitudes of endothelial force generation and matrix stiffness modulate capillary morphogenesis in vitro. Exp Cell Res. (2004) 297:574–84. doi: 10.1016/j.yexcr.2004.03.035
75. Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc Natl Acad Sci USA. (2017) 114:492–7. doi: 10.1073/pnas.1613855114
76. Sewell-Loftin MK, Bayer SVH, Crist E, Hughes T, Joison SM, Longmore GD, et al. Cancer-associated fibroblasts support vascular growth through mechanical force. Sci Rep. (2017) 7:12574. doi: 10.1038/s41598-017-13006-x
77. Sewell-Loftin MK, Katz JB, George SC, Longmore GD. Micro-strains in the extracellular matrix induce angiogenesis. Lab Chip. (2020) 20:2776–87. doi: 10.1039/D0LC00145G
78. Khaidakov M, Wang X, Mehta JL. Potential involvement of LOX-1 in functional consequences of endothelial senescence. PLoS ONE. (2011) 6:e20964. doi: 10.1371/journal.pone.0020964
79. Uryga AK, Bennett MR. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J Physiol. (2016) 594:2115–24. doi: 10.1113/JP270923
80. Kohn JC, Lampi MC, Reinhart-King CA. Age-related vascular stiffening: causes and consequences. Front Genet. (2015) 6:112. doi: 10.3389/fgene.2015.00112
81. Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. (2005) 25:932–43. doi: 10.1161/01.ATV.0000160548.78317.29
82. Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, et al. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med. (2011) 3:112ra22. doi: 10.1126/scitranslmed.3002761
83. Matsuo E, Okamoto T, Ito A, Kawamoto E, Asanuma K, Wada K, et al. Substrate stiffness modulates endothelial cell function via the YAP-Dll4-Notch1 pathway. Exp Cell Res. (2021) 408:112835. doi: 10.1016/j.yexcr.2021.112835
84. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA. (2016) 113:11525–30. doi: 10.1073/pnas.1613121113
85. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. (2020) 77:1745–70. doi: 10.1007/s00018-019-03351-7
86. Conte SM, Vale PR. Peripheral arterial disease. Heart Lung Circ. (2018) 27:427–32. doi: 10.1016/j.hlc.2017.10.014
87. Inampudi C, Akintoye E, Ando T, Briasoulis A. Angiogenesis in peripheral arterial disease. Curr Opin Pharmacol. (2018) 39:60–7. doi: 10.1016/j.coph.2018.02.011
88. Safar ME. Arterial stiffness and peripheral arterial disease. Adv Cardiol. (2007) 44:199–211. doi: 10.1159/000096731
89. Cho YI, Cho DJ, Rosenson RS. Endothelial shear stress and blood viscosity in peripheral arterial disease. Curr Atheroscler Rep. (2014) 16:404. doi: 10.1007/s11883-014-0404-6
90. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. (2016) 126:821–8. doi: 10.1172/JCI83083
91. Lowe GD, Fowkes FG, Dawes J, Donnan PT, Lennie SE, Housley E. Blood viscosity, fibrinogen, and activation of coagulation and leukocytes in peripheral arterial disease and the normal population in the Edinburgh Artery Study. Circulation. (1993) 87:1915–20. doi: 10.1161/01.CIR.87.6.1915
92. Wieczor R, Rosc D, Wieczor AM, Kulwas A. VASCULAR-1 and VASCULAR-2 as a new potential angiogenesis and endothelial dysfunction markers in peripheral arterial disease. Clin Appl Thromb Hemost. (2019) 25:1076029619877440. doi: 10.1177/1076029619877440
93. Rajagopalan S, Mohler ER III, Lederman RJ, Mendelsohn FO, Saucedo JF, Goldman CK, et al. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation. (2003) 108:1933–8. doi: 10.1161/01.CIR.0000093398.16124.29
94. Annex BH, Cooke JP. New directions in therapeutic angiogenesis and arteriogenesis in peripheral arterial disease. Circ Res. (2021) 128:1944–57. doi: 10.1161/CIRCRESAHA.121.318266
95. Samakova A, Gazova A, Sabova N, Valaskova S, Jurikova M, Kyselovic J. The PI3k/Akt pathway is associated with angiogenesis, oxidative stress and survival of mesenchymal stem cells in pathophysiologic condition in ischemia. Physiol Res. (2019) 68(Suppl 2):S131–8. doi: 10.33549/physiolres.934345
96. Mitsos S, Katsanos K, Koletsis E, Kagadis GC, Anastasiou N, Diamantopoulos A, et al. Therapeutic angiogenesis for myocardial ischemia revisited: basic biological concepts and focus on latest clinical trials. Angiogenesis. (2012) 15:1–22. doi: 10.1007/s10456-011-9240-2
97. Clarke SA, Richardson WJ, Holmes JW. Modifying the mechanics of healing infarcts: Is better the enemy of good? J Mol Cell Cardiol. (2016) 93:115–24. doi: 10.1016/j.yjmcc.2015.11.028
98. Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. (1996) 270(5 Pt 2):H1803–11. doi: 10.1152/ajpheart.1996.270.5.H1803
99. Richardson WJ, Clarke SA, Quinn TA, Holmes JW. Physiological implications of myocardial scar structure. Compr Physiol. (2015) 5:1877–909. doi: 10.1002/cphy.c140067
100. Uccioli L, Meloni M, Izzo V, Giurato L, Merolla S, Gandini R. Critical limb ischemia: current challenges and future prospects. Vasc Health Risk Manag. (2018) 14:63–74. doi: 10.2147/VHRM.S125065
101. Vrsalovic M, Vucur K. Diabetes and critical limb ischemia: the deadly duo in patients with symptomatic peripheral artery disease. Acta Clin Croat. (2016) 55:240–6. doi: 10.20471/acc.2016.55.02.09
102. Rissanen TT, Vajanto I, Hiltunen MO, Rutanen J, Kettunen MI, Niemi M, et al. Expression of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 (KDR/Flk-1) in ischemic skeletal muscle and its regeneration. Am J Pathol. (2002) 160:1393–403. doi: 10.1016/S0002-9440(10)62566-7
103. Tuomisto TT, Rissanen TT, Vajanto I, Korkeela A, Rutanen J, Yla-Herttuala S. HIF-VEGF-VEGFR-2, TNF-alpha and IGF pathways are upregulated in critical human skeletal muscle ischemia as studied with DNA array. Atherosclerosis. (2004) 174:111–20. doi: 10.1016/j.atherosclerosis.2004.01.015
104. Mendes-Pinto D, Ribeiro JM, Rodrigues-Machado MDG. Association between critical limb ischemia and arterial stiffness measured by brachial artery oscillometry. J Vasc Bras. (2019) 18:e20180073. doi: 10.1590/1677-5449.007318
105. Barc P, Antkiewicz M, Sliwa B, Fraczkowska K, Guzinski M, Dawiskiba T, et al. Double VEGF/HGF gene therapy in critical limb ischemia complicated by diabetes mellitus. J Cardiovasc Transl Res. (2021) 14:409–15. doi: 10.1007/s12265-020-10066-9
106. Powell RJ, Simons M, Mendelsohn FO, Daniel G, Henry TD, Koga M, et al. Results of a double-blind, placebo-controlled study to assess the safety of intramuscular injection of hepatocyte growth factor plasmid to improve limb perfusion in patients with critical limb ischemia. Circulation. (2008) 118:58–65. doi: 10.1161/CIRCULATIONAHA.107.727347
107. Miquerol L, Gertsenstein M, Harpal K, Rossant J, Nagy A. Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol. (1999) 212:307–22. doi: 10.1006/dbio.1999.9355
108. Cleaver O, Tonissen KF, Saha MS, Krieg PA. Neovascularization of the xenopus embryo. Dev Dyn. (1997) 210:66–77. doi: 10.1002/(SICI)1097-0177(199709)210:1<66::AID-AJA7>3.0.CO;2-#
109. Flamme I, Breier G, Risau W. Vascular endothelial growth factor (VEGF) and VEGF receptor 2 (flk-1) are expressed during vasculogenesis and vascular differentiation in the quail embryo. Dev Biol. (1995) 169:699–712. doi: 10.1006/dbio.1995.1180
110. Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. (1996) 380:435–9. doi: 10.1038/380435a0
111. Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. (1996) 380:439–42. doi: 10.1038/380439a0
112. Rouwkema J, Khademhosseini A. Vascularization and angiogenesis in tissue engineering: beyond creating static networks. Trends Biotechnol. (2016) 34:733–45. doi: 10.1016/j.tibtech.2016.03.002
113. Jain RK, Au P, Tam J, Duda DG, Fukumura D. Engineering vascularized tissue. Nat Biotechnol. (2005) 23:821–3. doi: 10.1038/nbt0705-821
114. Butt OI, Carruth R, Kutala VK, Kuppusamy P, Moldovan NI. Stimulation of peri-implant vascularization with bone marrow-derived progenitor cells: monitoring by in vivo EPR oximetry. Tissue Eng. (2007) 13:2053–61. doi: 10.1089/ten.2006.0225
115. Rouwkema J, Rivron NC, van Blitterswijk CA. Vascularization in tissue engineering. Trends Biotechnol. (2008) 26:434–41. doi: 10.1016/j.tibtech.2008.04.009
117. Freed LE, Guilak F, Guo XE, Gray ML, Tranquillo R, Holmes JW, et al. Advanced tools for tissue engineering: scaffolds, bioreactors, and signaling. Tissue Eng. (2006) 12:3285–305. doi: 10.1089/ten.2006.12.3285
118. Gui L, Boyle MJ, Kamin YM, Huang AH, Starcher BC, Miller CA, et al. Construction of tissue-engineered small-diameter vascular grafts in fibrin scaffolds in 30 days. Tissue Eng A. (2014) 20:1499–507. doi: 10.1089/ten.tea.2013.0263
119. Fan W, Li C, Qin X, Wang S, Da H, Cheng K, et al. Adipose stromal cell and sarpogrelate orchestrate the recovery of inflammation-induced angiogenesis in aged hindlimb ischemic mice. Aging Cell. (2013) 12:32–41. doi: 10.1111/acel.12014
120. Bachmann J, Ehlert E, Becker M, Otto C, Radeloff K, Blunk T, et al. Ischemia-like stress conditions stimulate trophic activities of adipose-derived stromal/stem cells. Cells. (2020) 9:1935–54. doi: 10.3390/cells9091935
121. Dou YQ, Kong P, Li CL, Sun HX, Li WW, Yu Y, et al. Smooth muscle SIRT1 reprograms endothelial cells to suppress angiogenesis after ischemia. Theranostics. (2020) 10:1197–212. doi: 10.7150/thno.39320
122. Chiaverina G, di Blasio L, Monica V, Accardo M, Palmiero M, Peracino B, et al. Dynamic interplay between pericytes and endothelial cells during sprouting angiogenesis. Cells. (2019) 8:1109–21. doi: 10.3390/cells8091109
123. Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
124. Chanakira A, Dutta R, Charboneau R, Barke R, Santilli SM, Roy S. Hypoxia differentially regulates arterial and venous smooth muscle cell proliferation via PDGFR-beta and VEGFR-2 expression. Am J Physiol Heart Circ Physiol. (2012) 302:H1173–84. doi: 10.1152/ajpheart.00411.2011
125. Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, Martins JR, et al. Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Rep. (2015) 13:1161–71. doi: 10.1016/j.celrep.2015.09.072
126. Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J Clin Invest. (2016) 126:4527–36. doi: 10.1172/JCI87343
127. Smyth LCD, Rustenhoven J, Scotter EL, Schweder P, Faull RLM, Park TIH, et al. Markers for human brain pericytes and smooth muscle cells. J Chem Neuroanat. (2018) 92:48–60. doi: 10.1016/j.jchemneu.2018.06.001
128. Shenoy AK, Jin Y, Luo H, Tang M, Pampo C, Shao R, et al. Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells. J Clin Invest. (2016) 126:4174–86. doi: 10.1172/JCI86623
129. Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, Stallcup WB. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn. (2001) 222:218–27. doi: 10.1002/dvdy.1200
130. Eglinger J, Karsjens H, Lammert E. Quantitative assessment of angiogenesis and pericyte coverage in human cell-derived vascular sprouts. Inflamm Regen. (2017) 37:2. doi: 10.1186/s41232-016-0033-2
131. Sainson RC, Harris AL. Regulation of angiogenesis by homotypic and heterotypic notch signalling in endothelial cells and pericytes: from basic research to potential therapies. Angiogenesis. (2008) 11:41–51. doi: 10.1007/s10456-008-9098-0
132. Labernadie A, Kato T, Brugues A, Serra-Picamal X, Derzsi S, Arwert E, et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. (2017) 19:224–37. doi: 10.1038/ncb3478
133. Eilken HM, Dieguez-Hurtado R, Schmidt I, Nakayama M, Jeong HW, Arf H, et al. Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nat Commun. (2017) 8:1574. doi: 10.1038/s41467-017-01738-3
134. Kim BG, Gao MQ, Kang S, Choi YP, Lee JH, Kim JE, et al. Mechanical compression induces VEGFA overexpression in breast cancer via DNMT3A-dependent miR-9 downregulation. Cell Death Dis. (2017) 8:e2646. doi: 10.1038/cddis.2017.73
135. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
136. De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A, Maggiolini M. HIF-1alpha/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. (2013) 15:R64. doi: 10.1186/bcr3458
137. Rosenfeld D, Landau S, Shandalov Y, Raindel N, Freiman A, Shor E, et al. Morphogenesis of 3D vascular networks is regulated by tensile forces. Proc Natl Acad Sci USA. (2016) 113:3215–20. doi: 10.1073/pnas.1522273113
138. Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. (2013) 15:677–87. doi: 10.1038/ncb2743
139. Gimbrone MA Jr., Garcia-Cardena G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. (2013) 22:9–15. doi: 10.1016/j.carpath.2012.06.006
140. Nayak L, Lin Z, Jain MK. “Go with the flow”: how Kruppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid Redox Signal. (2011). 15:1449–61. doi: 10.1089/ars.2010.3647
141. Tedgui A, Mallat Z. Anti-inflammatory mechanisms in the vascular wall. Circ Res. (2001) 88:877–87. doi: 10.1161/hh0901.090440
142. Hahn C, Orr AW, Sanders JM, Jhaveri KA, Schwartz MA. The subendothelial extracellular matrix modulates JNK activation by flow. Circ Res. (2009) 104:995–1003. doi: 10.1161/CIRCRESAHA.108.186486
143. Orr AW, Hahn C, Blackman BR, Schwartz MA. p21-activated kinase signaling regulates oxidant-dependent NF-kappa B activation by flow. Circ Res. (2008) 103:671–9. doi: 10.1161/CIRCRESAHA.108.182097
144. Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal. (2011) 15:1405–14. doi: 10.1089/ars.2010.3679
145. Urschel K, Garlichs CD, Daniel WG, Cicha I. VEGFR2 signalling contributes to increased endothelial susceptibility to TNF-alpha under chronic non-uniform shear stress. Atherosclerosis. (2011) 219:499–509. doi: 10.1016/j.atherosclerosis.2011.09.045
146. Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, et al. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. (2009) 457:1103–8. doi: 10.1038/nature07765
147. Payne S, De Val S, Neal A. Endothelial-specific cre mouse models. Arterioscler Thromb Vasc Biol. (2018) 38:2550–61. doi: 10.1161/ATVBAHA.118.309669
148. Carmeliet P, Collen D. Transgenic mouse models in angiogenesis and cardiovascular disease. J Pathol. (2000) 190:387–405. doi: 10.1002/(SICI)1096-9896(200002)190:3<387::AID-PATH595>3.0.CO;2-R
149. Hiratsuka S, Kataoka Y, Nakao K, Nakamura K, Morikawa S, Tanaka S, et al. Vascular endothelial growth factor A (VEGF-A) is involved in guidance of VEGF receptor-positive cells to the anterior portion of early embryos. Mol Cell Biol. (2005) 25:355–63. doi: 10.1128/MCB.25.1.355-363.2005
150. Oladipupo SS, Kabir AU, Smith C, Choi K, Ornitz DM. Impaired tumor growth and angiogenesis in mice heterozygous for Vegfr2 (Flk1). Sci Rep. (2018) 8:14724. doi: 10.1038/s41598-018-33037-2
151. Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L, et al. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev Cell. (2017) 42:462–78.e7. doi: 10.1016/j.devcel.2017.08.002
152. Pulkkinen HH, Kiema M, Lappalainen JP, Toropainen A, Beter M, Tirronen A, et al. BMP6/TAZ-Hippo signaling modulates angiogenesis and endothelial cell response to VEGF. Angiogenesis. (2021) 24:129–44. doi: 10.1007/s10456-020-09748-4
153. Abe Y, Watanabe M, Chung S, Kamm RD, Tanishita K, Sudo R. Balance of interstitial flow magnitude and vascular endothelial growth factor concentration modulates three-dimensional microvascular network formation. APL Bioeng. (2019) 3:036102. doi: 10.1063/1.5094735
154. Polacheck WJ, Li R, Uzel SG, Kamm RD. Microfluidic platforms for mechanobiology. Lab Chip. (2013) 13:2252–67. doi: 10.1039/c3lc41393d
155. Semino CE, Kamm RD, Lauffenburger DA. Autocrine EGF receptor activation mediates endothelial cell migration and vascular morphogenesis induced by VEGF under interstitial flow. Exp Cell Res. (2006) 312:289–98. doi: 10.1016/j.yexcr.2005.10.029
156. Sudo R, Chung S, Zervantonakis IK, Vickerman V, Toshimitsu Y, Griffith LG, et al. Transport-mediated angiogenesis in 3D epithelial coculture. FASEB J. (2009) 23:2155–64. doi: 10.1096/fj.08-122820
157. Brown A, Burke G, Meenan BJ. Modeling of shear stress experienced by endothelial cells cultured on microstructured polymer substrates in a parallel plate flow chamber. Biotechnol Bioeng. (2011) 108:1148–58. doi: 10.1002/bit.23022
158. Shirure VS, Lezia A, Tao A, Alonzo LF, George SC. Low levels of physiological interstitial flow eliminate morphogen gradients and guide angiogenesis. Angiogenesis. (2017) 20:493–504. doi: 10.1007/s10456-017-9559-4
159. Shirure VS, Sewell-Loftin MK, Lam SF, Todd TD, Hwang PY, George SC. Building better tumor models: organoid systems to investigate angiogenesis. In: Soker S, Skardal A, editors. Tumor Organoids. Cham: Springer International Publishing (2018). p. 117–48. doi: 10.1007/978-3-319-60511-1_7
160. Vazgiourakis VM, Zervou MI, Eliopoulos E, Sharma S, Sidiropoulos P, Franek BS, et al. Implication of VEGFR2 in systemic lupus erythematosus: a combined genetic and structural biological approach. Clin Exp Rheumatol. (2013) 31:97–102.
161. Cho SJ, Park MH, Han C, Yoon K, Koh YH. VEGFR2 alteration in Alzheimer's disease. Sci Rep. (2017) 7:17713. doi: 10.1038/s41598-017-18042-1
162. Smith RO, Ninchoji T, Gordon E, Andre H, Dejana E, Vestweber D, et al. Vascular permeability in retinopathy is regulated by VEGFR2 Y949 signaling to VE-cadherin. eLife. (2020) 9:e54056. doi: 10.7554/eLife.54056
163. Ruiz S, Zhao H, Chandakkar P, Papoin J, Choi H, Nomura-Kitabayashi A, et al. Correcting Smad1/5/8, mTOR, and VEGFR2 treats pathology in hereditary hemorrhagic telangiectasia models. J Clin Invest. (2020) 130:942–57. doi: 10.1172/JCI127425
164. Bruno L, Karagil S, Mahmood A, Elbediwy A, Stolinski M, Mackenzie FE. Mechanosensing and the hippo pathway in microglia: a potential link to Alzheimer's disease pathogenesis? Cells. (2021) 10:3144–58. doi: 10.3390/cells10113144
165. Jorge S, Chang S, Barzilai JJ, Leppert P, Segars JH. Mechanical signaling in reproductive tissues: mechanisms and importance. Reprod Sci. (2014) 21:1093–107. doi: 10.1177/1933719114542023
166. Lei Y, Rajabi S, Pedrigi RM, Overby DR, Read AT, Ethier CR. In vitro models for glaucoma research: effects of hydrostatic pressure. Invest Ophthalmol Vis Sci. (2011) 52:6329–39. doi: 10.1167/iovs.11-7836
167. Henderson K, Sligar AD, Le VP, Lee J, Baker AB. Biomechanical regulation of mesenchymal stem cells for cardiovascular tissue engineering. Adv Healthc Mater. (2017) 6:1700556. doi: 10.1002/adhm.201700556
168. Qiu J, Zheng Y, Hu J, Liao D, Gregersen H, Deng X, et al. Biomechanical regulation of vascular smooth muscle cell functions: from in vitro to in vivo understanding. J R Soc Interface. (2014) 11:20130852. doi: 10.1098/rsif.2013.0852
169. Huse M. Mechanical forces in the immune system. Nat Rev Immunol. (2017) 17:679–90. doi: 10.1038/nri.2017.74
170. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. (2006) 126:677–89. doi: 10.1016/j.cell.2006.06.044
171. Vempati P, Popel AS, Mac Gabhann F. Extracellular regulation of VEGF: isoforms, proteolysis, and vascular patterning. Cytokine Growth Factor Rev. (2014) 25:1–19. doi: 10.1016/j.cytogfr.2013.11.002
172. Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. (1992) 267:26031–7. doi: 10.1016/S0021-9258(18)35712-0
173. Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. (1993) 4:1317–26. doi: 10.1091/mbc.4.12.1317
174. Hall H, Hubbell JA. Matrix-bound sixth Ig-like domain of cell adhesion molecule L1 acts as an angiogenic factor by ligating alphavbeta3-integrin and activating VEGF-R2. Microvasc Res. (2004) 68:169–78. doi: 10.1016/j.mvr.2004.07.001
175. Gilde F, Fourel L, Guillot R, Pignot-Paintrand I, Okada T, Fitzpatrick V, et al. Stiffness-dependent cellular internalization of matrix-bound BMP-2 and its relation to Smad and non-Smad signaling. Acta Biomater. (2016) 46:55–67. doi: 10.1016/j.actbio.2016.09.014
176. Schmidt C, Pommerenke H, Durr F, Nebe B, Rychly J. Mechanical stressing of integrin receptors induces enhanced tyrosine phosphorylation of cytoskeletally anchored proteins. J Biol Chem. (1998) 273:5081–5. doi: 10.1074/jbc.273.9.5081
177. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. (2020) 19:1997–2007. doi: 10.3892/etm.2020.8454
178. Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells. (2020) 9:198–230. doi: 10.3390/cells9010198
179. Gallo S, Vitacolonna A, Bonzano A, Comoglio P, Crepaldi T. ERK: a key player in the pathophysiology of cardiac hypertrophy. Int J Mol Sci. (2019) 20:2164–84. doi: 10.3390/ijms20092164
180. McCain J. The MAPK (ERK) pathway: investigational combinations for the treatment of BRAF-mutated metastatic melanoma. P T. (2013) 38:96–108.
181. Yang JM, Bhattacharya S, West-Foyle H, Hung CF, Wu TC, Iglesias PA, et al. Integrating chemical and mechanical signals through dynamic coupling between cellular protrusions and pulsed ERK activation. Nat Commun. (2018) 9:4673. doi: 10.1038/s41467-018-07150-9
182. De Belly H, Stubb A, Yanagida A, Labouesse C, Jones PH, Paluch EK, et al. Membrane tension gates ERK-mediated regulation of pluripotent cell fate. Cell Stem Cell. (2021) 28:273–84.e6. doi: 10.1016/j.stem.2020.10.018
183. Tanabe Y, Koga M, Saito M, Matsunaga Y, Nakayama K. Inhibition of adipocyte differentiation by mechanical stretching through ERK-mediated downregulation of PPARgamma2. J Cell Sci. (2004) 117(Pt 16):3605–14. doi: 10.1242/jcs.01207
184. Hino N, Rossetti L, Marin-Llaurado A, Aoki K, Trepat X, Matsuda M, et al. ERK-mediated mechanochemical waves direct collective cell polarization. Dev Cell. (2020) 53:646–60.e8. doi: 10.1016/j.devcel.2020.05.011
185. Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF, et al. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature. (2017) 543:118–21. doi: 10.1038/nature21407
186. Fearnley GW, Smith GA, Abdul-Zani I, Yuldasheva N, Mughal NA, Homer-Vanniasinkam S, et al. VEGF-A isoforms program differential VEGFR2 signal transduction, trafficking and proteolysis. Biol Open. (2016) 5:571–83. doi: 10.1242/bio.017434
187. Almalki SG, Agrawal DK. ERK signaling is required for VEGF-A/VEGFR2-induced differentiation of porcine adipose-derived mesenchymal stem cells into endothelial cells. Stem Cell Res Ther. (2017) 8:113. doi: 10.1186/s13287-017-0568-4
188. Svensson S, Jirstrom K, Ryden L, Roos G, Emdin S, Ostrowski MC, et al. ERK phosphorylation is linked to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene. (2005) 24:4370–9. doi: 10.1038/sj.onc.1208626
189. Xie Y, Mansouri M, Rizk A, Berger P. Regulation of VEGFR2 trafficking and signaling by Rab GTPase-activating proteins. Sci Rep. (2019) 9:13342. doi: 10.1038/s41598-019-49646-4
190. Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. (1997) 13:513–609. doi: 10.1146/annurev.cellbio.13.1.513
191. Reinehr R, Sommerfeld A, Haussinger D. The Src family kinases: distinct functions of c-Src, Yes, and Fyn in the liver. Biomol Concepts. (2013) 4:129–42. doi: 10.1515/bmc-2012-0047
192. Petch LA, Bockholt SM, Bouton A, Parsons JT, Burridge K. Adhesion-induced tyrosine phosphorylation of the p130 src substrate. J Cell Sci. (1995) 108 (Pt 4):1371–9. doi: 10.1242/jcs.108.4.1371
193. Hall CL, Lange LA, Prober DA, Zhang S, Turley EA. pp60(c-src) is required for cell locomotion regulated by the hyaluronanreceptor RHAMM. Oncogene. (1996) 13:2213–24.
194. Twamley-Stein GM, Pepperkok R, Ansorge W, Courtneidge SA. The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc Natl Acad Sci USA. (1993) 90:7696–700. doi: 10.1073/pnas.90.16.7696
195. Mladenovic J, Anderson SM. Over-expression of c-src or v-src in bone marrow stromal cells stimulates hematopoiesis in long-term bone marrow culture. Blood. (1992) 80:3079–89. doi: 10.1182/blood.V80.12.3079.bloodjournal80123079
196. Moysenovich AM, Tatarskiy VV, Yastrebova MA, Bessonov IV, Arkhipova AY, Kolosov AS, et al. Akt and Src mediate the photocrosslinked fibroin-induced neural differentiation. Neuroreport. (2020) 31:770–5. doi: 10.1097/WNR.0000000000001482
197. Hutcheson JD, Ryzhova LM, Setola V, Merryman WD. 5-HT(2B) antagonism arrests non-canonical TGF-beta1-induced valvular myofibroblast differentiation. J Mol Cell Cardiol. (2012). 53:707–14. doi: 10.1016/j.yjmcc.2012.08.012
198. Schroer AK, Merryman WD. Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J Cell Sci. (2015) 128:1865–75. doi: 10.1242/jcs.162891
199. Takahashi M, Berk BC. Mitogen-activated protein kinase (ERK1/2) activation by shear stress and adhesion in endothelial cells. Essential role for a herbimycin-sensitive kinase. J Clin Invest. (1996) 98:2623–31. doi: 10.1172/JCI119083
200. Jalali S, Li YS, Sotoudeh M, Yuan S, Li S, Chien S, et al. Shear stress activates p60src-Ras-MAPK signaling pathways in vascular endothelial cells. Arterioscler Thromb Vasc Biol. (1998) 18:227–34. doi: 10.1161/01.ATV.18.2.227
201. Rezzola S, Di Somma M, Corsini M, Leali D, Ravelli C, Polli VAB, et al. VEGFR2 activation mediates the pro-angiogenic activity of BMP4. Angiogenesis. (2019) 22:521–33. doi: 10.1007/s10456-019-09676-y
202. Piroli ME, Blanchette JO, Jabbarzadeh E. Polarity as a physiological modulator of cell function. Front Biosci. (2019) 24:451–62. doi: 10.2741/4728
203. Butler MT, Wallingford JB. Planar cell polarity in development and disease. Nat Rev Mol Cell Biol. (2017) 18:375–88. doi: 10.1038/nrm.2017.11
204. Goggolidou P. Wnt and planar cell polarity signaling in cystic renal disease. Organogenesis. (2014) 10:86–95. doi: 10.4161/org.26766
205. Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. (2014) 5:e29846. doi: 10.4161/sgtp.29846
206. Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. (1999) 285:895–8. doi: 10.1126/science.285.5429.895
207. McGrail DJ, Kieu QM, Dawson MR. The malignancy of metastatic ovarian cancer cells is increased on soft matrices through a mechanosensitive Rho-ROCK pathway. J Cell Sci. (2014) 127(Pt 12):2621–6. doi: 10.1242/jcs.144378
208. Mertsch S, Thanos S. Opposing signaling of ROCK1 and ROCK2 determines the switching of substrate specificity and the mode of migration of glioblastoma cells. Mol Neurobiol. (2014) 49:900–15. doi: 10.1007/s12035-013-8568-6
209. Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. (2008) 95:5374–84. doi: 10.1529/biophysj.108.133116
210. Sun H, Breslin JW, Zhu J, Yuan SY, Wu MH. Rho and ROCK signaling in VEGF-induced microvascular endothelial hyperpermeability. Microcirculation. (2006) 13:237–47. doi: 10.1080/10739680600556944
211. Liu J, Gao HY, Wang XF. The role of the Rho/ROCK signaling pathway in inhibiting axonal regeneration in the central nervous system. Neural Regen Res. (2015) 10:1892–6. doi: 10.4103/1673-5374.170325
212. Yoneda A, Multhaupt HA, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. (2005) 170:443–53. doi: 10.1083/jcb.200412043
213. Yoneda A, Ushakov D, Multhaupt HA, Couchman JR. Fibronectin matrix assembly requires distinct contributions from Rho kinases I and -II. Mol Biol Cell. (2007) 18:66–75. doi: 10.1091/mbc.e06-08-0684
214. McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. (2004) 6:483–95. doi: 10.1016/S1534-5807(04)00075-9
215. Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, et al. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci USA. (2006) 103:14495–500. doi: 10.1073/pnas.0601911103
216. Civelekoglu-Scholey G, Orr AW, Novak I, Meister JJ, Schwartz MA, Mogilner A. Model of coupled transient changes of Rac, Rho, adhesions and stress fibers alignment in endothelial cells responding to shear stress. J Theor Biol. (2005) 232:569–85. doi: 10.1016/j.jtbi.2004.09.004
217. Struckhoff AP, Vitko JR, Rana MK, Davis CT, Foderingham KE, Liu CH, et al. Dynamic regulation of ROCK in tumor cells controls CXCR4-driven adhesion events. J Cell Sci. (2010) 123(Pt 3):401–12. doi: 10.1242/jcs.052167
218. Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, et al. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. (2015) 208:821–38. doi: 10.1083/jcb.201404140
219. Guilluy C, Swaminathan V, Garcia-Mata R, O'Brien ET, Superfine R, Burridge K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol. (2011) 13:722–7. doi: 10.1038/ncb2254
220. Ghosh K, Thodeti CK, Dudley AC, Mammoto A, Klagsbrun M, Ingber DE. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci USA. (2008) 105:11305–10. doi: 10.1073/pnas.0800835105
221. Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. (2001) 20:4639–47. doi: 10.1093/emboj/20.17.4639
222. Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci. (2009) 122(Pt 8):1059–69. doi: 10.1242/jcs.039446
223. Stiegler AL, Boggon TJ. p190RhoGAP proteins contain pseudoGTPase domains. Nat Commun. (2017) 8:506. doi: 10.1038/s41467-017-00483-x
224. Corry J, Mott HR, Owen D. Activation of STAT transcription factors by the Rho-family GTPases. Biochem Soc Trans. (2020) 48:2213–27. doi: 10.1042/BST20200468
225. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. (2014) 94:1287–312. doi: 10.1152/physrev.00005.2014
226. Moya IM, Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. (2019) 20:211–26. doi: 10.1038/s41580-018-0086-y
227. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. (2007) 21:2747–61. doi: 10.1101/gad.1602907
228. Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. (2008) 28:2426–36. doi: 10.1128/MCB.01874-07
229. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. (2011) 474:179–83. doi: 10.1038/nature10137
230. Yasuda D, Kobayashi D, Akahoshi N, Ohto-Nakanishi T, Yoshioka K, Takuwa Y, et al. Lysophosphatidic acid-induced YAP/TAZ activation promotes developmental angiogenesis by repressing Notch ligand Dll4. J Clin Invest. (2019) 129:4332–49. doi: 10.1172/JCI121955
231. Baker AB, Ettenson DS, Jonas M, Nugent MA, Iozzo RV, Edelman ER. Endothelial cells provide feedback control for vascular remodeling through a mechanosensitive autocrine TGF-beta signaling pathway. Circ Res. (2008) 103:289–97. doi: 10.1161/CIRCRESAHA.108.179465
232. Sehgal P, Kong X, Wu J, Sunyer R, Trepat X, Leckband D. Epidermal growth factor receptor and integrins control force-dependent vinculin recruitment to E-cadherin junctions. J Cell Sci. (2018) 131:62–73. doi: 10.1242/jcs.206656
233. Zhao FL, Li L, Guan LY, Yang H, Wu CH, Liu YY. Roles for GP IIb/IIIa and alpha v beta 3 integrins in MDA-MB-231 cell invasion and shear flow-induced cancer cell mechanotransduction. Cancer Lett. (2014) 344:62–73. doi: 10.1016/j.canlet.2013.10.019
234. Cipitria A, Salmeron-Sanchez M. Mechanotransduction and growth factor signalling to engineer cellular microenvironments. Adv Healthc Mater. (2017) 6:1700052. doi: 10.1002/adhm.201700052
235. Zuidema A, Wang W, Sonnenberg A. Crosstalk between cell adhesion complexes in regulation of mechanotransduction. Bioessays. (2020) 42:e2000119. doi: 10.1002/bies.202000119
236. Kuiper EJ, Hughes JM, Van Geest RJ, Vogels IM, Goldschmeding R, Van Noorden CJ, et al. Effect of VEGF-A on expression of profibrotic growth factor and extracellular matrix genes in the retina. Invest Ophthalmol Vis Sci. (2007) 48:4267–76. doi: 10.1167/iovs.06-0804
237. Muhamed I, Wu J, Sehgal P, Kong X, Tajik A, Wang N, et al. E-cadherin-mediated force transduction signals regulate global cell mechanics. J Cell Sci. (2016) 129:1843–54. doi: 10.1242/jcs.185447
238. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. (2007) 179:1311–23. doi: 10.1083/jcb.200704042
239. Giacomini MM, Travis MA, Kudo M, Sheppard D. Epithelial cells utilize cortical actin/myosin to activate latent TGF-beta through integrin alpha(v)beta(6)-dependent physical force. Exp Cell Res. (2012) 318:716–22. doi: 10.1016/j.yexcr.2012.01.020
240. Dey N, De P, Brian LJ. Evading anti-angiogenic therapy: resistance to anti-angiogenic therapy in solid tumors. Am J Transl Res. (2015) 7:1675–98. doi: 10.1038/bjc.2014.439
241. van Beijnum JR, Nowak-Sliwinska P, Huijbers EJ, Thijssen VL, Griffioen AW. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol Rev. (2015) 67:441–61. doi: 10.1124/pr.114.010215
242. Foley TR, Waldo SW, Armstrong EJ. Medical Therapy in Peripheral Artery Disease and Critical Limb Ischemia. Curr Treat Options Cardiovasc Med. (2016) 18:42. doi: 10.1007/s11936-016-0464-8
243. Ribatti D. The inefficacy of antiangiogenic therapies. J Angiogenes Res. (2010) 2:27. doi: 10.1186/2040-2384-2-27
244. Ribatti D. Tumor refractoriness to anti-VEGF therapy. Oncotarget. (2016) 7:46668–77. doi: 10.18632/oncotarget.8694
245. Sasich LD, Sukkari SR. The US FDAs withdrawal of the breast cancer indication for Avastin (bevacizumab). Saudi Pharm J. (2012) 20:381–5. doi: 10.1016/j.jsps.2011.12.001
246. Mansour SE, Browning DJ, Wong K, Flynn HW Jr., Bhavsar AR. The evolving treatment of diabetic retinopathy. Clin Ophthalmol. (2020) 14:653–78. doi: 10.2147/OPTH.S236637
247. Ngo DTM, Williams T, Horder S, Kritharides L, Vardy J, Mandaliya H, et al. Factors associated with adverse cardiovascular events in cancer patients treated with bevacizumab. J Clin Med. (2020) 9:2664–73. doi: 10.3390/jcm9082664
248. Scappaticci FA, Skillings JR, Holden SN, Gerber HP, Miller K, Kabbinavar F, et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J Natl Cancer Inst. (2007) 99:1232–9. doi: 10.1093/jnci/djm086
249. Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. (2009) 6:465–77. doi: 10.1038/nrclinonc.2009.94
250. Zhang Y, Wu S. Effects of fasudil on pulmonary hypertension in clinical practice. Pulm Pharmacol Ther. (2017) 46:54–63. doi: 10.1016/j.pupt.2017.08.002
251. Koch JC, Kuttler J, Maass F, Lengenfeld T, Zielke E, Bahr M, et al. Compassionate use of the ROCK inhibitor fasudil in three patients with amyotrophic lateral sclerosis. Front Neurol. (2020) 11:173. doi: 10.3389/fneur.2020.00173
252. Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, et al. Efficacy and safety of fasudil in patients with stable angina: a double-blind, placebo-controlled, phase 2 trial. J Am Coll Cardiol. (2005) 46:1803–11. doi: 10.1016/j.jacc.2005.07.047
253. Suzuki Y, Shibuya M, Satoh S, Sugimoto Y, Takakura K. A postmarketing surveillance study of fasudil treatment after aneurysmal subarachnoid hemorrhage. Surg Neurol. (2007) 68:126–31; discussion: 31–2. doi: 10.1016/j.surneu.2006.10.037
254. Wu DJ, Xu JZ, Wu YJ, Jean-Charles L, Xiao B, Gao PJ, et al. Effects of fasudil on early atherosclerotic plaque formation and established lesion progression in apolipoprotein E-knockout mice. Atherosclerosis. (2009) 207:68–73. doi: 10.1016/j.atherosclerosis.2009.04.025
255. Song Y, Chen X, Wang LY, Gao W, Zhu MJ. Rho kinase inhibitor fasudil protects against beta-amyloid-induced hippocampal neurodegeneration in rats. CNS Neurosci Ther. (2013) 19:603–10. doi: 10.1111/cns.12116
256. Hou Y, Zhou L, Yang QD, Du XP, Li M, Yuan M, et al. Changes in hippocampal synapses and learning-memory abilities in a streptozotocin-treated rat model and intervention by using fasudil hydrochloride. Neuroscience. (2012) 200:120–9. doi: 10.1016/j.neuroscience.2011.10.030
257. Huentelman MJ, Stephan DA, Talboom J, Corneveaux JJ, Reiman DM, Gerber JD, et al. Peripheral delivery of a ROCK inhibitor improves learning and working memory. Behav Neurosci. (2009) 123:218–23. doi: 10.1037/a0014260
258. Nohria A, Grunert ME, Rikitake Y, Noma K, Prsic A, Ganz P, et al. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ Res. (2006) 99:1426–32. doi: 10.1161/01.RES.0000251668.39526.c7
259. Sung YC, Jin PR, Chu LA, Hsu FF, Wang MR, Chang CC, et al. Delivery of nitric oxide with a nanocarrier promotes tumour vessel normalization and potentiates anti-cancer therapies. Nat Nanotechnol. (2019) 14:1160–9. doi: 10.1038/s41565-019-0570-3
260. Tolaney SM, Boucher Y, Duda DG, Martin JD, Seano G, Ancukiewicz M, et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc Natl Acad Sci USA. (2015) 112:14325–30. doi: 10.1073/pnas.1518808112
261. Mortezaee K. Normalization in tumor ecosystem: Opportunities and challenges. Cell Biol Int. (2021) 45:2017–30. doi: 10.1002/cbin.11655
262. Leikas AJ, Hassinen I, Hedman A, Kivela A, Yla-Herttuala S, Hartikainen JEK. Long-term safety and efficacy of intramyocardial adenovirus-mediated VEGF-D(DeltaNDeltaC) gene therapy eight-year follow-up of phase I KAT301 study. Gene Ther. (2021). doi: 10.1038/s41434-021-00295-1. [Epub ahead of print].
263. Husmann M, Dorffler-Melly J, Kalka C, Diehm N, Baumgartner I, Silvestro A. Successful lower extremity angioplasty improves brachial artery flow-mediated dilation in patients with peripheral arterial disease. J Vasc Surg. (2008) 48:1211–6. doi: 10.1016/j.jvs.2008.06.039
Keywords: angiogenesis, VEGFR-2, mechanobiology, mechanoregulation, endothelial cells
Citation: Miller B and Sewell-Loftin MK (2022) Mechanoregulation of Vascular Endothelial Growth Factor Receptor 2 in Angiogenesis. Front. Cardiovasc. Med. 8:804934. doi: 10.3389/fcvm.2021.804934
Received: 29 October 2021; Accepted: 10 December 2021;
Published: 11 January 2022.
Edited by:
Andrea Caporali, University of Edinburgh, United KingdomReviewed by:
Vijaykumar S. Meli, University of California, Irvine, United StatesJohanna Laakkonen, University of Eastern Finland, Finland
Copyright © 2022 Miller and Sewell-Loftin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary Kathryn Sewell-Loftin, mksewellloftin@uab.edu