An Overview of in vivo Functions of Chondroitin Sulfate and Dermatan Sulfate Revealed by Their Deficient Mice
 Shuji Mizumoto
Shuji Mizumoto Shuhei Yamada
Shuhei Yamada- Department of Pathobiochemistry, Faculty of Pharmacy, Meijo University, Nagoya, Japan
Chondroitin sulfate (CS), dermatan sulfate (DS) and heparan sulfate (HS) are covalently attached to specific core proteins to form proteoglycans in their biosynthetic pathways. They are constructed through the stepwise addition of respective monosaccharides by various glycosyltransferases and maturated by epimerases as well as sulfotransferases. Structural diversities of CS/DS and HS are essential for their various biological activities including cell signaling, cell proliferation, tissue morphogenesis, and interactions with a variety of growth factors as well as cytokines. Studies using mice deficient in enzymes responsible for the biosynthesis of the CS/DS and HS chains of proteoglycans have demonstrated their essential functions. Chondroitin synthase 1-deficient mice are viable, but exhibit chondrodysplasia, progression of the bifurcation of digits, delayed endochondral ossification, and reduced bone density. DS-epimerase 1-deficient mice show thicker collagen fibrils in the dermis and hypodermis, and spina bifida. These observations suggest that CS/DS are essential for skeletal development as well as the assembly of collagen fibrils in the skin, and that their respective knockout mice can be utilized as models for human genetic disorders with mutations in chondroitin synthase 1 and DS-epimerase 1. This review provides a comprehensive overview of mice deficient in CS/DS biosyntheses.
Introduction
Chondroitin sulfate (CS) and dermatan sulfate (DS) are covalently attached to core proteins to form proteoglycans (PGs). CS-PGs and DS-PGs are ubiquitously distributed in the extracellular matrix as well as on the cell surface (Rodén, 1980; Kjellén and Lindahl, 1991; Iozzo, 1998). Both glycosaminoglycans (GAGs) are linear polysaccharides. CS-PGs is abundantly distributed in cartilage (Rodén, 1980), whereas DS-PGs is predominantly distributed in skin, aorta, and blood vessel (Fransson et al., 1993). The backbone of CS is composed of repeating disaccharide units of D-glucuronic acid (GlcA) and N-acetyl-D-galactosamine (GalNAc) (Figure 1). DS is a stereoisomer of CS and consists of l-iduronic acid (IdoA) instead of GlcA and GalNAc (Figure 1). CS/DS chains are modified by sulfation at various hydroxy groups, which gives rise to structural diversity, thereby playing an important role in a variety of biological processes including interactions with various growth factors, cytokines, and morphogens, cell proliferation, tissue morphogenesis, and infections by viruses (Trowbridge and Gallo, 2002; Sugahara et al., 2003; Sugahara and Mikami, 2007; Malavaki et al., 2008; Yamada and Sugahara, 2008; Malmström et al., 2012; Thelin et al., 2013; Mizumoto et al., 2015; 2013; 2017; Mizumoto and Sugahara, 2013; Schaefer et al., 2017; Kosho et al., 2019). A variety of functions of CS/DS are thought to be dependent on sulfation modification (Sugahara and Mikami, 2007; Mizumoto et al., 2015). A, C, B, D, and E disaccharide units stand for the disaccharide (GlcA-GalNAc) units containing one or two sulfate groups in different combinations (Figure 1). If the GlcA residue has been epimerized to IdoA in each disaccharide unit, “i” is added to the codes, such as iA, iC, iB, iD, and iE (Figure 1). The A, iA, D, and E units are involved in infection of malaria, binding with heparin cofactor II, neurite outgrowth, and infection of herpes simplex virus, respectively (Maimone and Tollefsen 1991; Clement et al., 1998; Buffet et al., 1999; Bergefall et al., 2005). However, the functional domain in CS/DS does not appear to be composed of a single distinct saccharide sequence, but rather several heterogeneous sulfation patterns, the “wobble CS-DS motifs” (Purushothaman et al., 2012).
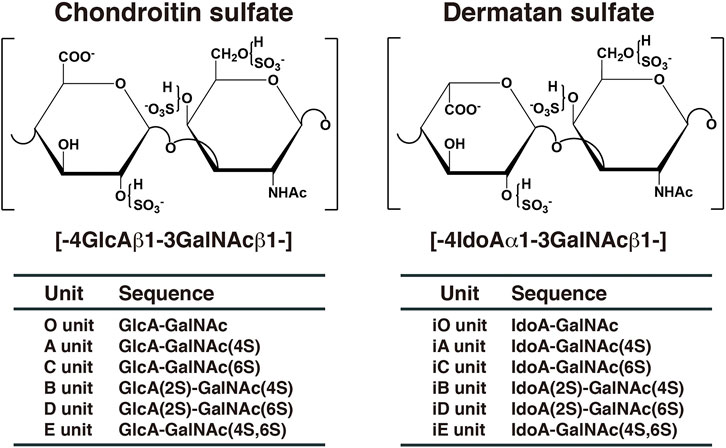
FIGURE 1. Typical repeating disaccharide units in CS and DS. CS consists of GlcA and GalNAc, whereas DS is a stereoisomer of CS including IdoA instead of GlcA. These sugar moieties are esterified by sulfate at various positions, as indicated in the figures.
Various glycosyltransferases, epimerases, sulfotransferases, and related enzymes in the biosynthesis of CS and DS have been identified and characterized (Figures 2, 3) (Kusche-Gullberg and Kjellén, 2003; Mikami and Kitagawa, 2013; Mizumoto, 2018). Moreover, functional analyses of CS and DS using model organisms such as nematodes, fruit flies, zebrafish, and mice have revealed that both are indispensable for normal development (Sugahara and Schwartz, 1979; Bernhardt and Schachner, 2000; Hwang et al., 2003; Mizuguchi et al., 2003; Sugahara et al., 2003; Takemae et al., 2003; Olson et al., 2006; Maccarana et al., 2009; Mizumoto et al., 2009; Li et al., 2010; Tian et al., 2010; Watanabe et al., 2010; Wilson et al., 2012; Takemura et al., 2020). Genetic disorders related to mutations in biosynthetic enzymes for CS/DS-biosynthesis were described in another review article (Mizumoto and Yamada, 2021). This review focuses on recent advances in studies on mice deficient in CS and DS biosynthetic enzymes.
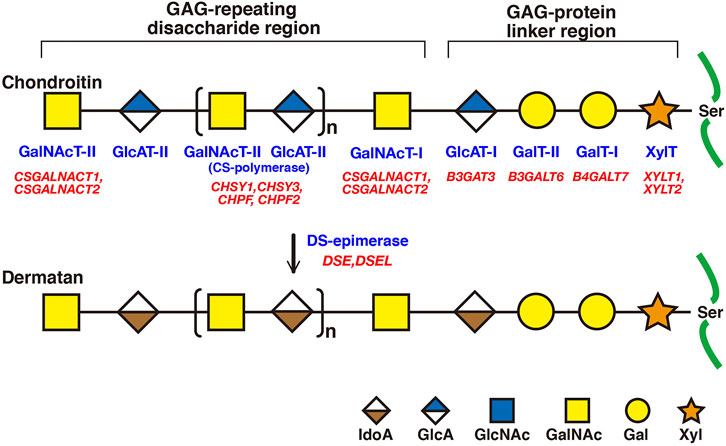
FIGURE 2. Biosynthetic assembly of CS and DS backbones by various glycosyltransferases. Schematic presentation of the biosynthesis of CS and DS backbones. All glycosyltransferases require a corresponding UDP-sugar, such as UDP-Xyl, -Gal, -GlcA, and -GalNAc, as a donor substrate. After specific core proteins have been translated, synthesis of the common GAG-protein linkage region, GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-, is evoked by XylT, which transfers a Xyl residue from UDP-Xyl to the specific serine residue(s) at the GAG attachment sites. The linker region tetrasaccharide is subsequently constructed by GalT-I, GalT-II, and GlcAT-I. The first β1-4-linked GalNAc residue is then transferred to the GlcA residue in the linker region by GalNAcT-I, which initiates the assembly of the chondroitin backbone, thereby resulting in the formation of the repeating disaccharide region, [-4GlcAβ1-3GalNAcβ1-]n, by CS-polymerase. DS-epimerase converts GlcA into IdoA by epimerizing the C-5 carboxy group in the chondroitin precursor, thereby resulting in the formation of the repeating disaccharide region of dermatan precusor, [-4IdoAα1-3GalNAcβ1-]n. Each enzyme and its coding gene are described under the respective sugar symbols.
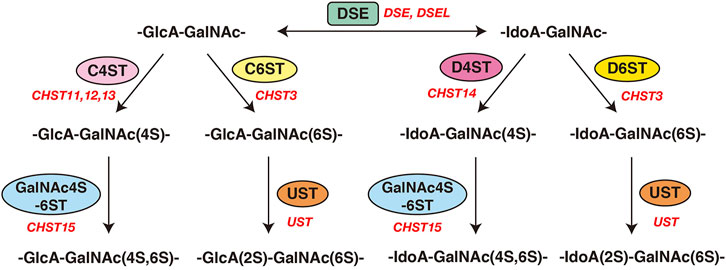
FIGURE 3. Modification of CS and DS by sulfotransferases and epimerases. Modification pathways of CS and DS. After formation of the CS/DS backbones, each sugar residue is modified by sulfation, catalyzed by sulfotransferases, as indicated in the figure. C4ST or C6ST transfers a sulfate group from PAPS to the C-4 or C-6 position of the GalNAc residues in the chondroitin chain, respectively. D4ST transfers a sulfate group from PAPS to the C-4 position of the GalNAc residues in dermatan. Further sulfation reactions are catalyzed by GalNAc4S-6ST or UST, which is required for formation of the disulfated disaccharide units indicated, respectively.
Biosyntheses of CS and DS
Biosyntheses of Donor Substrates for GAGs and Transporters of Uridine 5′-Diphosphate -Sugars, Sulfate Ions, and 3′-Phosphoadenosine 5′-Phosphosulfate
Most glycosyltransferases utilize uridine 5′-diphosphate (UDP)-sugars as the donor substrates, including: UDP-Glc, UDP-GlcNAc, UDP-GlcA, UDP-Gal, UDP-GalNAc, and UDP-Xyl, where Glc, GlcNAc, GlcA, Gal, GalNAc, and Xyl, represent D-glucose, N-acetyl-D-glucosamine, D-glucuronic acid, D-galactose, N-acetyl-D-galactosamine, and D-xylose, respectively. UDP-GlcA is formed by the action of UDP-Glc dehydrogenase on UDP-Glc in the cytosol (Table 1) (Spicer et al., 1998). UDP-Xyl is formed by the action of UDP-GlcA decarboxylase/UDP-xylose synthase in the endoplasmic reticulum and Golgi apparatus (Moriarity et al., 2002). These UDP-sugars mainly synthesized in the cytosol, except for UDP-Xyl, are incorporated into the endoplasmic reticulum and Golgi lumen through the corresponding nucleotide sugar transporters (Berninsone and Hirschberg, 2000; Orellana et al., 2016; Parker and Newstead, 2019).
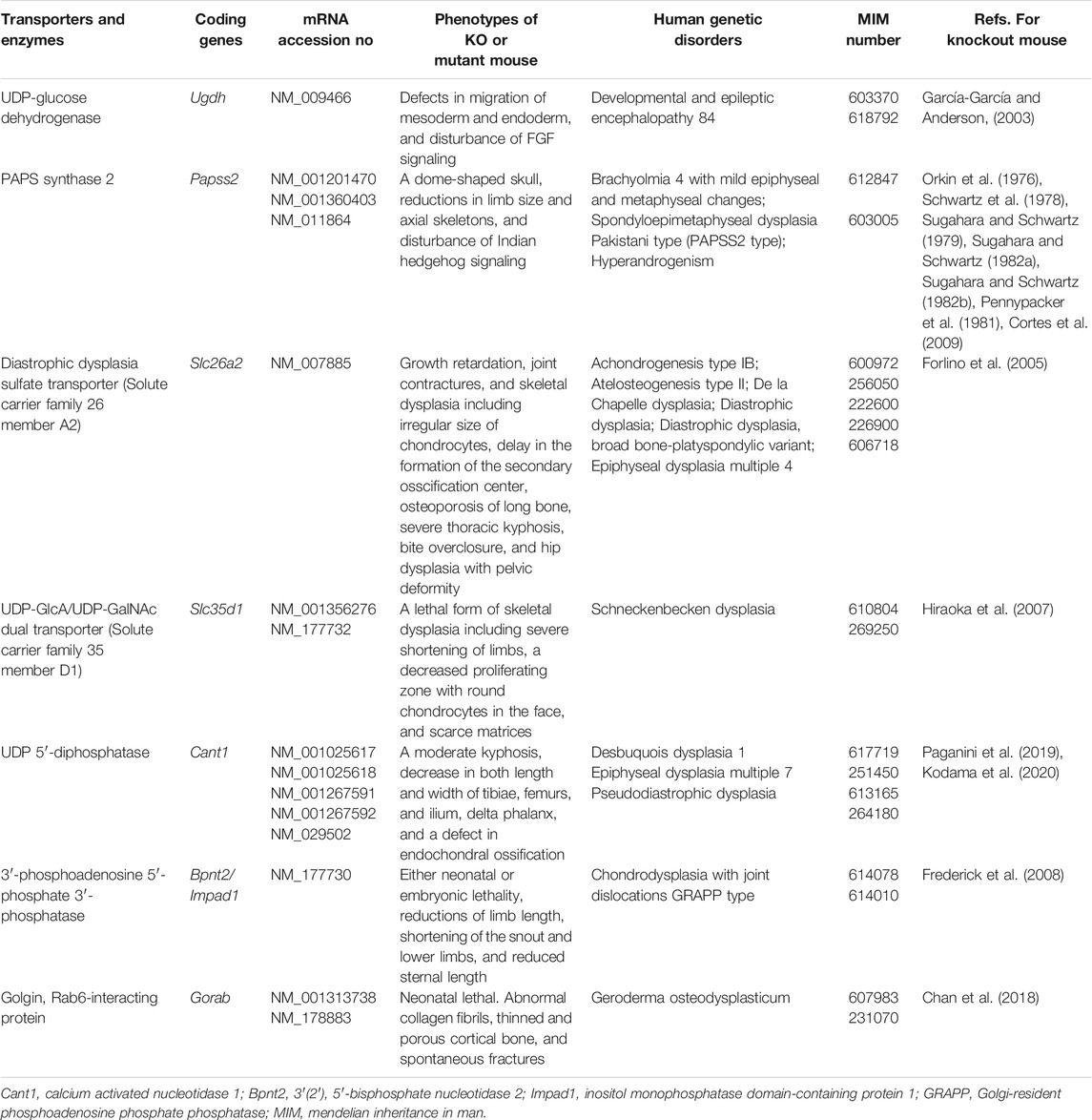
TABLE 1. Transporters for UDP-sugars and sulfate, biosynthetic enzymes for PAPS and UDP-GlcA, and related proteins. Among the several transporters and biosynthetic enzymes involved in PAPS and UDP-sugars, GAG biosynthesis-related genes are listed here.
Various GAG sulfotransferases catalyze the transfer of a sulfate group from 3′-phosphoadenosine 5′-phosphosulfate (PAPS), as a donor substrate, to respective acceptor substrates (Kusche-Gullberg and Kjellén, 2003). PAPS synthase (PAPSS) has two enzymatic domains, adenosine 5′-phosphosulfate kinase and ATP sulfurylase domains, in N- and C-terminals, respectively (Venkatachalam, 2003) (Table 1). PAPS is formed from inorganic sulfate, which is incorporated into the cytosol through the sulfate transporter at the plasma membrane and ATP (Hästbacka et al., 1994).
Backbones of CS and DS
CS and DS polysaccharides are covalently attached to specific serine residues in core proteins through the common GAG-protein linker region tetrasaccharide GlcAβ1-3Galβ1-3Galβ1-4xylose(Xyl)β1-O- (Figure 2) (Lindahl and Rodén, 1972; Kjellén and Lindahl, 1991; Sugahara and Kitagawa 2000). The transfer of a Xyl residue from UDP-Xyl to specific serine residues in the newly synthesized core proteins of PGs in the endoplasmic reticulum and/or cis-Golgi compartments is initiated by β-xylosyltransferase (XylT) encoded by XYLT1 or XYLT2 (Figure 2; Table 2) (Götting et al., 2000; Pönighaus et al., 2007). It should be noted that human genes, which were described by all upper capital, were utilized in this section, because enzymatic activity of glycosyltransferases, epimerase, and sulfotransferases, which are responsible for biosynthesis of CS/DS, had been measured using recombinant human enzymes. β4-Galactosyltransferase-I (GalT-I) encoded by B4GALT7, then transfers a Gal residue from UDP-Gal to Xyl-O-serine in the core proteins (Almeida et al., 1999; Okajima et al., 1999). β3-Galactosyltransferase-II (GalT-II) encoded by B3GALT6 transfers the second Gal residue from UDP-Gal to Gal-Xyl-O-serine (Bai et al., 2001). Thereafter, β3-glucuronyltransferase-I (GlcAT-I) encoded by B3GAT3, transfers a GlcA residue from UDP-GlcA to Gal-Gal-Xyl-O-serine (Figure 2; Table 2) (Kitagawa et al., 1998).
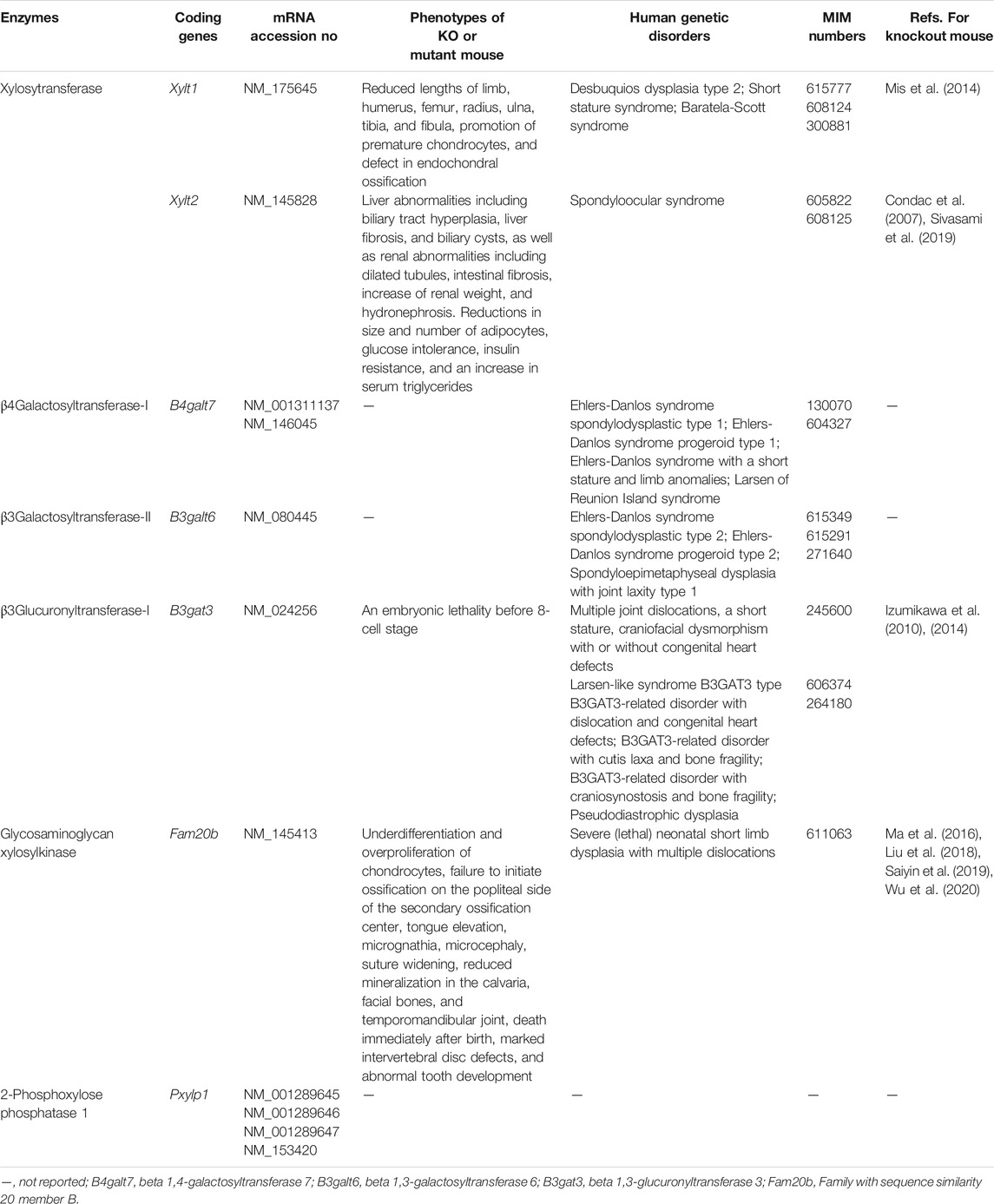
TABLE 2. Biosynthetic enzymes of the GAG-linkage region tetrasaccharide.
Several modifications occur such as 2-O-phosphorylation and 2-O-dephosphorylation of Xyl and Xyl-2-O-phosphate residues by Xyl kinase and Xyl-2-O-phosphate phosphatase encoded by FAM20B and PXYLP1, respectively (Koike et al., 2009; 2014). Furthermore, sulfation at the C6 position of the first Gal and at C4 or C6 of the second Gal residues has been identified (Sugahara and Kitagawa, 2000). Chondroitin 6-O-sulfotransferase 1 (C6ST1) encoded by CHST3 transfers a sulfate group from PAPS to Gal residues on the linker region tetrasaccharide GlcA-Gal-Gal-Xyl in vitro (Kitagawa et al., 2008). These modifications affect the glycosyltransferase reactions of GalT-I, GlcAT-I, CSGALNACT1, and may regulate the formation of CS/DS chains (Gulberti et al., 2005; Tone et al., 2008; Izumikawa et al., 2015).
Initiation of the repeating disaccharide region in the CS chain, [–4GlcAβ1–3GalNAcβ1–]n, is evoked by the transfer of the first GalNAc residue from UDP-GalNAc to the GlcA residue in the linker region tetrasaccharide, GlcA-Gal-Gal-Xyl-O-, by β4-N-acetylgalactosaminyltransferase-I (GalNAcT-I) encoded by CSGALNACT1 or CSGALNACT2 (Figure 2; Table 3) (Uyama et al., 2002; 2003). Chain elongation of CS occurs by the alternative addition of GlcA and GalNAc residues by CS-β3-glucuronyltransferase-II (CS-GlcAT-II) and GalNAcT-II, respectively (Figure 2; Table 3) (Mikami and Kitagawa, 2013). Chondroitin synthase (CHSY) encoded by CHSY1 or CHSY3 has a dual enzymatic activity of both CS-GlcAT-II and GalNAcT-II, which may be exerted in N- and C-terminal domains, respectively (Kitagawa et al., 2001b; Izumikawa et al., 2007). Chondroitin-polymerizing factor (CHPF) encoded by CHPF or CHPF2 is able to construct the repeating disaccharide region of CS by forming an enzyme complex with CHSY (Kitagawa et al., 2003; Izumikawa et al., 2008). CHPF2 has both CS-GlcAT-II and GalNAcT-II activities; thereby, CHPF2 was designated as CHSY (Izumikawa et al., 2008). After or during construction of the non-sulfated disaccharide region of CS, the chondroitin backbone, it is modified by sulfation by the respective sulfotransferase including uronyl 2-O-sulfotransferase (UST) encoded by UST (Kobayashi et al., 1999), chondroitin 4-O-sulfotransferase (C4ST) encoded by CHST11, CHST12, or CHST13 (Hiraoka et al., 2000; Yamauchi et al., 2000; Kang et al., 2002), C6ST encoded by CHST3 (Fukuta et al., 1995; 1998), and GalNAc 4-O-sulfate 6-O-sulfotransferase (GalNAc4S-6ST) encoded by CHST15 (Ohtake et al., 2001) (Figure 3; Table 3).
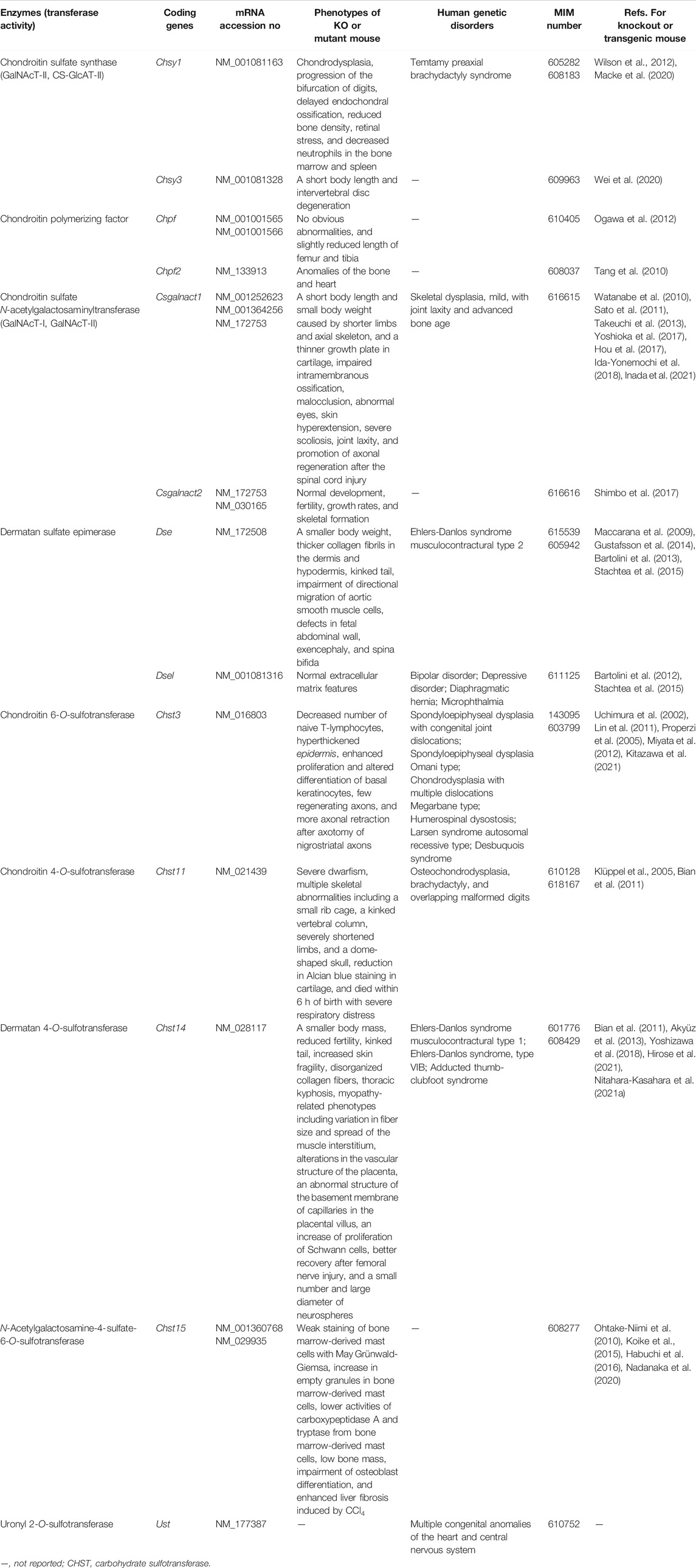
TABLE 3. Biosynthetic enzymes of CS and DS chains.

TABLE 4. Outstanding questions and perspectives for functions of glycosyltransferases, sulfotransferases, and epimerase involving CS/DS-biosynthesis.
Formation of the repeating disaccharide region, [–4IdoAβ1–3GalNAcβ1–]n, of DS chains occurs by epimerization of the C5 position of GlcA residues in a chondroitin precursor backbone, which is catalyzed by DS-epimerase encoded by DSE or DSEL (Figure 2) (Maccarana et al., 2006; Pacheco et al., 2009). The dermatan chains are modified by sulfation catalyzed by UST and dermatan 4-O-sulfotransferase (D4ST) encoded by UST and CHST14, which transfer the sulfate from PAPS to the C2 position of IdoA and C4 position of GalNAc residues, respectively (Kobayashi et al., 1999; Evers et al., 2001; Mikami et al., 2003) (Figure 3; Table 3).
Catabolism of Donor Substrates for CS/DS Biosynthesis
After glycosyltransferase reaction, the reaction product, UDP, derived from UDP-sugar is hydrolyzed into uridine 5′-monophosphate (UMP) by nucleoside 5′-diphosphatase, which is encoded by calcium-activated nucleotidase 1 (CANT1), in the endoplasmic reticulum and Golgi apparatus (Table 1) (Failer et al., 2002; Smith et al., 2002). UMP is exported to the cytosol by nucleotide sugar transporters, which are antiporters for UDP-sugars and UMP, from the Golgi apparatus and/or endoplasmic reticulum (Parker and Newstead, 2019).
After the sulfotransferase reaction, the reaction product, adenosine-3′, 5′-bisphosphate (PAP), derived from PAPS is hydrolyzed into adenosine 5′-phosphate (5′-AMP) by the Golgi-resident PAP 3′-phosphatase, which is encoded by 3′(2′), 5′-bisphosphate nucleotidase 2 (BPNT2)/inositol monophosphatase domain containing 1 (IMPAD1) (Table 1) (Frederick et al., 2008). The 5′-AMP may be exported to the cytosol by unidentified transporters from the Golgi apparatus and/or endoplasmic reticulum.
Knockout and Mutant Mice of Biosynthetic Enzymes of CS/DS and Its Donor Substrates as Well as Nucleotide Sugar Transporters
Ugdh
UDP-Glc dehydrogenase (UGDH) is an oxidoreductase that converts UDP-Glc to UDP-GlcA in the cytosol (Spicer et al., 1998). The mutant mice lazy mesoderm have a mutation in Ugdh, which was introduced by ethyl-nitrosourea, and show a phenotype of embryogenesis arrest during gastrulation with defects in migration of the mesoderm and endoderm (García-García and Anderson, 2003). Furthermore, no CS or heparan sulfate (HS) were detected in the mutant using respective antibodies against them (García-García and Anderson, 2003). HS is also linear polysaccharide of GAG family, and composed of repeating disaccharide unit, [-4GlcAβ1–4GlcNAcα1-]n, which is covalently attached to the specific core proteins, forming PGs (Supplemental Figure S1) (Kjellén and Lindahl, 1991). HS and HS-PGs play essential roles in signal transduction, tissue morphogenesis, early development, and tumor progression (Bishop et al., 2007). The disturbance of FGF signaling has been demonstrated in the Ugdh mutant, resulting in a similar phenotype to those of Fgf8 and Fgfr1 mutants (Yamaguchi et al., 1994; Sun et al., 1999). The interaction of not only HS but also CS with FGFs and their receptors has been shown to be required for signal transduction (Esko and Selleck, 2002; Bishop et al., 2007; Mizumoto et al., 2015). Thus, the phenotype of the Ugdh mutant might be caused by defects in HS and/or CS.
Papss2
PAPS synthase (PAPSS) is a dual enzyme with both adenosine 5′-phosphosulfate kinase and ATP sulfurylase activities, catalyzed by its N- and C-terminal domains, respectively (Fuda et al., 2002; Venkatachalam, 2003). The Papss2 mutant, brachymorphic mouse, which is generated by N-ethyl-N-nitrosourea, and has the substitution Gly79Arg, shows a normal life span, a dome-shaped skull, and reductions in limb as well as axial skeletons, thereby leading to brachymorphism (Schwartz et al., 1978; Sugahara and Schwartz, 1979, 1982a, 1982b; Pennypacker et al., 1981). Moreover, the mutant mice produce lower sulfated CS but not HS in the growth plate cartilage, and show disturbed Indian hedgehog signaling due to abnormal distribution in the extracellular matrix, which results in a reduction in chondrocyte proliferation (Orkin et al., 1976; Cortes et al., 2009). These findings suggest that the sulfation in CS side chains of PG(s), such as aggrecan, modulates Indian hedgehog signaling.
Slc26a2
The sulfate transporter is encoded by SLC26A2, which incorporates a sulfate anion into the cytosol at the plasma membrane (Hästbacka et al., 1994; Satoh et al., 1998; Seidler and Nikolovska, 2019). The incorporated sulfate is activated to adenosine-phosphosulfate and then to PAPS by PAPS synthase (Venkatachalam, 2003). An Slc26a2 knock-in mouse with an Ala386Val substitution in the eighth transmembrane domain of Slc26a2, whose mutation was detected in a patient with diastrophic dysplasia characterized by a short stature, cleft plate, and deformity of the external ear and thumb (Rossi and Superti-Furga, 2001), was characterized by growth retardation, joint contracture, and skeletal dysplasia including an irregular size of chondrocytes, delay in the formation of the secondary osscification center and osteoporosis of long bones, severe thoracic kyphosis, bite overclosure, and hip dysplasia with pelvic deformity (Forlino et al., 2005). Furthermore, the proportion of a non-sulfated disaccharide unit, GlcA-GalNAc, was higher than that of the wild-type in cartilage and bone, but not skin (Forlino et al., 2005). These findings suggest that abnormalities of proliferation and differentiation of chondrocytes contribute to reduced bone growth, and lead to similar phenotypes to probands of human diastrophic dysplasia. Thus, this mutant mouse is a useful model to explore the pathogenic and therapeutic approaches for human diastrophic dysplasia.
Slc35d1
UDP-GlcA/UDP-GalNAc dual transporter encoded by solute carrier family 35 member D1 (SLC35D1) incorporates both UDP-GlcA and UDP-GalNAc from the cytosol into endoplasmic reticulum (Muraoka et al., 2001). The Slc35d1-deficient mouse showed a lethal form of skeletal dysplasia associated with severe shortening of limbs, abnormal facial structures, a decreased proliferating zone with round chondrocytes, scarce matrices, and reduced CS but not HS in long bones (Hiraoka et al., 2007). Furthermore, schneckenbecken dysplasia characterized by perinatally lethal skeletal dysplasia is caused by mutations in SLC35D1 (Hiraoka et al., 2007). These findings indicate that CS chains and/or CS-PGs are indispensable for early embryonic as well as skeletal development, and that the mutant mouse can be utilized to explore the pathogenic and therapeutic approaches for human schneckenbecken dysplasia.
Knockout and Mutant Mice of Biosynthetic Enzymes for CS/DS Backbones
Xylt1 and Xylt2
XYLT1 encoded by XYLT1 transfers Xyl to specific serine residues in core proteins of PGs from UDP-Xyl as a donor substrate in the Golgi apparatus (Figure 2) (Götting et al., 2000; Schön et al., 2006; Pönighaus et al., 2007). The Xylt1 mutant pug, which is generated by N-ethyl-N-nitrosourea, and has the substitution Trp932Arg, showed lower XYLT activity in chondrocytes from the mutant than the wild-types, thereby decreasing the production of GAGs in cartilage (Mis et al., 2014). It should be noted that a defect in XYLT1 may affect the biosyntheses of not only CS/DS but also HS, because the linker region tetrasaccharide GlcA-Gal-Gal-Xyl- is common to CS, DS, and HS (Supplemental Figure S1). Moreover, pug mutants showed phenotypes including reduced limb, humerus, femur, radius, ulna, tibia, and fibula lengths, and the normal proliferation as well as promotion of premature maturation of chondrocytes, which suggests a general defect in endochondral ossification, resulting in dwarfism. These skeletal abnormalities may be caused by an up-regulation of Indian hedgehog signaling but not FGF signaling (Mis et al., 2014). In fact, mutations in human XYLT1 cause Desbuquois dysplasia type 2 characterized by severe pre- and postnatal growth retardation, a short stature, joint laxity, and the dislocation of large joints (Bui et al., 2014). Thus, the pug mutant mouse is available to help understand the pathogenic mechanism and development of treatment for human Desbuquois dysplasia type 2.
XYLT2 encoded by XYLT2 also transfers Xyl to specific serine residues in core proteins of PGs from UDP-Xyl as a donor substrate in the Golgi apparatus (Götting et al., 2000; Schön et al., 2006; Pönighaus et al., 2007). The Xylt2-deficient mouse exhibited liver abnormalities including biliary tract hyperplasia, liver fibrosis, and biliary cysts, as well as renal abnormalities including dilated tubules, intestinal fibrosis, increase of the renal weight, and hydronephrosis (Condac et al., 2007). Furthermore, it was demonstrated that there is an 86% reduction in HS disaccharides from the liver of Xylt2-deficient mice compared with wild-type mice, and a lack of the GAG side chain of decorin, which is a DSPG, in both the liver and kidney of Xylt2-deficient mice. The defect in XYLT2 may affect the biosyntheses of not only CS/DS but also HS, because the linker region tetrasaccharide, GlcA-Gal-Gal-Xyl-, is common to CS, DS, and HS (Supplemental Figure S1). However, normal levels of renal CS as well as HS in Xylt2-deficient mice were detedcted (Condac et al., 2007). These findings suggest that the residual HS observed in liver from Xylt2-deficient mice may be sufficient for hepatocellular differentiation as well as proliferation, but not maturation, and that renal development requires decorin, the DS side chain, or other DSPGs. Homozygous mutations in XYLT2 cause spondyloocular syndrome that is characterized by retinal detachment, amblyopia, nystagmus, hearing loss, heart septal defects, bone fragility, and mild learning difficulties (Munns et al., 2015). However, patients with predicted null mutations in XYLT2 did not show polycystic disease. Hence, XYLT1 may compensate for the loss-of-function mutation of XYLT2 in the human liver as well as kidney.
The Xylt2-deficient mouse also showed reductions in the size and number of adipocytes, glucose intolerance, and insulin resistance, as well as an increase in serum triglycerides as compared with wild-type mice (Sivasami et al., 2019). Moreover, elevations of interleukin-6 and interleukin-1β, which are proinflammatory M1 cytokines, and the upregulation of TGFβ signaling that inhibits adipogenesis in preadipocyte cells, result in the inflammation of adipose tissues. It was demonstrated that adipose-derived stem cells showed impaired adipogenic differentiation in Xylt2-deficient mice, and that maturation of endothelium from gonadal fat tissue was reduced, thereby increasing adipogenic precursors. These findings suggest that the GAG decrease caused by a defect in XYLT2 leads to reduced steady state adipose tissue stores, which is a unique lipodystrophic model.
Fam20b
Xyl 2-O-kinase encoded by FAM20B transfers a phosphate group to the Xyl residue in the linkage region from ATP as a donor substrate in the Golgi apparatus (Koike et al., 2009). Conditional knockout (cKO) of Fam20b (Osr2-Cre;Fam20Bflox/flox) in the joint cartilage, palate mesenchyme, and metanephric mesenchyme-derived glomeruli tissues, showed that chondrocytes overproliferated but underdifferentiated, and failed to initiate ossification on the popliteal side of the secondary ossification center (Ma et al., 2016). Furthermore, the gain-of-functions of bone morphogenetic protein (BMP) as well as WNT, and the down-regulation of Indian hedgehog, which coordinates chondrocyte proliferation and maturation, were detected in the cartilage of Fam20b cKO (Ma et al., 2016). These phenotypes lead to chondrosarcoma in the knee joint and marked defects of postnatal ossification in long bones. However, no significant changes in FGF and TGF-β signaling in Fam20b cKO mice were detected. Taken together, the FAM20B-catalyzed PGs are essential for chondrocyte differentiation and maturation, as well as subsequent ossification.
Wnt1-Cre;Fam20bflox/flox cKO mice, which were deficient in Fam20b in the neural crest and midbrain, died immediately after birth due to complete cleft palates (Liu et al., 2018). Moreover, the Fam20b cKO mice showed tongue elevation, micrognathia, microcephaly, suture widening, and reduced mineralization in the calvaria, facial bones, and temporomandibular joint (Liu et al., 2018). These findings suggest that GAG side chains of PGs formed by catalysis of FAM20B are necessary for the morphogenesis and mineralization of the craniofacial complex.
Col1a1-Cre;Fam20Bflox/flox cKO mice, which were deficient in Fam20b in osteoblasts, showed apparent postnatal growth retardation, a shorter tail and spine, and the spinal curvature, resulting in severe kyphosis (Saiyin et al., 2019). Furthermore, Fam20B cKO mice showed marked intervertebral disc defects associated with malformation of the peripheral annulus fibrosus, which resulted from the fibrosus tissue transforming to cartilage-like tissue. Not only CS but also HS were reduced in the annulus fibrosus from Fam20B cKO mice. TGF-β signaling required for the development and maintenance of the annulus fibrosus and intervertebral disc, was not activated in Fam20B cKO mice. MAPK signaling was also modified in cKO mice, i.e., increases in phospho-P38 and phospho-ERK but decreases in phospho-JNK (Saiyin et al., 2019). These findings indicate that FAM20B-mediated PGs may play an essential role in annulus fibrosus development through regulating TGF-β and MAPK signaling pathways.
K14-Cre;Fam20Bflox/flox cKO mice, which were deficient in Fam20b in the dental epithelium, showed supernumerary tooth formation. Reductions in CS and HS in the dental epithelium attenuated FGFR2b as well as WNT signalings in the initial stage and later cap stage, respectively, of tooth development (Wu et al., 2020). These findings suggest that FAM20B-catalyzed GAG biosynthesis on PGs regulates the number of murine teeth through FGFR2b signaling in the initial stage of tooth development.
B3gat3
GlcAT-I encoded by B3GAT3 transfers the 4th sugar residue in the linker region tetrasaccharide GlcA-Gal-Gal-Xyl from UDP-GlcA to Gal-Gal-Xyl-O-serine (Figure 2) (Kitagawa et al., 1998). The B3gat3-deficient mice showed embryonic lethality before the 8-cell stage due to the failure of cytokinesis (Izumikawa et al., 2010). Moreover, neither CS nor HS was detected in blastocysts from B3gat3-deficient mice (Izumikawa et al., 2010). The defect in B3GAT3 may affect the biosynthesis of not only CS/DS but also HS, because the linker region tetrasaccharide GlcA-Gal-Gal-Xyl- is common to CS, DS, and HS (Supplemental Figure S1). Interestingly, treatment of 2-cell embryos with chondroitinase, which is a bacterial eliminase acting specifically on CS, resulted in embryonic lethality between 2- and 8-cell stages, but treatment with heparitinase, a bacterial eliminase acting specifically on HS, showed no lethality (Izumikawa et al., 2010). Ext1- or Ext2-deficient mice that lack HS developed normally until embryonic day 6.5 (Lin et al., 2000; Stickens et al., 2005). EXT1 and EXT2 have both HS-GlcAT-II and α-1,4N-acetylglucosaminyltransferase-II activities, which are required for biosynthesis of HS chains (Lind et al., 1998; McCormick et al., 1998) (Supplemental Figure S1). Caenorhabditis elegans synthesizes chondroitin, non-sulfated CS, which is required for normal cell division and cytokinesis in an early developmental stage (Mizuguchi et al., 2003; Izumikawa et al., 2004). These findings suggest that abnormal cytokinesis in B3gat3-deficient mice may be attributed to deficiency in CS, but not HS.
Embryonic stem cells derived from B3gat3-deficient mice completely lost both CS and HS, and failed to differentiate into multiple lineages (Izumikawa et al., 2014). Degradation of CS on wild-type embryonic stem cells by treatment with chondroitinase had effects on the formation of embryonic bodies, which is in vitro differentiation by free-floating aggregates of the embryonic stem cells, whereas treatment with heparitinase showed no effects on the development of embryonic bodies. Furthermore, the exogeneous addition of CS-A or CS-E polysaccharides to embryonic bodies derived from B3gat3-deficient mice rescued the differentiation of these cells into primitive endodermal cells in a culture assay (Izumikawa et al., 2014). The interaction of CS with E-cadherin regulates the Rho signaling pathway, which leads to the control of differentiation of embryonic stem cells (Izumikawa et al., 2014). These findings suggest that CS contributes to the integrity of embryonic stem cells via interaction with E-cadherin.
Csgalnact1 and Csgalnact2
N-Acetylgalactosaminyltransferase (GalNAcT) encoded by CSGALNACT1 or CSGALNACT2 transfers a GalNAc residue from UDP-GalNAc to GlcA-Gal-Gal-Xyl-O-serine and [GlcA-GalNAc]n (Figure 2) (Uyama et al., 2002; 2003). Csgalnact1-deficient mice showed a short body length and small body weight, caused by shortening of the limbs and axial skeleton, and a thinner growth plate in cartilage than wild-type mice (Watanabe et al., 2010; Sato et al., 2011). The level of CS disaccharides in the cartilage from the Csgalnact1-deficient mice was reduced to half of that in the wild-type (Watanabe et al., 2010; Sato et al., 2011). These findings indicate that CSGALNACT1 and/or CS-PG is necessary for the differentiation and maturation of cartilage.
Csgalnact1-deficient mice also showed impaired intramembranous ossification, resulting in a shorter face, and higher and broader calvaria (Ida-Yonemochi et al., 2018). Protein levels of Wnt3a and β-catenin were decreased in the mesenchymal tissues of calvaria, and collagen fibers were irregular, thick, and aggregated in the calvaria and scalp from Csgalnact1-deficient mice, which causes skull abnormalities (Ida-Yonemochi et al., 2018). Furthermore, Csgalnact1-deficient mice were characterized by malocclusion, abnormal eyes, skin hyperextension, severe scoliosis, joint laxity, and reduction of CS in skin, muscle, tendon, and bone, which are similar to the hallmarks of Ehlers-Danlos syndrome in humans. Loss of CSGALNACT1 may cause disturbance of DS biosynthesis, because chondroitin is a precursor for DS. Musculocontractural Ehlers-Danlos syndrome is caused by a defect in DS (Malfait et al., 2017; 2020).
Csgalnact1-deficient mice showed better recovery after spinal cord injury than wild-type mice, based on a footfall test, footprint test, and electromyography, because of the promotion of axonal regeneration (Takeuchi et al., 2013). On the other hand, Csgalnact2-deficient mice have not been demonstrated to show such promotional activity. After spinal cord injury, the biosynthesis of CS is promoted and resultant CS inhibits axonal regeneration as a barrier-forming molecule (Carulli et al., 2005). However, the promotion of CS biosynthesis is lower in Csgalnact1-deficient mice than in wild-type mice (Takeuchi et al., 2013). Interestingly, an increase of HS was detected in association with up-regulations of Ext1, Ext2, and Extl3 mRNAs that encode glycosyltransferases responsible for HS biosynthesis (Takeuchi et al., 2013). HS has been reported to promote axonal growth and regulate axon guidance (Yamaguchi, 2001). Thus, the decrease and increase of CS and HS, respectively, in Csgalnact1-deficient mice resulted in better recovery from spinal cord injury than in wild-type mice.
CS-PG is a major component in perineuronal nets, which are unique extracellular matrix structures that wrap around neurons during development and control plasticity in the central nervous system (Sorg et al., 2016). Csgalnact1-deficient mice showed a significant decrease in CS in the cerebrum, diencephalon, spinal cord, and visual cortex (Yoshioka et al., 2017). Furthermore, Csgalnact1-deficient mice showed a significantly greater total distance traveled than wild-type mice in the open field test, which measures voluntary activity in a novel environment. Csgalnact1-deficient mice manifested much larger responses than wild-type mice in an acoustic startle test, which can measure reflex movement in response to a sudden loud sound stimulus (Yoshioka et al., 2017). These findings suggest that CS generated by CSGALNACT1 may affect the formation of perineuronal nets as well as behaviors of mice.
Csgalnact1-deficient mice were characterized by a reduction in CS in the visual cortical area and impaired ocular plasticity, which is caused by a decrease of Otx2 accumulation (Hou et al., 2017). CS binds to Otx2 in perineuronal nets, and promotes uptake of Otx2 into parvalbumin-expressing basket cells, thereby terminating the critical period for plasticity (Miyata and Kitagawa, 2015). These findings indicate that CS and CS-PGs are required for the critical period for plasticity in the visual cortex.
Csgalnact1-deficient mice with experimentally induced autoimmune encephalomyelitis showed milder symptoms including lower cell infiltration, proliferation, and productions of interleukin-6 and interferon-γ than those in the wild-type (Inada et al., 2021). These findings suggest that CS side chains of PGs may be associated with autoimmune encephalomyelitis and potential therapeutic targets for neuroimmunological diseases.
Csgalnact2-deficient mice exhibited normal development, fertility, growth rates, and skeletal formation (Shimbo et al., 2017). These findings suggest that loss of functions of CSGALNACT2 might be compensated for by CSGALNACT1.
Mice with double KO of Csgalnact1 and Csgalnact2 died during the postnatal stage due to respiratory failure (Shimbo et al., 2017). Furthermore, the double KO mice exhibited severer phenotypes including short humeral and tibial lengths compared with Csgalnact1-or Csgalnact2-deficient mice. The total CS disaccharides in rib cartilage from Csgalnact1-KO, Csgalnact2-KO, and double KO mice were reduced to ∼74, ∼99, and ∼40%, compared with that of the wild-type, respectively (Shimbo et al., 2017).
Approximately 80% of Col2a1-Cre; Csgalnact1flox/—; Csgalnact2flox/— double cKO mice, which were deficient in both Csgalnact1 and Csgalnact2 in chondrocytes, immediately died after birth because of respiratory failure, and the remaining ∼20% of the double KO mice could start spontaneous respiration (Shimbo et al., 2017). They were characterized by a lower body weight, severer dwarfism, and lower proliferation of chondrocytes than control mice.
These data indicate that CS synthesized by CSGALNACT1 and CSGALNACT2, may be required for pulmonary and skeletal development during embryogenesis.
Chsy1
GalNAcT-II and glucuronyltransferase-II (GlcAT-II) encoded by CHSY1 transfer GalNAc and GlcA residues from UDP-GalNAc and UDP-GlcA to [GlcA-GalNAc]n or [GalNAc-GlcA]n, respectively (Figure 2) (Kitagawa et al., 2001b). Chsy1-deficient mice were characterized by chondrodysplasia, progression of the bifurcation of digits, delayed endochondral ossification, and reduced bone density (Wilson et al., 2012). Furthermore, a decrease in 4-O-sulfation and increases in 6-O-sulfation as well as non-sulfated GalNAc residues were detected in the cartilage of Chsy1-deficient mice. The up-regulation of transcriptional target of Hedgehog, Gli1, was detected in embryonic fibroblast cultures from Chsy1-deficient mice (Wilson et al., 2012). Moreover, a brachymorphic mouse with mutation in Papss2 also showed low sulfated CS in the cartilage, and its Hedgehog signaling was attenuated (Orkin et al., 1976; Cortes et al., 2009). These findings indicate that CS and Hedgehog protein may coordinately modulate bone development.
Small with kinky tail (skt) mutant mice spontaneously arose at the Jackcon Laboratory with recessive mutation (Lane, 1988). The skt mutant was caused by a 27-kb deletion containing Chsy1 (Macke et al., 2020). The skt mutant mice showed reduced CS in the retina as well as hippocampus compared with heterozygous deficient mice, an increase in a number of empty spaces surrounding cells in the cornu ammonis 1, 2, and 3 hippocampal subfields compared with control mice, decreased neutrophils in bone marrow as well as macrophages in both the bone marrow and spleen, and age-dependent retinal changes including progressive photoreceptor cell degeneration with an increase of glial fibrillary acidic protein, considered to be a sign of retinal stress (Macke et al., 2020). In contrast, frequencies of monocytic cells and lymphocytic cells such as T-cells, B-cells, and natural killer cells, did not appear to be consistently altered in the skt mutant mice compared with heterozygous controls. These findings suggest that CS constructed by CHSY1 regulates the development of the hippocampus, retina, neutrophils, and macrophages.
Chsy3
CHSY3 also has a dual enzymatic activity with β1,3-GlcA transferase and β1,4-GalNAc transferase on its amino- and carboxy-terminal sides, respectively (Yada et al., 2003a; Izumikawa et al., 2007). Chsy3-deficient mice showed a shorter body length than the wild-type after 4 weeks old, a reduction of CS in disc tissues, and intervertebral disc degeneration such as a narrowed disc height, loss of the nucleus pulposus, and unclear demarcation between the nucleus pulposus and annulus fibrosus (Wei et al., 2020). Furthermore, the Hippo signaling pathway, which is regulated by a kinase of the Sterile-20 family and activates the suppressor Warts (Zheng and Pan, 2019), was significantly downregulated. The activation of Yap1, which is a transcriptional coactivator as well as a negative regulator of the Hippo pathway, and is involved in intervertebral disc degeneration (Chen et al., 2019), was mainly affected in nucleus pulposus cells from Chsy3-deficient mice (Wei et al., 2020). These findings suggest that CS activates Yap signaling and spontaneous intervertebral disc degeneration.
Chpf
Chondroitin polymerizing factor encoded by CHPF exhibits an enzymatic activity to polymerize the disaccharide region of CS in concert with CHSY1 in vitro (Kitagawa et al., 2003). Since CHPF has a dual enzymatic activity of β1,3-GlcA transferase and β1,4-GalNAc transferase, it was also designated as CHSY2 (Yada et al., 2003b). Although Chpf-deficient mice showed no obvious abnormalities, the femur and tibia lengths were slightly reduced, and the chain length of CS was shorter in cartilage than in wild-type mice (Ogawa et al., 2012). These findings indicate that other CHSY family proteins, CHPF2, CHSY1, and/or CHSY3, might compensate for the activity of CHPF.
Chpf2
CHPF2 also has a dual enzymatic activity of β1,3-GlcA transferase and β1,4-GalNAc transferase, and has been designated as CHSY3 or CSGlcA-T (Gotoh et al., 2002; Izumikawa et al., 2008). Chpf2-deficient mice have been registered in the knockout mouse library, and their anomalies in the bone and heart were reported without detailed analyses (Tang et al., 2010). Further investigation is required for elucidation of the in vivo function of CHPF2.
Chst3
C6ST1 encoded by carbohydrate sulfotransferase 3 (CHST3) transfers a sulfate group from PAPS to the C-6 hydroxy group of GalNAc residues in the CS repeating disaccharide region, [GlcA-GalNAc]n (Figure 3) (Fukuta et al., 1995, 1998; Uchimura et al., 1998). Chst3-deficient mice showed a loss of 6-O-sulfated disaccharide units such as the C-unit, GlcA–GalNAc(6-O-sulfate), and D-unit, GlcA(2-O-sulfate)–GalNAc(6-O-sulfate), in the spleen, cartilage, and brain (Uchimura et al., 2002), although brain development seems to be normal in Chst3-deficient mice. Chst3 was not expressed in the thymus (Uchimura et al., 1998), where naive T-cells differentiate, and the proportion of CD4+/CD8– and CD4–/CD8+ cells in the thymus from Chst3-deficient mice did not change (Uchimura et al., 2002). However, the number of naive T-lymphocytes decreased (Uchimura et al., 2002). These findings indicate that survival, retention, and/or emigration of naive T lymphocytes was affected in the spleen of the Chst3-deficient mice, rather than that of thymocytes.
After axotomy of nigrostriatal axons, Chst3-deficient mice exhibited fewer regenerating axons and more axonal retraction than wild-type mice (Lin et al., 2011), although repair of the median and ulnar nerves was similar between wild-type and Chst3-deficient mice after peripheral nerve injury. Increases in the expression of Chst3 and proportion of the 6-O-sulfated structure have been demonstrated in glial scars after cortical injury (Properzi et al., 2005). These findings suggest that the suppression of 6-O-sulfation in CS after injury of the central nervous system prevents axons to regenerate.
Chst3-transgenic mice with an increase in 6-O-sulfation of the brain CS showed loss of perineuronal nets in the brain, leading to the continuance of the critical period for cortical plasticity (Miyata et al., 2012). Furthermore, Otx2, which is a homeoprotein and regulates ocular dominance plasticity via its effects on maturation of parvalbumin-expressing interneurons (Sugiyama et al., 2008), diffused and reduced at the surrounding parvalbumin-expressing interneurons in Chst3-transgenic mice (Miyata et al., 2012). These findings indicate that 6-O-sulfation of CS at perineuronal nets in the brain regulates the critical period for cortical plasticity by maturation of parvalbumin-expressing interneurons.
Chst3-deficient mice presented with a hyperthickened epidermis, enhanced proliferation, and altered differentiation of basal keratinocytes, thereby impairing the epidermal permeability barrier function (Kitazawa et al., 2021). Furthermore, the 6-O-sulfated CS directly binds to epidermal growth factor receptor (EGFR), leading to the blockade of EGFR signaling (Kitazawa et al., 2021). The Chst3-deficient mice had a thicker epidermis and increased levels of acute inflammation including erythema, scaling, and skin induration, compared with wild-type mice when psoriasis was induced by imiquimod (Kitazawa et al., 2021). These findings indicate that the 6-O-sulfated CS repress proliferation of keratinocytes and progression of psoriasis in the skin.
Chst11
C4ST1 encoded by carbohydrate sulfotransferase 11 (CHST11) transfers a sulfate group from PAPS to the C-4 hydroxy group of GalNAc residues in the CS repeating disaccharide region, [GlcA-GalNAc]n (Figure 3) (Hiraoka et al., 2000; Yamauchi et al., 2000). Chst11-deficient mice showed a more than 90% reduction of the 4-O-sulfated disaccharide unit in the growth plate compared with the wild-type (Klüppel et al., 2005). Furthermore, they exhibited severe dwarfism, multiple skeletal abnormalities including a small rib cage, a kinked vertebral column, severely shortened limbs, a dome-shaped skull, reduction in Alcian blue staining in cartilage, and fatality within 6 h of birth with severe respiratory distress (Klüppel et al., 2005). In the Chst11-deficient embryos, chondrocyte differentiation was affected during morphogenesis of the cartilage growth plate because of upregulation of TGFβ signaling with concomitant downregulation of BMP signaling, but not Indian hedgehog signaling (Klüppel et al., 2005), although mesenchymal aggregation and cartilage primordium formation were normal. These findings suggest that CS 4-O-sulfation and C4ST1 are required for embryonic development and morphogenesis of the cartilage growth plate by modulation of signaling pathways.
Chst15
GalNAc4S-6ST encoded by carbohydrate sulfotransferase 15 (CHST15) transfers a sulfate group from PAPS to the C-6 hydroxy group of GalNAc4-O-sulfate residues in the CS repeating disaccharide region, [GlcA-GalNAc(4-O-sulfate)]n (Figure 3) (Ohtake et al., 2001). Chst15-deficient mice showed complete loss of GalNAc 4- and 6-O-disulfated structure (E-unit) in CS/DS from the tissues examined, including the cerebrum, cerebellum, heart, lung, liver, spleen, kidney, thymus, stomach, small intestine, large intestine, mesentery, testis, whole embryo, and bone marrow-derived mast cells, suggesting that GalNAc4S-6ST encoded by Chst15 is the sole enzyme responsible for the biosynthesis of GalNAc 4- and 6-O-disulfated structure (Ohtake-Niimi et al., 2010). Furthermore, Chst15-deficient mice were fertile, showed normal development, exhibited weak staining of bone marrow-derived mast cells with May Grünwald-Giemsa, showed an increase of empty granules in bone marrow-derived mast cells, and presented lower activities of carboxypeptidase A as well as tryptase from bone marrow-derived mast cells (Ohtake-Niimi et al., 2010). These findings suggest that GalNAc 4- and 6-O-disulfated structure in CS/DS-PGs may be involved in the storage of these proteases in the granules of mast cells.
Chst15-deficient mice also exhibited impairment of osteoblast differentiation leading to be low bone mass (Koike et al., 2015). Liver fibrosis induced by CCl4 was enhanced in these mice (Habuchi et al., 2016). These findings indicate that GalNAc4S-6ST and/or E-disaccharide unit-containing CS, [GlcA-GalNAc(4-, 6-O-disulfates)], may be a therapeutic target for osteopenia, osteoporosis, and fibrosis. However, GalNAc 4- and 6-O-disulfated structure was not necessary for binding with semaphoring 3A in the perineuronal nets of brain (Nadanaka et al., 2020).
Dse and Dsel
DS-epimerase encoded by DSE or DSEL converts GlcA into IdoA by C5-epimerization of GlcA residues in the CS repeating disaccharide region, [GlcA-GalNAc]n (Figure 2) (Maccarana et al., 2006; Pacheco et al., 2009). Dse-deficient mice exhibited a smaller body weight, reductions in IdoA-containing structures in the skin, thicker collagen fibrils in the dermis and hypodermis, kinked tails, impairment of directional migration of aortic smooth muscle cells, and defects in the fetal abdominal wall, exencephaly, and spina bifida (Maccarana et al., 2009; Bartolini et al., 2013; Gustafsson et al., 2014). Dse and/or DS may be indispensable for normal development and formation of collagen fibrils.
Dsel-deficient mice had no anatomical, histological, or morphological abnormalities (Bartolini et al., 2012). Furthermore, Dsel-deficient mice exhibited reduced epimerase activity in the skin (24% reduction), lung (34%), liver (38%), spleen (44%), kidney (55%), and brain (89%) compared with those in the wild-type mouse tissues (Bartolini et al., 2012). Consistent with this result, IdoA contents of CS/DS chains from the neonatal brain and kidney were reduced to 87 and 62% of wild-type mice, respectively (Bartolini et al., 2012). Brain from Dsel-deficient mice showed normal extracellular matrix features by immunohistological staining. DSE may compensate for the function of DSEL.
Double knockout mice of Dse and Dsel exhibited perinatal lethality with an umbilical hernia, exencephaly, a kinked tail, and complete loss of DS, suggesting that DS plays an important role in embryonic development as well as perinatal survival (Stachtea et al., 2015).
Chst14
D4ST1 encoded by carbohydrate sulfotransferase 14 (CHST14) transfers a sulfate group from PAPS to the C-4 hydroxy group of GalNAc residues in the repeating disaccharide region of DS, [IdoA-GalNAc]n (Figure 3) (Evers et al., 2001; Mikami et al., 2003). Chst14-deficient mice showed a smaller body mass, reduced fertility, kinked tail, and increased skin fragility compared with wild-type littermates (Akyüz et al., 2013). Moreover, in Chst14-deficient mouse skin, the amount of DS was markedly decreased with elevation of the level of CS, which is a precursor chain of DS. These phenotypes of Chst14-deficient mice were considerably similar to those of Dse-deficient mice (Maccarana et al., 2006). In addition to both enzymes involving the biosynthesis of DS, it has been reported that 4-O-sulfated GalNAc residues in DS chains prevent back-epimerization by DSE in vitro (Malmström, 1984). Furthermore, DSE and CHST14 forms heterocomplex, but not DSEL, which is necessary to build longer IdoA-containing chains (Tykesson et al., 2018). Therefore, the cooperation of both enzymes by heterocomplex is required for the formation of repeating disaccharide, [GalNAc(4S)–IdoA], in DS.
Its skin tensile strength was significantly decreased compared with that in wild-type mice, and the collagen fibrils were oriented in various directions to form disorganized collagen fibers in the reticular layer (Hirose et al., 2021). Rod-shaped linear GAG chains were found to be attached at one end to collagen fibrils and protruded outside of the fibrils in the Chst14-deficient mice, in contrast to those being round and wrapping the collagen fibrils in wild-type mice (Hirose et al., 2021). These findings suggest that the DS side chain of decorin is necessary for assembly of decorin-PG with collagen, and maintenance of the skin strength.
CRISPR/Cas9-genome engineered Chst14-deficient mice exhibited common growth impairment and skin fragility similar to the conventional knockout mice of Chst14 (Nitahara-Kasahara et al., 2021a). In addition, CRISPR/Cas9-genome engineered Chst14-deficient mice showed decreased DS in muscle, thoracic kyphosis, and myopathy-related phenotypes including variation in fiber size and spread of the muscle interstitium, as well as diffuse localization of decorin in the spread endomysium of skeletal muscle, which caused the lower grip strength and decreased exercise capacity, compared with wild-type and heterozygous mutant mice (Nitahara-Kasahara et al., 2021a; 2021b). The CRISPR/Cas9-engineered Chst14-mutant mouse is a useful model for musculocontractural Ehlers-Danlos syndrome caused by mutations in CHST14 (Dündar et al., 2009; Malfait et al., 2010; Miyake et al., 2010; Voermans et al., 2012; Kosho et al., 2019; Malfait et al., 2020).
Chst14-deficient mice are sometimes perinatally lethal (Yoshizawa et al., 2018). Their placenta showed immaturity such as a reduced weight of the placenta, alteration in the vascular structure with ischemic and/or necrotic-like change, an abnormal structure of the basement membrane of capillaries in the placental villus, and significantly decreased DS (Yoshizawa et al., 2018). These findings suggest that DS may be essential for placental vascular development.
Cultured Schwann cells from dorsal roots and nerves, cerebellar neurons, and motoneurons of Chst14-deficient mice exhibited longer cell processes compared with those from wild-type cells (Akyüz et al., 2013). Schwann cells from Chst14-deficient mice had a higher proliferation rate. Moreover, the values for the foot-base and heel-tail angles in Chst14-deficient mice showed better recovery than those in wild-type mice at each time-point between 1 and 12 weeks after femoral nerve injury (Akyüz et al., 2013). These findings indicate that Chst14 partially controls inhibitory functions during neural development and recovery from nerve injury.
Neurospheres from Chst14-deficient, but not Chst11-deficient mice exhibited fewer numbers and larger diameters than those from wild-types (Bian et al., 2011). This was caused by impairments of self-renewal and proliferation, but neither apotosis nor migration, of neural stem cells in vitro as well as in vivo (Bian et al., 2011). The expression level of GLAST but not Nestin, which are markers of radial glial cells and neurons, respectively, was increased in neurospheres from Chst14-deficient mice. These findings suggest that DS-PGs play important roles in the proliferation and differentiation of neural stem cells.
Knockout and Mutant Mice of Catabolism of the Reaction Products of Donor Substrates, UDP and PAP
Cant1
Most glycosyltransferases utilize UDP-sugar as a donor substrate, which is converted to UDP after the reaction in the endoplasmic reticulum or Golgi apparatus. The UDP is hydrolyzed to UMP by 5′-diphosphatase encoded by CANT1 (Failer et al., 2002; Smith et al., 2002). Cant1-deficient mice exhibited moderate kyphosis, a decrease in both the length and width of tibiae, femurs, and ilium, delta phalanx, a defect in endochondral ossification, and reduction in GAGs in chondrocytes (Paganini et al., 2019). Furthermore, the phenotypes of the Cant1-knockout mouse were similar to those of a Cant1 knock-in mouse with an Arg302His substitution in the catalytic domain (Huber et al., 2009), which corresponds to the human mutation in patients with Desbuquois dysplasia characterized by a short stature, round face, progressive scoliosis, and joint laxity (Paganini et al., 2019). Cant1-deficient mice generated by the CRISPR/Cas9 system also exhibited a lower body weight, short stature, thoracic kyphosis, delta phalanx, reduction in GAG content in growth plate cartilage, and impairment of differentiation of chondrocytes (Kodama et al., 2020).
These findings suggest that CANT1 and/or hydrolysis of UDP to UMP may be necessary for the metabolism of GAGs and that it affects the maturation of chondrocytes in the cartilage growth plate. Accumulation of UDP may inhibit the activity of glycosyltransferases involved in the biosynthesis of GAGs. The lack of UMP may inhibit the incorporation of UDP-sugars from the cytosol into the endoplasmic reticulum and Golgi apparatus through antiporters, nucleotide sugar transporters. Further biochemical analyses of the cellular pathways will be crucial in order to elucidate the molecular basis of CANT1 deficiency as well as Desbuquois dysplasia.
Bpnt2
Most sulfotransferases utilize PAPS as a donor substrate, which is converted to PAP after the reaction in the cytosol as well as Golgi apparatus. PAP is hydrolyzed to 5′-AMP by PAP 3′-phosphatase encoded by BPNT1 and BPNT2/IMPAD1 in the cytosol and Golgi apparatus, respectively (Frederick et al., 2008; Hudson et al., 2013). The gene trap Bpnt2-deficient mice are neonatally or embryonically lethal, and showed reduction of the limb length, shortening of the snout and lower limbs, reduced sternal length, and diminished rib spacing (Frederick et al., 2008). Furthermore, a marked decrease in chondroitin 4-O-sulfate and an increase in non-sulfated chondroitin were detected in the cartilage, lung, and embryos of Bpnt2-deficient mice. Although significant changes in the amount and sulfation modification of HS were not observed in the embryos from mutant mice, the degree of sulfation of HS was slightly decreased in the lung (Frederick et al., 2008). These findings indicate that BPNT2 and/or hydrolysis of PAP to 5′-AMP may be necessary for the metabolism of sulfation of GAGs and that it affects skeletal development. The accumulation of PAP may inhibit sulfotransferases involved in the biosynthesis of GAGs. The lack of 5′-AMP may inhibit the incorporation of PAPS from the cytosol into Golgi apparatus through an unidentified antiporter(s).
Knockout Mice of Golgins
Gorab
Golgins comprise a family of vesicle-tethering proteins at the Golgi apparatus (Witkos and Lowe, 2017; Lowe, 2019). The vesicle-bound cargo tethers to the Golgi apparatus, which triggers membrane fusion. Various golgins are localized to distinct regions of the Golgi apparatus, and their ability to tether transported vesicles selectively is necessary for the specificity of vesicle traffic in the secretory pathway. Because the biosynthesis of GAG side chains on PGs is achieved in the endoplasmic reticulum and Golgi apparatus, some golgins are most likely involved in the transport of PGs.
GORAB encodes a Rab6-interacting Golgi protein, and its mutations cause human genetic disorder, gerodermia osteodysplastica, which is characterized by skin laxity and early-onset osteoporosis (Hennies et al., 2008). Mutant mice of Gorab have been generated, with fully and conditionally inactivated mesenchymal progenitor cells (Prx1-cre), pre-osteoblasts (Runx2-cre), and late osteoblasts/osteocytes (Dmp1-cre), respectively (Chan et al., 2018). The Gorab full-knockout mice (GorabNull) were neonatal lethal, and showed disorganized collagen fibrils (Chan et al., 2018). The Gorab conditional-knockout mice, GorabPrx1 and GorabRunx2, exhibited thinned, porous cortical bone and spontaneous fractures (Chan et al., 2018), which were also observed in a patient with gerodermia osteodysplastica (Hennies et al., 2008). Furthermore, the level of DS, but not CS or HS, was decreased in skin and cartilage from GorabNull mutants. The glycanation of DS-proteoglycans, biglycan and decorin, in skin and bone may be reduced (Chan et al., 2018). The Golgi apparatus compartment of cultured fibroblasts from GorabNull mutants showed the accumulation of decorin core protein, but a reduced level of DS, indicating that the newly synthesized decorin core protein accumulates within the Golgi apparatus due to the impairment of DS biosynthesis. However, it remains unclear whether there are anomalies in the transport of decorin core protein or DS-biosynthetic enzymes including DSE as well as D4ST1 to the Golgi apparatus. Taken together, these findings suggest that mutation and/or deficiency of Gorab primarily perturbs pre-osteoblasts, and that gerodermia osteodysplastica might be affected by biosynthesis of the DS side chain in proteoglycans and/or transport of decorin core protein in the Golgi compartment.
Conclusions and Perspectives
Mice deficient in glycosyltransferases or sulfotransferases involved in the biosynthesis of CS/DS demonstrated abnormalities of bone, skin, and nervous systems. These knockout mice with deficiency of Chst11, Chst3, and Chst15 have revealed that A, C, and E units in CS chains play essential roles in chondrocyte differentiation, T-cell differentiation, and storage of proteases in mast cells, respectively. Furthermore, Chst14-knockout mice revealed that DS-containing iA unit, but not CS-containing A unit, bundles collagen fibrils in skin, which might be dependent on the structural and conformational alteration of CS and DS chains (Casu et al., 1988; Hirose et al., 2021). These findings indicate that specific sulfation modifications as well as conformation of uronic acid in CS/DS are essential for connective tissue and neuronal development.
Recent advances in studies on human genetic disorders in connective tissues have also clarified the biological significance of CS/DS side chains of PGs (Mizumoto et al., 2013, 2017; Mizumoto, 2018; Kosho et al., 2019; Malfait et al., 2020). The clinical halmarks in human diseases caused by deficiency in the biosynthetic enzymes of CS/DS are not always consistent with the phenotypes of knockout mice with deficiency of the corresponding enzymes. This contradiction may be due to residual enzymatic activity in human patients. However, the phenotypes of some null-mutant mice are consistent with human clinical symptoms in patients with mutations in the corresponding gene. Further studies on molecular pathogeneses involving CS and DS chains of PGs are necessary to develop therapeutics and new drugs against these diseases (Table 4).
The biosynthesis of CS/DS-PGs is up-regulated in both tumor stroma and neoplastic cells, resulting in the abundant accumulation of these components in the tumor stroma adjacent to neoplastic cells (Fukatsu et al., 1988; Iozzo et al., 1989; ten Dam et al., 2009; Thelin et al., 2012). Consistent with these observations, up-regulations of gene expressions including glycosyltransferases, epimerases, and sulfotransferases responsible for the biosynthesis of CS/DS (Huang et al., 2021). These findings indicate that CS/DS-PGs contribute to the functions and phenotypes of tumor cells as effectors or modulator macromolecules (ten Dam et al., 2007; Bi et al., 2008; Li et al., 2008; Sugahara et al., 2008; Mizumoto et al., 2012). However, there is little or no report regarding tumor biology of CS/DS using the knockout mice. Further studies on the molecular mechanisms underlying pathological conditions involving CS/DS-PGs using the knockout mice will provide insights into new therapeutic approaches for tumor development (Table 4).
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported in part by a Grant-in Aid for Scientific Research (C) from the Japan Society for the Promotion of Science, Japan (19K07054 to SM; 21K06552 to SY). Grant-in Aid for Research Center for Pathogenesis of Intractable Diseases from the Research Institute of Meijo University (SM and SY).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Medical English Service for editing the English text of the draft of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.764781/full#supplementary-material
References
Akyüz, N., Rost, S., Mehanna, A., Bian, S., Loers, G., Oezen, I., et al. (2013). Dermatan 4-O-Sulfotransferase1 Ablation Accelerates Peripheral Nerve Regeneration. Exp. Neurol. 247, 517–530. doi:10.1016/j.expneurol.2013.01.025
Almeida, R., Levery, S. B., Mandel, U., Kresse, H., Schwientek, T., Bennett, E. P., et al. (1999). Cloning and Expression of a Proteoglycan UDP-Galactose:β-Xylose β1,4-Galactosyltransferase I. J. Biol. Chem. 274, 26165–26171. doi:10.1074/jbc.274.37.26165
Bai, X., Zhou, D., Brown, J. R., Crawford, B. E., Hennet, T., and Esko, J. D. (2001). Biosynthesis of the Linkage Region of Glycosaminoglycans. J. Biol. Chem. 276, 48189–48195. doi:10.1074/jbc.M107339200
Bartolini, B., Thelin, M. A., Rauch, U., Feinstein, R., Oldberg, A., Malmström, A., et al. (2012). Mouse Development Is Not Obviously Affected by the Absence of Dermatan Sulfate Epimerase 2 in Spite of a Modified Brain Dermatan Sulfate Composition. Glycobiology 22, 1007–1016. doi:10.1093/glycob/cws065
Bartolini, B., Thelin, M. A., Svensson, L., Ghiselli, G., van Kuppevelt, T. H., Malmström, A., et al. (2013). Iduronic Acid in Chondroitin/dermatan Sulfate Affects Directional Migration of Aortic Smooth Muscle Cells. PLoS One 8, e66704. doi:10.1371/journal.pone.0066704
Bergefall, K., Trybala, E., Johansson, M., Uyama, T., Naito, S., Yamada, S., et al. (2005). Chondroitin Sulfate Characterized by the E-Disaccharide Unit Is a Potent Inhibitor of Herpes Simplex Virus Infectivity and Provides the Virus Binding Sites on gro2C Cells. J. Biol. Chem. 280, 32193–32199. doi:10.1074/jbc.M503645200
Bernhardt, R. R., and Schachner, M. (2000). Chondroitin Sulfates Affect the Formation of the Segmental Motor Nerves in Zebrafish Embryos. Dev. Biol. 221, 206–219. doi:10.1006/dbio.2000.9673
Berninsone, P. M., and Hirschberg, C. B. (2000). Nucleotide Sugar Transporters of the Golgi Apparatus. Curr. Opin. Struct. Biol. 10, 542–547. doi:10.1016/s0959-440x(00)00128-7
Bi, X., Tong, C., Dockendorff, A., Bancroft, L., Gallagher, L., Guzman, G., et al. (2008). Genetic Deficiency of Decorin Causes Intestinal Tumor Formation through Disruption of Intestinal Cell Maturation. Carcinogenesis 29, 1435–1440. doi:10.1093/carcin/bgn141
Bian, S., Akyüz, N., Bernreuther, C., Loers, G., Laczynska, E., Jakovcevski, I., et al. (2011). Dermatan Sulfotransferase Chst14/D4st1, but Not Chondroitin Sulfotransferase Chst11/C4st1, Regulates Proliferation and Neurogenesis of Neural Progenitor Cells. J. Cell Sci. 124, 4051–4063. doi:10.1242/jcs.088120
Bishop, J. R., Schuksz, M., and Esko, J. D. (2007). Heparan Sulphate Proteoglycans fine-tune Mammalian Physiology. Nature 446, 1030–1037. doi:10.1038/nature05817
Buffet, P. A., Gamain, B., Scheidig, C., Baruch, D., Smith, J. D., Hernandez-Rivas, R., et al. (1999). Plasmodium Falciparum Domain Mediating Adhesion to Chondroitin Sulfate A: a Receptor for Human Placental Infection. Proc. Natl. Acad. Sci. 96, 12743–12748. doi:10.1073/pnas.96.22.12743
Bui, C., Huber, C., Tuysuz, B., Alanay, Y., Bole-Feysot, C., Leroy, J. G., et al. (2014). XYLT1 Mutations in Desbuquois Dysplasia Type 2. Am. J. Hum. Genet. 94, 405–414. doi:10.1016/j.ajhg.2014.01.020
Carulli, D., Laabs, T., Geller, H. M., and Fawcett, J. W. (2005). Chondroitin Sulfate Proteoglycans in Neural Development and Regeneration. Curr. Opin. Neurobiol. 15, 116–120. doi:10.1016/j.conb.2005.01.014
Casu, B., Petitou, M., Provasoli, M., and Sinaÿ, P. (1988). Conformational Flexibility: a New Concept for Explaining Binding and Biological Properties of Iduronic Acid-Containing Glycosaminoglycans. Trends Biochem. Sci. 13, 221–225. doi:10.1016/0968-0004(88)90088-6
Chan, W. L., Steiner, M., Witkos, T., Egerer, J., Busse, B., Mizumoto, S., et al. (2018). Impaired Proteoglycan Glycosylation, Elevated TGF-β Signaling, and Abnormal Osteoblast Differentiation as the Basis for Bone Fragility in a Mouse Model for Gerodermia Osteodysplastica. Plos Genet. 14, e1007242. doi:10.1371/journal.pgen.1007242
Chen, J., Mei, Z., Huang, B., Zhang, X., Liu, J., Shan, Z., et al. (2019). IL‐6/YAP1/β‐catenin Signaling Is Involved in Intervertebral Disc Degeneration. J. Cell. Physiol. 234, 5964–5971. doi:10.1002/jcp.27065
Clement, A. M., Nadanaka, S., Masayama, K., Mandl, C., Sugahara, K., and Faissner, A. (1998). The DSD-1 Carbohydrate Epitope Depends on Sulfation, Correlates with Chondroitin Sulfate D Motifs, and Is Sufficient to Promote Neurite Outgrowth. J. Biol. Chem. 273, 28444–28453. doi:10.1074/jbc.273.43.28444
Condac, E., Silasi-Mansat, R., Kosanke, S., Schoeb, T., Towner, R., Lupu, F., et al. (2007). Polycystic Disease Caused by Deficiency in Xylosyltransferase 2, an Initiating Enzyme of Glycosaminoglycan Biosynthesis. Proc. Natl. Acad. Sci. 104, 9416–9421. doi:10.1073/pnas.0700908104
Cortes, M., Baria, A. T., and Schwartz, N. B. (2009). Sulfation of Chondroitin Sulfate Proteoglycans Is Necessary for Proper Indian Hedgehog Signaling in the Developing Growth Plate. Development 136, 1697–1706. doi:10.1242/dev.030742
Dündar, M., Müller, T., Zhang, Q., Pan, J., Steinmann, B., Vodopiutz, J., et al. (2009). Loss of Dermatan-4-Sulfotransferase 1 Function Results in Adducted Thumb-Clubfoot Syndrome. Am. J. Hum. Genet. 85, 873–882. doi:10.1016/j.ajhg.2009.11.010
Esko, J. D., and Selleck, S. B. (2002). Order Out of Chaos: Assembly of Ligand Binding Sites in Heparan Sulfate. Annu. Rev. Biochem. 71, 435–471. doi:10.1146/annurev.biochem.71.110601.135458
Evers, M. R., Xia, G., Kang, H.-G., Schachner, M., and Baenziger, J. U. (2001). Molecular Cloning and Characterization of a Dermatan-specific N-Acetylgalactosamine 4-O-Sulfotransferase. J. Biol. Chem. 276, 36344–36353. doi:10.1074/jbc.M105848200
Failer, B. U., Braun, N., and Zimmermann, H. (2002). Cloning, Expression, and Functional Characterization of a Ca2+-dependent Endoplasmic Reticulum Nucleoside Diphosphatase. J. Biol. Chem. 277, 36978–36986. doi:10.1074/jbc.M201656200
Ferreira, C. R., Xia, Z.-J., Clément, A., Parry, D. A., Davids, M., Taylan, F., et al. Undiagnosed Diseases Network, Scottish Genome Partnership (2018). A Recurrent De Novo Heterozygous COG4 Substitution Leads to Saul-Wilson Syndrome, Disrupted Vesicular Trafficking, and Altered Proteoglycan Glycosylation. Am. J. Hum. Genet. 103, 553–567. doi:10.1016/j.ajhg.2018.09.003
Forlino, A., Piazza, R., Tiveron, C., Della Torre, S., Tatangelo, L., Bonafè, L., et al. (2005). A Diastrophic Dysplasia Sulfate Transporter (SLC26A2) Mutant Mouse: Morphological and Biochemical Characterization of the Resulting Chondrodysplasia Phenotype. Hum. Mol. Genet. 14, 859–871. doi:10.1093/hmg/ddi079
Fransson, L-. A., Cheng, F., Yoshida, K., Heinegård, D., Malmström, A., and Schmidtchen, A. (1993). “Patterns of Epimerization and Sulphation in Dermatan Sulphate Chains,” in Dermatan Sulphate Proteoglycans: Chemistry, Biology, Chemical Pathology. Editor J. E. Scott (London: Portland Press), 11–25.
Frederick, J. P., Tafari, A. T., Wu, S.-M., Megosh, L. C., Chiou, S.-T., Irving, R. P., et al. (2008). A Role for a Lithium-Inhibited Golgi Nucleotidase in Skeletal Development and Sulfation. Proc. Natl. Acad. Sci. 105, 11605–11612. doi:10.1073/pnas.0801182105
Fuda, H., Shimizu, C., Lee, Y. C., Akita, H., and Strott, C. A. (2002). Characterization and Expression of Human Bifunctional 3′-phosphoadenosine 5′-phosphosulphate Synthase Isoforms. Biochem. J. 365, 497–504. doi:10.1042/BJ20020044
Fukatsu, T., Sobue, M., Nagasaka, T., Ohiwa, N., Fukata, S., Nakashima, N., et al. (1988). Immunohistochemical Localization of Chondroitin Sulphate and Dermatan Sulphate Proteoglycans in Tumour Tissues. Br. J. Cancer 57, 74–78. doi:10.1038/bjc.1988.12
Fukuta, M., Kobayashi, Y., Uchimura, K., Kimata, K., and Habuchi, O. (1998). Molecular Cloning and Expression of Human Chondroitin 6-sulfotransferase. Biochim. Biophys. Acta (Bba) - Gene Struct. Expr. 1399, 57–61. doi:10.1016/s0167-4781(98)00089-x
Fukuta, M., Uchimura, K., Nakashima, K., Kato, M., Kimata, K., Shinomura, T., et al. (1995). Molecular Cloning and Expression of Chick Chondrocyte Chondroitin 6-sulfotransferase. J. Biol. Chem. 270, 18575–18580. doi:10.1074/jbc.270.31.18575
García-García, M. J., and Anderson, K. V. (2003). Essential Role of Glycosaminoglycans in Fgf Signaling during Mouse Gastrulation. Cell 114, 727–737. doi:10.1016/s0092-8674(03)00715-3
Gotoh, M., Yada, T., Sato, T., Akashima, T., Iwasaki, H., Mochizuki, H., et al. (2002). Molecular Cloning and Characterization of a Novel Chondroitin Sulfate Glucuronyltransferase that Transfers Glucuronic Acid toN-Acetylgalactosamine. J. Biol. Chem. 277, 38179–38188. doi:10.1074/jbc.M202601200
Götting, C., Kuhn, J., Zahn, R., Brinkmann, T., and Kleesiek, K. (2000). Molecular Cloning and Expression of Human UDP-D-Xylose:Proteoglycan Core Protein β-d-Xylosyltransferase and its First Isoform XT-II. J. Mol. Biol. 304, 517–528. doi:10.1006/jmbi.2000.4261
Gulberti, S., Lattard, V., Fondeur, M., Jacquinet, J.-C., Mulliert, G., Netter, P., et al. (2005). Phosphorylation and Sulfation of Oligosaccharide Substrates Critically Influence the Activity of Human β1,4-Galactosyltransferase 7 (GalT-I) and β1,3-Glucuronosyltransferase I (GlcAT-I) Involved in the Biosynthesis of the Glycosaminoglycan-Protein Linkage Region of Proteoglycans. J. Biol. Chem. 280, 1417–1425. doi:10.1074/jbc.M411552200
Gustafsson, R., Stachtea, X., Maccarana, M., Grottling, E., Eklund, E., Malmström, A., et al. (2014). Dermatan Sulfate Epimerase 1 Deficient Mice as a Model for Human Abdominal wall Defects. Birth Defects Res. A: Clin. Mol. Teratology 100, 712–720. doi:10.1002/bdra.23300
Habuchi, H., Ushida, T., and Habuchi, O. (2016). Mice Deficient in N-Acetylgalactosamine 4-sulfate 6-O-Sulfotransferase Exhibit Enhanced Liver Fibrosis and Delayed Recovery from Fibrosis in Carbon Tetrachloride-Treated Mice. Heliyon 2, e00138. doi:10.1016/j.heliyon.2016.e00138
Hästbacka, J., de la Chapelle, A., Mahtani, M. M., Clines, G., Reeve-Daly, M. P., Daly, M., et al. (1994). The Diastrophic Dysplasia Gene Encodes a Novel Sulfate Transporter: Positional Cloning by fine-structure Linkage Disequilibrium Mapping. Cell 78, 1073–1087. doi:10.1016/0092-8674(94)90281-x
Hennies, H. C., Kornak, U., Zhang, H., Egerer, J., Zhang, X., Seifert, W., et al. (2008). Gerodermia Osteodysplastica Is Caused by Mutations in SCYL1BP1, a Rab-6 Interacting Golgin. Nat. Genet. 40, 1410–1412. doi:10.1038/ng.252
Hiraoka, N., Nakagawa, H., Ong, E., Akama, T. O., Fukuda, M. N., and Fukuda, M. (2000). Molecular Cloning and Expression of Two Distinct Human Chondroitin 4-O-Sulfotransferases that Belong to the HNK-1 Sulfotransferase Gene Family. J. Biol. Chem. 275, 20188–20196. doi:10.1074/jbc.M002443200
Hiraoka, S., Furuichi, T., Nishimura, G., Shibata, S., Yanagishita, M., Rimoin, D. L., et al. (2007). Nucleotide-sugar Transporter SLC35D1 Is Critical to Chondroitin Sulfate Synthesis in Cartilage and Skeletal Development in Mouse and Human. Nat. Med. 13, 1363–1367. doi:10.1038/nm1655
Hirose, T., Mizumoto, S., Hashimoto, A., Takahashi, Y., Yoshizawa, T., Nitahara-Kasahara, Y., et al. (2021). Systematic Investigation of the Skin in Chst14−/− Mice: A Model for Skin Fragility in Musculocontractural Ehlers-Danlos Syndrome Caused by CHST14 Variants (mcEDS-CHST14). Glycobiology 31, 137–150. doi:10.1093/glycob/cwaa058
Hou, X., Yoshioka, N., Tsukano, H., Sakai, A., Miyata, S., Watanabe, Y., et al. (2017). Chondroitin Sulfate Is Required for Onset and Offset of Critical Period Plasticity in Visual Cortex. Sci. Rep. 7, 12646. doi:10.1038/s41598-017-04007-x
Huang, Y.-F., Mizumoto, S., and Fujita, M. (2021). Novel Insight into Glycosaminoglycan Biosynthesis Based on Gene Expression Profiles. Front. Cel Dev. Biol. 9, 709018. doi:10.3389/fcell.2021.709018
Huber, C., Oulès, B., Bertoli, M., Chami, M., Fradin, M., Alanay, Y., et al. (2009). Identification of CANT1 Mutations in Desbuquois Dysplasia. Am. J. Hum. Genet. 85, 706–710. doi:10.1016/j.ajhg.2009.10.001
Hudson, B. H., Frederick, J. P., Drake, L. Y., Megosh, L. C., Irving, R. P., and York, J. D. (2013). Role for Cytoplasmic Nucleotide Hydrolysis in Hepatic Function and Protein Synthesis. Proc. Natl. Acad. Sci. 110, 5040–5045. doi:10.1073/pnas.1205001110
Hwang, H.-Y., Olson, S. K., Esko, J. D., and Robert Horvitz, H. (2003). Caenorhabditis elegans Early Embryogenesis and Vulval Morphogenesis Require Chondroitin Biosynthesis. Nature 423, 439–443. doi:10.1038/nature01634
Ida-Yonemochi, H., Morita, W., Sugiura, N., Kawakami, R., Morioka, Y., Takeuchi, Y., et al. (2018). Craniofacial Abnormality with Skeletal Dysplasia in Mice Lacking Chondroitin Sulfate N-Acetylgalactosaminyltransferase-1. Sci. Rep. 8, 17134. doi:10.1038/s41598-018-35412-5
Inada, R., Miyamoto, K., Tanaka, N., Moriguchi, K., Kadomatsu, K., Takeuchi, K., et al. (2021). Chondroitin Sulfate N-Acetylgalactosyltransferase-1 Knockout Shows Milder Phenotype in Experimental Autoimmune Encephalomyelitis Than in Wild Type. Glycobiology 31, 260–265. doi:10.1093/glycob/cwaa072
Iozzo, R. V. (1998). Matrix Proteoglycans: from Molecular Design to Cellular Function. Annu. Rev. Biochem. 67, 609–652. doi:10.1146/annurev.biochem.67.1.609
Iozzo, R. V., Sampson, P. M., and Schmitt, G. K. (1989). Neoplastic Modulation of Extracellular Matrix: Stimulation of Chondroitin Sulfate Proteoglycan and Hyaluronic Acid Synthesis in Co-cultures of Human colon Carcinoma and Smooth Muscle Cells. J. Cell. Biochem. 39, 355–378. doi:10.1002/jcb.240390403
Izumikawa, T., Kanagawa, N., Watamoto, Y., Okada, M., Saeki, M., Sakano, M., et al. (2010). Impairment of Embryonic Cell Division and Glycosaminoglycan Biosynthesis in Glucuronyltransferase-I-Deficient Mice. J. Biol. Chem. 285, 12190–12196. doi:10.1074/jbc.M110.100941
Izumikawa, T., and Kitagawa, H. (2015). Amino Acid Sequence Surrounding the Chondroitin Sulfate Attachment Site of Thrombomodulin Regulates Chondroitin Polymerization. Biochem. Biophysical Res. Commun. 460, 233–237. doi:10.1016/j.bbrc.2015.03.016
Izumikawa, T., Kitagawa, H., Mizuguchi, S., Nomura, K. H., Nomura, K., Tamura, J.-i., et al. (2004). Nematode Chondroitin Polymerizing Factor Showing Cell-/organ-specific Expression Is Indispensable for Chondroitin Synthesis and Embryonic Cell Division. J. Biol. Chem. 279, 53755–53761. doi:10.1074/jbc.M409615200
Izumikawa, T., Koike, T., Shiozawa, S., Sugahara, K., Tamura, J.-i., and Kitagawa, H. (2008). Identification of Chondroitin Sulfate Glucuronyltransferase as Chondroitin Synthase-3 Involved in Chondroitin Polymerization. J. Biol. Chem. 283, 11396–11406. doi:10.1074/jbc.M707549200
Izumikawa, T., Sato, B., and Kitagawa, H. (2014). Chondroitin Sulfate Is Indispensable for Pluripotency and Differentiation of Mouse Embryonic Stem Cells. Sci. Rep. 4, 3701. doi:10.1038/srep03701
Izumikawa, T., Sato, B., Mikami, T., Tamura, J.-i., Igarashi, M., and Kitagawa, H. (2015). GlcUAβ1-3Galβ1-3Galβ1-4Xyl(2-O-phosphate) Is the Preferred Substrate for Chondroitin N-Acetylgalactosaminyltransferase-1. J. Biol. Chem. 290, 5438–5448. doi:10.1074/jbc.M114.603266
Izumikawa, T., Uyama, T., Okuura, Y., Sugahara, K., and Kitagawa, H. (2007). Involvement of Chondroitin Sulfate Synthase-3 (Chondroitin Synthase-2) in Chondroitin Polymerization through its Interaction with Chondroitin Synthase-1 or Chondroitin-Polymerizing Factor. Biochem. J. 403, 545–552. doi:10.1042/BJ20061876
Kang, H.-G., Evers, M. R., Xia, G., Baenziger, J. U., and Schachner, M. (2002). Molecular Cloning and Characterization of Chondroitin-4-O-Sulfotransferase-3. J. Biol. Chem. 277, 34766–34772. doi:10.1074/jbc.M204907200
Kitagawa, H., Izumikawa, T., Uyama, T., and Sugahara, K. (2003). Molecular Cloning of a Chondroitin Polymerizing Factor that Cooperates with Chondroitin Synthase for Chondroitin Polymerization. J. Biol. Chem. 278, 23666–23671. doi:10.1074/jbc.M302493200
Kitagawa, H., Taoka, M., Tone, Y., and Sugahara, K. (2001a). Human Glycosaminoglycan Glucuronyltransferase I Gene and a Related Processed Pseudogene: Genomic Structure, Chromosomal Mapping and Characterization. Biochem. J. 358, 539–546. doi:10.1042/0264-6021:358053910.1042/bj3580539
Kitagawa, H., Tone, Y., Tamura, J.-i., Neumann, K. W., Ogawa, T., Oka, S., et al. (1998). Molecular Cloning and Expression of Glucuronyltransferase I Involved in the Biosynthesis of the Glycosaminoglycan-Protein Linkage Region of Proteoglycans. J. Biol. Chem. 273, 6615–6618. doi:10.1074/jbc.273.12.6615
Kitagawa, H., Tsutsumi, K., Ikegami-Kuzuhara, A., Nadanaka, S., Goto, F., Ogawa, T., et al. (2008). Sulfation of the Galactose Residues in the Glycosaminoglycan-Protein Linkage Region by Recombinant Human Chondroitin 6-O-Sulfotransferase-1. J. Biol. Chem. 283, 27438–27443. doi:10.1074/jbc.M803279200
Kitagawa, H., Uyama, T., and Sugahara, K. (2001b). Molecular Cloning and Expression of a Human Chondroitin Synthase. J. Biol. Chem. 276, 38721–38726. doi:10.1074/jbc.M106871200
Kitazawa, K., Nadanaka, S., Kadomatsu, K., and Kitagawa, H. (2021). Chondroitin 6-sulfate Represses Keratinocyte Proliferation in Mouse Skin, Which Is Associated with Psoriasis. Commun. Biol. 4, 114. doi:10.1038/s42003-020-01618-5
Kjellén, L., and Lindahl, U. (1991). Proteoglycans: Structures and Interactions. Annu. Rev. Biochem. 60, 443–475. doi:10.1146/annurev.bi.60.070191.002303
Klüppel, M., Wight, T. N., Chan, C., Hinek, A., and Wrana, J. L. (2005). Maintenance of Chondroitin Sulfation Balance by Chondroitin-4-Sulfotransferase 1 Is Required for Chondrocyte Development and Growth Factor Signaling during Cartilage Morphogenesis. Development 132, 3989–4003. doi:10.1242/dev.01948
Kobayashi, M., Sugumaran, G., Liu, J., Shworak, N. W., Silbert, J. E., and Rosenberg, R. D. (1999). Molecular Cloning and Characterization of a Human Uronyl 2-sulfotransferase that Sulfates Iduronyl and Glucuronyl Residues in Dermatan/chondroitin Sulfate. J. Biol. Chem. 274, 10474–10480. doi:10.1074/jbc.274.15.10474
Kodama, K., Takahashi, H., Oiji, N., Nakano, K., Okamura, T., Niimi, K., et al. (2020). CANT1 Deficiency in a Mouse Model of Desbuquois Dysplasia Impairs Glycosaminoglycan Synthesis and Chondrocyte Differentiation in Growth Plate Cartilage. FEBS Open Bio 10, 1096–1103. doi:10.1002/2211-5463.12859
Koike, T., Izumikawa, T., Sato, B., and Kitagawa, H. (2014). Identification of Phosphatase that Dephosphorylates Xylose in the Glycosaminoglycan-Protein Linkage Region of Proteoglycans. J. Biol. Chem. 289, 6695–6708. doi:10.1074/jbc.M113.520536
Koike, T., Izumikawa, T., Tamura, J.-I., and Kitagawa, H. (2009). FAM20B Is a Kinase that Phosphorylates Xylose in the Glycosaminoglycan-Protein Linkage Region. Biochem. J. 421, 157–162. doi:10.1042/BJ20090474
Koike, T., Mikami, T., Shida, M., Habuchi, O., and Kitagawa, H. (2015). Chondroitin Sulfate-E Mediates Estrogen-Induced Osteoanabolism. Sci. Rep. 5, 8994. doi:10.1038/srep08994
Kosho, T., Mizumoto, S., Watanabe, T., Yoshizawa, T., Miyake, N., and Yamada, S. (2019). Recent Advances in the Pathophysiology of Musculocontractural Ehlers-Danlos Syndrome. Genes 11, 43. doi:10.3390/genes11010043
Kusche-Gullberg, M., and Kjellén, L. (2003). Sulfotransferases in Glycosaminoglycan Biosynthesis. Curr. Opin. Struct. Biol. 13, 605–611. doi:10.1016/j.sbi.2003.08.002
Li, F., ten Dam, G. B., Murugan, S., Yamada, S., Hashiguchi, T., Mizumoto, S., et al. (2008). Involvement of Highly Sulfated Chondroitin Sulfate in the Metastasis of the Lewis Lung Carcinoma Cells. J. Biol. Chem. 283, 34294–34304. doi:10.1074/jbc.M806015200
Li, Y., Laue, K., Temtamy, S., Aglan, M., Kotan, L. D., Yigit, G., et al. (2010). Temtamy Preaxial Brachydactyly Syndrome Is Caused by Loss-Of-Function Mutations in Chondroitin Synthase 1, a Potential Target of BMP Signaling. Am. J. Hum. Genet. 87, 757–767. doi:10.1016/j.ajhg.2010.10.003
Lin, R., Rosahl, T. W., Whiting, P. J., Fawcett, J. W., and Kwok, J. C. F. (2011). 6-Sulphated Chondroitins Have a Positive Influence on Axonal Regeneration. PLoS One 6, e21499. doi:10.1371/journal.pone.0021499
Lin, X., Wei, G., Shi, Z., Dryer, L., Esko, J. D., Wells, D. E., et al. (2000). Disruption of Gastrulation and Heparan Sulfate Biosynthesis in EXT1-Deficient Mice. Dev. Biol. 224, 299–311. doi:10.1006/dbio.2000.9798
Lind, T., Tufaro, F., McCormick, C., Lindahl, U., and Lidholt, K. (1998). The Putative Tumor Suppressors EXT1 and EXT2 Are Glycosyltransferases Required for the Biosynthesis of Heparan Sulfate. J. Biol. Chem. 273, 26265–26268. doi:10.1074/jbc.273.41.26265
Lindahl, U., and Rodén, L. (1972). in Carbohydrate-protein Linkages in Proteoglycans of Animal, Plant and Bacterial Origin in Glycoproteins: Their Composition, Structure And Function. Editor A. Gottschalk (Amsterdam: Elsevier), 491–517.
Liu, X., Li, N., Zhang, H., Liu, J., Zhou, N., Ran, C., et al. (2018). Inactivation of Fam20b in the Neural Crest-Derived Mesenchyme of Mouse Causes Multiple Craniofacial Defects. Eur. J. Oral Sci. 126, 433–436. doi:10.1111/eos.12563
Lowe, M. (2019). The Physiological Functions of the Golgin Vesicle Tethering Proteins. Front. Cell Dev. Biol. 7, 94. doi:10.3389/fcell.2019.00094
Ma, P., Yan, W., Tian, Y., Wang, J., Feng, J. Q., Qin, C., et al. (20162016). Inactivation of Fam20b in Joint Cartilage Leads to Chondrosarcoma and Postnatal Ossification Defects. Sci. Rep. 6, 29814. doi:10.1038/srep29814
Maccarana, M., Kalamajski, S., Kongsgaard, M., Magnusson, S. P., Oldberg, A., and Malmström, A. (2009). Dermatan Sulfate Epimerase 1-deficient Mice Have Reduced Content and Changed Distribution of Iduronic Acids in Dermatan Sulfate and an Altered Collagen Structure in Skin. Mol. Cell. Biol. 29, 5517–5528. doi:10.1128/MCB.00430-09
Maccarana, M., Olander, B., Malmström, J., Tiedemann, K., Aebersold, R., Lindahl, U., et al. (2006). Biosynthesis of Dermatan Sulfate. J. Biol. Chem. 281, 11560–11568. doi:10.1074/jbc.M513373200
Macke, E. L., Henningsen, E., Jessen, E., Zumwalde, N. A., Landowski, M., Western, D. E., et al. (2020). Loss of Chondroitin Sulfate Modification Causes Inflammation and Neurodegeneration in Skt Mice. Genetics 214, 121–134. doi:10.1534/genetics.119.302834
Maimone, M. M., and Tollefsen, D. M. (1991). Structure of a Dermatan Sulfate Hexasaccharide that Binds to Heparin Cofactor II with High Affinity. J. Biol. Chem. 265, 18263–18271. doi:10.1016/s0021-9258(18)98760-0
Malavaki, C., Mizumoto, S., Karamanos, N., and Sugahara, K. (2008). Recent Advances in the Structural Study of Functional Chondroitin Sulfate and Dermatan Sulfate in Health and Disease. Connect. Tissue Res. 49, 133–139. doi:10.1080/03008200802148546
Malfait, F., Castori, M., Francomano, C. A., Giunta, C., Kosho, T., and Byers, P. H. (2020). The Ehlers-Danlos Syndromes. Nat. Rev. Dis. Primers 6, 64. doi:10.1038/s41572-020-0194-9
Malfait, F., Francomano, C., Byers, P., Belmont, J., Berglund, B., Black, J., et al. (2017). The 2017 International Classification of the Ehlers-Danlos Syndromes. Am. J. Med. Genet. 175, 8–26. doi:10.1002/ajmg.c.31552
Malfait, F., Syx, D., Vlummens, P., Symoens, S., Nampoothiri, S., Hermanns-Lê, T., et al. (2010). Musculocontractural Ehlers-Danlos Syndrome (Former EDS Type VIB) and Adducted Thumb Clubfoot Syndrome (ATCS) Represent a Single Clinical Entity Caused by Mutations in the Dermatan-4-Sulfotransferase 1 Encoding CHST14 Gene. Hum. Mutat. 31, 1233–1239. doi:10.1002/humu.21355
Malmström, A., Bartolini, B., Thelin, M. A., Pacheco, B., and Maccarana, M. (2012). Iduronic Acid in Chondroitin/Dermatan Sulfate. J. Histochem. Cytochem. 60, 916–925. doi:10.1369/0022155412459857
Malmström, A. (1984). Biosynthesis of Dermatan Sulfate. II. Substrate Specificity of the C-5 Uronosyl Epimerase. J. Biol. Chem. 259, 161–165. doi:10.1016/s0021-9258(17)43635-0
McCormick, C., Leduc, Y., Martindale, D., Mattison, K., Esford, L., Dyer, A., et al. (1998). The Putative Tumour Suppressor EXT1 Alters the Expression of Cell-Surface Heparan Sulfate. Nat. Genet. 19, 158–161. doi:10.1038/514
Mikami, T., and Kitagawa, H. (2013). Biosynthesis and Function of Chondroitin Sulfate. Biochim. Biophys. Acta (Bba) - Gen. Subjects 1830, 4719–4733. doi:10.1016/j.bbagen.2013.06.006
Mikami, T., Mizumoto, S., Kago, N., Kitagawa, H., and Sugahara, K. (2003). Specificities of Three Distinct Human Chondroitin/Dermatan N-Acetylgalactosamine 4-O-Sulfotransferases Demonstrated Using Partially Desulfated Dermatan Sulfate as an Acceptor. J. Biol. Chem. 278, 36115–36127. doi:10.1074/jbc.M306044200
Mis, E. K., Liem, K. F., Kong, Y., Schwartz, N. B., Domowicz, M., and Weatherbee, S. D. (2014). Forward Genetics Defines Xylt1 as a Key, Conserved Regulator of Early Chondrocyte Maturation and Skeletal Length. Dev. Biol. 385, 67–82. doi:10.1016/j.ydbio.2013.10.014
Miyake, N., Kosho, T., Mizumoto, S., Furuichi, T., Hatamochi, A., Nagashima, Y., et al. (2010). Loss-of-function Mutations of CHST14 in a New Type of Ehlers-Danlos Syndrome. Hum. Mutat. 31, 966–974. doi:10.1002/humu.21300
Miyata, S., and Kitagawa, H. (2015). Mechanisms for Modulation of Neural Plasticity and Axon Regeneration by Chondroitin Sulphate. J. Biochem. 157, 13–22. doi:10.1093/jb/mvu067
Miyata, S., Komatsu, Y., Yoshimura, Y., Taya, C., and Kitagawa, H. (2012). Persistent Cortical Plasticity by Upregulation of Chondroitin 6-sulfation. Nat. Neurosci. 15, 414–422. doi:10.1038/nn.3023
Mizuguchi, S., Uyama, T., Kitagawa, H., Nomura, K. H., Dejima, K., Gengyo-Ando, K., et al. (2003). Chondroitin Proteoglycans Are Involved in Cell Division of Caenorhabditis elegans. Nature 423, 443–448. doi:10.1038/nature01635
Mizumoto, S. (2018). Defects in Biosynthesis of Glycosaminoglycans Cause Hereditary Bone, Skin, Heart, Immune, and Neurological Disorders. Trends Glycosci. Glycotechnol. 30, E67–E89. doi:10.4052/tigg.1812.2e
Mizumoto, S., Ikegawa, S., and Sugahara, K. (2013). Human Genetic Disorders Caused by Mutations in Genes Encoding Biosynthetic Enzymes for Sulfated Glycosaminoglycans. J. Biol. Chem. 288, 10953–10961. doi:10.1074/jbc.R112.437038
Mizumoto, S., Kosho, T., Yamada, S., and Sugahara, K. (2017). Pathophysiological Significance of Dermatan Sulfate Proteoglycans Revealed by Human Genetic Disorders. Pharmaceuticals 10, 34. doi:10.3390/ph10020034
Mizumoto, S., Mikami, T., Yasunaga, D., Kobayashi, N., Yamauchi, H., Miyake, A., et al. (2009). Chondroitin 4-O-Sulfotransferase-1 Is Required for Somitic Muscle Development and Motor Axon Guidance in Zebrafish. Biochem. J. 419, 387–399. doi:10.1042/BJ20081639
Mizumoto, S., and Sugahara, K. (2013). Glycosaminoglycans Are Functional Ligands for Receptor for Advanced Glycation End-Products in Tumors. FEBS J. 280, 2462–2470. doi:10.1111/febs.12156
Mizumoto, S., Takahashi, J., and Sugahara, K. (2012). Receptor for Advanced Glycation End Products (RAGE) Functions as Receptor for Specific Sulfated Glycosaminoglycans, and Anti-RAGE Antibody or Sulfated Glycosaminoglycans Delivered In Vivo Inhibit Pulmonary Metastasis of Tumor Cells. J. Biol. Chem. 287, 18985–18994. doi:10.1074/jbc.M111.313437
Mizumoto, S., and Yamada, S. (2021). Congenital Disorders of Deficiency in Glycosaminoglycan Biosynthesis. Front. Genet. 12, 717535. doi:10.3389/fgene.2021.717535
Mizumoto, S., Yamada, S., and Sugahara, K. (2015). Molecular Interactions between Chondroitin-Dermatan Sulfate and Growth Factors/receptors/matrix Proteins. Curr. Opin. Struct. Biol. 34, 35–42. doi:10.1016/j.sbi.2015.06.004
Moriarity, J. L., Hurt, K. J., Resnick, A. C., Storm, P. B., Laroy, W., Schnaar, R. L., et al. (2002). UDP-glucuronate Decarboxylase, a Key Enzyme in Proteoglycan Synthesis. J. Biol. Chem. 277, 16968–16975. doi:10.1074/jbc.M109316200
Munns, C. F., Fahiminiya, S., Poudel, N., Munteanu, M. C., Majewski, J., Sillence, D. O., et al. (2015). Homozygosity for Frameshift Mutations in XYLT2 Result in a Spondylo-Ocular Syndrome with Bone Fragility, Cataracts, and Hearing Defects. Am. J. Hum. Genet. 96, 971–978. doi:10.1016/j.ajhg.2015.04.017
Muraoka, M., Kawakita, M., and Ishida, N. (2001). Molecular Characterization of Human UDP-Glucuronic acid/UDP-N-Acetylgalactosamine Transporter, a Novel Nucleotide Sugar Transporter with Dual Substrate Specificity. FEBS Lett. 495, 87–93. doi:10.1016/s0014-5793(01)02358-4
Nadanaka, S., Miyata, S., Yaqiang, B., Tamura, J.-i., Habuchi, O., and Kitagawa, H. (2020). Reconsideration of the semaphorin-3A Binding Motif Found in Chondroitin Sulfate Using Galnac4s-6st-Knockout Mice. Biomolecules 10, 1499. doi:10.3390/biom10111499
Nitahara-Kasahara, Y., Mizumoto, S., Inoue, Y. U., Saka, S., Posadas-Herrera, G., Nakamura-Takahashi, A., et al. (2021a). Muscle Pathophysiology in Mouse Models of Musculocontractural Ehlers-Danlos Syndrome Due to CHST14 Mutations (mcEDS-CHST14), Generated through CRISPR/Cas9-mediated Genomic Editing. Dis. Model. Mech. In press.
Nitahara-Kasahara, Y., Posadas-Herrera, G., Mizumoto, S., Nakamura-Takahashi, A., Inoue, Y. U., Inoue, T., et al. (2021b). Myopathy Associated with Dermatan Sulfate-Deficient Decorin and Myostatin in Musculocontractural Ehlers-Danlos Syndrome: a Mouse Model Investigation. Front. Cell Dev. Biol. 9, 695021. doi:10.3389/fcell.2021.695021
Ogawa, H., Hatano, S., Sugiura, N., Nagai, N., Sato, T., Shimizu, K., et al. (2012). Chondroitin Sulfate Synthase-2 Is Necessary for Chain Extension of Chondroitin Sulfate but Not Critical for Skeletal Development. PLoS One 7, e43806. doi:10.1371/journal.pone.0043806
Ohtake, S., Ito, Y., Fukuta, M., and Habuchi, O. (2001). Human N-Acetylgalactosamine 4-sulfate 6-O-Sulfotransferase cDNA Is Related to Human B Cell Recombination Activating Gene-Associated Gene. J. Biol. Chem. 276, 43894–43900. doi:10.1074/jbc.M104922200
Ohtake-Niimi, S., Kondo, S., Ito, T., Kakehi, S., Ohta, T., Habuchi, H., et al. (2010). Mice Deficient in N-Acetylgalactosamine 4-sulfate 6-O-Sulfotransferase Are Unable to Synthesize Chondroitin/dermatan Sulfate Containing N-Acetylgalactosamine 4,6-bissulfate Residues and Exhibit Decreased Protease Activity in Bone Marrow-Derived Mast Cells. J. Biol. Chem. 285, 20793–20805. doi:10.1074/jbc.M109.084749
Okajima, T., Yoshida, K., Kondo, T., and Furukawa, K. (1999). Human Homolog of Caenorhabditis elegans Sqv-3 Gene Is Galactosyltransferase I Involved in the Biosynthesis of the Glycosaminoglycan-Protein Linkage Region of Proteoglycans. J. Biol. Chem. 274, 22915–22918. doi:10.1074/jbc.274.33.22915
Olson, S. K., Bishop, J. R., Yates, J. R., Oegema, K., and Esko, J. D. (2006). Identification of Novel Chondroitin Proteoglycans in Caenorhabditis elegans: Embryonic Cell Division Depends on CPG-1 and CPG-2. J. Cell Biol. 173, 985–994. doi:10.1083/jcb.200603003
Orellana, A., Moraga, C., Araya, M., and Moreno, A. (2016). Overview of Nucleotide Sugar Transporter Gene Family Functions across Multiple Species. J. Mol. Biol. 428, 3150–3165. doi:10.1016/j.jmb.2016.05.021
Orkin, R. W., Pratt, R. M., and Martin, G. R. (1976). Undersulfated Chondroitin Sulfate in the Cartilage Matrix of Brachymorphic Mice. Dev. Biol. 50, 82–94. doi:10.1016/0012-1606(76)90069-5
Pacheco, B., Malmström, A., and Maccarana, M. (2009). Two Dermatan Sulfate Epimerases Form Iduronic Acid Domains in Dermatan Sulfate. J. Biol. Chem. 284, 9788–9795. doi:10.1074/jbc.M809339200
Paganini, C., Monti, L., Costantini, R., Besio, R., Lecci, S., Biggiogera, M., et al. (2019). Calcium Activated Nucleotidase 1 (CANT1) Is Critical for Glycosaminoglycan Biosynthesis in Cartilage and Endochondral Ossification. Matrix Biol. 81, 70–90. doi:10.1016/j.matbio.2018.11.002
Parker, J. L., and Newstead, S. (2019). Gateway to the Golgi: Molecular Mechanisms of Nucleotide Sugar Transporters. Curr. Opin. Struct. Biol. 57, 127–134. doi:10.1016/j.sbi.2019.03.019
Pennypacker, J. P., Kimata, K., and Brown, K. S. (1981). Brachymorphic Mice (Bm/bm): A Generalized Biochemical Defect Expressed Primarily in Cartilage. Dev. Biol. 81, 280–287. doi:10.1016/0012-1606(81)90291-8
Poönighaus, C., Ambrosius, M., Casanova, J. C., Prante, C., Kuhn, J., Esko, J. D., et al. (2007). Human Xylosyltransferase II Is Involved in the Biosynthesis of the Uniform Tetrasaccharide Linkage Region in Chondroitin Sulfate and Heparan Sulfate Proteoglycans. J. Biol. Chem. 282, 5201–5206. doi:10.1074/jbc.M611665200
Properzi, F., Carulli, D., Asher, R. A., Muir, E., Camargo, L. M., van Kuppevelt, T. H., et al. (2005). Chondroitin 6-sulphate Synthesis Is Up-Regulated in Injured CNS, Induced by Injury-Related Cytokines and Enhanced in Axon-Growth Inhibitory Glia. Eur. J. Neurosci. 21, 378–390. doi:10.1111/j.1460-9568.2005.03876.x
Purushothaman, A., Sugahara, K., and Faissner, A. (2012). Chondroitin Sulfate "wobble Motifs" Modulate Maintenance and Differentiation of Neural Stem Cells and Their Progeny. J. Biol. Chem. 287, 2935–2942. doi:10.1074/jbc.R111.298430
Rodén, L. (1980). “Structure and Metabolism of Connective Tissue Proteoglycans,” in The Biochemistry Of Glycoproteins And Proteoglycans. Editor W. J. Lennarz (New York: Plenum Press), 267–371. doi:10.1007/978-1-4684-1006-8_7
Rossi, A., and Superti-Furga, A. (2001). Mutations in the Diastrophic Dysplasia Sulfate Transporter (DTDST) Gene (SLC26A2): 22 Novel Mutations, Mutation Review, Associated Skeletal Phenotypes, and Diagnostic Relevance. Hum. Mutat. 17, 159–171. doi:10.1002/humu.1
Saiyin, W., Li, L., Zhang, H., Lu, Y., and Qin, C. (2019). Inactivation of FAM20B Causes Cell Fate Changes in Annulus Fibrosus of Mouse Intervertebral Disc and Disc Defects via the Alterations of TGF-β and MAPK Signaling Pathways. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1865, 165555. doi:10.1016/j.bbadis.2019.165555
Sato, T., Kudo, T., Ikehara, Y., Ogawa, H., Hirano, T., Kiyohara, K., et al. (2011). Chondroitin Sulfate N-Acetylgalactosaminyltransferase 1 Is Necessary for normal Endochondral Ossification and Aggrecan Metabolism. J. Biol. Chem. 286, 5803–5812. doi:10.1074/jbc.M110.159244
Satoh, H., Susaki, M., Shukunami, C., Iyama, K.-I., Negoro, T., and Hiraki, Y. (1998). Functional Analysis of Diastrophic Dysplasia Sulfate Transporter. J. Biol. Chem. 273, 12307–12315. doi:10.1074/jbc.273.20.12307
Schaefer, L., Tredup, C., Gubbiotti, M. A., and Iozzo, R. V. (2017). Proteoglycan Neofunctions: Regulation of Inflammation and Autophagy in Cancer Biology. FEBS J. 284, 10–26. doi:10.1111/febs.13963
Schön, S., Prante, C., Bahr, C., Kuhn, J., Kleesiek, K., and Götting, C. (2006). Cloning and Recombinant Expression of Active Full-Length Xylosyltransferase I (XT-I) and Characterization of Subcellular Localization of XT-I and XT-II. J. Biol. Chem. 281, 14224–14231. doi:10.1074/jbc.M510690200
Schwartz, N. B., Ostrowski, V., Brown, K. S., and Pratt, R. M. (1978). Defective PAPS-Synthesis in Epiphyseal Cartilage from Brachymorphic Mice. Biochem. Biophysical Res. Commun. 82, 173–178. doi:10.1016/0006-291x(78)90592-2
Seidler, U., and Nikolovska, K. (2019). Slc26 Family of Anion Transporters in the Gastrointestinal Tract: Expression, Function, Regulation, and Role in Disease. Compr. Physiol. 9, 839–872. doi:10.1002/cphy.c180027
Shimbo, M., Suzuki, R., Fuseya, S., Sato, T., Kiyohara, K., Hagiwara, K., et al. (2017). Postnatal Lethality and Chondrodysplasia in Mice Lacking Both Chondroitin Sulfate N-Acetylgalactosaminyltransferase-1 and -2. PLoS One 12, e0190333. doi:10.1371/journal.pone.0190333
Sivasami, P., Poudel, N., Munteanu, M. C., Hudson, J., Lovern, P., Liu, L., et al. (2019). Adipose Tissue Loss and Lipodystrophy in Xylosyltransferase II Deficient Mice. Int. J. Obes. 43, 1783–1794. doi:10.1038/s41366-019-0324-1
Smith, T. M., Hicks-Berger, C. A., Kim, S., and Kirley, T. L. (2002). Cloning, Expression, and Characterization of a Soluble Calcium-Activated Nucleotidase, a Human Enzyme Belonging to a New Family of Extracellular Nucleotidases. Arch. Biochem. Biophys. 406, 105–115. doi:10.1016/s0003-9861(02)00420-4
Sorg, B. A., Berretta, S., Blacktop, J. M., Fawcett, J. W., Kitagawa, H., Kwok, J. C. F., et al. (2016). Casting a Wide Net: Role of Perineuronal Nets in Neural Plasticity. J. Neurosci. 36, 11459–11468. doi:10.1523/JNEUROSCI.2351-16.2016
Spicer, A. P., Kaback, L. A., Smith, T. J., and Seldin, M. F. (1998). Molecular Cloning and Characterization of the Human and Mouse UDP-Glucose Dehydrogenase Genes. J. Biol. Chem. 273, 25117–25124. doi:10.1074/jbc.273.39.25117
Stachtea, X. N., Tykesson, E., van Kuppevelt, T. H., Feinstein, R., Malmström, A., Reijmers, R. M., et al. (2015). Dermatan Sulfate-free Mice Display Embryological Defects and Are Neonatal Lethal Despite normal Lymphoid and Non-lymphoid Organogenesis. PLoS One 10, e0140279. doi:10.1371/journal.pone.0140279
Stickens, D., Zak, B. M., Rougier, N., Esko, J. D., and Werb, Z. (2005). Mice Deficient in Ext2 Lack Heparan Sulfate and Develop Exostoses. Development 132, 5055–5068. doi:10.1242/dev.02088
Sugahara, K., and Kitagawa, H. (2000). Recent Advances in the Study of the Biosynthesis and Functions of Sulfated Glycosaminoglycans. Curr. Opin. Struct. Biol. 10, 518–527. doi:10.1016/s0959-440x(00)00125-1
Sugahara, K., and Mikami, T. (2007). Chondroitin/dermatan Sulfate in the central Nervous System. Curr. Opin. Struct. Biol. 17, 536–545. doi:10.1016/j.sbi.2007.08.015
Sugahara, K., Mikami, T., Uyama, T., Mizuguchi, S., Nomura, K., and Kitagawa, H. (2003). Recent Advances in the Structural Biology of Chondroitin Sulfate and Dermatan Sulfate. Curr. Opin. Struct. Biol. 13, 612–620. doi:10.1016/j.sbi.2003.09.011
Sugahara, K. N., Hirata, T., Tanaka, T., Ogino, S., Takeda, M., Terasawa, H., et al. (2008). Chondroitin Sulfate E Fragments Enhance CD44 Cleavage and CD44-dependent Motility in Tumor Cells. Cancer Res. 68, 7191–7199. doi:10.1158/0008-5472.CAN-07-6198
Sugahara, K., and Schwartz, N. B. (1979). Defect in 3'-phosphoadenosine 5'-phosphosulfate Formation in Brachymorphic Mice. Proc. Natl. Acad. Sci. 76, 6615–6618. doi:10.1073/pnas.76.12.6615
Sugahara, K., and Schwartz, N. B. (1982b). Defect in 3'-phosphoadenosine 5'-phosphosulfate Synthesis in Brachymorphic Mice. II. Tissue Distribution of the Defect. Arch. Biochem. Biophys. 214, 602–609. doi:10.1016/0003-9861(82)90065-0
Sugahara, K., and Schwartz, N. B. (1982a). Defect in 3′-phosphoadenosine 5′-phosphosulfate Synthesis in Brachymorphic Mice. Arch. Biochem. Biophys. 214, 589–601. doi:10.1016/0003-9861(82)90064-9
Sugiyama, S., Di Nardo, A. A., Aizawa, S., Matsuo, I., Volovitch, M., Prochiantz, A., et al. (2008). Experience-dependent Transfer of Otx2 Homeoprotein into the Visual Cortex Activates Postnatal Plasticity. Cell 134, 508–520. doi:10.1016/j.cell.2008.05.054
Sun, X., Meyers, E. N., Lewandoski, M., and Martin, G. R. (1999). Targeted Disruption of Fgf8 Causes Failure of Cell Migration in the Gastrulating Mouse Embryo. Genes Dev. 13, 1834–1846. doi:10.1101/gad.13.14.1834
Takemae, H., Ueda, R., Okubo, R., Nakato, H., Izumi, S., Saigo, K., et al. (2003). Proteoglycan UDP-Galactose:β-Xylose β1,4-Galactosyltransferase I Is Essential for Viability in Drosophila Melanogaster. J. Biol. Chem. 278, 15571–15578. doi:10.1074/jbc.M301123200
Takemura, M., Noborn, F., Nilsson, J., Bowden, N., Nakato, E., Baker, S., et al. (2020). Chondroitin Sulfate Proteoglycan Windpipe Modulates Hedgehog Signaling in Drosophila. Mol. Biol. Cell 31, 813–824. doi:10.1091/mbc.E19-06-0327
Takeuchi, K., Yoshioka, N., Higa Onaga, S., Watanabe, Y., Miyata, S., Wada, Y., et al. (2013). Chondroitin Sulphate N-Acetylgalactosaminyl-Transferase-1 Inhibits Recovery from Neural Injury. Nat. Commun. 4, 2740. doi:10.1038/ncomms3740
Tang, T., Li, L., Tang, J., Li, Y., Lin, W. Y., Martin, F., et al. (2010). A Mouse Knockout Library for Secreted and Transmembrane Proteins. Nat. Biotechnol. 28, 749–755. doi:10.1038/nbt.1644
ten Dam, G. B., van de Westerlo, E. M. A., Purushothaman, A., Stan, R. V., Bulten, J., Sweep, F. C. G. J., et al. (2007). Antibody GD3G7 Selected against Embryonic Glycosaminoglycans Defines Chondroitin Sulfate-E Domains Highly Up-Regulated in Ovarian Cancer and Involved in Vascular Endothelial Growth Factor Binding. Am. J. Pathol. 171, 1324–1333. doi:10.2353/ajpath.2007.070111
ten Dam, G. B., Yamada, S., Kobayashi, F., Purushothaman, A., Westerlo, E. M. A., Bulten, J., et al. (2009). Dermatan Sulfate Domains Defined by the Novel Antibody GD3A12, in normal Tissues and Ovarian Adenocarcinomas. Histochem. Cell Biol. 132, 117–127. doi:10.1007/s00418-009-0592-2
Thelin, M. A., Bartolini, B., Axelsson, J., Gustafsson, R., Tykesson, E., Pera, E., et al. (2013). Biological Functions of Iduronic Acid in Chondroitin/dermatan Sulfate. FEBS J. 280, 2431–2446. doi:10.1111/febs.12214
Thelin, M. A., Svensson, K. J., Shi, X., Bagher, M., Axelsson, J., Isinger-Ekstrand, A., et al. (2012). Dermatan Sulfate Is Involved in the Tumorigenic Properties of Esophagus Squamous Cell Carcinoma. Cancer Res. 72, 1943–1952. doi:10.1158/0008-5472.CAN-11-1351
Tian, J., Ling, L., Shboul, M., Lee, H., O'Connor, B., Merriman, B., et al. (2010). Loss of CHSY1, a Secreted FRINGE Enzyme, Causes Syndromic Brachydactyly in Humans via Increased NOTCH Signaling. Am. J. Hum. Genet. 87, 768–778. doi:10.1016/j.ajhg.2010.11.005
Tone, Y., Pedersen, L. C., Yamamoto, T., Izumikawa, T., Kitagawa, H., Nishihara, J., et al. (2008). 2-O-phosphorylation of Xylose and 6-O-Sulfation of Galactose in the Protein Linkage Region of Glycosaminoglycans Influence the Glucuronyltransferase-I Activity Involved in the Linkage Region Synthesis. J. Biol. Chem. 283, 16801–16807. doi:10.1074/jbc.M709556200
Trowbridge, J. M., and Gallo, R. L. (2002). Dermatan Sulfate: New Functions from an Old Glycosaminoglycan. Glycobiology 12, 117R–125R. doi:10.1093/glycob/cwf066
Tykesson, E., Hassinen, A., Zielinska, K., Thelin, M. A., Frati, G., Ellervik, U., et al. (2018). Dermatan Sulfate Epimerase 1 and Dermatan 4-O-Sulfotransferase 1 Form Complexes that Generate Long Epimerized 4-O-Sulfated Blocks. J. Biol. Chem. 293, 13725–13735. doi:10.1074/jbc.RA118.003875
Uchimura, K., Kadomatsu, K., Fan, Q.-W., Muramatsu, H., Kurosawa, N., Kaname, T., et al. (1998). Mouse Chondroitin 6-sulfotransferase: Molecular Cloning, Characterization and Chromosomal Mapping. Glycobiology 8, 489–496. doi:10.1093/glycob/8.5.489
Uchimura, K., Kadomatsu, K., Nishimura, H., Muramatsu, H., Nakamura, E., Kurosawa, N., et al. (2002). Functional Analysis of the Chondroitin 6-sulfotransferase Gene in Relation to Lymphocyte Subpopulations, Brain Development, and Oversulfated Chondroitin Sulfates. J. Biol. Chem. 277, 1443–1450. doi:10.1074/jbc.M104719200
Uyama, T., Kitagawa, H., Tamura, J.-i., and Sugahara, K. (2002). Molecular Cloning and Expression of Human ChondroitinN-Acetylgalactosaminyltransferase. J. Biol. Chem. 277, 8841–8846. doi:10.1074/jbc.M111434200
Uyama, T., Kitagawa, H., Tanaka, J., Tamura, J.-i., Ogawa, T., and Sugahara, K. (2003). Molecular Cloning and Expression of a Second Chondroitin N-Acetylgalactosaminyltransferase Involved in the Initiation and Elongation of Chondroitin/dermatan Sulfate. J. Biol. Chem. 278, 3072–3078. doi:10.1074/jbc.M209446200
Venkatachalam, K. V. (2003). Human 3′-phosphoadenosine 5′-phosphosulfate (PAPS) Synthase: Biochemistry, Molecular Biology and Genetic Deficiency. IUBMB Life (International Union Biochem. Mol. Biol. Life) 55, 1–11. doi:10.1080/1521654031000072148
Voermans, N. C., Kempers, M., Lammens, M., van Alfen, N., Janssen, M. C., Bönnemann, C., et al. (2012). Myopathy in a 20-Year-Old Female Patient with D4ST-1 Deficient Ehlers-Danlos Syndrome Due to a Homozygous CHST14 Mutation. Am. J. Med. Genet. 158A, 850–855. doi:10.1002/ajmg.a.35232
Watanabe, Y., Takeuchi, K., Higa Onaga, S., Sato, M., Tsujita, M., Abe, M., et al. (2010). Chondroitin Sulfate N-Acetylgalactosaminyltransferase-1 Is Required for normal Cartilage Development. Biochem. J. 432, 47–55. doi:10.1042/BJ20100847
Wei, L., Cao, P., Xu, C., Zhong, H., Wang, X., Bai, M., et al. (2020). Chondroitin Synthase‐3 Regulates Nucleus Pulposus Degeneration through Actin‐induced YAP Signaling. FASEB J. 34, 16581–16600. doi:10.1096/fj.202001021R
Wilson, D. G., Phamluong, K., Lin, W. Y., Barck, K., Carano, R. A. D., Diehl, L., et al. (2012). Chondroitin Sulfate Synthase 1 (Chsy1) Is Required for Bone Development and Digit Patterning. Dev. Biol. 363, 413–425. doi:10.1016/j.ydbio.2012.01.005
Witkos, T. M., and Lowe, M. (2017). Recognition and Tethering of Transport Vesicles at the Golgi Apparatus. Curr. Opin. Cell Biol. 47, 16–23. doi:10.1016/j.ceb.2017.02.003
Wu, J., Tian, Y., Han, L., Liu, C., Sun, T., Li, L., et al. (2020). FAM20B-catalyzed Glycosaminoglycans Control Murine Tooth Number by Restricting FGFR2b Signaling. BMC Biol. 18, 87. doi:10.1186/s12915-020-00813-4
Yada, T., Gotoh, M., Sato, T., Shionyu, M., Go, M., Kaseyama, H., et al. (2003a). Chondroitin Sulfate Synthase-2. J. Biol. Chem. 278, 30235–30247. doi:10.1074/jbc.M303657200
Yada, T., Sato, T., Kaseyama, H., Gotoh, M., Iwasaki, H., Kikuchi, N., et al. (2003b). Chondroitin Sulfate Synthase-3. J. Biol. Chem. 278, 39711–39725. doi:10.1074/jbc.M304421200
Yamada, S., and Sugahara, K. (2008). Potential Therapeutic Application of Chondroitin Sulfate/dermatan Sulfate. Curr. Drug Discov. Technol. 5, 289–301. doi:10.2174/157016308786733564
Yamaguchi, T. P., Harpal, K., Henkemeyer, M., and Rossant, J. (1994). Fgfr-1 Is Required for Embryonic Growth and Mesodermal Patterning during Mouse Gastrulation. Genes Dev. 8, 3032–3044. doi:10.1101/gad.8.24.3032
Yamaguchi, Y. (2001). Heparan Sulfate Proteoglycans in the Nervous System: Their Diverse Roles in Neurogenesis, Axon Guidance, and Synaptogenesis. Semin. Cell Dev. Biol. 12, 99–106. doi:10.1006/scdb.2000.0238
Yamauchi, S., Mita, S., Matsubara, T., Fukuta, M., Habuchi, H., Kimata, K., et al. (2000). Molecular Cloning and Expression of Chondroitin 4-sulfotransferase. J. Biol. Chem. 275, 8975–8981. doi:10.1074/jbc.275.12.8975
Yoshioka, N., Miyata, S., Tamada, A., Watanabe, Y., Kawasaki, A., Kitagawa, H., et al. (2017). Abnormalities in Perineuronal Nets and Behavior in Mice Lacking CSGalNAcT1, a Key Enzyme in Chondroitin Sulfate Synthesis. Mol. Brain 10, 47. doi:10.1186/s13041-017-0328-5
Yoshizawa, T., Mizumoto, S., Takahashi, Y., Shimada, S., Sugahara, K., Nakayama, J., et al. (2018). Vascular Abnormalities in the Placenta of Chst14−/− Fetuses: Implications in the Pathophysiology of Perinatal Lethality of the Murine Model and Vascular Lesions in Human CHST14/D4ST1 Deficiency. Glycobiology 28, 80–89. doi:10.1093/glycob/cwx099
Keywords: chondroitin sulfate, dermatan sulfate, epimerase, glycosyltransferase, knockout mouse, proteoglycan, sulfotransferase, transporter
Citation: Mizumoto S and Yamada S (2021) An Overview of in vivo Functions of Chondroitin Sulfate and Dermatan Sulfate Revealed by Their Deficient Mice. Front. Cell Dev. Biol. 9:764781. doi: 10.3389/fcell.2021.764781
Received: 26 August 2021; Accepted: 09 November 2021;
Published: 24 November 2021.
Edited by:
Ana Cuenda, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Krishna Mohan Sepuru, Howard Hughes Medical Institute (HHMI), United StatesMauro Sergio Pavao, Federal University of Rio de Janeiro, Brazil
Hiroshi Kitagawa, Kobe Pharmaceutical University, Japan
Göran Larson, University of Gothenburg, Sweden
Chiara Schiraldi, Università della Campania Luigi Vanvitelli, Italy
Copyright © 2021 Mizumoto and Yamada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuji Mizumoto, mizumoto@meijo-u.ac.jp