
Autophagy-related mechanisms for treatment of multiple myeloma
 0
0 ,
, Abstract
Multiple myeloma (MM) is a type of hematological cancer that occurs when B cells become malignant. Various drugs such as proteasome inhibitors, immunomodulators, and compounds that cause DNA damage can be used in the treatment of MM. Autophagy, a type 2 cell death mechanism, plays a crucial role in determining the fate of
Keywords
INTRODUCTION
Multiple myeloma (MM) is associated with the proliferation of a single plasma B cell variant in the bone marrow and the secretion of monoclonal immunoglobulins[1]. MM is the second most common type of hematological malignancy worldwide, which accounts for approximately 10% of cases[2]. The mean age of diagnosis is 69 years, and the disease diagnosis is based on the monoclonal M protein produced by malignant B cells[3]. Myeloma cells are plasma cells that secrete immunoglobulins and usually synthesize IgG or IgA[4]. As plasmacytomas increase, monoclonal gammopathy of undetermined significance (MGUS), a premalignant and painless disease, emerges. Subsequently, asymptomatic smoldering myeloma (SMM) and eventually symptomatic MM appear. 1% of cases with MGUS and 10% of cases with SMM might convert to MM[5]. Rapidly accumulating malignant B cells cause bone destruction, anemia, hypercalcemia, and renal failure[6]. In the clinical treatment of the disease, proteasome inhibitors, steroids, monoclonal antibodies, DNA damage, and immunomodulatory compounds are used in combination[7]. Most of these therapies focus on killing MM cells. Commonly, there are three different death mechanisms in cells: apoptosis
The mechanisms of autophagy
Autophagy is a recycling activity that is employed to maintain the survival of other cells and to maintain body homeostasis in case of stress such as nutrient deficiency and reactive oxygen compounds (ROS)[14]. In tumorigenesis, autophagy can work in a bidirectional manner by increasing disease progression or causing cancer cell death[15]. In addition, differentiations in autophagic balance might lead to drug resistance in cancer cells. Commonly autophagy-related genes are under-expressed or contain deletions in cancer cells[16]. In recent years, it has been declared that the loss of PTEN, a tumor suppressor, leads to a decrease in autophagy and thus plays a key role in shaping the fate of tumor cells[17]. The formation of autophagosomes primarily serves as a recycling process, not exclusively aimed at cancer cells. However, it works in tandem with the immune system in its specific function related to cancer[18]. Many autophagy-related proteins (ATG) are involved in the regulation of autophagy. The mechanisms of autophagy can be summarized into five items[19], as shown in Figure 1.

Figure 1. Schematic of the stages of autophagy. (A) Phagophore formation triggered by Beclin-1/VPS34 in response to a stress signal; (B) Multimerization resulting from ATG5/ATG12 conjugation and interaction with ATG16; (C) Embedding in the phagophore membrane by LC3 engraving; (D) Autophagosome formation by ATG4-directed LC3-II/ATG8 recycling; (E) Fragmentation of molecules following association with lysosome. ATG: Autophagy-related proteins.
The initiation of phagophore formation is determined by the interaction between Beclin1 and Bcl-2, which is triggered by the phosphorylation of Bcl-2 by JNK-1. This results in the dissociation of Bcl-2 from Beclin1, which allows for the initiation of autophagy by the Beclin1/Vps34 complex. Recently, the role of Beclin1 and UV radiation resistance-associated gene (UVRAG) as a tumor suppressor and autophagy trigger in cancer was emphasized[20]. mTOR plays a pivotal role in regulating autophagy by inhibiting ATG1/ULK1-2 from the early stages. mTOR interacts with various proteins to form mTORC1/2 complexes, which activate different transduction pathways. Essentially, mTOR serves as the linchpin governing the balance between cell growth and the induction of autophagy. mTOR is inhibited by REDD1 and AMPK through Rheb under conditions of hypoxia or low ATP [Figure 1]. In addition, mTOR is activated via the IRS1, PI3K/AKT, TSC1/TSC2, and Rheb signaling pathways. Although three variants of autophagy have been proposed so far [microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy], degradation by lysosomal fusion is inevitable. Macroautophagy is characterized by the formation of an autophagosome, which consists of lipid bilayers so that large proteins and organelles can be broken down and recycled by the lysosome[19]. In microautophagy, the cell membrane spontaneously folds inwards to form a tube. The end of the tube buds and connects with the lysosome through vesicles[16]. Unlike others, CMA is the selective degradation of misfolded proteins by lysosomes via chaperones. The KFERQ motif in misfolded proteins is recognized by Hsc70 and forms a complex[21]. This complex is pulled in and destroyed by LAMP-2A in the lysosome membrane without the need for vesicles. Finally, they are degraded into the fundamental units by lytic enzymes (protease, lipase, and hydrolase) in the lysosome and are released to the cytoplasm for synthesizing new complex molecules.
Autophagy is a crucial factor in the circumstance of the disease, as MM cells are plasma cells that synthesize a large amount of immunoglobulin. Therefore, it is important to assess how autophagy functions, considering its dual impact as both a suppressor and supporter of the tumor[15]. ATG5, an important autophagy-related indicator, is cleaved upon exposure to Bortezomib and initiates autophagy in MM cells[22]. It is known that inhibiting autophagy in conjunction with proteasome inhibitors, which are effective in treating MM, presents a promising therapeutic avenue. Therefore, it is necessary to elucidate the fundamental mechanisms of autophagy at the forefront of MM research. In this review, autophagy mechanisms and autophagy modulators in MM disease were comparatively reviewed. This review emphasizes the significance of incorporating autophagy alongside appropriate medications and timing as a strategy to significantly extend the lifespan of MM patients.
AUTOPHAGY-RELATED MECHANISMS IN MM
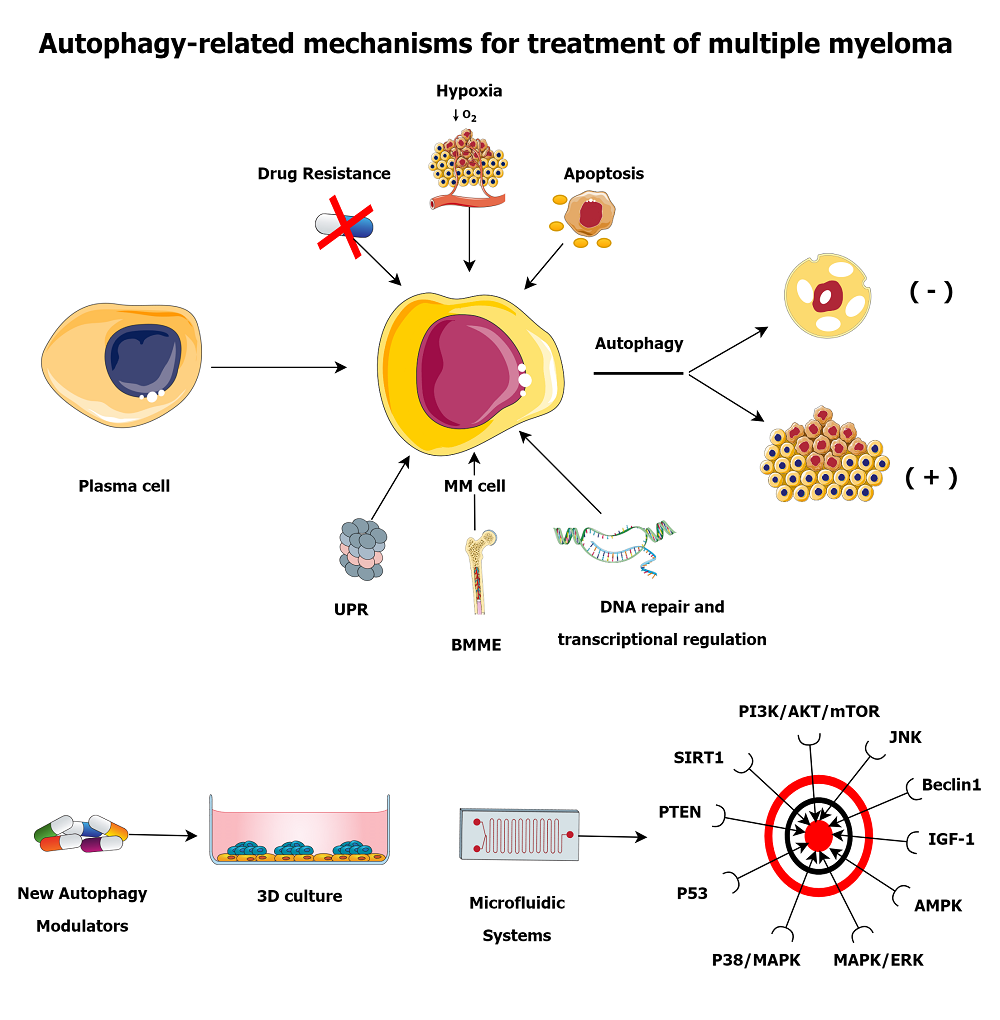
MM cells use multiple mechanisms to evade cell death, including proteasome, unfolded protein response (UPR), and autophagy. MM cells that can escape from death develop resistance to drugs, which makes available therapies useless. The viability of MM cells is based on the proteasome-mediated degradation of misfolded proteins and non-functional newly synthesized proteins[23]. Therefore, autophagy can maintain the continuity of MM cells by averting the toxic stack of misfolded proteins. A basal level of autophagy is essential for the life cycle of MM cells. When these mechanisms collapse, apoptosis is finally induced by immune cells for the integrity and health of the organism[18]. Autophagy may be involved in the progression of MM from malignant plasma cells. Exemplarily, the enhancement of autophagy in plasma cells of MGUS and MM patients was only in the direction of MM[24]. A study conducted with biopsies of 89 patients with MM reported that patients with Beclin1 and LC3 biomarkers had a longer life expectancy[25]. It was revealed that elevated IFNγ in neutrophils isolated from MGUS and MM patients triggers JAK2/STAT3 signaling to form an autophagic survival system[26]. Recently, CD46, IKBKE, PARK2, ULK4, ATG5, and CDKN2A were considered in a three-cohort meta-analysis study that investigated the contribution of polymorphisms in autophagy-related genes to MM disease risk[27]. Considering all these, autophagy is related not only to MM progression or regression but also to MM development at the beginning. We can examine the mechanisms thanks to the fact that autophagy is effective in MM under the headings of UPR, bone marrow microenvironment (BMME), drug resistance, hypoxia, DNA repair and transcriptional regulation, and apoptosis.
UPR
The folding of proteins takes place in the endoplasmic reticulum (ER). Misfolded proteins are ubiquitinated by a system including ER chaperones and are degraded by the proteasome[28]. Besides contributing to the degradation of p62/SQSTM1 aggregated proteins via proteasomes, it is known that p62 can sustain cell survival by inducing autophagy[29]. Additionally, recent studies have shown that SQSTM1/p62 plays a crucial role in supporting proteostasis and is not just involved in working with the proteasome to degrade Ub-protein aggregates[30,31]. SQSTM1/p62 is a vital component of the autophagic reserve that helps the cell adapt to proteasome stress induced by proteasome inhibitors with the modification of the interactome[30]. In this sense, it can be interpreted that p62 functions in the regulation of autophagy. Autophagy plays a significant part in the transformation of B cells into plasma cells, and ATG5 mediates these tasks[32]. Mice with Atg5f/f were observed to have a higher ER stress with increased immunoglobulin synthesis compared to wild type[33]. Therefore, autophagy plays a role in guiding plasma cells to the bone marrow for adaptive immune responses. Myeloma cells are under constant ER stress due to the high content of immunoglobulins they synthesize and can easily induce the UPR[34]. Inhibition of the proteasome with chemotherapeutics makes the damaged proteins roll up, turn toxic, and trigger UPR[35]. UPR restores homeostasis and performs in conjunction with autophagy. Thus, autophagy is upregulated and initiates a survival mechanism to eliminate UPR.
The UPR proteolytic system is controlled by the signaling mechanism of IRE1/XBP-1, PERK/ATF4, and ATF6/cleaved ATF6. IRE1, PERK, and ATF6 are separated from GRP78, to which they are attached when UPR is triggered, which accounts for a signal transduction cascade[36]. The PERK signaling pathway conduces to the UPR system by suppressing protein synthesis. IRE1 participates both by augmentation of misfolded protein impairment and by enhancement of protein folding together with ATF6. The function of XBP-1 is of great importance in the autophagy mechanism in UPR and MM cells. IRE1 fulfills the cell viability role of autophagy by regulating itself and XBP-1 at the mRNA level[37]. Directing the maturation of plasma cells, XBP-1 is involved in autophagy through ATG5[38]. However, XBP-1 facilitates IRE1 binding to the BiP chaperone to downregulate the UPR function in B cells[39]. IRE1a/XBP-1 balance is critical in MM cells as it reduces ER stress[40]. Furthermore, it was reported that inhibition of XBP-1 splicing by IRE1 may be a treatment modality for MM cells[41]. Additionally, it was demonstrated that there is a correlation between elevated spliced XBP-1 levels and Bortezomib resistance[35]. XBP-1 is also involved in the regulation of ATG5 and Beclin1 via eIF2AK3 for the activation of autophagy[42]. Moreover, XBP-1 functions as a transcription factor in the signaling cascade between IRE1 and ATF6 cleavage in UPR[43]. XBP-1 influences the expression of genes grave in B cell differentiation, such as BAFF, APRIL, BCMA, and TACI[44]. In addition, these genes also affect NF-κB expression. In MM cells, the NF-κB pathway leads to the expression of cytokines, cell adhesion molecules, and cell growth and survival factors. NF-κB, which also mediates XBP-1 expression, contributes to the induction of UPR and autophagy in cancer cells[45]. It was previously mentioned that Beclin1 and LC3 work together in autophagosome formation. NF-κB raises the possibility of autophagy by augmenting Beclin1 expression[46]. PERK obstructs the phosphorylation of eIF2B, which leads to a significant reduction in the production of ATF4 and protein synthesis[47]. This mechanism serves as a crucial intervention point for regulating protein synthesis in cells. PERK can operate to diminish the oxidative stress response that may be motivated by unfolded proteins by activating NRF2[48]. In this sense, it plays a binary part in up or down-regulating UPR in autophagy. PERK labors with ATF4/6 to activate the transcription factor CHOP and to reduce ER stress[49]. From this point of view, PERK primarily functions to sustain cell life, but it can also operate cell death mechanisms in response to the strong signals it receives. In an experiment study where IRE1, ATF6, and PERK were individually knocked out using RNAi, it was indicated that PERK functions as a sensor of autophagy in MM cells[50]. An inhibitor that targets the PERK pathway via eIF2AK3 in MM cells holds great potential in inducing cell death and providing effective therapy[51]. This approach must be further explored to unlock its full potential in treating MM.
BMME
The BMME consists of cells and supporting elements. BMME stimulates a reputable part in the differentiation of B cells and their transformation into cancer[52]. As a result of the interaction of MM cells with stromal cells, IL-6, VEGF, and IGF-1 are secreted, and signaling pathways that cause tumor aggression, such as PI3K/AKT/mTOR, MEK/ERK, JAK/STAT3, and NF-κB, are activated [Figure 2]. Intercellular interactions between stromal cells and MM cells in the bone marrow and relationships with extracellular matrix (ECM) elements determine the fate of MM cells. Furthermore, adhesion molecules such as TNF-α, ICAM-1, LFA-1, and VLA-4 generated by stromal cells are responsible for the interaction of MM cells and bone marrow stromal cells (BMSCs). Stimulation of adhesion molecules for expression is mediated by

Figure 2. Causes and consequences of autophagy in MM cells within BMME. In the bone marrow, stromal cells secrete IL-6, VEGF, and IGF-1 due to intercellular interactions with MM cells. IL-6 stimulates UPR and autophagy through XBP-1. Apoptosis triggers autophagy via the JAK/STAT3 and MAPK/ERK pathways or IL-6. Autophagy and survival are promoted by the interaction of BCMA and APRIL through NF-κB and XBP-1. Adhesion molecules such as TNF-α, ICAM-1, LFA-1, and VLA-4 develop drug resistance by inducing autophagy via NF-κB. PI3K/AKT/mTOR inhibition promotes autophagy in drug-resistant cells. UPR upregulates HIF1-α through XBP-1 processing, which triggers autophagy through AMPK and mTOR. BMME: Bone marrow microenvironment; MM: multiple myeloma; UPR: unfolded protein response.
Angiogenesis is an important element in the transition of the disease from MGUS to MM[62]. Although it is a distinguishing fact that angiogenesis occurs only in MM, various cytokines play a significant role in the process[63]. Pro-angiogenic cytokines are released by interactions among MM cells, stromal cells, and endothelial cells. Studies reported that MM cells secrete higher levels of bFGF, HGF, and VEGF than plasma cells, which promotes angiogenesis[64]. In a recent study, it was concluded that endothelial cells from patients with MM expressed higher levels of EGFR and its ligand HB-EGF than those from MGUS patients[65]. When EGFR is activated, it promotes blood vessel growth and assists endothelial cells in surviving. Blockade of HB-EGF-EGFR signaling limited the angiogenic potential of bone marrow endothelial cells and hampered tumor growth in an MM mouse model[65]. These findings suggest that HB-EGF-EGFR signaling could be a potential target in anti-angiogenic therapy for MM. Another study aimed to investigate the effect of combining Bortezomib and Hydroxychloroquine on plasma and endothelial cells isolated from patients with MM and MGUS[66]. The results showed that the combined treatment had different effects on MM cells and endothelial cells[66]. LC3B and p62 expressions induced autophagy in MM cells[66], and it was suggested that Bortezomib should be used in combination with an anti-angiogenic drug to enhance its effectiveness. Considering the dual role of autophagy in MM, it initially acts as a tumor suppressor, but may also promote tumor growth, survival, and therapy resistance. The relationship between EGFR and autophagy is intricate. In fact, inhibiting EGFR could trigger autophagy as a survival mechanism. Thus, by gaining a deeper understanding of the connection among EGFR, autophagy, and bone marrow angiogenesis, we can potentially develop novel treatments for MM.
Drug resistance
Drug resistance is one of the major issues in MM and causes relapse and refractory of the disease. MM cells can avoid the toxic effects of chemotherapeutics via autophagy and thus develop drug resistance[14]. Bortezomib, which is frequently used in the treatment of MM and ratified by the FDA, triggers autophagy via UPR by inhibiting proteasomes[67]. Bortezomib resistance occurs frequently in MM, and different approaches are sought for treatment. Caspase-10 endorses the survival and resistance of MM cells by regulating autophagy[15]. Through induction of IRF4 in MM, caspase-10 cleaves BCLAF1, which competes with Beclin-1 for binding to Bcl-2 and inhibits autophagy[68]. Therefore, the sensitivity of MM cells to Bortezomib can be restored by developing caspase-10 inhibitors. The association of Bortezomib resistance with P-gp and drug resistance-related proteins in MM cells was demonstrated[69]. The increase in ATF4 expression after Bortezomib treatment in breast cancer cells[70] suggests that Bortezomib resistance can be overcome by combined suppression of autophagy and proteasomes. In a recent study on Bortezomib-resistant MM patients and cell lines, it was mentioned that upregulation of CMA may be a mechanism leading to resistance formation and targeting this mechanism could be a therapeutic target[71]. It was revealed that the CMA pathway was induced due to the ER stress caused by Bortezomib, and the expression of LAMP2A, which is an indispensable part of this pathway, was boosted[71]. However, it was stated that inhibition of the CMA pathway restored Bortezomib sensitivity, and the dual combination was more cytotoxic[71]. It was revealed that the Profilin-1 gene contributes to Bortezomib resistance in MM by boosting autophagy through Beclin1[72]. The combination of anti-β2M monoclonal antibodies with Bortezomib in MM cells promoted cellular death even in resistant cells by inhibition of autophagy[73]. Bortezomib response decreased in MM cells as a result of suppression of NEDD4L, which induced autophagy by binding the 19S proteasome[74]. A mechanism for the NEK2/USP7/Beclin-1 complex was identified in autophagy-induced Bortezomib resistance in MM patients[75]. As a result of Bortezomib treatment applied to MM cells, autophagy was incited by GRP78 in cells[76]. Furthermore, it was displayed that HSPA5 expression was upregulated in Bortezomib-resistant cells[76]. It was declared that transfection of mir145-3p into MM cells suppresses HDAC4 and mTORC1 and causes sensitivity to Bortezomib due to autophagy[77].
Autophagy functions in the development of resistance to many chemotherapeutics used in the treatment of MM. In a related study, by using primary patient samples and Dexamethasone-resistant MM cell lines, Beclin1 and LC3/ATG8 expressions were found to be inversely proportional to the p62 level[78]. In this case, the authors claimed that autophagy leads to drug resistance in MM, and Beclin1/p62 is a biomarker. Additionally, antiestrogen binding site ligands regulated the cholesterol mechanism via the PI3K/AKT/mTOR pathway, inducing autophagy and overcoming Dexamethasone resistance[79]. The relationship of Carfilzomib resistance with autophagy in MM cells and the contribution of the KLF4 transcription factor were obvious[80]. KLF4 attaches to the p62 gene promoter and upregulates its expression. It was emphasized that Doxorubicin-induced resistance in MM cells is mediated by DEPTOR, and autophagy plays a main role in this resistance[81].
Hypoxia
Tumor cells have a high metabolic rate and require large amounts of nutrients and oxygen. The needs of the rapidly growing tumor mass cannot be met at the same rate through the vessels, and a hypoxic environment develops. Cancer cells can continue to proliferate by inducing HIF1-α and adapting to these unfavorable conditions. HIF1-α triggers autophagy by inhibiting Bcl-2/Beclin1 and upregulating ATGs through BNIP3 and its ligand[82]. However, tumor cells can initiate autophagy independently of HIF1-α through the activation of mTOR or AMPK[83]. Since BMME has a low pO2 level compared to blood, MM cells can easily adapt to the hypoxia environment[84]. The hypoxia state around the MM cell provokes the induction of UPR and autophagy due to the aggregation of the unfolded protein[85] because many proteins need O2 in order to be folded properly. Due to hypoxia-induced O2 deficiency, sufficient ATP cannot be produced from glucose. In this case, MM cells can also induce starvation-induced autophagy[86]. It was reported that upregulation of Hexokinase-2 in hypoxia conditions intensifies autophagy in MM cells and introduces it as an antiapoptotic feature[87]. UPR initiates hypoxia by upregulating the expression of HIF1-α and VEGF via XBP-1[88]. In addition, activation of mTORC1, an autophagy-related factor, was found to increase susceptibility to hypoxia in MM cells[89].
DNA repair and transcriptional regulation
Dysfunctional DNA repair pathways facilitate the progression and metastasis of MM disease from its initial stages to the development of drug-resistant phenotypes, because autophagy can be induced through DNA repair mechanisms[90]. The activation of autophagy via the mTORC1/TSC2/ULK1 pathway is triggered by the ATM-mediated phosphorylation of AMPK[91,92]. Additionally, ATM enables the inhibition of mTOR through the activation of Che-1 via REDD1 and DEPTOR[93]. Furthermore, stress-induced Sestrin2 binds to GATOR2, thereby suppressing mTORC1[94]. NF-κB, also activated by ATM, leads to the activation of Beclin1 and induction of autophagy[95]. ROS-induced DNA damage assists in the suppression of AMPK and triggering of autophagy by activation of PARP1[96]. ATM can also induce autophagy through the regulation of p53. p53 induces the initiation of autophagy through the upregulation of PTEN and AMPK[97]. Moreover, DAPK regulated by p53 facilitates autophagy through its connection with Beclin1 phosphorylation and VPS34 activation[98].
Histone deacetylases (HDACs) are crucial transcription regulators and can control functional proteins in signaling pathways. HDAC6 is critical in MM due to its connection with UPR. Because of its deficiency, autophagy can be induced by forming aggresomes. Therefore, it is a promising target for patient treatment. HMGB1 is a DNA-binding protein that contributes to the functionality of nucleosomes and causes tumor progression. HMGB1 expression is increased during the treatment of various cancers with different chemotherapeutics[99]. HMGB1 is involved in autophagy and DNA damage repair, and the knockdown of HMGB1 in MM cells restores mTOR-mediated Dexamethasone sensitivity[100]. Additionally, it was reported that HMGB1 expression increased in Bortezomib-resistant MM cells, and the combination of Bortezomib and Lycorine reversed the resistance[101]. Furthermore, it was emphasized that MALAT1, a lncRNA, caused the upregulation of HMGB1 in MM cells, and the knockdown of MALAT1 caused a decrease in the expressions of Beclin1 and LC3B together with HMGB1 in MM[102]. In light of these findings, it is clear that targeting HMGB1 in drug-resistant MM patients is an essential and promising therapeutic approach.
Apoptosis
Apoptosis against cellular stress is the first type of cell death pathway and is used to eliminate situations where autophagy cannot be overcome. Depending on the stress state of the cell, these death mechanisms can block or trigger each other. Apoptosis in MM cells can be induced by IL-6, VEGF, IGF1, SDF1, and FGF factors via the JAK/STAT3 and MAPK/ERK pathways[16]. It was reported that drug-resistant MM cells induce apoptosis via the Apo2L/TRAIL pathway[103]. The expression of Mcl-1, a proliferation marker of MM cells, increased by JAK/STAT3 and VEGF, resulting in resistance to apoptosis[104]. The importance of the Bcl-2/Bax ratio in MM apoptosis is indisputable[105], and the increase in Bcl-XL protein leads to the inhibition of apoptosis via IL-6. NF-κb’s involvement in MM cell apoptosis, metastasis, and drug resistance was investigated[106]. Autophagy helps to alleviate metabolic stress, but when apoptosis and autophagy are both impaired, it can lead to necrotic cell death[13]. Therefore, autophagy plays a role in suppressing tumors by reducing metabolic stress and preventing necrotic cell death, along with apoptosis[13].
The similarity of proteins in the apoptosis and autophagy pathways is a well-known phenomenon in MM, which highlights the complex interplay between these cellular processes[18]. As an example, Beclin1 executes its function as a tumor suppressor through regulation by AMBRA1, Bif1, and UVRAG. Both siRNA-mediated inhibition of Beclin-1 and autophagy inhibition by 3-methyladenine and Chloroquine resulted in apoptosis of MM cells[107]. Furthermore, inhibition of autophagy with 3-methyladenine augmented Oridonin-dependent apoptosis by regulating ROS and SIRT1 in MM cells[108]. Beclin1 inhibition extinguishes both autophagy and apoptosis, because Bcl-2 inhibits apoptosis by MOMP inhibition and stops autophagy by tethering Beclin1 or AMBRA1[109]. It was claimed that HMGB1 disturbs the equilibration between Beclin1 and Bcl-2 in the resistance to Bortezomib in MM[101]. AMBRA1 induces autophagy and suppresses apoptosis[110]. UVRAG induces autophagy by activating Beclin1, while Bif1 induces tumor suppression via autophagy[111]. In addition, a high expression of p38/MAPK engenders autophagy, while a low expression causes apoptosis[112]. STAT3 regulates many autophagy-related genes, such as FOXO1/3, by sequestering eIF2AK2 to fulfill this function[113]. It was suggested that the nutritional status marker mTORC1 could be responsible for drug resistance in MM via the AKT/ERK signaling pathway[114]. In a study with MM patients and cell lines, it appeared that gene expressions related to the PI3K/AKT/mTOR pathway and autophagy were higher in resistant groups[115]. Additionally, it was also unveiled that inhibition of PI3K/AKT/mTOR signaling promotes autophagy and apoptosis[115]. Autophagy can also induce apoptosis by aggregation of caspase 8[116]. Although p53 is an important biomarker in cancer, its role in the control of cell cycle and apoptosis in MM was clarified[105].
Recent studies reveal that MM cells can reshape cellular traffic mechanisms to combat ER stress. FAM46C, which is mutated in more than 10% of MM patients, is frequently emphasized. In a study on MM cells, it was reported that reactivation of FAM46C triggered apoptosis and ER stress by forming a complex with FNDC3A, an ER-associated protein[117]. The FAM46C and FNDC3A complex increases the exocytosis of lysosomes, which causes changes in cellular traffic and secretion[117]. This leads to aggregate accumulation and negative regulation of autophagy in MM cells[117]. Eventually, MM cell-specific FAM46C/FNDC3A-mediated tumor suppression occurs by induction of apoptosis[117]. Another study highlights the tumor suppressor and therapeutic role of FAM46C[118]. FAM46 interaction with FNDC3 proteins stabilizes ER-targeted protein mRNAs and increases ER growth and Ig secretion[118]. However, when the proteasomal balance is disrupted, p62 associates with FAM46C and prevents its interaction with FNDC3 proteins[118]. The p62/FAM46C/FNDC3 ensures the survival of MM cells, and targeting this pathway with proteasome inhibitors appears as a mechanism that enables selective cell death.
THERAPEUTICS FOR AUTOPHAGY IN MM
Autophagy plays a crucial role in the development of MM, a disease where plasma cells produce large quantities of immunoglobulin. Combining autophagy inhibition with proteasome inhibitors is an effective MM treatment. Since autophagy can work in both directions, strategies to induce or inhibit autophagy could be helpful in the treatment of MM. Benefits can be obtained by inhibiting the tumor mass-preserving effect of autophagy or by directly triggering autophagy in MM cells. Therefore, suppressing the PI3K/AKT/mTOR signaling pathway to induce autophagy in MM cells may be a therapeutic option. The PI3K/AKT/mTOR signaling pathway is associated with p38/MAPK, PTEN, p53, SIRT1, IGF-1 and ROS. Since the MAPK/ERK signaling pathway is in connection with mTOR, its suppression in MM may inhibit autophagy. JNK may have a tumor suppressor task due to its capacity to regulate the function of Beclin1. Ca2+ itself, as a signaling molecule, can trigger autophagy through its ability to activate Beclin1 and AMPK. The above-mentioned signaling pathways can be regulated with drugs to achieve the desired autophagy benefit in MM. For this reason, Table 1 lists drugs that can inhibit or induce autophagy by various mechanisms, especially in MM. Table 2 provides an overview of the most promising autophagy modulators clinically tested in MM patients.
Autophagy modulators and functioning mechanisms in MM
| Mechanism | Drug | Effect | Ref. |
| AMPK activators | Metformin | Activation | [119-121] |
| Spermidine | [122] | ||
| AMPK and mTOR inhibitors | Resveratrol | Activation | [123,124] |
| Autophagy flux inhibitors | Elaiophylin | Inhibition | [125] |
| 4-Acetylantroquinonol B | [126] | ||
| Autophagy inducer | Tat-Beclin1 peptide | Activation | [127] |
| BTK inhibitors | Ibrutinib | Activation | [128] |
| Ca2-ATPase inhibitors | Thapsigargin | Inhibition | [129,130] |
| Class I PI3K inhibitors | CH5132799 | Activation | [131] |
| GDC-0941 | [132] | ||
| Class III PI3K inhibitors | 3-Methyladenine | Inhibition | [107,108,133,134] |
| Wortmannin | [135] | ||
| LY294002 | [136] | ||
| HDAC inhibitors | Vorinostat | Activation | [137,138] |
| Lysosomal alkalizers | Chloroquine | Inhibition | [107,139,140] |
| Hydroxychloroquine | [66,134] | ||
| Lys05 | [141] | ||
| Lysosome membrane permeabilization | Thymoquinone | Inhibition | [142] |
| mTORC1/2 inhibitors | Rapamycin | Inhibition | [124,134] |
| Everolimus | [143,144] | ||
| Deforolimus | [145] | ||
| Temsirolimus | [146] | ||
| AZD8055 | Activation | [147] | |
| Torin1 | [148] | ||
| PP242 | [149] | ||
| PI3K/AKT inhibitors | Perifosine | Activation | [150] |
| PI3K/mTOR inhibitors | Apitolisib | Activation | [151] |
| Protease inhibitor | Pepstatin A | Inhibition | [152] |
| E64d | [68,153] | ||
| Vacuolar H-ATPase inhibitors | Bafilomycin A1 | Inhibition | [140,154] |
| Concanamycin A | [155] | ||
| VPS34 kinase inhibitors | Paclitaxel | Inhibition | [156,157] |
Summary of autophagy therapeutics in MM clinical trial
| Title | Therapeutics | Phase | Publication | Identifier |
| A pilot study of infusional Cyclophosphamide and pulse Dexamethasone with Rapamycin or Hydroxychloroquine in patients with relapsed or refractory multiple myeloma | Hydroxychloroquine Rapamycin | Early phase 1 | - | NCT01396200 |
| A Phase I/II trial of Hydroxychloroquine added to Bortezomib for relapsed/refractory myeloma | Bortezomib Hydroxychloroquine | Phase 1 | [158] | NCT00568880 |
| A Phase I dose escalation study of Hydroxychloroquine with infusional Cyclophosphamide, pulse Dexamethasone, and Rapamycin in patients with relapsed or refractory multiple myeloma | Cyclophosphamide Dexamethasone Hydroxychloroquine Sirolimus | Phase 1 | - | NCT01689987 |
| A Phase II, trial of Chloroquine in combination with VELCADE and Cyclophosphamide in patients with relapsed and refractory myeloma | Velcade Cyclophosphamide Chloroquine | Phase 2 | [139] | NCT01438177 |
| A Phase 1B/2 multicenter, open label, dose-escalation study to determine the maximum tolerated dose, safety, and efficacy of ACY-1215 (RICOLINOSTAT) in combination with Pomalidomide and low-dose Dexamethasone in patients with relapsed and refractory multiple myeloma | Ricolinostat Pomalidomide Dexamethasone | Phase 1 Phase 2 | - | NCT01997840 |
| A Phase 1/2, open-label, multicenter study of ACY-1215 administered orally as monotherapy and in combination with Bortezomib and Dexamethasone for the treatment of relapsed or relapsed/refractory multiple myeloma | Ricolinostat | Phase 1 Phase 2 | [159] | NCT01323751 |
| A Phase 1/2, open-label, multicenter study of ACY-1215 (Ricolinostat) in combination with Lenalidomide and Dexamethasone for the treatment of relapsed or relapsed/refractory multiple myeloma | Ricolinostat Lenalidomide Dexamethasone | Phase 1 | [160] | NCT01583283 |
| An open-label phase I study of the safety and efficacy of RAD001 in combination with Lenalidomide in the treatment of subjects with relapsed and relapsed/refractory multiple myeloma | Everolimus Lenalidomide | Phase 1 | [161] | NCT00729638 |
| Phase II trial of RAD001 in relapsed/refractory multiple myeloma | Everolimus | Phase 2 | - | NCT00618345 |
| An open-label phase 1 study of Metformin and Nelfinavir in combination with Bortezomib in patients with relapsed and/or refractory multiple myeloma | Bortezomib Metformin Nelfinavir | Phase 1 | - | NCT03829020 |
| Phase II trial, open label, clinical activity of Metformin in combination with high-dose of Dexamethasone (HDdexa) in patients with relapsed/refractory multiple myeloma | Metformin Dexamethasone | Phase 2 | [120] | NCT02967276 |
Lysosomal alkalizers
Lysosomes are important organelles of autophagy, and changes in pH levels prevent the activity of lysosomal enzymes and the fusion of autophagosomes with lysosomes. As mentioned before, the life of MM cells is dependent on the protein synthesis cycle. Therefore, the combination of lysosomal alkalizers and proteasome inhibitors or alkylating agents is a powerful therapeutic option for the selective killing of MM cells. It is known that Hydroxychloroquine, which is widely used in the treatment of malaria, can inhibit autophagy in cancer cells[162]. The combination of Bortezomib and Hydroxychloroquine increased cellular cytotoxicity via autophagy in MM patient samples[66]. In the Phase 1 study of MM patients, the same treatment combination not only improved disease symptoms but also reduced side effects[158]. In Phase II trials, whose Chloroquine was administered in combination with Bortezomib and Cyclophosphamide, a curative effect was shown for relapsed and refractory MM patients[139].
HDAC inhibitors
The performance of the HDAC6 inhibitor Panobinostat added to the MM treatment protocol in Phase 3 trials is clear[163] because HDAC6 inhibitors work together with UPR in toxic protein aggregation to promote autophagy. Additionally, HATs and HDACs can control the acetylation status of autophagy-related genes. An example of HDAC inhibitors inducing autophagy is the inactivation of mTOR by Vorinostat. Autophagy is suppressed by phosphorylating ULK1 through mTOR. The HDAC inhibitor Vorinostat triggers autophagy in tumor cells through the production of ROS[164]. The combination of Suberoylanilide hydroxamic acid (Vorinostat), 17-allylamino-17-demethoxy-geldanamycin (Tanespimycin), and Clonazepam overcame the complication of peripheral neuropathy by inducing autophagy via HSP70 or LAMP-2A[165]. Moreover, the efficacy of the combination of Clarithromycin, a high-spectrum antibiotic, in MM and various cancers has been previously reported[166]. Clarithromycin’s suppression of autophagy by reducing cytokines such as IL-6, which is an important factor in MM development and drug resistance, makes it a possible candidate. It was shown that the combination of Bortezomib, Lenalidomide, and Clarithromycin would also be suitable for therapy without Dexamethasone in MM patients with diabetes[167]. The combination of Vorinostat, Bortezomib, and Clarithromycin in MM cells causes cell death and upregulation of CHOP genes that stimulate ER stress[137]. In this sense, combinations of proteasome inhibitors and next-generation HDAC6 inhibitors may serve as therapeutic options to increase autophagy in MM cells.
Vacuolar H-ATPase inhibitors
Vacuolar type H-ATPases are pumps responsible for regulating intracellular organelle pH. Inhibiting these pumps with specific inhibitors blocks autophagy and prevents cell metastasis. The combined effect of Bortezomib and Bafilomycin A1 in MM cells is more cytotoxic than their use alone, which reveals the link between UPR, autophagy, and ER stress[154]. Furthermore, a combination of Ixazomib and autophagy inhibitors Bafilomycin A and Chloroquine increases cytotoxicity and cell death via JNK[140]. Therefore, the combination of vacuolar-type H-ATPase inhibitors with proteasome inhibitors stands as a potential therapeutic option. Additionally, inhibition of autophagic flux is also a treatment option. In a recent study, the treatment of p53 mutant MM cells with Elaiophylin suppressed autophagy and resulted in cell death by apoptosis[125].
AMPK activators and mTOR inhibitors
AMPK plays a critical role in responding to stress caused by metabolic disorders. It helps in maintaining the balance between anabolic and catabolic reactions, which is crucial for the stability of the cell and energy preservation. In cancer treatment, AMPK activators are used to trigger autophagy and cell death. In peripheral neuropathy, one of the best-known side effects of Bortezomib treatment in patients, pathological changes occur in Schwann cells. Metformin, which is used as an antidiabetic drug, in combination with Bortezomib, suppressed GRP78 and promoted autophagy[120]. Furthermore, Metformin was recently shown to induce apoptosis and necrosis in MM cells and mouse models[119]. Additionally, Metformin applied to MM cell lines and mouse models promoted cell proliferation arrest and autophagy by AMPK and mTORC1/2 regulation[121]. Thus, AMPK activators are a promising option for inducing autophagy by selectively targeting MM cells.
mTOR plays a cornerstone role in autophagy by regulating proliferation and metabolism. Resveratrol induces cell cycle arrest, apoptosis, and autophagy. In a study conducted to prove this, it was emphasized that Resveratrol induces autophagy and apoptosis in MM cells by inhibiting AMPK and mTOR[123].
PI3K/AKT/mTOR inhibitors
The PI3K/AKT/mTOR signaling pathway promotes the survival and proliferation of tumor cells. Therefore, inhibition of one or more proteins on the signaling pathway causes tumor regression. A report on testing autophagy-regulating drugs in MM cells stated that Rapamycin can induce apoptosis and autophagy, Hydroxychloroquine can inhibit autophagy and induce apoptosis, and 3-methyladenine can only inhibit autophagy[134]. Furthermore, the combination of 3-methyladenine and VEGF inhibitor Bevacizumab in MM cells promoted apoptosis[133]. In a recent study, NVP-BEZ235 was discovered to induce autophagy through the mTOR2-Akt-FOXO3a-BNIP3 signaling pathway in MM cell lines, mouse models, and primary samples[168]. Targeting the PI3K/AKT/mTOR signaling pathway, which is central to cell life and death choices, is a good strategy and a powerful therapeutic option for clinicians.
Other drugs or compounds
Glycosphingolipids located outside the cell membrane are important in cell adhesion, cell-cell interactions, and oncogenesis. In MM and MGUS preclinical models, it was shown that Eliglustat can inhibit autophagy through the glycosphingolipid mechanism, thereby maintaining osteoclastogenesis and preventing bone loss[169]. The tumor burden-reducing effect of the combination of hexokinase-2 inhibitor 3-bromopyruvate and Bortezomib was published[87]. It was reported that Solamargine alone induces autophagy, and its combination with Bortezomib may be a good treatment strategy[170]. The induction of autophagy in MM cells by inhibitors of the isoprenoid biosynthesis pathway is a potential target for developing new therapeutic drugs[171].
Dihydroartemisinin induced autophagy and apoptosis via WNT/β-catenin and P38/MAPK in MM cell lines and mouse models[172]. Hsp90 inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin triggered autophagy over mTOR inhibition in MM cells[173]. Clioquinol engendered autophagy by causing mTOR downregulation in MM and leukemia cells[174].
Trifluoperazine, an antipsychotic drug, caused the inhibition of autophagy by NUPR1 overexpression in MM cells and led to the induction of apoptosis[175]. Betulinic acid performs its cytotoxicity in MM cells by inducing autophagy or apoptosis via the PP2A switch[176]. It was reported that Fingolimod regulates apoptosis by inducing autophagy in MM cells[177].
Drug discovery
Currently, there is no known treatment option to be considered definitive for targeting autophagy in cancer. This is because traditional cell cultures do not fully describe the complex interaction between cancer cells, other cells, and the microenvironment. However, 3D cell cultures are more effective in depicting the tumor microenvironment than traditional cultures, and they better reflect the differentiation and proliferation of tumor cells[178]. Microfluidic systems are biomedical platforms that enable the controlled distribution of small amounts of liquids, which facilitates the simultaneous execution of multiple tests[179]. These devices allow for more accurate and quicker studies of the tumor microenvironment, metastasis, and drug resistance[1]. They also enable drug screening to be carried out rapidly and precisely, which assists in identifying new drug candidates with more efficiency[180].
CONCLUSION
MM is a disease of plasma cells and is very sensitive to protein turnover processes. Autophagy, a mechanism linked to cell death, contributes to the progression of the disease by activating the UPR system in MM cells. Proteasome inhibitors, commonly employed in MM, induce UPR and ER stress. However, inhibiting proteasomes not only triggers autophagy but also leads to drug resistance. Therefore, there is a close interplay among UPR, autophagy, apoptosis, and drug resistance. The combination of proteasome inhibitors such as Bortezomib and Carfilzomib, which are frequently used in the treatment of MM, and autophagy inhibitors can effectively induce cell death. Additionally, agents that inhibit heat shock proteins also show therapeutic potential in MM by initiating autophagy. Furthermore, as in all cancer processes, autophagy in MM is affected by BMME, DNA repair, and transcriptional regulation, crucial aspects in the development of MM disease. The addition of autophagy inhibitors to agents that cause DNA damage will prevent MM cells from evading death. The widespread use of HDAC6 inhibitors in treating MM cells deactivates the UPR protection and induces autophagy. Clinically, PI3K/AKT/mTOR inhibitors prove effective by restricting MM cell growth, making cell death inevitable. Furthermore, even in normal cells, stress-causing starvation and hypoxia conditions trigger autophagy. Consequently, VEGF and EGFR inhibitors, which will prevent MM cells from feeding, and HIF1-α inhibitors, which will cause oxygen deprivation, become beneficial. Moreover, small molecules that target genes involved in the autophagy mechanism also have a therapeutic potential. There is a considerable volume of both completed and ongoing clinical trials exploring this avenue. However, a clinically promising drug has not yet been released, and there is no standardized autophagy treatment yet. In this regard, leveraging 3D and microfluidic systems holds the potential to develop disease models. These platforms have the capability to generate results more rapidly than traditional clinical trials. Additionally, reconstructing disease models with 3D models and microfluidic systems can lead to the identification of new autophagy modulators[1,178]. Moreover, it is possible to conduct tests on new agents before administering them to the patient and to develop appropriate strategies that can be tailored according to the patient's needs. This enables the utilization of autophagy, similar to apoptosis, in halting or even eradicating the progression of MM.
DECLARATIONS
Authors’ contributions
Conceptualized, did the literature review, designed the review, and drafted the manuscript: Kozalak G
Edited the manuscript, supervised the process, and critically revised the article: Koşar A
Both authors read and agreed with the published version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This research was funded by TUBITAK (Turkish Scientific and Technological Council) grant number 121C417.
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2023.
REFERENCES
1. Kozalak G, Bütün İ, Toyran E, Koşar A. Review on Bortezomib resistance in multiple myeloma and potential role of emerging technologies. Pharmaceuticals 2023;16:111.
2. Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol 2020;95:548-67.
3. Yang T, Liu X, Kumar SK, Jin F, Dai Y. Decoding DNA methylation in epigenetics of multiple myeloma. Blood Rev 2022;51:100872.
4. Gkoliou G, Agathangelidis A, Karakatsoulis G, et al. Differences in the immunoglobulin gene repertoires of IgG versus IgA multiple myeloma allude to distinct immunopathogenetic trajectories. Front Oncol 2023;13:1123029.
5. Ho M, Patel A, Hanley C, et al. Exploiting autophagy in multiple myeloma. J Cancer Metastasis Treat 2019;5:70.
6. Mateos MV, Ludwig H, Bazarbachi A, et al. Insights on multiple myeloma treatment strategies. Hemasphere 2019;3:e163.
7. Pinto V, Bergantim R, Caires HR, Seca H, Guimarães JE, Vasconcelos MH. Multiple myeloma: available therapies and causes of drug resistance. Cancers 2020;12:407.
8. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018;25:486-541.
9. Yan G, Elbadawi M, Efferth T. Multiple cell death modalities and their key features (Review). World Acad Sci J 2020;2:39-48.
11. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol 2021;18:1106-21.
12. Newton K, Wickliffe KE, Dugger DL, et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019;574:428-31.
13. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006;10:51-64.
14. Yun Z, Zhichao J, Hao Y, et al. Targeting autophagy in multiple myeloma. Leuk Res 2017;59:97-104.
15. Carroll RG, Martin SJ. Autophagy in multiple myeloma: what makes you stronger can also kill you. Cancer Cell 2013;23:425-6.
16. Bashiri H, Tabatabaeian H. Autophagy: a potential therapeutic target to tackle drug resistance in multiple myeloma. Int J Mol Sci 2023;24:6019.
17. Aquila S, Santoro M, Caputo A, Panno ML, Pezzi V, De Amicis F. The tumor suppressor PTEN as molecular switch node regulating cell metabolism and autophagy: implications in immune system and tumor microenvironment. Cells 2020;9:1725.
18. Al-Odat OS, Guirguis DA, Schmalbach NK, et al. Autophagy and apoptosis: current challenges of treatment and drug resistance in multiple myeloma. Int J Mol Sci 2022;24:644.
19. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010;221:3-12.
20. Song Y, Quach C, Liang C. UVRAG in autophagy, inflammation, and cancer. Autophagy 2020;16:387-8.
21. Aman Y, Schmauck-Medina T, Hansen M, et al. Autophagy in healthy aging and disease. Nat Aging 2021;1:634-50.
22. Jaganathan S, Malek E, Vallabhapurapu S, Vallabhapurapu S, Driscoll JJ. Bortezomib induces AMPK-dependent autophagosome formation uncoupled from apoptosis in drug resistant cells. Oncotarget 2014;5:12358-70.
23. Nikesitch N, Lee JM, Ling S, Roberts TL. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin Transl Immunology 2018;7:e1007.
24. Harmon KA, Roman S, Lancaster HD, et al. Structural and ultrastructural analysis of the multiple myeloma cell niche and a patient-specific model of plasma cell dysfunction. Microsc Microanal 2022;28:254-64.
25. Jung G, Roh J, Lee H, et al. Autophagic markers BECLIN 1 and LC3 are associated with prognosis of multiple myeloma. Acta Haematol 2015;134:17-24.
26. Puglisi F, Parrinello NL, Giallongo C, et al. Plasticity of high-density neutrophils in multiple myeloma is associated with increased autophagy via STAT3. Int J Mol Sci 2019;20:3548.
27. Clavero E, Sanchez-Maldonado JM, Macauda A, et al. Polymorphisms within autophagy-related genes as susceptibility biomarkers for multiple myeloma: a meta-analysis of three large cohorts and functional characterization. Int J Mol Sci 2023;24:8500.
29. Kumar AV, Mills J, Lapierre LR. Selective autophagy receptor p62/SQSTM1, a pivotal player in stress and aging. Front Cell Dev Biol 2022;10:793328.
30. Milan E, Perini T, Resnati M, et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015;11:1161-78.
31. Auner HW, Cenci S. Recent advances and future directions in targeting the secretory apparatus in multiple myeloma. Br J Haematol 2015;168:14-25.
32. Conway KL, Kuballa P, Khor B, et al. ATG5 regulates plasma cell differentiation. Autophagy 2013;9:528-37.
33. Pengo N, Scolari M, Oliva L, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 2013;14:298-305.
34. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006;107:4907-16.
35. Borjan B, Kern J, Steiner N, Gunsilius E, Wolf D, Untergasser G. Spliced XBP1 levels determine sensitivity of multiple myeloma cells to proteasome inhibitor Bortezomib independent of the unfolded protein response mediator GRP78. Front Oncol 2019;9:1530.
36. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci 2019;6:11.
37. Cairrão F, Santos CC, Le Thomas A, Marsters S, Ashkenazi A, Domingos PM. Pumilio protects Xbp1 mRNA from regulated Ire1-dependent decay. Nat Commun 2022;13:1587.
38. Zheng W, Xie W, Yin D, Luo R, Liu M, Guo F. ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling. Cell Commun Signal 2019;17:42.
39. Park SM, Kang TI, So JS. Roles of XBP1s in transcriptional regulation of target genes. Biomedicines 2021;9:791.
40. Harnoss JM, Le Thomas A, Shemorry A, et al. Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc Natl Acad Sci U S A 2019;116:16420-9.
41. Mimura N, Fulciniti M, Gorgun G, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012;119:5772-81.
42. Dang J, Bian X, Ma X, et al. ORMDL3 facilitates the survival of splenic B cells via an ATF6α-endoplasmic reticulum stress-Beclin1 autophagy regulatory pathway. J Immunol 2017;199:1647-59.
43. Sharma RB, Darko C, Alonso LC. Intersection of the ATF6 and XBP1 ER stress pathways in mouse islet cells. J Biol Chem 2020;295:14164-77.
44. Sintes J, Gentile M, Zhang S, et al. mTOR intersects antibody-inducing signals from TACI in marginal zone B cells. Nat Commun 2017;8:1462.
45. Hu R, Warri A, Jin L, et al. NF-κB signaling is required for XBP1 (unspliced and spliced)-mediated effects on antiestrogen responsiveness and cell fate decisions in breast cancer. Mol Cell Biol 2015;35:379-90.
46. Verzella D, Pescatore A, Capece D, et al. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis 2020;11:210.
47. Shi Z, Yu X, Yuan M, et al. Activation of the PERK-ATF4 pathway promotes chemo-resistance in colon cancer cells. Sci Rep 2019;9:3210.
48. Jang JE, Eom JI, Jeung HK, et al. PERK/NRF2 and autophagy form a resistance mechanism against G9a inhibition in leukemia stem cells. J Exp Clin Cancer Res 2020;39:66.
49. White-Gilbertson S, Hua Y, Liu B. The role of endoplasmic reticulum stress in maintaining and targeting multiple myeloma: a double-edged sword of adaptation and apoptosis. Front Genet 2013;4:109.
50. Michallet AS, Mondiere P, Taillardet M, Leverrier Y, Genestier L, Defrance T. Compromising the unfolded protein response induces autophagy-mediated cell death in multiple myeloma cells. PLoS One 2011;6:e25820.
51. Bagratuni T, Patseas D, Mavrianou-Koutsoukou N, et al. Characterization of a PERK kinase inhibitor with anti-myeloma activity. Cancers 2020;12:2864.
52. Allegra A, Casciaro M, Barone P, Musolino C, Gangemi S. Epigenetic crosstalk between malignant plasma cells and the tumour microenvironment in multiple myeloma. Cancers 2022;14:2597.
53. Cippitelli M, Stabile H, Kosta A, et al. Role of NF-κB signaling in the interplay between multiple myeloma and mesenchymal stromal cells. Int J Mol Sci 2023;24:1823.
54. Gu J, Huang X, Zhang Y, Bao C, Zhou Z, Jin J. Cytokine profiles in patients with newly diagnosed multiple myeloma: survival is associated with IL-6 and IL-17A levels. Cytokine 2021;138:155358.
55. Sanchez E, Gillespie A, Tang G, et al. Soluble B-cell maturation antigen mediates tumor-induced immune deficiency in multiple myeloma. Clin Cancer Res 2016;22:3383-97.
56. Yan Y, Chen X, Wang X, et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J Exp Clin Cancer Res 2019;38:171.
57. Frassanito MA, De Veirman K, Desantis V, et al. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2016;30:640-8.
58. Liu Z, Xu J, He J, et al. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget 2015;6:34329-41.
59. Cea M, Cagnetta A, Fulciniti M, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012;120:3519-29.
60. Spaan I, Raymakers RA, van de Stolpe A, Peperzak V. Wnt signaling in multiple myeloma: a central player in disease with therapeutic potential. J Hematol Oncol 2018;11:67.
61. Liu YS, Liu XB, Qiu YY, Lan T, Chen Y. Molecular mechanism of Wnt signal pathway in multiple myeloma cell line H929 cell autophagy. Eur Rev Med Pharmacol Sci 2018;22:3327-32.
62. Pereira IEG, da Costa Cacemiro M, Cominal JG, Barretto GD, de Castro FA. Role of endothelial cells in hematological malignancies. EJMO 2022;6:210-8.
63. Tenreiro MM, Correia ML, Brito MA. Endothelial progenitor cells in multiple myeloma neovascularization: a brick to the wall. Angiogenesis 2017;20:443-62.
64. Wang X, Zhang Z, Yao C. Angiogenic activity of mesenchymal stem cells in multiple myeloma. Cancer Invest 2011;29:37-41.
65. Rao L, Giannico D, Leone P, et al. HB-EGF-EGFR signaling in bone marrow endothelial cells mediates angiogenesis associated with multiple myeloma. Cancers 2020;12:173.
66. Di Lernia G, Leone P, Solimando AG, et al. Bortezomib treatment modulates autophagy in multiple myeloma. J Clin Med 2020;9:552.
67. Muz B, Azab F, De La Puente P, Ghobrial IM, Vij R, Azab AK. Hypoxia induces drug resistance in multiple myeloma. Blood 2013;122:1852.
68. Lamy L, Ngo VN, Emre NCT, et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013;23:435-49.
69. Kozalak G, Öksüzoğlu E. Efficacy of multi-drug resistance transporters and glutathione S-transferase P-1 at developing bortezomib resistance in multiple myeloma cell lines. Lat Am J Pharm 2021;40:2709-16. Available from: https://www.latamjpharm.org/resumenes/40/11/LAJOP_40_11_1_23.pdf. [Last accessed on 22 Dec 2023]
70. Milani M, Rzymski T, Mellor HR, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 2009;69:4415-23.
71. Nikesitch N, Rebeiro P, Ho LL, et al. The role of chaperone-mediated autophagy in Bortezomib resistant multiple myeloma. Cells 2021;10:3464.
72. Lu Y, Wang Y, Xu H, Shi C, Jin F, Li W. Profilin 1 induces drug resistance through Beclin1 complex-mediated autophagy in multiple myeloma. Cancer Sci 2018;109:2706-16.
73. Zhang M, He J, Liu Z, et al. Anti-β2-microglobulin monoclonal antibodies overcome bortezomib resistance in multiple myeloma by inhibiting autophagy. Oncotarget 2015;6:8567-78.
74. Huang X, Cao W, Yao S, et al. NEDD4L binds the proteasome and promotes autophagy and bortezomib sensitivity in multiple myeloma. Cell Death Dis 2022;13:197.
75. Xia J, He Y, Meng B, et al. NEK2 induces autophagy-mediated bortezomib resistance by stabilizing Beclin-1 in multiple myeloma. Mol Oncol 2020;14:763-78.
76. Abdel Malek MAY, Jagannathan S, Malek E, et al. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 2015;6:3098-110.
77. Wu H, Liu C, Yang Q, et al. MIR145-3p promotes autophagy and enhances bortezomib sensitivity in multiple myeloma by targeting HDAC4. Autophagy 2020;16:683-97.
78. Tucci M, Stucci S, Savonarola A, et al. An imbalance between Beclin-1 and p62 expression promotes the proliferation of myeloma cells through autophagy regulation. Exp Hematol 2014;42:897-908.e1.
79. Sola B, Poirot M, de Medina P, et al. Antiestrogen-binding site ligands induce autophagy in myeloma cells that proceeds through alteration of cholesterol metabolism. Oncotarget 2013;4:911-22.
80. Riz I, Hawley TS, Hawley RG. KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to carfilzomib resistance in multiple myeloma models. Oncotarget 2015;6:14814-31.
81. Pan YZ, Wang X, Bai H, Wang CB, Zhang Q, Xi R. Autophagy in drug resistance of the multiple myeloma cell line RPMI8226 to doxorubicin. Genet Mol Res 2015;14:5621-9.
82. Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 2009;29:2570-81.
83. Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ 2008;15:1572-81.
84. Gastelum G, Veena M, Lyons K, et al. Can targeting hypoxia-mediated acidification of the bone marrow microenvironment kill myeloma tumor cells? Front Oncol 2021;11:703878.
85. Ikeda S, Tagawa H. Impact of hypoxia on the pathogenesis and therapy resistance in multiple myeloma. Cancer Sci 2021;112:3995-4004.
86. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer cell metabolism in hypoxia: role of HIF-1 as key regulator and therapeutic target. Int J Mol Sci 2021;22:5703.
87. Ikeda S, Abe F, Matsuda Y, Kitadate A, Takahashi N, Tagawa H. Hypoxia-inducible hexokinase-2 enhances anti-apoptotic function via activating autophagy in multiple myeloma. Cancer Sci 2020;111:4088-101.
88. Bhaskar A, Tiwary BN. Hypoxia inducible factor-1 alpha and multiple myeloma. Int J Adv Res 2016;4:706-15.
89. Darawshi O, Muz B, Naamat SG, et al. An mTORC1 to HRI signaling axis promotes cytotoxicity of proteasome inhibitors in multiple myeloma. Cell Death Dis 2022;13:969.
90. Eliopoulos AG, Havaki S, Gorgoulis VG. DNA damage response and autophagy: a meaningful partnership. Front Genet 2016;7:204.
91. Alexander A, Kim J, Walker CL. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010;6:672-3.
92. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132-41.
93. Desantis A, Bruno T, Catena V, et al. Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J 2015;34:1214-30.
94. Kim JS, Ro SH, Kim M, et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci Rep 2015;5:9502.
95. Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 2009;29:2594-608.
96. Rodríguez-Vargas JM, Ruiz-Magaña MJ, Ruiz-Ruiz C, et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 2012;22:1181-98.
97. Feng Z, Hu W, de Stanchina E, et al. The regulation of AMPK β1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res 2007;67:3043-53.
98. Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ 2012;19:788-97.
99. Wang L, Zhang H, Sun M, Yin Z, Qian J. High mobility group box 1-mediated autophagy promotes neuroblastoma cell chemoresistance. Oncol Rep 2015;34:2969-76.
100. Guo X, He D, Zhang E, et al. HMGB1 knockdown increases MM cell vulnerability by regulating autophagy and DNA damage repair. J Exp Clin Cancer Res 2018;37:205.
101. Roy M, Liang L, Xiao X, et al. Lycorine downregulates HMGB1 to inhibit autophagy and enhances Bortezomib activity in multiple myeloma. Theranostics 2016;6:2209-24.
102. Gao D, Lv AE, Li HP, Han DH, Zhang YP. LncRNA MALAT-1 elevates HMGB1 to promote autophagy resulting in inhibition of tumor cell apoptosis in multiple myeloma. J Cell Biochem 2017;118:3341-8.
103. Balsas P, López-Royuela N, Galán-Malo P, Anel A, Marzo I, Naval J. Cooperation between Apo2L/TRAIL and Bortezomib in multiple myeloma apoptosis. Biochem Pharmacol 2009;77:804-12.
104. Al-Odat O, von Suskil M, Chitren RJ, et al. Mcl-1 inhibition: managing malignancy in multiple myeloma. Front Pharmacol 2021;12:699629.
105. Öksüzoğlu E, Kozalak G. Inhibition of apoptosis may lead to the development of bortezomib resistance in multiple myeloma cancer cells. Turkish J Biochem 2021;46:65-71.
106. Roy P, Sarkar UA, Basak S. The NF-κB activating pathways in multiple myeloma. Biomedicines 2018;6:59.
107. Hoang B, Benavides A, Shi Y, Frost P, Lichtenstein A. Effect of autophagy on multiple myeloma cell viability. Mol Cancer Ther 2009;8:1974-84.
108. Zeng R, Chen Y, Zhao S, Cui GH. Autophagy counteracts apoptosis in human multiple myeloma cells exposed to oridonin in vitro via regulating intracellular ROS and SIRT1. Acta Pharmacol Sin 2012;33:91-100.
109. Strappazzon F, Vietri-Rudan M, Campello S, et al. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J 2011;30:1195-208.
110. Sun WL. Ambra1 in autophagy and apoptosis: implications for cell survival and chemotherapy resistance. Oncol Lett 2016;12:367-74.
111. Mohammadi K, Salimi M, Angaji SA, Saniotis A, Mahjoobi F. Association study of Bif-1 gene expression with histopathological characteristics and hormone receptors in breast cancer. BMC Womens Health 2022;22:471.
112. Sui X, Kong N, Ye L, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett 2014;344:174-9.
114. Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk Lymphoma 2018;59:2524-34.
115. Fu YF, Liu X, Gao M, Zhang YN, Liu J. Endoplasmic reticulum stress induces autophagy and apoptosis while inhibiting proliferation and drug resistance in multiple myeloma through the PI3K/Akt/mTOR signaling pathway. Oncotarget 2017;8:61093-106.
116. Jin Z, Li Y, Pitti R, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009;137:721-35.
117. Manfrini N, Mancino M, Miluzio A, et al. FAM46C and FNDC3A are multiple myeloma tumor suppressors that act in concert to impair clearing of protein aggregates and autophagy. Cancer Res 2020;80:4693-706.
118. Fucci C, Resnati M, Riva E, et al. The interaction of the tumor suppressor FAM46C with p62 and FNDC3 proteins integrates protein and secretory homeostasis. Cell Rep 2020;32:108162.
119. Wu Z, Wu L, Zou L, Wang M, Liu X. Metformin induces myeloma cells necrosis and apoptosis and it is considered for therapeutic use. J Chemother 2023;35:131-41.
120. Jagannathan S, Abdel-Malek MAY, Malek E, et al. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia 2015;29:2184-91.
121. Wang Y, Xu W, Yan Z, et al. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Cancer Res 2018;37:63.
122. Hibino S, Eto S, Hangai S et al. Tumor cell-derived spermidine is an oncometabolite that suppresses TCR clustering for intratumoral CD8+ T cell activation. Proc Natl Acad Sci U S A 2023;120:e2305245120.
123. Ma R, Yu D, Peng Y, et al. Resveratrol induces AMPK and mTOR signaling inhibition-mediated autophagy and apoptosis in multiple myeloma cells. Acta Biochim Biophys Sin 2021;53:775-83.
124. Jin HG, Wu GZ, Wu GH, Bao YG. Combining the mammalian target of rapamycin inhibitor, rapamycin, with resveratrol has a synergistic effect in multiple myeloma. Oncol Lett 2018;15:6257-64.
125. Wang G, Zhou P, Chen X, et al. The novel autophagy inhibitor elaiophylin exerts antitumor activity against multiple myeloma with mutant TP53 in part through endoplasmic reticulum stress-induced apoptosis. Cancer Biol Ther 2017;18:584-95.
126. Wu CH, Ou CH, Yen IC, Lee SY. 4-acetylantroquinonol B inhibits osteoclastogenesis by inhibiting the autophagy pathway in a simulated microgravity model. Int J Mol Sci 2020;21:6971.
127. Li N, Zhang X, Chen J, Gao S, Wang L, Zhao Y. Perturbation of autophagy by a Beclin 1-targeting stapled peptide induces mitochondria stress and inhibits proliferation of pancreatic cancer cells. Cancers 2023;15:953.
128. Ailawadhi S, Parrondo RD, Moustafa MA, et al. Ibrutinib, lenalidomide and dexamethasone in patients with relapsed and/or refractory multiple myeloma: phase I trial results. Hematol Oncol 2022;40:695-703.
129. Domenger A, Choisy C, Baron L, et al. The Sec61 translocon is a therapeutic vulnerability in multiple myeloma. EMBO Mol Med 2022;14:e14740.
130. Shi Y, Yang Y, Hoang B, et al. Retracted article: Therapeutic potential of targeting IRES-dependent c-myc translation in multiple myeloma cells during ER stress. Oncogene 2016;35:1015-24.
131. Suder A. Final results of phase I study of the oral class I PI3K inhibitor CH5132799 in patients with advanced solid tumours. Ann Oncol 2013;24:i33.
132. Kikuchi H, Amofa E, Mcenery M, et al. Inhibition of PI3K class IA kinases using GDC-0941 overcomes cytoprotection of multiple myeloma cells in the osteoclastic bone marrow microenvironment enhancing the efficacy of current clinical therapeutics. Cancers 2023;15:462.
133. Attar-Schneider O, Drucker L, Zismanov V, Tartakover-Matalon S, Rashid G, Lishner M. Bevacizumab attenuates major signaling cascades and eIF4E translation initiation factor in multiple myeloma cells. Lab Invest 2012;92:178-90.
134. Wang JZ, Chen JM, Zeng ZY, Qiu DB. [Effects of autophagy regulating drugs on proliferation, apoptosis and autophagy of multiple myeloma cells]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2018;26:817-23.
135. Hsu J, Shi Y, Krajewski S, et al. The AKT kinase is activated in multiple myeloma tumor cells. Blood 2001;98:2853-5.
136. Chen P, Wen X, Wang B, et al. PI3K/Akt inhibitor LY294002 potentiates homoharringtonine antimyeloma activity in myeloma cells adhered to stromal cells and in SCID mouse xenograft. Ann Hematol 2018;97:865-75.
137. Moriya S, Komatsu S, Yamasaki K, et al. Targeting the integrated networks of aggresome formation, proteasome, and autophagy potentiates ER stress-mediated cell death in multiple myeloma cells. Int J Oncol 2015;46:474-86.
138. Waldschmidt JM, Keller A, Ihorst G, et al. Safety and efficacy of vorinostat, bortezomib, doxorubicin and dexamethasone in a phase I/II study for relapsed or refractory multiple myeloma (VERUMM study: vorinostat in elderly, relapsed and unfit multiple myeloma). Haematologica 2018;103:e473-9.
139. Montanari F, Lu M, Marcus S, Saran A, Malankar A, Mazumder A. A phase II trial of chloroquine in combination with Bortezomib and Cyclophosphamide in patients with relapsed and refractory multiple myeloma. Blood 2014;124:5775.
140. Salimi A, Schroeder KM, Schemionek-Reinders M, et al. Targeting autophagy increases the efficacy of proteasome inhibitor treatment in multiple myeloma by induction of apoptosis and activation of JNK. BMC Cancer 2022;22:735.
141. McAfee Q, Zhang Z, Samanta A, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A 2012;109:8253-8.
142. Fatfat Z, Fatfat M, Gali-Muhtasib H. Therapeutic potential of thymoquinone in combination therapy against cancer and cancer stem cells. World J Clin Oncol 2021;12:522-43.
143. Günther A, Baumann P, Burger R, et al. Activity of everolimus (RAD001) in relapsed and/or refractory multiple myeloma: a phase I study. Haematologica 2015;100:541-7.
144. Liu H, Liu Y, Fu R, Shao Z. Everolimus shows synergistic anti-myeloma effects with bortezomib via the AKT/mTOR pathway. Blood 2017;130:5396.
145. Rizzieri DA, Feldman E, Dipersio JF, et al. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 2008;14:2756-62.
146. Ghobrial IM, Weller E, Vij R, et al. Weekly bortezomib in combination with temsirolimus in relapsed or relapsed and refractory multiple myeloma: a multicentre, phase 1/2, open-label, dose-escalation study. Lancet Oncol 2011;12:263-72.
147. Cirstea D, Santo L, Hideshima T, et al. Delineating the mTOR kinase pathway using a dual TORC1/2 inhibitor, AZD8055, in multiple myeloma. Mol Cancer Ther 2014;13:2489-500.
148. Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009;137:873-86.
149. Lamanuzzi A, Saltarella I, Desantis V, et al. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget 2018;9:20563-77.
150. Richardson PG, Nagler A, Ben-Yehuda D, et al. Randomized, placebo-controlled, phase 3 study of perifosine combined with bortezomib and dexamethasone in patients with relapsed, refractory multiple myeloma previously treated with bortezomib. EJHaem 2020;1:94-102.
151. Dolly SO, Wagner AJ, Bendell JC, et al. Phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2016;22:2874-84.
152. Sternke-Hoffmann R, Pauly T, Norrild RK, et al. Widespread amyloidogenicity potential of multiple myeloma patient-derived immunoglobulin light chains. BMC Biol 2023;21:21.
153. Savvidou I, Khong TT, Spencer A. The role of autophagy in multiple myeloma progression. Blood 2017;130:5355.
154. Kawaguchi T, Miyazawa K, Moriya S, et al. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress. Int J Oncol 2011;38:643-54.
155. Liu J, Hamrouni A, Wolowiec D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007;110:296-304.
156. Gazitt Y, Shaughnessy P, Rothenberg M. A phase II trial with gemcitabine and paclitaxel for the treatment of refractory and relapsed multiple myeloma patients. Oncol Rep 2006;16:877-84.
157. Veldhoen RA, Banman SL, Hemmerling DR, et al. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene 2013;32:736-46.
158. Vogl DT, Stadtmauer EA, Tan KS, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014;10:1380-90.
159. Vogl DT, Raje N, Jagannath S, et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with Bortezomib and Dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res 2017;23:3307-15.
160. Yee AJ, Bensinger WI, Supko JG, et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol 2016;17:1569-78.
161. Yee AJ, Hari P, Marcheselli R, et al. Outcomes in patients with relapsed or refractory multiple myeloma in a phase I study of everolimus in combination with lenalidomide. Br J Haematol 2014;166:401-9.
162. Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol 2009;625:220-33.
163. San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol 2014;15:1195-206.
164. Li J, Liu R, Lei Y, et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010;6:711-24.
165. Watanabe T, Nagase K, Chosa M, Tobinai K. Schwann cell autophagy induced by SAHA, 17-AAG, or clonazepam can reduce bortezomib-induced peripheral neuropathy. Br J Cancer 2010;103:1580-7.
166. Van Nuffel AM, Sukhatme V, Pantziarka P, Meheus L, Sukhatme VP, Bouche G. Repurposing drugs in oncology (ReDO)-clarithromycin as an anti-cancer agent. Ecancermedicalscience 2015;9:513.
167. Takemori N, Imai G, Hoshino K, Ooi A, Kojima M. A novel combination of bortezomib, lenalidomide, and clarithromycin produced stringent complete response in refractory multiple myeloma complicated with diabetes mellitus - clinical significance and possible mechanisms: a case report. J Med Case Rep 2018;12:40.
168. Ma Y, Jin Z, Yu K, Liu Q. NVP-BEZ235-induced autophagy as a potential therapeutic approach for multiple myeloma. Am J Transl Res 2019;11:87-105.
169. Leng H, Zhang H, Li L, et al. Modulating glycosphingolipid metabolism and autophagy improves outcomes in pre-clinical models of myeloma bone disease. Nat Commun 2022;13:7868.
170. Han Q, Bai H, Xu Y, et al. Solamargine induces autophagy-mediated apoptosis and enhances bortezomib activity in multiple myeloma. Clin Exp Pharmacol Physiol 2022;49:674-85.
171. Dykstra KM, Allen C, Born EJ, Tong H, Holstein SA. Mechanisms for autophagy modulation by isoprenoid biosynthetic pathway inhibitors in multiple myeloma cells. Oncotarget 2015;6:41535-49.
172. Wu X, Liu Y, Zhang E, et al. Dihydroartemisinin modulates apoptosis and autophagy in multiple myeloma through the P38/MAPK and Wnt/β-catenin signaling pathways. Oxid Med Cell Longev 2020;2020:6096391.
173. Palacios C, Martín-Pérez R, López-Pérez AI, Pandiella A, López-Rivas A. Autophagy inhibition sensitizes multiple myeloma cells to 17-dimethylaminoethylamino-17-demethoxygeldanamycin-induced apoptosis. Leuk Res 2010;34:1533-8.
174. Cao B, Li J, Zhou X, et al. Clioquinol induces pro-death autophagy in leukemia and myeloma cells by disrupting the mTOR signaling pathway. Sci Rep 2014;4:5749.
175. Li A, Chen X, Jing Z, Chen J. Trifluoperazine induces cellular apoptosis by inhibiting autophagy and targeting NUPR1 in multiple myeloma. FEBS Open Bio 2020;10:2097-106.
176. Zhou H, Luo W, Zeng C, et al. PP2A mediates apoptosis or autophagic cell death in multiple myeloma cell lines. Oncotarget 2017;8:80770-89.
177. Liao A, Hu R, Zhao Q, et al. Autophagy induced by FTY720 promotes apoptosis in U266 cells. Eur J Pharm Sci 2012;45:600-5.
178. Choi JR, Kozalak G, di Bari I, et al. In vitro human cancer models for biomedical applications. Cancers 2022;14:2284.
179. Mehta P, Rahman Z, Ten Dijke P, Boukany PE. Microfluidics meets 3D cancer cell migration. Trends Cancer 2022;8:683-97.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kozalak G, Koşar A. Autophagy-related mechanisms for treatment of multiple myeloma. Cancer Drug Resist 2023;6:838-57. http://dx.doi.org/10.20517/cdr.2023.108
AMA Style
Kozalak G, Koşar A. Autophagy-related mechanisms for treatment of multiple myeloma. Cancer Drug Resistance. 2023; 6(4): 838-57. http://dx.doi.org/10.20517/cdr.2023.108
Chicago/Turabian Style
Kozalak, Gül, Ali Koşar. 2023. "Autophagy-related mechanisms for treatment of multiple myeloma" Cancer Drug Resistance. 6, no.4: 838-57. http://dx.doi.org/10.20517/cdr.2023.108
ACS Style
Kozalak, G.; Koşar A. Autophagy-related mechanisms for treatment of multiple myeloma. Cancer Drug Resist. 2023, 6, 838-57. http://dx.doi.org/10.20517/cdr.2023.108
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 11
likes
Like This Article 11
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.