Article Text

Abstract
Background Checkpoint inhibitors targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) have demonstrated clinical efficacy in advanced melanoma, but only a subset of patients with inflamed tumors are responsive. Talimogene laherparepvec (T-VEC), a modified herpes simplex virus type 1 (HSV-1) expressing granulocyte-macrophage colony-stimulating factor (GM-CSF), is a first-in-class oncolytic immunotherapy approved for the treatment of melanoma and has been shown to inflame the tumor microenvironment. To evaluate the potential and mechanisms of T-VEC to elicit systemic antitumor immunity and overcome resistance to checkpoint inhibitors in murine tumor models, OncoVEXmGM-CSF was developed similarly to T-VEC, except the human GM-CSF transgene was replaced with murine GM-CSF. Previous work had demonstrated that OncoVEXmGM-CSF generated systemic antitumor immunity dependent on CD8+ T cells in an immune checkpoint-sensitive tumor cell model.
Methods A novel B16F10 syngeneic tumor model with both HSV-1−permissive subcutaneous tumors and HSV-1−refractory experimental lung metastasis was used to study the local and systemic effects of OncoVEXmGM-CSF treatment alone or in combination with checkpoint inhibitors.
Results Intratumoral injection of OncoVEXmGM-CSF in combination with an anti-CTLA-4 or anti-PD-1 blocking antibody led to increased tumor growth inhibition, a reduction in the number of lung metastases, and prolonged animal survival. OncoVEXmGM-CSF induced both neoantigen-specific and tumor antigen-specific T-cell responses. Furthermore, cured mice from the combination treatment of OncoVEXmGM-CSF and anti-CTLA-4 antibody rejected tumor rechallenges.
Conclusions These data support the concept that T-VEC and checkpoint inhibition may be an effective combination to treat patients with advanced melanoma.
- Immunotherapy
- Oncolytic Virotherapy
- T-Lymphocytes
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. The data required to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Materials will be made available to the scientific community by completion of a material transfer agreement. Requests can be made at www.amgen.com/partners/academic-collaborations/new-requests.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Talimogene laherparepvec (T-VEC) is a first-in-class modified herpes simplex virus type 1 (HSV-1) oncolytic immunotherapy that is approved for the treatment of patients with advanced melanoma. OncoVEXmGM-CSF, an oncolytic virus that encodes mouse granulocyte-macrophage colony-stimulating factor, enables preclinical evaluation of T-VEC in murine models. OncoVEXmGM-CSF causes direct tumor lysis, potentiates durable systemic tumor antigen-specific T-cell responses, and can be combined with cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockade in models sensitive to checkpoint inhibition (A20 B-cell lymphoma and CT-26 colorectal carcinoma).
WHAT THIS STUDY ADDS
We aimed to address the unmet need for combination therapies in melanoma by developing a novel checkpoint-refractory B16F10 melanoma model with both HSV-1 permissive subcutaneous tumors and HSV-1−refractory experimental lung metastasis. We demonstrated that OncoVEXmGM-CSF in combination with an anti-CTLA-4 or anti-programmed cell death protein 1 blocking antibody can sensitize tumors to immune therapy, leading to reduced subcutaneous tumor volume, improved survival, and generation of a tumor-antigen specific systemic immune response that inhibits the growth of distant (lung) metastases.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These data support the concept that oncolytic viral therapy and checkpoint inhibition may be an effective combination to treat patients with melanoma who have primary or acquired resistance to immunotherapy.
Background
Oncolytic immunotherapy is an emerging cancer treatment option characterized by the use of genetically modified viruses to target cancer cells and generate a systemic antitumor immune response.1 Talimogene laherparepvec (T-VEC) (IMLYGIC, Amgen, Thousand Oaks, California, USA) is a first-in-class oncolytic immunotherapy derived from herpes simplex virus type 1 (HSV-1) that is approved for the treatment of patients with advanced melanoma.2 T-VEC was engineered to target and kill cancer cells by direct lysis and to stimulate an adaptive antitumor immune response.3 The HSV-1 ICP34.5 gene was deleted, conferring tumor-selective replication and substantial reduction in the potential for neurovirulence.3–5 The ICP47 gene was also deleted, supporting proper antigen presentation and allowing for earlier and enhanced expression of US11, thereby increasing selective replication in tumor cells.3 Lastly, an expression cassette of human granulocyte-macrophage colony-stimulating factor (GM-CSF) was inserted to provide local GM-CSF production and enhance systemic antitumor immunity.3
In the OPTiM phase 3 clinical trial in advanced melanoma, T-VEC was delivered through intratumoral injection into cutaneous, subcutaneous, and/or nodal lesions. The response rate of injected lesions was 64%. By contrast, the response rate was 34% for uninjected distant non-visceral lesions and 15% for visceral lesions.6 Therefore, it is imperative to improve the systemic activity of T-VEC. In addition, further mechanistic studies of how T-VEC elicits systemic antitumor immunity would help guide the development of more effective therapies.
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) are co-inhibitory receptor molecules expressed on the surface of activated T cells, and the interactions with their cognate ligands result in a suppression of T-cell receptor signaling and reduced T-cell function.7–9 In tumors, upregulated CTLA-4 or PD-1 expression inhibits the cytotoxicity of CD8+ T cells against tumors and functions as immune checkpoints.10 11 Checkpoint inhibitors, such as anti-CTLA-4 and anti-PD-1 blocking antibodies, have transformed cancer treatment resulting in long-term survival of patients with locally advanced and metastatic disease.12 However, only a fraction of patients with melanoma respond to such therapies.13 Emerging clinical studies reveal that patients with more T-cell infiltration in their tumors or higher programmed cell death ligand 1 (PD-L1) expression at baseline are more likely to benefit from checkpoint blockade, whereas patients with few or no T cells in their tumors tend not to respond.14 15 Because T-VEC has demonstrated the ability to alter the tumor microenvironment, recruiting immune cells (including T cells) into injected and uninjected tumors,16 it provides a complementary mechanism of action and is considered a potential combination therapeutic option for patients who respond poorly to immune checkpoint inhibitors. A phase 2 clinical trial has shown that T-VEC in combination with ipilimumab, an anti-CTLA-4 blocking antibody, significantly improved overall response rate (ORR) over ipilimumab alone in patients with unresectable melanoma.17 Similarly, a recent phase 1 study demonstrated that T-VEC in combination with pembrolizumab, an anti-PD-1 antagonistic antibody, achieved superior ORR and complete response rate over pembrolizumab alone.18 Most recently, a phase 3 trial of T-VEC and pembrolizumab combination therapy evaluated patients with advanced unresectable melanoma (NCT02263508).19 Although the authors did not observe a statistically significant difference in the median overall survival (OS) and progression-free survival (PFS) between the treatment arms, there was a numerical difference of 5.8 months favoring the T-VEC-pembrolizumab arm and was similar to that observed in the KEYNOTE-006 trial for the combined pembrolizumab group.20 Nevertheless, the highest unmet need is the population of immunotherapy-refractory patients where effective treatment options are lacking. Combining intratumoral oncolytic virus therapy with checkpoint inhibitors is an encouraging strategy and positive results were observed in a phase 2 trial of T-VEC plus pembrolizumab in patients with advanced melanoma who progressed on prior anti-PD-1. T-VEC plus pembrolizumab treatment showed ORRs of 40.0%–46.7% in patients with advanced melanoma who progressed on prior anti-PD-1 in the adjuvant setting while ORRs of 0%–6.7% were observed in patients who progressed on prior PD-1 in the locally recurrent or metastatic setting.21
To enable further preclinical studies of T-VEC in murine syngeneic tumor models, OncoVEXmGM-CSF was created by substituting the human GM-CSF transgene with murine GM-CSF.3 Previous studies in a bilateral mouse syngeneic A20 tumor model have demonstrated that OncoVEXmGM-CSF treatment led to the cure of all A20 tumors that were directly injected and half of the contralateral uninjected tumors, with evidence of direct tumor cell lysis and the detection of HSV-1 only in the injected tumors.16 Using adoptive T-cell transfer and depletion of CD8+ T-cell approaches, the authors further demonstrated that the antitumor efficacy is mediated by cytotoxic CD8+ T cells, and the combination treatment with an anti-CTLA-4 blocking antibody significantly improved systemic efficacy, leading to the curing of 90% of contralateral A20 tumors. These preclinical results have provided initial evidence that CD8+ T cells are the central player and critical mediator of the antitumor activity of OncoVEXmGM-CSF and partly explained the enhanced clinical activity observed in the phase 2 trial of T-VEC in combination with ipilimumab in patients with melanoma.17
These encouraging results have been demonstrated with multiple oncolytic viruses, suggesting that this is potentially a class effect.22–24 As a consequence, numerous clinical studies are ongoing to evaluate the combination of oncolytic viruses and checkpoint inhibitors.25 Despite this optimism, major questions about the combinability of these reagents and their associated mechanism of action remain. The objective of this study was to evaluate whether OncoVEXmGM-CSF could improve outcomes in a checkpoint resistant tumor model and to elucidate the underlying mechanisms. We developed a novel melanoma model with both subcutaneously implanted tumors and experimental lung metastasis. Using this experimental model, we examined systemic immunity, effector memory, and tumor antigen-specific T-cell responses with OncoVEXmGM-CSF alone and in combination with anti-CTLA-4 or anti-PD-1 blocking antibody. The data presented here demonstrate that OncoVEXmGM-CSF expands and potentiates durable systemic tumor antigen-specific T-cell immunity and overcomes resistance to checkpoint inhibition in a checkpoint inhibitor-resistant model.
Materials and methods
Study design
In vivo studies were designed to evaluate antitumor activity, immune memory, and tumor-specific T-cell responses elicited after treatment with OncoVEXmGM-CSF alone or in combination with checkpoint inhibitors. Sample sizes (10–15 animals/cohort) were selected to detect relevant effect size with 80% statistical power. Mice implanted with tumor cells subcutaneously were randomized (deterministic) when tumor sizes reached an average of 100 mm3 to achieve a consistent tumor volume distribution and average tumor volume between cohorts. Confounders such as order of treatments and measurements were not controlled. Each animal study used a vehicle-treated group as a negative control. No data were excluded from any of the experiments and the studies were not blinded. Each of the experiments described in figures 1–5were replicated with comparable results. The immunohistochemistry data described in figure 6 was from a singular experiment.
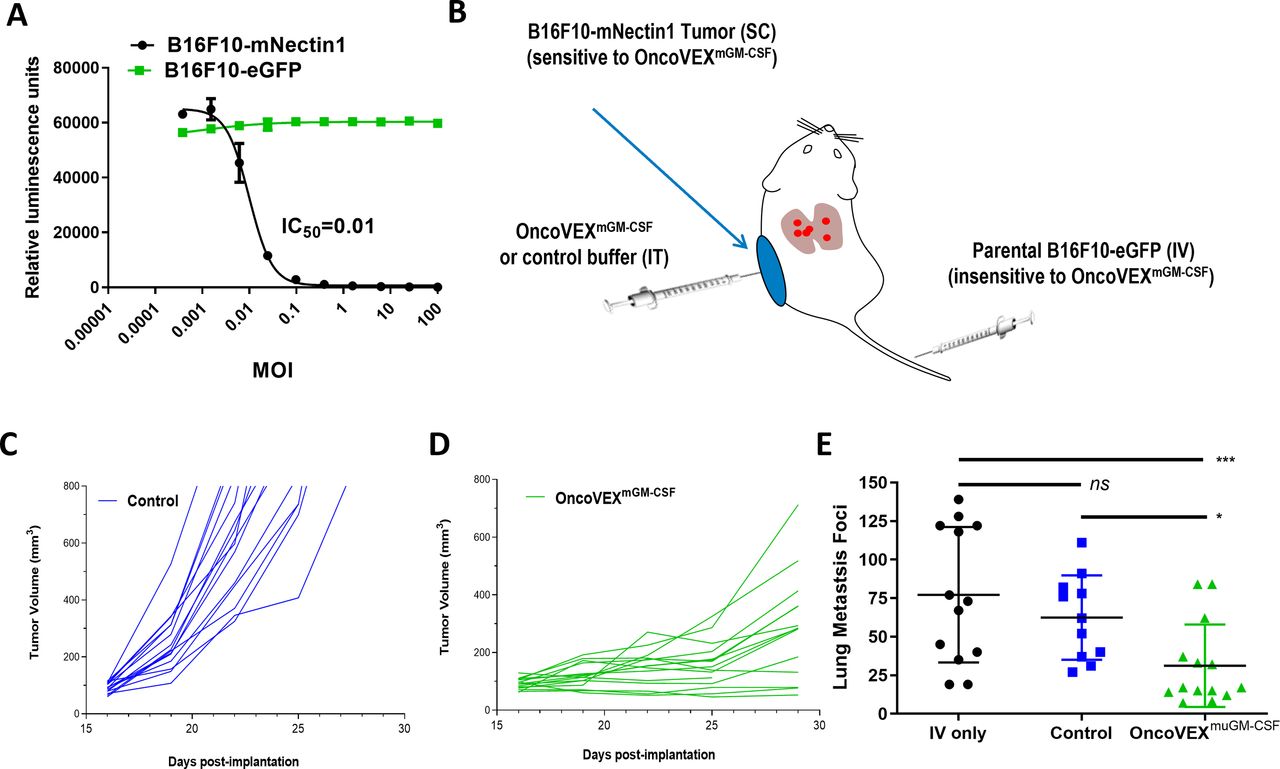
Development of the B16F10 mouse melanoma model with experimental lung metastasis. (A) Oncolytic activity of OncoVEXmGM-CSF against murine melanoma cell line B16F10 expressing either eGFP (B16F10-eGFP) or mouse nectin1 (B16F10-mNectin1). Relative luminescence units represent the number of live cells following treatment for 72 hours in vitro. (B) Schematic of the B16F10 SC tumor model with experimental lung metastases. (C) SC tumor volumes of mice treated with vehicle alone; each line represents an individual B16F10-mNectin1 tumor. (D) SC tumor volumes of mice treated with OncoVEXmGM-CSF; each line represents an individual B16F10-mNectin1 tumor. (E) Comparison of the number of surface lung metastases in naive mice (no SC tumor), vehicle-treated (with SC tumor), and OncoVEXmGM-CSF-treated mice on day 28 of study. Data are presented as individual tumor volumes (C,D) or individual lung metastasis foci (E) for each treatment group; n=15 (C,D) and n=11–14 (E). The significance of surface lung metastasis data was determined using a Kruskal-Wallis test followed by Mann-Whitney post hoc analysis with false discovery rate correction. ns p>0.05, *p<0.05, and ***p<0.001. eGFP, enhanced green fluorescent protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; IC50, half-maximal inhibitory concentration; IT, intratumoral; IV, intravenous; MOI, multiplicity of infection; ns, not significant; SC, subcutaneous.
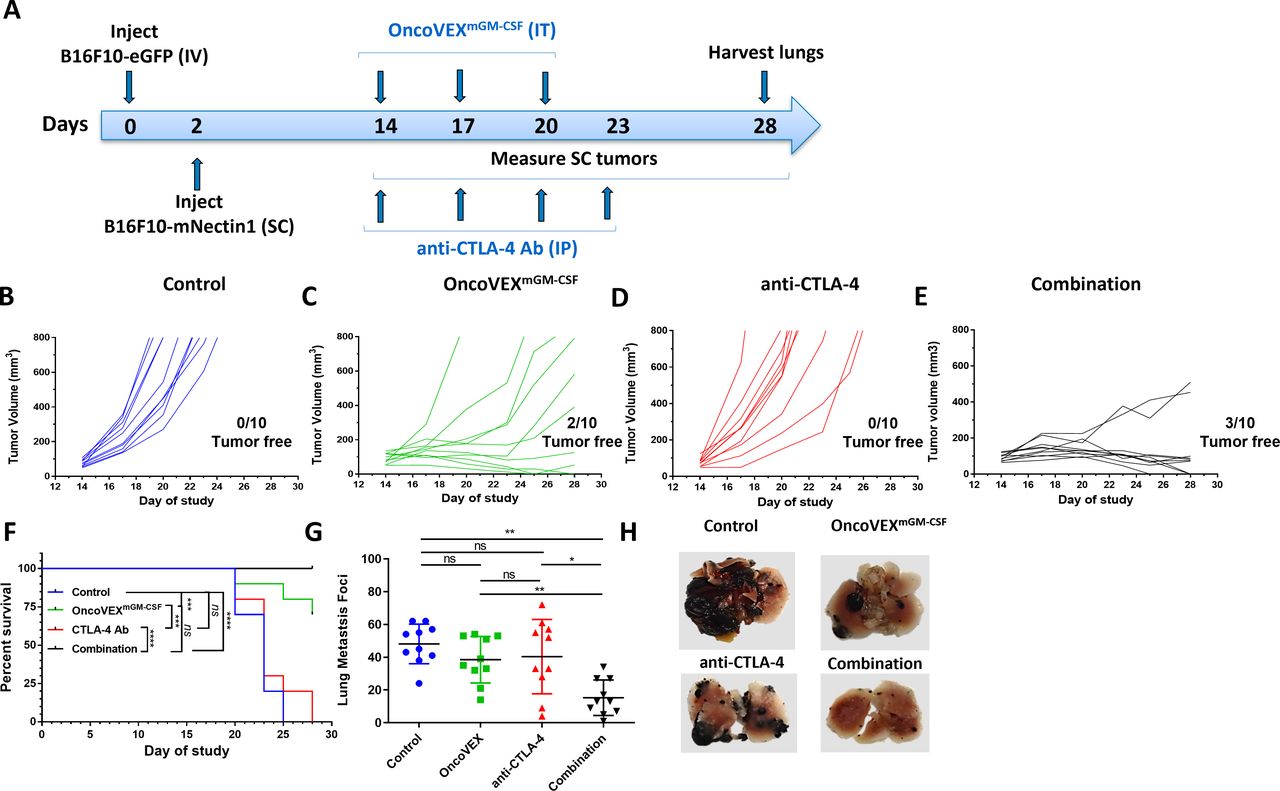
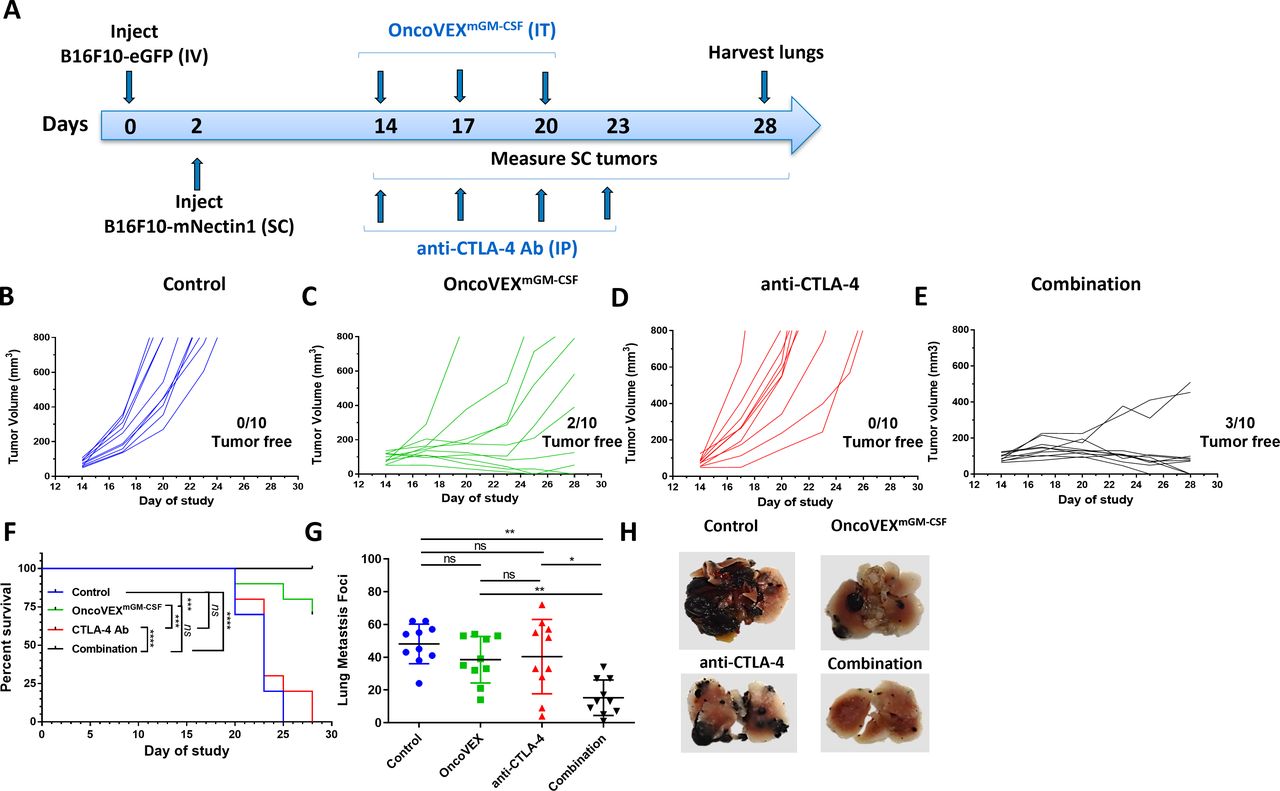
OncoVEXmGM-CSF treatment alone or in combination with an anti-CTLA-4 antibody led to a significant reduction in both SC tumor volume and lung metastases. (A) Timeline indicating the timing and route of administration for each cell line, OncoVEXmGM-CSF, and anti-CTLA-4 antibody. SC tumor volumes from individual mice throughout the study treated with (B) vehicle (control), (C) OncoVEXmGM-CSF alone, (D) anti-CTLA-4 antibody alone, and (E) the combination of OncoVEXmGM-CSF and anti-CTLA-4 antibody. (F) Kaplan-Meier analysis of median survival of mice from each treatment group. Events were recorded when tumor volume exceeded 800 mm3. (G) Enumeration of surface lung metastases on day 28 of study. (H) Representative photographs of lungs from mice from each treatment group on day 28. Data are presented as individual mouse tumor volumes (B–E) or individual mouse lung metastasis foci (H) for each treatment group; n=10. Significance within the Kaplan-Meier analysis was determined using a Mantel-Cox log-rank test to compare curves. The significance of lung metastasis foci data was determined using a Kruskal-Wallis test followed by Mann-Whitney post hoc analysis with false discovery rate correction. ns p>0.05, *p<0.05 and **p<0.01, ***p<0.001 and ****p<0.0001. Ab, antibody; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; GM-CSF, granulocyte-macrophage colony-stimulating factor; IP, intraperitoneal; IT, intratumoral; IV, intravenous; ns, not significant; SC, subcutaneous.

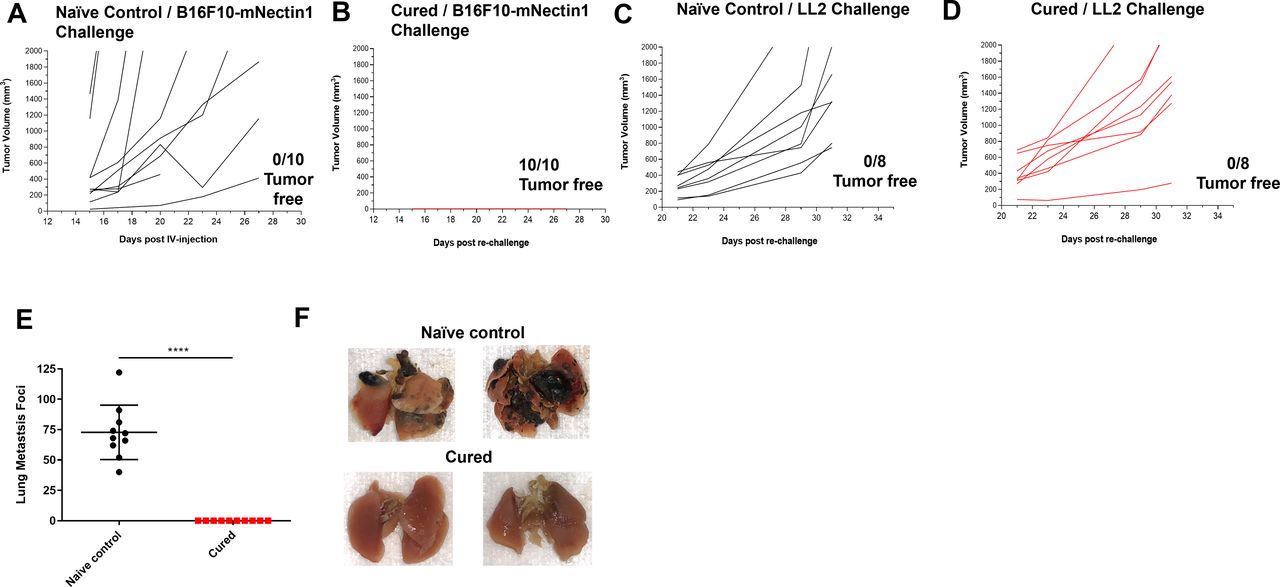
OncoVEXmGM-CSF treatment in combination with anti-CTLA-4 antibody led to tumor-specific systemic immunity. Tumor volumes from treatment-naive age-matched control mice (A, C) and mouse subcutaneous B16F10-mNecin1 tumors that had previously been cured with the combination of OncoVEXmGM-CSF and anti-CTLA-4 antibody (B, D) after subcutaneous rechallenge with B16F10-mNectin1 cells (A, B) or LL2 cells (C, D). (E) Age-matched naive control mice and previously cured mice were rechallenged intravenously with B16F10-eGFP cells, and lung metastases were enumerated 28 days later. (F) Representative photographs of lungs from naive age-matched control and previously cured mice 28 days after being rechallenged intravenously with B16F10-eGFP cells. Data are presented as individual mouse tumor volumes (A–D) or individual mouse lung metastasis foci (E) for each treatment group; n=10. The significance of lung metastasis foci data was determined using a Kruskal-Wallis test followed by Mann-Whitney post hoc analysis with false discovery rate correction. ****p<0.0001. CTLA-4, cytotoxic T-lymphocyte-associated protein 4; eGFP, enhanced green fluorescent protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; IV, intravenous.

OncoVEXmGM-CSF treatment alone or in combination with anti-PD-1 antibody led to a significant reduction in both SC tumor volume and lung metastases. (A) Timeline indicating the timing and route of administration for each cell line, OncoVEXmGM-CSF, and anti-PD-1 antibody. SC tumor volumes from (B) control mice, (C) mice treated with OncoVEXmGM-CSF alone, (D) anti-PD-1 antibody alone, or (E) the combination of OncoVEXmGM-CSF plus anti-PD-1 antibody throughout the study. (F) Kaplan-Meier analysis of median survival of mice treated with vehicle, OncoVEXmGM-CSF, anti-PD-1 antibody, or the combination of OncoVEXmGM-CSF plus anti-PD-1 antibodies. (G) Enumeration of surface lung metastases in mice treated with vehicle, OncoVEXmGM-CSF, anti-PD-1 antibody, or the combination of OncoVEXmGM-CSF plus anti-PD-1 antibody. Events were recorded when tumor volume exceeded 800 mm3. Data are presented as individual mouse lung metastasis foci (G) or individual mouse tumor volumes (B-E) or for each treatment group; n=10. Significance within the Kaplan-Meier analysis was determined using a Mantel-Cox log-rank test to compare curves. The significance of lung metastasis foci data was determined using a Kruskal-Wallis test followed by Mann-Whitney post hoc analysis with false discovery rate correction. Ab, antibody; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; eGFP, enhanced green fluorescent protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; IP, intraperitoneal; IT, intratumoral; IV, intravenous; ns, not significant; PD-1, programmed cell death protein 1; SC, subcutaneous; ns p>0.05; *p≤0.05; **p≤0.001; ***p≤0.0001; ****p<0.0001.

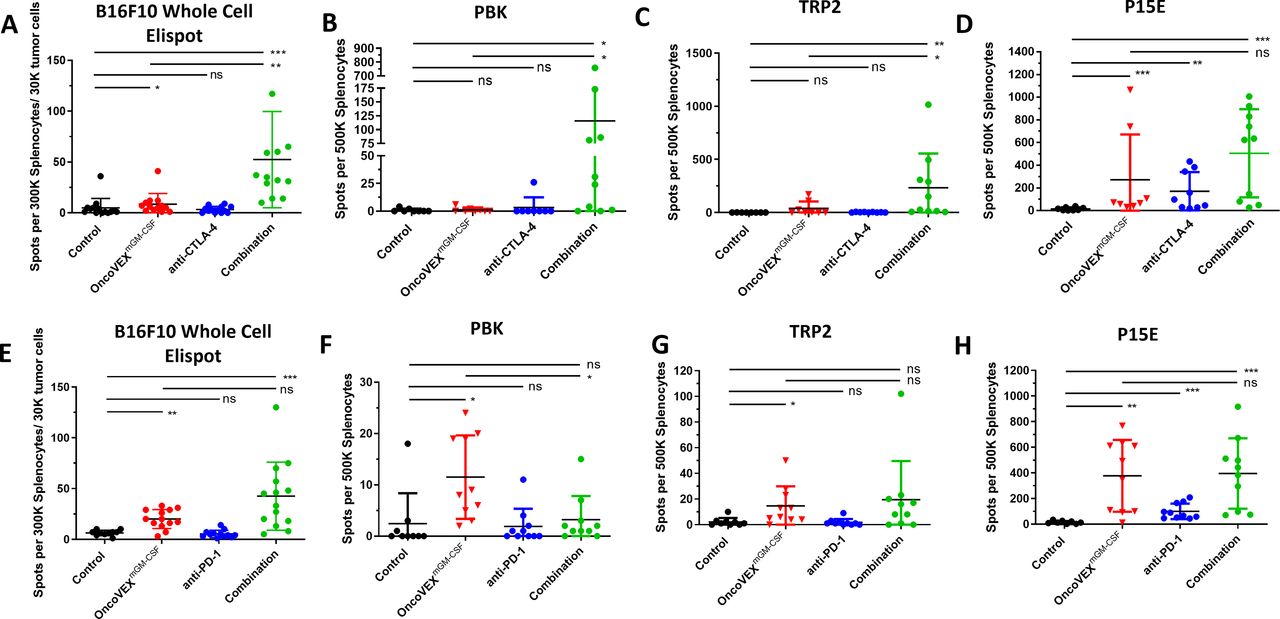
OncoVEXmGM-CSF treatment alone or in combination with an anti-CTLA-4 or anti-PD-1 antibody elicited specific T-cell responses against tumor antigens and neoantigens. Splenocytes were isolated from mice at the end of each efficacy model (combination with CTLA-4 antibody schema (figure 2A) and combination with anti-PD-1 Ab schema (figure 4A) and evaluated for reactivity to either B16F10-eGFP tumor cells (whole-cell ELISpot assay) or tumor antigens in a peptide restimulation ELISpot assay. The number of interferon-γ positive spots per 3×105 splenocytes mixed with 3×104 B16F10-eGFP tumor cells is shown (A, E). The number of interferon-γ positive spots per 5×105 splenocytes is shown for neoantigen PBK (B, F), tumor antigen TRP2 (C, G) and tumor antigen P15E (D, H). Results from the anti-CTLA-4 antibody model (A−D) and the anti-PD-1 model (E−H) are shown. Ab, antibody; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; eGFP, enhanced green fluorescent protein; ELISpot, enzyme-linked immunospot; GM-CSF, granulocyte-macrophage colony-stimulating factor; ns, not significant; ns, not significant; PBK, PDZ-binding kinase; PD-1, programmed cell death protein 1; PDZ, post-synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1); TRP2, tyrosinase-related protein 2; T-VEC, talimogene laherparepvec. *p≤0.05; **p≤0.001; ***p≤0.0001; ****p<0.0001.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
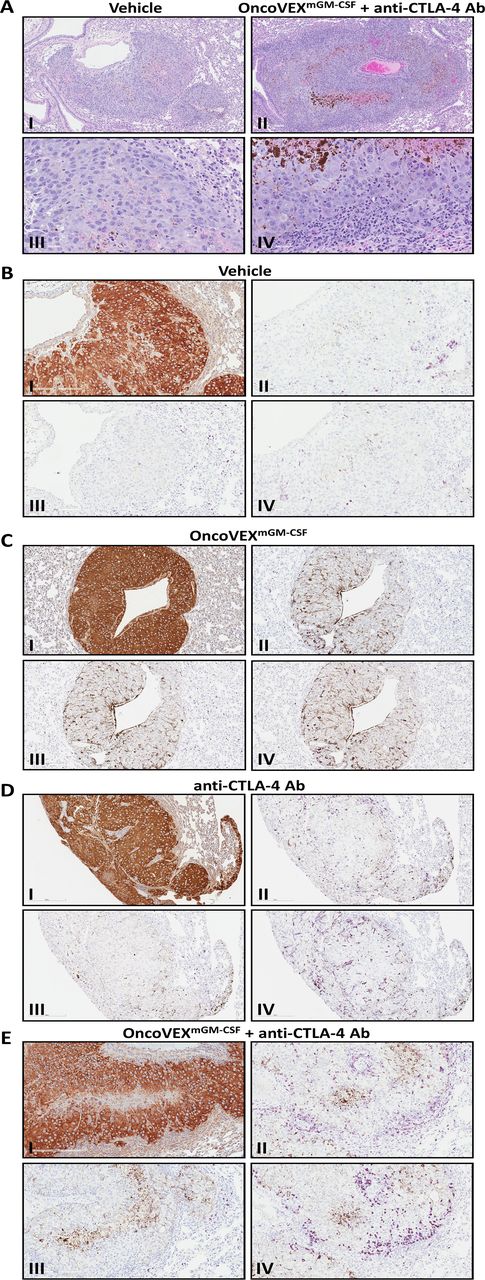
OncoVEXmGM-CSF in combination with anti-CTLA-4 antibody leads to increased immune infiltrate and evidence of necrosis in lung metastasis. (A) Low magnification (100×; I and II) and high magnification (400×; III and IV) representative photomicrographs of B16F10-eGFP tumors from vehicle-treated mice (I and III) and the combination of OncoVEXmGM-CSF plus an anti-CTLA-4 antibody in the lung stained by hematoxylin and eosin. (B–D) Representative photomicrographs of B16F10-eGFP tumors in mice administered vehicle (B), OncoVEXmGM-CSF (C), or anti-CTLA-4 antibody (D) (200× magnification). The B16F10-eGFP tumor cells are immunopositive in the anti-MART1 immunohistochemical assay (I; intense brown=immunopositive). There are scant CD3+T cells (II; purple=immunopositive) and rare B220+B cells (III; purple=immunopositive) restricted to the tumor periphery, and scant F4/80+macrophages (IV; purple=immunopositive) within the tumors and at the tumor periphery. (E) Representative photomicrographs of a B16F10-eGFP tumor in a mouse coadministered OncoVEXmGM-CSF and anti-CTLA-4 antibody (200× magnification). The B16F10-eGFP tumor cells are immunopositive in the anti-MART1 immunohistochemical assay (I; intense brown=immunopositive). There are dense CD3+T cells (II; purple=immunopositive) and F4/80+macrophages (IV; purple=immunopositive) at the tumor periphery and fewer CD3+T cells and F4/80+macrophages within the tumor. B220+B cells (III; purple=immunopositive) are scant and restricted to the tumor periphery. Brown melanin pigment is variably present in the histologic sections, notably in regions of central necrosis. Ab, antibody; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; eGFP, enhanced green fluorescent protein; GM-CSF, granulocyte-macrophage colony-stimulating factor.
Oncolytic viruses, cell lines, and in vitro viability assay
The engineering of the oncolytic virus T-VEC (IMLYGIC, Amgen) has been described previously.3 The design of OncoVEXmGM-CSF is similar to T-VEC, except that the human GM-CSF transgene has been replaced with murine GM-CSF.3 Mouse syngeneic tumor cell lines B16F10 (melanoma) and LL2 (Lewis lung carcinoma) were obtained from the American Type Culture Collection (Manassas, Virginia, USA) and were cultured as indicated (online supplemental table 1). B16F10-enhanced green fluorescent protein (eGFP) and B16F10-mouse nectin1 (mNectin1) cell-line pools were generated by lentiviral transduction and drug selection. Cells were plated in a 96-well plate at 7000 cells per well and incubated overnight at 37°C. OncoVEXmGM-CSF was added in 1:4 serial dilutions starting at 10 or 100 multiplicity of infection (MOI). After incubating for 72 hours, the number of cells remaining in each well was determined using an ATPlite assay (PerkinElmer, Waltham, Massachusetts, USA).
Supplemental material
Animal care and use
Female C57Bl/6 mice (Charles River Laboratories, Wilmington, Massachusetts, USA) 6–8 weeks old were maintained in accordance with the Guide for the Care and Use of Laboratory Animals26 and housed at Association for Assessment and Accreditation of Laboratory Animal Care International–accredited facilities in ventilated microisolator housing with corncob bedding (or equivalent) with five animals per cage. An Institutional Animal Care and Use Committee approved all non-clinical protocols. Access to sterile pelleted feed and reverse osmosis-purified water via automatic watering system or water bottle was provided ad libitum. Studies were conducted at Amgen.
Tumor growth evaluation in the B16F10 subcutaneous and lung metastasis model
Experimental lung metastases were established by delivering 5×104 B16F10-enhanced green fluorescent protein (eGFP) cells intravenously. Subcutaneous tumors were established by implanting 3×105 B16F10-mNectin1 cells on the rear flank. When subcutaneous tumors reached approximately 100 mm3, mice were randomized into treatment groups and administered three intratumoral injections of OncoVEXmGM-CSF every third day. Anti-CTLA-4 antibody (100 µg, intraperitoneal injection; clone 9D9, mIgG2a) or anti-PD-1 antibody (300 µg, intraperitoneal injection; clone 29F1A12, mIgG1) was administered on the same schedule, for a total of four doses. Control animals received vehicle control on the same dosing schedule. Tumor volumes were measured with digital calipers two times per week until the defined endpoint when lungs were excised and surface metastasis foci were counted using a dissecting microscope.
Immunohistochemistry
B16F10-eGFP lungs with tumors were harvested at necropsy, inflated with 10% neutral-buffered formalin (NBF) under constant pressure, and placed in NBF. After 48 hours of fixation, tumors were routinely processed to paraffin blocks. Serial histologic sections (4 µm) were stained with hematoxylin and eosin or used in immunohistochemistry assays. Immunohistochemistry assays were performed on an intelliPATH Automated Staining System (Biocare Medical, Irvine, California, USA) using antibodies directed against melanoma antigen recognized by T cells 1 (MART-1; melan-A) for melanocytes (Novus Biologicals, Littleton, Colorado, USA), CD3 for T cells (Bio-Rad, Hercules, California, USA), F4/80 for macrophages (Bio-Rad), and B220 for B cells (Novus Biologicals) (online supplemental table 2). Rat and rabbit isotype controls were included as negative controls. Histologic slides with tissue sections were baked at 60°C, deparaffinized with xylene and rehydrated in descending concentrations of ethanol. Pretreatment for epitope unmasking was performed by heat under pressure using Biocare Heat-induced Epitope Retrieval buffers and default settings for the Biocare Decloaking Chamber NxGen (Biocare Medical). Slides were treated with 10% hydrogen peroxide to reduce excess melanin pigment. Endogenous protein and peroxidase were blocked with Serum Free Rodent Block M (Biocare Medical) and Peroxidazed 1 (Biocare Medical). Slides were then incubated with the primary antibody or isotype control followed by a biotinylated secondary antibody (Vector) and HRP Polymer (Dako, Carpinteria, California, USA). The chromogenic substrate 3,3’-diaminobenzidine (Dako) was used for visualization of MART-1 (brown), and aminoethyl carbazole (Biocare Medical) was used for visualization of immune cell markers (purple). All slides were routinely counterstained with hematoxylin for visualization of cell nuclei (blue). After staining, slides were dehydrated in 100% ethanol, cleared in xylene, coverslipped, and scanned using an Aperio AT2 scanner (Leica Biosystems). Sections of normal mouse spleen and lymph were included as tissue controls to confirm appropriate staining patterns of immune cells. Light microscopic analysis of histologic sections was performed using a Nikon Eclipse 90i (Nikon, Japan) by a pathologist certified by the American College of Veterinary Pathologists. Staining intensity was qualitatively scored according to the following grading scheme: 0, none to rare immunopositive cell infiltrate; 1+, scant immunopositive cell infiltrate; 2+, mild immunopositive cell infiltrate; and 3+, heavy immunopositive cell infiltrate.
Tumor rechallenge model
Treatment-naive control mice (control group) and treated mice (test group) whose subcutaneous tumors had resolved after treatment with OncoVEXmGM-CSF and anti-CTLA-4 antibodies were rested for 60 days and then rechallenged with 3×105 B16F10-eGFP or MC38 tumor cells either subcutaneously on the flank or intravenously (B16F10-eGFP only, 1×105 cells). Tumor growth was then evaluated for the following 4–5 weeks. Lungs were collected from intravenous-challenged mice on day 28 for enumeration of lung metastases.
Identification of B16F10 tumor neoantigens
Whole-exome sequencing was performed on B16F10 mouse tumor cells by MedGenome (Foster City, California, USA). Briefly, coding variants were called relative to the reference mouse genome to identify non-synonymous variants. Computational algorithms were then used to filter these mutations and select those that are likely to be expressed, presented by antigen-presenting cells with high affinity for major histocompatibility complex (MHC) class I, and bind to the T-cell receptor. These 9-mer sequences were synthesized into peptides for use in enzyme-linked immunospot (ELISpot) assays to measure the immune response against them.
ELISpot assays
Antigen-specific T-cell responses were assessed using an interferon-γ (IFN-γ) ELISpot assay (Cellular Technology, Shaker Heights, Ohio, USA). Splenocytes were isolated from individual mice from each treatment group (including vehicle control) and used in both peptide restimulation and whole-cell ELISpot assays. Briefly, 5–8×105 splenocytes were incubated with control peptides (GFP) or tumor antigen peptides (online supplemental table 3) at a final concentration of 1 µM for 18–20 hours at 37°C. The P15E peptide is a component of the endogenous murine leukemia virus transmembrane envelope protein; the 9-mer from this peptide is presented in the context of MHC Kb.27 For whole-cell assays, 2.5×105 splenocytes were mixed with 5×104 B16F10 or MC38 tumor cells and incubated for 20 hours at 37°C. A CTLS6 FluoroSpot analyzer (CTL, Shaker Heights, Ohio, USA) was used to enumerate the total number of spots, representing the number of tumor or tumor-antigen specific T cells.
Statistical analysis
Non-linear regression analysis to determine half-maximal inhibitory concentration (IC50) values was performed by GraphPad Prism software (v8.4.3). Kaplan-Meier analysis of median survival time of mice treated with OncoVEXmGM-CSF alone or in combination with anti-CTLA-4 antibodies was used to assess in vivo efficacy. Events were recorded when tumor volume exceeded 800 mm3. Significance was assessed using a Mantel-Cox log-rank test to compare curves (GraphPad Prism, v8.4.3). The significance of lung metastasis foci and ex vivo T-cell reactivities was evaluated using a Kruskal-Wallis test followed by Mann-Whitney post hoc analysis with false discovery rate correction. The p value cut-off of 0.05 was used to determine significant differences between groups.
Results
Expression of mNectin1 on B16F10 cells confers sensitivity to OncoVEXmGM-CSF in vitro and in vivo
B16F10 murine melanoma cells are not susceptible to HSV-1 infection due to the lack of expression of HSV-1 entry receptors. To evaluate the response of B16F10 to OncoVEXmGM-CSF administration both in vitro and in vivo, we generated a cell line stably expressing the HSV-1 entry receptor murine Nectin1 (termed B16F10-mNectin1). Unlike the parental B16F10, mNectin1-expressing cells were killed by OncoVEXmGM-CSF in a dose-dependent manner in a 72-hour viability assay. The IC50 for the B16F10-mNectin1 was 0.01 MOI; by contrast, control cells stably expressing the eGFP (termed B16F10-eGFP) were insensitive to OncoVEXmGM-CSF treatment even at an MOI of 100 (figure 1A).
To evaluate B16F10 response to OncoVEXmGM-CSF in vivo, we established a novel tumor model to evaluate both local and systemic efficacy simultaneously. In previous experiments, contralateral subcutaneous tumors were implanted to evaluate if an antitumor effect could be observed in injected and uninjected tumors.3 16 However, both tumors were implanted subcutaneously on opposite flanks and while no virus was observed in the uninjected tumor, both tumors expressed HSV-1 entry receptor. To further investigate that OncoVEXmGM-CSF could have a systemic antitumor effect that spanned organs and was independent of virus entry or cell binding, we generated a novel tumor model system. We also selected B16F10 as this was derived from a melanoma, the indication for which T-VEC is approved (compared with the relatively treatment-sensitive models A20 and CT26 published previously).3 16 Mice were implanted subcutaneously with OncoVEXmGM-CSF-sensitive B16F10-mNectin1 cells and intravenously with OncoVEXmGM-CSF-insensitive B16F10-eGFP cells (figure 1B). A control group of mice were inoculated intravenously with B16F10-eGFP cells only to evaluate the potential impact of subcutaneous tumor growth on the establishment of experimental lung metastasis. Either vehicle control or OncoVEXmGM-CSF was injected into the established subcutaneous tumors only. Subcutaneous tumor volume was monitored two times per week until day 29 when the study was terminated so that the lungs could be evaluated for metastatic tumor burden. Compared with vehicle-treated mice (figure 1C), mice treated with OncoVEXmGM-CSF alone showed a growth delay in subcutaneous tumors (figure 1D). Moreover, a significant decrease was seen in the number of surface lung metastases observed with OncoVEXmGM-CSF treatment compared with intravenous-only control (p=0.007) or vehicle-treated mice (p=0.016; figure 1E). No significant difference was noted between the intravenous-only and vehicle-control groups (ns, p=0.500).
OncoVEXmGM-CSF in combination with anti-CTLA-4 blocking antibody reduces subcutaneous tumor volume and systemic HSV-1−refractory lung metastasis foci and improves survival
Previous studies using bilateral syngeneic A20 and CT-26 tumor models have demonstrated that local intratumoral delivery of OncoVEXmGM-CSF results in oncolysis at injection site and that the antitumor efficacy in non-injected contralateral tumors depends on a CD8+ T cell-mediated systemic response.16 Using the B16F10 model described above, we studied the local (subcutaneous tumor) and systemic (lung metastases) efficacy of OncoVEXmGM-CSF in combination with a blocking anti-CTLA-4 antibody (figure 2A). In comparison with vehicle control (figure 2B), treatment with anti-CTLA-4 antibody alone resulted in no change in subcutaneous tumor growth (n=0/10 tumor-free for each; figure 2D). However, treatment with OncoVEXmGM-CSF alone (figure 2C) or OncoVEXmGM-CSF in combination with anti-CTLA-4 antibody (figure 2E) resulted in significant tumor reduction; two mice in the OncoVEXmGM-CSF alone group and three mice in the combination group became tumor-free (figure 2C,E). Median OS was significantly improved with OncoVEXmGM-CSF alone and in combination with anti-CTLA-4 antibody compared with vehicle-treated and CTLA-4 antibody-treated mice (OncoVEXmGM-CSF alone, undefined vs 23 days, p=0.0003; combination, undefined vs 23 days, p<0.0001; figure 2F). A significant decrease was seen in the number of surface lung metastasis foci observed with the combination of OncoVEXmGM-CSF and anti-CTLA-4 antibody treatment compared with vehicle control (p=0.003) or each treatment as a monotherapy (combination vs OncoVEXmGM-CSF, p=0.007; combination vs CTLA-4 antibody, p=0.004; figure 2G). Representative images of lungs from each group are shown in figure 2H. Combination treatment with OncoVEXmGM-CSF and anti-CTLA-4 antibody led to not only fewer surface lung metastases but also smaller lesions. As expected, OncoVEXmGM-CSF shows a significant impact on tumor progress in the injected (subcutaneous) tumor. However, the effect on lung metastases is not significant based on the number of foci (although the size of the foci did appear to be affected; figure 2H). Anti-CTLA-4 antibody as monotherapy did not significantly impact the subcutaneous tumor or the number of lung metastatic foci. Given the improvement in effect on metastatic foci number with combination treatment, we hypothesize that OncoVEXmGM-CSF treatment creates a tumor microenvironment that is more amenable to treatment with anti-CTLA-4 antibody. Together, these data demonstrated that OncoVEXmGM-CSF treatment in combination with anti-CTLA-4 antibody achieved superior efficacy compared with either agent used as a monotherapy.
OncoVEXmGM-CSF in combination with anti-CTLA-4 blocking antibody leads to increased immune infiltrate and necrosis in distant HSV-1−refractory lung metastasis
To evaluate the systemic effect on tumor and tumor microenvironment, lungs from vehicle control or OncoVEXmGM-CSF and anti-CTLA-4 antibody-treated animals were analyzed by immunohistochemistry for immune infiltrate. Increased inflammatory cell infiltration was observed in lung metastases following OncoVEXmGM-CSF and CTLA-4 blockade, suggesting an enhanced antitumor immune response in the uninjected distant lung metastasis. Representative photomicrographs of HSV-1 non-permissive B16F10-eGFP tumors formed in the lung are shown in figure 6. Compared with tumors in mice treated with vehicle controls (figure6A; I and III), or OncoVEXmGM-CSF or CTLA-4 monotherapy (not shown), tumors in mice administered both OncoVEXmGM-CSF and CTLA-4 (figure 6A; II and IV) were surrounded with a dense inflammatory cell infiltrate. As shown in figure 6B,C (I; intense brown=immunopositive), the B16F10-eGFP tumor cells are immunopositive for anti-MART1 immunohistochemical staining. In the vehicle-treated group (figure 6B; II, III, and IV; purple=immunopositive), there were scant CD3+T cells and rare B220+B cells restricted to the tumor periphery, and scant F4/80+macrophages within the tumor and at the tumor periphery. By contrast, in the OncoVEXmGM-CSF and CTLA-4 combination treatment group (figure 6C; II, III, and IV; purple=immunopositive), there were dense CD3+T cells and F4/80+macrophages at the tumor periphery and fewer CD3+T cells and F4/80+macrophages within the tumor. B220+B cells were scant and restricted to the tumor periphery. Brown melanin pigment is present in the histologic sections, notably in regions of central necrosis.
OncoVEXmGM-CSF in combination with anti-CTLA-4 blocking antibody results in durable immune memory
To evaluate treatment longevity and durability, tumor rechallenge was conducted using the B16F10 tumor model. Mice with established B16F10-mNectin1 subcutaneous tumors were treated with OncoVEXmGM-CSF in combination with an anti-CTLA-4 antibody (online supplemental figure 1A). Of those treated with the combination, mice that had complete tumor regression by day 30 (approximately 65% of mice; online supplemental figure 1B) and age-matched naive controls were rested for 2 months and then rechallenged with either B16F10-mNectin1 or LL2 cells via subcutaneous injection on the flank or with B16F10-eGFP via intravenous injection (online supplemental figure 1A). For mice that underwent rechallenge via subcutaneous injection with B16F10-eGFP cells, tumor growth was prevented in cured mice (n=10/10 tumor-free; figure 3B) but not in naive age-matched control mice (n=0/10 tumor-free; figure 3A). Mice were also rechallenged subcutaneously with C57Bl/6-derived LL2 cells, leading to tumor outgrowth in all age-matched control and cured mice (0/8 tumor-free in both groups; figure 3C,D). The mice were rechallenged with B16F10-eGFP cells intravenously, and complete rejection of lung metastasis (0 surface lung metastases in all 10 mice) was observed in the previously cured mice, compared with control naive mice, which developed numerous lung metastases after intravenous injection (mean of 73±22 surface lung metastases; figure 3E); sample lung images from each group are shown in figure 3F.
OncoVEXmGM-CSF alone or in combination with anti-PD-1 blocking antibody reduces subcutaneous tumor volume and systemic lung metastasis and improves survival
Using a model system similar to what was used to study the combination with anti-CTLA-4 antibody, we evaluated the local (subcutaneous tumor) and systemic (lung metastases) efficacy of OncoVEXmGM-CSF alone and in combination with an anti-PD-1 blocking antibody (figure 4A). When we evaluated subcutaneous tumors versus vehicle control (figure 4B), treatment with anti-PD-1 antibody alone resulted in no change in tumor growth (n=0/10 tumor-free for each; figure 4D). However, treatment with OncoVEXmGM-CSF alone (figure 4C) or in combination with anti-PD-1 antibody (figure 4E) resulted in tumor reduction; two mice in the OncoVEXmGM-CSF alone group and eight mice in the combination group became tumor-free (figure 4C,E). Median OS was significantly improved with OncoVEXmGM-CSF alone and in combination with anti-PD-1 antibody compared with both vehicle-treated and anti-PD-1 antibody-treated mice (OncoVEXmGM-CSF alone, undefined vs 21 days, p<0.0001) and combination (undefined vs 21 days, p<0.0001; Figure 4F). A significant decrease was observed in the number of surface lung metastases with OncoVEXmGM-CSF treatment alone (p=0.03) and in combination with anti-PD-1 antibody treatment compared with vehicle control (p=0.0001). We also observed a significant decrease in surface lung metastases using anti-PD-1 antibody as a monotherapy compared with control (p=0.005) or the combination of OncoVEXmGM-CSF and anti-PD-1 antibody (p=0.03; Figure 4G).
OncoVEXmGM-CSF alone or in combination with anti-CTLA-4 blockade or anti-PD-1 antibody elicits specific T-cell responses against tumor antigens and neoantigens
Given the increased efficacy observed in subcutaneous tumors and lung metastases, we aimed to measure increases in tumor-specific and neoantigen-specific T-cell responses. Using the same treatment schema as the efficacy models (figures 2A, 4A), splenocytes were isolated and evaluated for reactivity to B16F10-eGFP tumor cells as well as tumor antigens in a peptide restimulation ELISpot assay. We screened a total of 59 peptides (online supplemental table 3) including mutated tumor neoantigens, endogenous retroviral antigen p15E,28 and self-antigens29 and discovered significant reactivity to three of them (figure 5). Treatment with OncoVEXmGM-CSF alone or in combination with anti-CTLA-4 antibody led to a statistically significant increase in reactivity to B16F10-eGFP tumor cells (p=0.013 and p=0.0005; Figure 5A). Treatment with the combination of OncoVEXmGM-CSF and anti-CTLA-4 antibody led to a statistically significant increase in reactivity to neoantigen PDZ binding kinase (PBK; p=0.04; Figure 5B) and self-antigen tyrosinase-related protein 2 (TRP2; p=0.002; Figure 5C) compared with vehicle, OncoVEXmGM-CSF, or anti-CTLA-4 antibody alone. Reactivity to viral antigen P15E was increased with OncoVEXmGM-CSF, CTLA-4 antibody, and the combination of both (figure 5D). Treatment with OncoVEXmGM-CSF alone or in combination with anti-PD-1 antibody led to a statistically significant increase in reactivity to B16F10-eGFP tumor cells (p=0.002 and p=0.0009; Figure 5E). Treatment with OncoVEXmGM-CSF alone led to a statistically significant increase in reactivity to neoantigen PBK (p=0.020; Figure 5F) and self-antigen TRP2 (p=0.020; Figure 5G). Reactivity to viral antigen P15E was significantly increased with OncoVEXmGM-CSF alone (p=0.002). The combination of OncoVEXmGM-CSF and anti-PD-1 antibody was significantly higher than control (p=0.0008) but not compared with OncoVEXmGM-CSF alone (p=0.97; Figure 5H).
Discussion
T-VEC has demonstrated single-agent activity in advanced melanoma in injected lesions as well as distant uninjected visceral lesions, although with a lower rate of response in uninjected visceral lesions.6 Early-stage clinical studies revealed that in combination with checkpoint inhibitors, such as anti-CTLA-4 monoclonal antibody (mAb; ipilimumab) and anti-PD-1 mAb (pembrolizumab), T-VEC increases the ORR compared with either agent alone.17 18 In a recent phase 3 trial, the combination of T-VEC plus pembrolizumab did not significantly improve PFS or OS compared with pembrolizumab treatment alone in immunotherapy-naïve patients with advanced melanoma (NCT02263508).19 The basis for the lack of benefit is not clearly understood. However, we have seen promising clinical results in patients with melanoma refractory to previous PD-L1 that have been treated with T-VEC plus pembrolizumab.21
Previous preclinical studies have shown that OncoVEXmGM-CSF can modify the tumor microenvironment to boost durable antitumor immunity in a T cell-dependent manner.3 16 These studies support the general notion that oncolytic viruses attack tumors through direct local oncolysis and subsequent systemic immunity. The exact nature of the antitumor immune response elicited by oncolytic viruses has not been fully investigated, including the repertoire of antitumor immune cells and how systemic antitumor immunity is established after local injection.
Consistent with the approved indication of T-VEC, we developed a unique murine syngeneic melanoma tumor model, known to be resistant to immunotherapy with checkpoint inhibitors. This allowed simultaneous interrogation of the antitumor effect of OncoVEXmGM-CSF at the local injection site (driven by oncolysis and immune activation as a direct consequence of viral infection) as well as in distant lung metastatic lesions (driven by a systemic antitumor immune response). This strategy allowed us to rule out the possibility that efficacy in distant, uninjected tumors occurs due to viral infection. Because only the subcutaneously implanted B16F10-mNectin1 tumors are permissive to OncoVEXmGM-CSF infection and killing and the metastatic lung lesions seeded by the parental B16F10-eGFP cells are refractory to OncoVEXmGM-CSF infection, the reduced foci number in the lungs and the extended survival of mice treated by injection of OncoVEXmGM-CSF into the established subcutaneous tumors provide the first definitive evidence that OncoVEXmGM-CSF not only kills injected tumors but also elicits a systemic immune response to inhibit the growth of distant metastases.
The B16F10 murine syngeneic tumor cells have low levels of MHC-I antigen presentation at cell surface30 and are infiltrated with low numbers of T cells when grown as subcutaneous tumors in vivo,31 representing the immunologically cold tumor phenotype observed in human cancers that tend to be refractory checkpoint inhibitor treatment. B16F10 has been widely reported to be resistant to checkpoint inhibitors, including anti-CTLA-4, anti-PD-1, and anti-PD-L1 mAbs when given as monotherapy treatments.32–34 Because previous studies revealed that OncoVEXmGM-CSF treatment induces massive infiltration of various immune cells, including T cells into injected and non-injected tumors, and increases the expression of CTLA-4, PD-1, and PD-L1 in tumor tissues,16 we evaluated whether OncoVEXmGM-CSF treatment could overcome resistance to checkpoint inhibitors. In addition, CTLA-4 blockade has been shown to prime new tumor neoantigens,35 as opposed to PD-1 blockade which is more effective in reversing the exhaustion phenotype observed in mouse models and human patients.36 We hypothesized that the priming mechanisms driven by CTLA-4 blockade and OncoVEXmGM-CSF may be synergistic. These studies demonstrated that compared with OncoVEXmGM-CSF alone, the combination of OncoVEXmGM-CSF with anti-CTLA-4 blocking antibody led to further reduction in subcutaneous tumor volume and lung metastatic growth and prolonged the survival of mice significantly. Interestingly, we also saw a similar effect when OncoVEXmGM-CSF was combined with PD-1, suggesting that in the B16F10 mouse model both additional priming and/or the reversal of exhaustion is sufficient to drive efficacy. Moreover, the cured mice from OncoVEXmGM-CSF and anti-CTLA-4 combination treatment rejected the rechallenge with the HSV-1 non-permissive B16F10 cells but not unrelated LL2 tumors, demonstrating the acquisition of durable tumor-specific immune memory. The superior combination efficacy observed in the current study is consistent with our previous findings with OncoVEXmGM-CSF and anti-CTLA-4 co-treatment of A20 B cell lymphoma and CT-26 colorectal carcinoma (both immunologically hot tumors) in the bilateral setting, and further extends to OncoVEXmGM-CSF and anti-PD-1 combination treatment for the first time in a non-inflamed tumor model. Together, these results suggest that OncoVEXmGM-CSF can overcome resistance to anti-CTLA-4 or anti-PD-1 in immunologically cold tumors.
Our previous preclinical studies revealed that CD8+ T cells are required and necessary to mediate antitumor efficacy of OncoVEXmGM-CSF in A20 syngeneic tumor model.16 Because the B16F10-eGFP metastatic lung lesions in the current study are refractory to OncoVEXmGM-CSF infection and the cured mice from OncoVEXmGM-CSF and anti-CTLA-4 combination treatment rejected a rechallenge with B16F10 cells both subcutaneously and in the lungs, we sought to identify tumor antigens recognized by CD8+ T cells. Based on exosome sequencing and MHC-I neoantigen prediction algorithm, we identified and evaluated 56 neoantigens as well as well-known endogenous tumor antigens presented by B16F10. Using ELISpot assay with splenic T cells from treated mice, we found that OncoVEXmGM-CSF treatment expanded CD8+ T-cell response to melanoma self-antigen TRP2 significantly, whereas neither anti-CTLA-4 nor anti-PD-1 generated IFN-γ−producing T cells beyond control group. OncoVEXmGM-CSF also expanded the T-cell population that can recognize the endogenous retroviral antigen gp70 (P15E peptide presented by MHC-I) expressed by tumors from C57BL/6 strain. Moreover, we also discovered that OncoVEXmGM-CSF elicited CD8+ T-cell responses against B16F10-specific neoantigen PBK. Of note, we also observed that in combination with anti-CTLA-4 antibody, the number of PBK and TRP2 antigen-reactive T-cell populations in the spleen were markedly higher than either agent alone could achieve. By contrast, the combination of anti-PD-1 and OncoVEXmGM-CSF did not further expand T-cell populations recognizing these two tumor antigens. This could be due to the fact that we only examined CD8+ T-cell responses in the spleen, where the T cells are not exhausted or dysfunctional yet; thus, anti-PD-1 does not have a strong effect. Alternatively, anti-CTLA-4 may have a stronger effect on T-cell priming in conjunction with OncoVEXmGM-CSF treatment. Taken together, these findings reveal that OncoVEXmGM-CSF treatment expands the tumor-specific CD8+ T-cell repertoire that could traffic and infiltrate distant tumors, leading to efficacy and systemic antitumor immunity.
T-VEC has been combined with both an anti-PD-1 antibody and an anti-CTLA-4 antibody. The phase 1b data from the combination of T-VEC with pembrolizumab suggested that there was an increase in CD8+ T cells in the tumor microenvironment and increased responses to anti-PD-1.18 Despite failing to demonstrate a clinical benefit of the combination with pembrolizumab in immunotherapy-naïve patients with advanced melanoma, recent results from a phase 2 trial of T-VEC plus pembrolizumab demonstrated an objective response rate of 40.0% or greater in patients with advanced melanoma who progressed on prior anti-PD-1 in the adjuvant setting.21
While the data generated in preclinical models is encouraging and useful, such models have their limitations. In our experimental model, all the animals share the same MHC haplotype and genetic background. In addition, all the tumors share the same genetic mutations that those MHC alleles are capable of binding and presenting to immune cells. Unlike most human tumors, mouse syngeneic models often also express immunodominant viral epitopes (such as P15E described here) that may overestimate the antigenicity of human neoantigens. While the underlying mechanisms of antitumor immunity may be explored and validated in preclinical models such as those described here, a fuller understanding of the translatability of such findings will enhance the benefit to patients.
In summary, using this newly developed B16F10 model system, we discovered that OncoVEXmGM-CSF expands and potentiates durable systemic tumor antigen-specific T-cell immunity and overcomes resistance to checkpoint inhibition in a checkpoint inhibitor-resistant model. These data, which align with findings reported in previous clinical trials, support the concept that OncoVEXmGM-CSF and other oncolytic viruses may synergize with immune modulators to improve patient outcomes.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. The data required to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Materials will be made available to the scientific community by completion of a material transfer agreement. Requests can be made at www.amgen.com/partners/academic-collaborations/new-requests.
Ethics statements
Patient consent for publication
Ethics approval
The studies were approved by our International Care and Use Committee (IACUC), protocol # 2009-00046.
Acknowledgments
The authors thank James Balwit, MS, CMPP, Meghan Johnson, PhD, and Vicky Kanta, PhD (ICON, Blue Bell, Pennsylvania, USA), whose work was funded by Amgen, and Kristina Y Aguilera, PhD (Amgen) for medical writing assistance in the preparation of this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JE, JW, PJB, JD, JQ, and KSC provided conception, design, data analysis and interpretation, writing, and review. JZ and PM provided conception, design, execution, and data analysis and interpretation. JZ performed the in vitro cytotoxicity assays. JE performed the in vivo studies. JW provided immunohistochemistry analysis. JZ, KSC, JE, and PM designed and performed the ELISpot assays. Each of the authors commented on the manuscript. KSC accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.