Article Text

Abstract
Background Despite the success of HER2-targeted therapy in achieving prolonged survival in approximately 50% of treated individuals, treatment resistance is still an important challenge for HER2+ breast cancer (BC) patients. The influence of both adaptive and innate immune responses on the therapeutic outcomes of HER2+BC patients has been extensively demonstrated.
Methods Long non-coding RNAs expressed in non-pathological complete response (pCR) HER2 positive BC were screened and validated by RNA-seq. Survival analysis were made by Kaplan-Meier method. Cell death assay and proliferation assay were performed to confirm the phenotype of LINC00624. RT-qPCR and western blot were used to assay the IFN response. Xenograft mouse model were used for in vivo confirmation of anti-neu treatment resistance. RNA pull-down and immunoblot were used to confirm the interaction of ADAR1 and LINC00624. ADAR1 recombinant protein were purified from baculovirus expression system. B16-OVA cells were used to study antigen presentation both in vitro and in vivo. Flow cytometry was used to determine the tumor infiltrated immune cells of xenograft model. Antisense oligonucleotides (ASOs) were used for in vivo treatment.
Results In this study, we found that LINC00624 blocked the antitumor effect of HER2- targeted therapy both in vitro and in vivo by inhibiting type I interferon (IFN) pathway activation. The double-stranded RNA-like structure of LINC00624 can bind and be edited by the adenosine (A) to inosine (I) RNA-editing enzyme adenosine deaminase RNA specific 1 (ADAR1), and this editing has been shown to release the growth inhibition and attenuate the innate immune response caused by the IFN response. Notably, LINC00624 promoted the stabilization of ADAR1 by inhibiting its ubiquitination-induced degradation triggered by β-TrCP. In contrast, LINC00624 inhibited major histocompatibility complex (MHC) class I antigen presentation and limited CD8+T cell infiltration in the cancer microenvironment, resulting in immune checkpoint blockade inhibition and anti-HER2 treatment resistance mediated through ADAR1.
Conclusions In summary, these results suggest that LINC00624 is a cancer immunosuppressive lncRNA and targeting LINC00624 through ASOs in tumors expressing high levels of LINC00624 has great therapeutic potential in future clinical applications.
- Breast Neoplasms
- Immunity, Innate
- Antigen Presentation
Data availability statement
Data relevant to the study are included in the article or uploaded as online supplemental information.
Data availability statement
Original data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The efficacy of antitumor treatment relies on IFN response. ADAR1 inhibits the overactivation of double-stranded RNA sensors such as RIG-I and MDA5, therefore mitigate the alert system and thus shape a ‘cold’ tumor microenvironment.
WHAT THIS STUDY ADDS
We found an immunosuppressive lncRNA LINC00624 restrains the activation of IFN pathway through stabilizing ADAR1. LINC00624 also relies on the A-to-I RNA editing ability of ADAR1 to inhibit MHC class I antigen presentation and limited CD8+T cell infiltration in the cancer microenvironment, resulting in immune checkpoint blockade inhibition and anti-HER2 treatment resistance.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
LINC00624 could be a biomarker for anti-HER2 and immune therapy. Targeting LINC00624 through antisense oligonucleotides could show great therapeutic potential for future clinical use.
Introduction
The human innate immune system has evolved a well-designed mechanism for providing the first line of defense against viral infection. When viral double-stranded RNAs (dsRNAs) are sensed by cytosolic pattern recognition receptors, such as RIG-I and MDA5,1 IFNs are secreted by most human cells, and they trigger the expression of IFN stimulated genes (ISGs). ISGs stimulate antigen presentation pathways, which lead to the recruitment of immune cells and facilitate antiviral responses.1 2 In addition, immune systems are well adapted to avoid eliciting damage in normal tissue3 4 when mistranscribed RNAs are expressed by cells or released after physiological cell death.5 Tumors can evade T cell-mediated antitumor immunity by decreasing IFN or MHC class I antigen induction triggered by the dsRNA or cytoplasmic DNA products of aberrant transcription or mitosis.6 7 Therefore, the efficacy of antitumor treatment also relies on autonomous autocrine of type I IFN in tumor cells.8 In addition to traditional cytotoxic drugs, tyrosine kinase inhibitors (TKIs) and humanized antibodies can enhance type I IFN and antigen presentation pathway activation.9–11 Therefore, the regulation of the innate immune response is critical for therapeutic efficacy in breast cancer (BC).
Adenosine deaminase RNA specific 1 (ADAR1) is a regulator of the innate immune response. ADAR1 catalyzes the conversion of adenosine (A) to inosine (I) in a dsRNA substrate and destroys the dsRNA structure, and therefore, ADAR1 inhibits the overactivation of dsRNA sensors such as RIG-I and MDA5.7 12 13 The involvement of ADAR1 RNA-editing in cancer development and immune therapy failure has been established.7 12 13 Loss of ADAR1 in melanoma promotes antigen presentation and reverses cellular resistance to immune checkpoint blockade.14 Therefore, ADAR1 can mitigate the alert system and thus shape a ‘cold’ tumor microenvironment.
Human epidermal growth factor receptor 2 (HER2, also known as ErbB-2 or Neu) is an oncogene overexpressed in 20%–30% of BCs.15 Humanized monoclonal antibodies such as trastuzumab and TKIs such as lapatinib can prolong the survival of BC patients. However, treatment resistance is still an important issue for at least 50% of patients.16 17 It has been reported that HER2 suppresses the innate immune response and antitumor immunity.9 11 BC cell lines from transgenic mice expressing HER2 express low levels of MHC class I antigens.18 Blocking IFN receptor 1 (IFNAR1) weakens the therapeutic efficacy of anti-HER2 monoclonal antibodies.19 Interestingly, HER2-positive (HER2+) BC patients with higher ISG scores or more tumor-infiltrating lymphocytes have a better outcome after anti-HER2 treatment.20 21 These findings suggest that the activation of the innate immune response, particularly with respect to IFNs and the antigen presentation pathway, can enhance HER2+BC treatment, and the underlying mechanisms should be further clarified.
To discover new regulators that can affect HER2+BC treatment outcomes, we focused on long non-coding RNAs (lncRNAs), which might be involved in disease progression and therapeutic resistance. We compared lncRNA expression in tumors before neoadjuvant chemotherapy between HER2+BC patients in the pathological complete response (pCR) and non-pCR groups and found that LINC00624 was enriched in the non-pCR group. Overexpression of LINC00624 inhibited the IFN-related innate immune response and MHC class I antigen presentation, which subsequently induced cancer cell proliferation and blocked the antitumor effect of lapatinib and trastuzumab. We found that the ADAR1 Editing Region (AER) of LINC00624 could be edited in which adenosine was modified to inosine by ADAR. The edited LINC00624 stabilized ADAR1 and further suppressed the IFN induced expression of ISGs. Our data support the supposition that LINC00624 plays a critical role in ADAR regulation and may serve as an antitumor target in future BC combination treatments.
Results
LINC00624 promotes treatment resistance in HER2+ BC
To screen for lncRNAs involved in the modulation of the HER2-targeted treatment response, 20 core needle biopsy specimens taken from primary tumors in patients with HER2+BC before neoadjuvant therapy were retrospectively collected and subjected to RNA sequencing. The cohort was classified into the pCR and non-pCR groups based on the outcomes determined on pathological evaluation. We analyzed the expression profiles and found that LINC00624 was the most significantly increased long non-coding gene in the non-pCR group after the exclusion of pseudogenes (figure 1A,B). When the sample size was expanded to 100, LINC00624 was still significantly higher in non-pCR group (figure 1C). To further determine the potential function of LINC00624 in BC pathogenesis, we analyzed two independent clinical patient datasets. In early-stage BC patient samples in The Cancer Genome Atlas database and a cohort of 319 RNA samples in our center, high LINC00624 expression was significantly correlated with poor disease-free survival and overall survival (figure 1D, online supplemental figure 1B, and online supplemental tables 1 and 2). Among the patients characterized by molecular subtype, patients with HER2+or luminal A BC with high LINC00624 expression showed worse outcomes than those with low LINC00624 expression (figure 1D, online supplemental figure 1A, B), suggesting that LINC00624 may be involved in the progression of BC. Based on the expression difference in the HER2+BC pCR and non-pCR groups, we focused on the role of LINC00624 in HER2+BC.

LINC00624 promotes treatment resistance in HER2+BC. (A) HER2+BC patients core needle biopsy specimen before neoadjuvant treatment were divided into pCR (n=10) and non-pCR (n=10) groups according to pathology evaluation after surgery. The heatmap summarizes differentially expressed RNAs between pCR and non-pCR group. (B) The volcano plots showed the fold changes (FC) and p values in non-pCR tumors versus pCR tumors. The most differentially expressed lncRNAs were shown. (C) Fragments per kb of transcript per million mapped reads (FPKM) of LINC00624 in HER2+BC neoadjuvant treatment cohort. Patients were divided in to pCR or non-pCR group according to the pathological evaluation after surgery. The expression of LINC00624 were divided into high or low expression group with the cut-off value FPKM=0.5, separated by the dotted line. Statistical analysis was performed using Fisher’s exact tests. (D) Disease-free survival (DFS) plot of BC patients in a consecutive cohort receiving adjuvant treatment. Patients were divided into high and low LINC00624 groups according to RNA expression in the primary tumor. Statistical analysis was performed using two-sided log-rank tests. Left, all the molecular subtypes were pooled and shown. Right, HER2 enrichment subtype were shown. (E) RNA FISH with LINC00624 probe showed LINC00624 was mainly located in the cell nucleus of SK-BR-3 cells. Cell nucleus was stained with DAPI. 18S RNA was probed as positive control. (F) Cell proliferation assay of WT and LINC00624 KO cells in SK-BR-3 and BT-474 cells. n=6 for each time point. Statistical analysis was performed using two-sided t-test at the end point. (G) Inhibition rate of WT and LINC00624 KO SK-BR-3 and BT-474 cells in response to lapatinib. (H) Inhibition rate of pCDH and LINC00624 overexpression BT-474 cells in response to trastuzumab. (I) Tumor growth curve and tumor size of BT-474 pCDH or LINC00624 cells receiving anti-neu or isotype control in nude mice. n=5 animals in each group. Statistical analysis was performed using two-sided t-test for the tumor volume at the end point. *p<0.05. All data are mean±SE. (F–H) n=3 biological independent samples, similar results were obtained from two more independent experiments. BC, breast cancer; FISH, fluorescence in situ hybridization; KO, knockout; pCR, pathological complete response; WT, wild type.
To further characterize LINC00624, rapid amplification of cDNA ends was performed to obtain the full-length sequence of LINC00624. We found that LINC00624 was transcribed from chromosome 1q21.1-1q21.2 as four isoforms, all of which carrying intergenic regions between BCL9 and CHD1L in BC cell lines (online supplemental file 1). In contrast to full-length isoform 1, isoform-specific internal deletions in exon 4 were found in isoforms 2, 3, and 4 (online supplemental figure 1C,D). However, we did not observe the 3088 nt transcript (RefSeq Accession: NR_038423) annotated in the National Center for Bioinformation database; the 3088 nt sequence overlaps with the CHD1L gene locus. Full-length isoform 1 (RefSeq Accession: NR_038423) was the most abundant among all isoforms, and the other three isoforms were expressed in low abundance in the examined BC cell lines (online supplemental figure 1E). According to phylogenetic codon substitution frequencies and Coding Potential Assessing Tool,22 23 LINC00624 showed no coding ability (online supplemental figure 1F,G).
Fluorescence in situ hybridization assays showed that LINC00624 was mainly located in the cell nucleus of HER2+BC cell lines (figure 1E). Cytoplasmic and nuclear RNA purification also confirmed that the four isoforms were mainly located in the cell nucleus (online supplemental figure 2A). The BT-474 and SK-BR-3 ectopic overexpression cell lines were constructed with isoform 1 of LINC00624 by lentiviral infection, and knockout (KO) cells were generated by using CRISPR-cas9 to delete part of the promoter region and exons 1–2, which abolished the expression of all the isoforms (online supplemental figure 2B,C).
To illustrate the biological function of LINC00624, we found overexpressed LINC00624 accelerated cell growth (figure 1F and online supplemental figure 2D). Furthermore, cells with LINC00624 overexpression were resistant to lapatinib and trastuzumab treatment (figure 1G,H), while KO cells were more sensitive (online supplemental figure 2E). We also employed a BT-474 xenograft tumor model to evaluate the oncogenic functions of LINC00624 in vivo. We found LINC00624 promoted tumor growth and the resistantance to anti-HER2/neu treatment (figure 1I). In summary, these data suggest that LINC00624 may promote the treatment resistance of HER2+BC.
LINC00624 inhibits the innate immune response by inhibiting type I IFN signaling
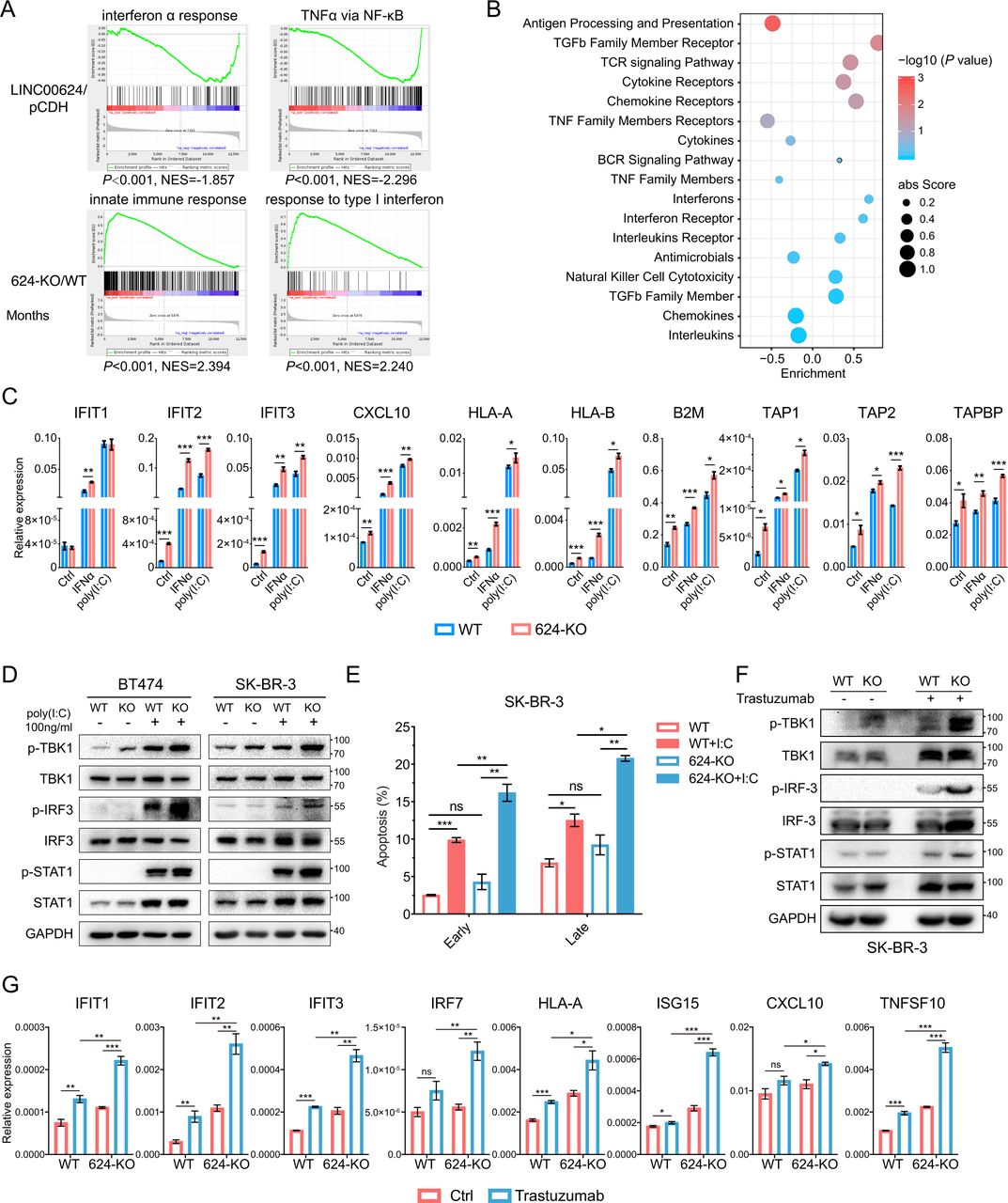
To further investigate the mechanism involved in LINC00624 signaling in BC, we performed RNA-seq with pCDH/LINC00624-expressing cells and WT/LINC00624-KO cells and analyzed candidate genes and pathways regulated by LINC00624. Interestingly, we found that LINC00624 expression was negatively correlated with the interferon α response, TNFα via NF-κB, and the innate immune response (figure 2A), and hallmarks related to IFN pathways were enriched in LINC00624-KO cells (online supplemental figure 3A), implying a role for LINC00624 in regulating the type I IFN response and antigen presentation. Then, we analyzed the involvement of LINC00624 in immune reactions with ImmLnc, a public database used for investigating the immune-related function of lncRNAs.24 In this analysis, LINC00624 exhibited a strong negative correlation with antigen processing and presentation (figure 2B). Consistently, RT-qPCR detection also showed the induction of ISGs by IFNα was increased significantly in LINC00624-KO SK-BR-3 cells, as well as MHC class I pathway-related genes (figure 2C). Then, we used polyinosinic:polycytidylic acid (poly(I:C)), a synthetic analog of dsRNA that can activate cytosolic RNA sensors to stimulate inflammatory signaling pathways.25 We found that LINC00624 inhibited the induction of ISGs by dsRNAs (figure 2C, online supplemental figure 3B), and the phosphorylation of STAT1 was inhibited on LINC00624 overexpression (online supplemental figure 3C). Then, we evaluated the signaling pathway in the dsRNA-triggered IFN response. We found that the phosphorylation of TBK1/IRF3/STAT1 was increased in LINC00624-KO cells (figure 2D). A previous study has reported that the type I IFN-generated antiviral response causes cell growth arrest and apoptosis.3 We found that overexpression of LINC00624 attenuated the cell apoptosis caused by the stimulation of dsRNA sensors (figure 2E, online supplemental figure 3D,E). Thus, we believe LINC00624 is a potential immunosuppressive lncRNA. Furthermore, treatment with poly(I:C) or IFNα induced the expression of LINC00624 (online supplemental figure 3F), indicating that LINC00624 is an ISG and can serve as a negative feedback regulator in the IFN signaling pathway.

LINC00624 inhibits the innate immune response by inhibiting type I IFN signaling. (A) GSEA analysis showed the indicated gene signatures in different LINC00624 expression SK-BR-3 cells. Top, LINC00624 overexpression versus pCDH; Bottom, LINC00624-KO vs WT. (B) LINC00624 related immune pathways were analyzed by ImmLnc Database. Enrichment score and p value were used for graphing. (C) WT or LINC00624-KO BT-474 cells were treated with 5 ng/mL IFNα for 4 hours or transfected poly(I:C) for 24 hours. RNA levels of ISGs and antigen presentation related genes were analyzed by RT-qPCR. GAPDH was used as reference gene. (D) WT or LINC00624 KO cells were treated with transfected poly(I:C) for 24 hours with indicated concentration. The levels of the indicated proteins were determined by immunoblot. The experiment was performed twice with similar results. (E) The percentage of early and late apoptosis were determined after 1 µg/mL transfected poly(I:C) in SK-BR-3 WT and LINC00624-KO cells. Statistical analysis was performed using two-sided t-test. (F, G) WT or LINC00624-KO SK-BR-3 cells were treated with 20 µg/mL trastuzumab for 3 days. (F) The levels of the indicated proteins were determined by immunoblot. The experiment was performed twice with similar results. (G) RNA levels of ISGs and antigen presentation related genes were analyzed by RT-qPCR. GAPDH antibodies were used as reference gene. (C, E, G) n=3 biological independent samples. Statistical analyses were performed using two-sided t-test. *p<0.05, **p<0.01, ***p<0.001. Data are shown as mean±SE. GSEA, Gene Set Enrichment Analysis; ISGs, IFN stimulated genes; NES, Normalized Enrichment Score; KO, knockout; WT, wild type.
Previous studies have reported that HER2 amplification leads to the impairment of IFN pathway activation and antitumor immune responses through inhibition of TBK1 phosphorylation.9 Moreover, HER-2/neu overexpression is associated with a reduction in MHC class I molecules at the cell surface, possibly induced through IFN response inhibition.18 26 In our study, treatment with the anti-HER2 antibody trastuzumab markedly induced ISG and antigen presentation-related gene expression in a HER2-driven BC cell line while has little effect in HER2 negative cell lines (online supplemental figure 3G,H). In addition, the LINC00624 level was increased (online supplemental figure 3H), suggesting that LINC00624 was possibly elevated by treatment-induced IFN activation. To determine whether LINC00624 could inhibit the induction of the type I IFN response after anti-HER2 treatment, we compared the expression of ISGs and antigen presentation-related genes after trastuzumab treatment of wild-type and LINC00624-KO cells. In the LINC00624-depleted cells, the number of ISG transcripts increased significantly in response to HER2 blockade compared with that in the wild-type cells (figure 2F,G). These results indicate that LINC00624 inhibits the anti-HER2-induced cell inflammatory response, which further contributes to treatment resistance.
LINC00624 is bound to and edited by ADAR1
To understand the underlying mechanism of LINC00624 in innate immune response blockade, we performed an RNA pull-down assay to explore its potential protein partners (figure 3A). We found that LINC00624 bound several RNA-binding proteins (online supplemental table 3). Among them, ADAR1, an A-to-I RNA-editing protein that can inhibit the innate immune response and is related to type I IFN response regulation, attracted our attention. ADAR1 has two isoforms: the longer ADAR1 p150 is expressed from an interferon (IFN)-inducible promoter and both nuclear and cytoplasmic, while the shorter p110 is constitutively expressed and mainly nuclear. The p110 could be translated from an alternative ATG start codon within the transcript of p150.12 27 We first confirmed the interaction between ADAR1 and LINC00624 (online supplemental figure 4A). As the expression of p150 isoform is relatively low under normal conditions, we found LINC00624 mainly binds p110 in BC cells. It has been reported that ADAR1 is a major RNA editor, catalyzing the deamination of A to generate I, which is prevalent across the whole transcriptome.13 By disrupting the secondary structure of self-generated or virus-produced dsRNAs through RNA editing, ADAR1 hinders the innate immune response, especially the type I IFN pathway-related response, which is activated by multiple RNA sensors in the presence of dsRNAs.13 28 Therefore, we speculated that the interaction between ADAR1 and LINC00624 might contribute to tumor progression and innate immune response repression.

LINC00624 is bound to and edited by ADAR1. (A) Silver staining of RNA pull-down proteins followed by SDS-PAGE. Biotin-labeled LINC00624 sense and antisense full-length (FL) RNA were incubated with SK-BR-3 whole cell lysates. ADAR1 was identified a specific band. (B) RNA pull-down assay with FL LINC00624 sense, antisense, iso 2 of LINC00624 (iso2), isoform3 with additional AER region (iso3+AER), and isoform 3 only (iso3) biotin labeled RNAs in 293 T cells. Top, immunoblot of ADAR1 and GAPDH antibodies were shown. Bottom, the input RNAs was confirmed by electrophoresis. The experiment was performed twice with similar results. (C) Predicted secondary structure of full length LINC00624 by RNAfold Web server. Three segments were truncated and annotated as S1, S2 and S3. The AER region was in S3. (D) Left, RNA pull-down assay was performed with Alu (a positive control that ADAR1 bound cloned from an Alu located in PHACTR4), LINC00624 FL, and the three segments. Right, S3, and S3 without AER (S3-AER) biotin labeled RNA was used. Immunoblot of ADAR1 was shown on the top. The input RNAs were confirmed by electrophoresis. The experiment was performed twice with similar results. (E) RNA electrophoretic mobility shift assay (EMSA) was performed with recombinant ADAR1 and in vitro transcribed biotin labeled AER sequence was used. (F) Left, Schematic view of ADAR1 p110 truncations with 3xFLAG tag used for RNA pull-down with S3 biotin labeled RNA. Right, Immunoblot of FLAG. Input of cell lysates expressing ADAR1 p110 truncations and RNA pull-down elutes were immunoblotted by FLAG antibody. (G) RNA pull-down of ADAR1 p110 truncations with S3 biotin labeled RNA. Up, Schematic view of ADAR p110 truncations with dsRBDs deletion. Down, Immunoblot of FLAG. Input of cell lysates expressing ADAR1 truncations and RNA pull-down elutes were shown. (H, I) LINC00624 AER was edited by ADAR1 in vivo. (H) AER was A-to-I edited in BT-474 cells. The editing ratio was increased after poly(I:C) treatment for 24 hours or IFNα treatment for 4 hours. (I) Up, schematic view of LINC00624 region domain. Down, Sanger sequencing data of AER region were shown. ADAR1 p110 overexpressed, WT, and ADAR1-KO cells of BT-474 were compared. (J) Recombinant ADAR1 could edit in vitro transcribed (IVT) LINC00624. Recombinant ADAR1 was incubated with IVT LINC00624 FL or AER. Sanger sequencing results were shown. For (A, B, D–J, the experiment was performed twice with similar results. dsRBD, double-stranded RNA-binding domains.
Next, we investigated the binding affinity between ADAR1 and different isoforms of LINC00624. Isoform 1 and isoform 2 showed a high affinity for ADAR1, while isoform 3 negligibly bound ADAR1, which suggested that isoform 3 missed a structure critical for LINC00624 and ADAR1 binding (figure 3B). To further elaborate the ADAR1-binding sequence on LINC00624, we employed the RNAfold tool to predict the secondary structure of LINC00624.29 We found a folded dsRNA-like structure (an AER) transcribed from inverted repeats on both sides of the S3 segment of LINC00624 (figure 3C); this fragment in full-length LINC00624 was absent in isoform 3. Then, we truncated LINC00624 according to its secondary structure and found that only S3 binds ADAR1 in human BC cells, and that the AER region-deletion mutation in S3 caused isoform 3 to lose its ADAR1-binding ability, suggesting that the AER region is the major domain contributing to ADAR1 binding (figure 3D). This hypothesis was confirmed through RNA electrophoretic mobility shift assay showing that the AER domain was shifted after incubation with recombinant ADAR1 protein (figure 3E and online supplemental figure 4B).
As reported, ADAR1 contains three dsRNA-binding domains (dsRBDs) that are involved in RNA binding under different circumstances (figure 3F).30 31 The A-to-I editase is located in the C-terminus and is the functional group for RNA editing.13 30–32 Therefore, we truncated the three dsRBDs and editase separately to map the ADAR1 domains involved in LINC00624 binding. The RNA pull-down assay showed that dsRBD3 of ADAR1 was essential to ADAR1 interaction with LINC00624 (figure 3G). As dsRBD3 domain is shared between ADAR1 p110 and p150,13 LINC00624 could be bound by both of the isoforms. As previously reported, the KKxxK motif in the dsRBD region is critical for dsRNA binding.33 We mutated KKxxK to EExxA in dsRBD3. Consistently, LINC00624 failed to bind the ADAR1 mutant with EExxA in dsRBD3 (online supplemental figure 4C).
As A-to-I RNA editing can be catalyzed by ADAR enzymes, which converted ‘A’s in dsRNA structures into ‘I’s via hydrolytic deamination, we asked whether LINC00624 could be edited by ADAR1. As expected, we found that the AER structure of LINC00624, which was bound by ADAR1, was edited in BC cell lines, and the portion that was edited was further increased after poly(I:C) or IFNα treatment (figure 3H). This finding was consistent with observations of BC tissues (online supplemental figure 4D). In addition, the frequency of AER region editing events was reduced in ADAR1-KO BT-474 cells, confirming that ADAR1 is the major editase involved in LINC00624 A-to-I substitution (figure 3I). This discovery also explained the multiple inconsistent A-to-G mutations found in the cDNA of LINC00624 when we cloned LINC00624 extracted from human cell lines. To confirm the A-to-I editing ability of ADAR1 on LINC00624, we incubated recombinant ADAR1 with transcribed LINC00624 in vitro. The AER region was also edited at the same sites as those in the regions examined in vivo (figure 3J). These data demonstrated that LINC00624 could be bound and edited by ADAR1.
LINC00624 promotes ADAR1 stabilization
As LINC00624 interacted with ADAR1, we hypothesized that LINC00624 might affect ADAR1 function. First, to determine whether the RNA-editing events of ADAR1 were affected by LINC00624 in BC cells, the Alu-Editing Index (AEI) score, a normalized measure based on hyperediting of Alu elements that allows comparison of editing activity across tissues and tumors, was used to evaluate the editase activity in cancer cells.34 35 The AEI score has been validated with experimental data obtained with both clinical samples and cell lines, and an increased AEI score is correlated with higher ADAR1 activity.34 35 We first validated the correlation between the AEI score and ADAR1 expression by assessing the AEI score in ADAR1-WT and ADAR1-KO cells. The AEI score was indeed higher in the ADAR1-WT cells than in the ADAR1-KO cells, as we expected (online supplemental figure 4E). Interestingly, the AEI score was decreased in LINC00624-depleted SK-BR-3 cells compared with that in wild-type cells (figure 4A), supporting the idea that LINC00624 can enhance the RNA-editing ability of ADAR1.
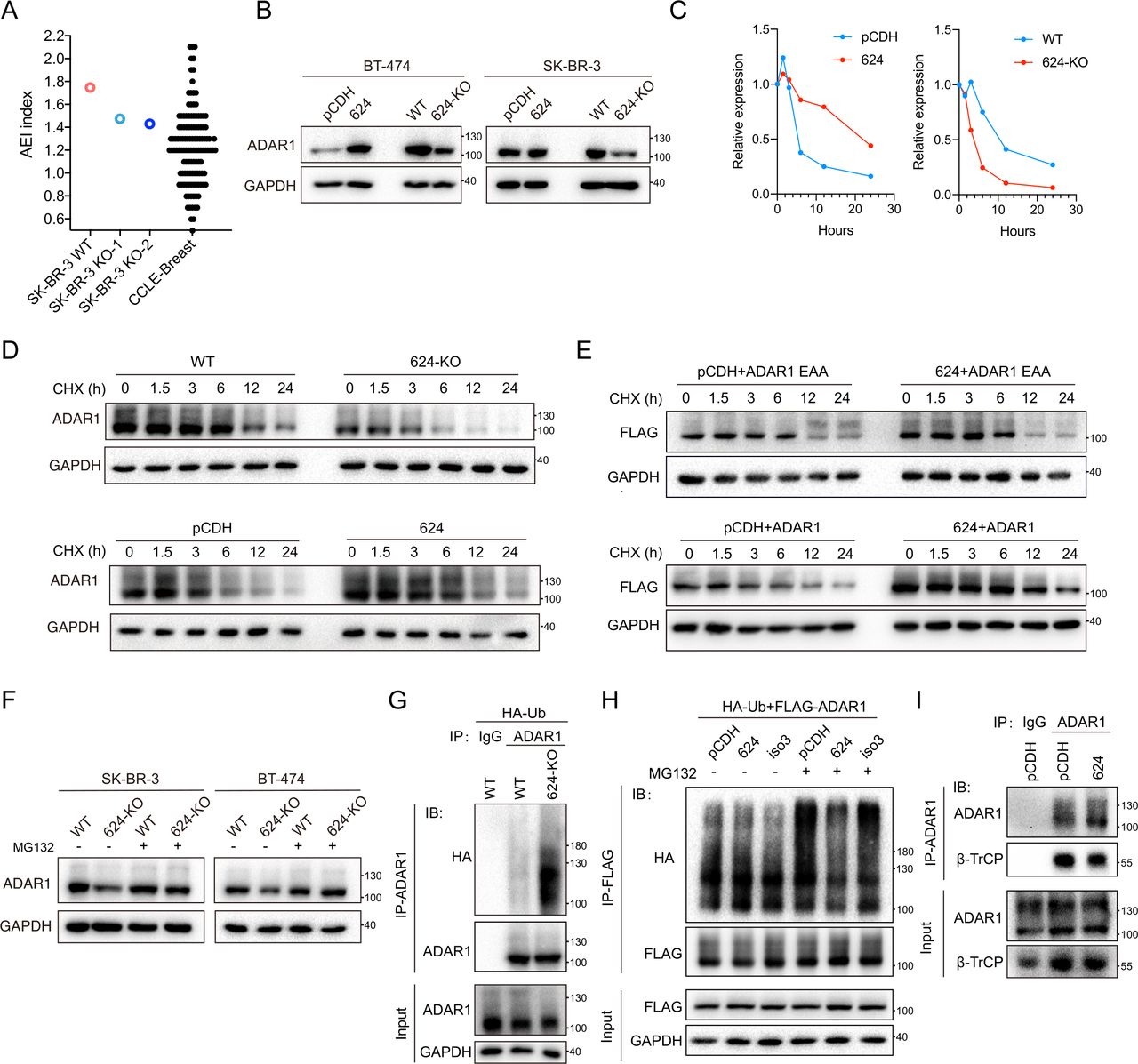
LINC00624 promotes ADAR1 expression. (A) AEI score represents A-to-I RNA editing in cells. The AEI score of WT and LINC00624 KO SK-BR-3 cells were shown. AEI score of breast cancer cell lines from CCLE was used as reference control. (B), ADAR1 protein expression in LINC00624 overexpression and 624-KO cells was examined by immunoblot. (C, D) Expression kinetics of ADAR1 with differentially expressed LINC00624 in SK-BR-3 cells. (C) Relative expression of ADAR1 normalized to GAPDH from (D) was shown. (D) Immunoblot was carried out in cells treated with the transcription inhibitor CHX (200 µg/mL). (E) Expression kinetics of ADAR1-p110-FLAG, and ADAR1-p110-FLAG with EAA mutation in 293 T cells. Cells were treated with CHX and collected at the time points as indicated. (F) WT or LINC00624-KO SK-BR-3 and BT-474 cells were treated with MG132 as indicated. The expression of ADAR1 was determined. (G) WT and LINC00624-KO SK-BR-3 cells were transfected with HA-Ubiquitin (HA-Ub). ADAR1 were immunoprecipitated (IP) with anti-ADAR1 antibody. IgG isotype control antibody was used. Immunoblot of the HA, ADAR1 and GAPDH was shown as indicated. (H) 293 T cells were transfected with FLAG-ADAR1-p110, HA-Ub, and LINC00624 isoforms as indicated (pCDH control vector, LINC00624, or isoform3 (iso3)). Treated with or without MG132 for 4 hours, cells were harvested and immunoprecipitated (IP) with anti-FLAG. Immunoblot (IB) of HA, FLAG, and GAPDH was shown. (I) SK-BR-3 cells with pCDH control or LINC00624 overexpression were immunoprecipitated (IP) with anti-ADAR1 or IgG isotype control. ADAR1 and β-TrCP were detected by immunoblot (IB). For (B–I) the experiments were performed twice with similar results. AEI, Alu-Editing Index; KO, knockout; WT, wild type.
We next sought to determine whether LINC00624 promoted ADAR1 RNA-editing ability by regulating ADAR1 expression. The mRNA level of ADAR1 was stable on exposure to different LINC00624 levels (online supplemental figure 4F). However, the protein expression of ADAR1 was correlated with the level of LINC00624 in BT474 and SK-BR-3 cells (figure 4B). In SK-BR-3 cells, the half-life of ADAR1 was prolonged significantly after LINC00624 overexpression, indicating that LINC00624 could stabilize the ADAR1 protein (figure 4C,D). KO of LINC00624 promoted the degradation of ADAR1 (figure 4C,D). As LINC00624 isoform 3 binds only weakly to ADAR1, we reconstituted either LINC00624 (isoform 1) or isoform 3 in LINC00624-KO cells. As expected, LINC00624 isoform 1, but not isoform 3, restored ADAR1 protein expression (online supplemental figure 4G). In addition, ectopic overexpression of LINC00624 in 293 T cells promoted the stability of coexpressed ADAR1, while the half-life of ADAR1 with EExxA (EAA) mutation in dsRBD3 remained unchanged with or without LINC00624 (figure 4E). These results confirmed the stability of ADAR1 depended on LINC00624 binding.
A previous study has showed that ADAR1 is degraded through the ubiquitin-proteasome pathway in human cells.36 Similarly, we found that the proteasomal degradation of ADAR1 enhanced by on LINC00624-KO was blocked by MG132 (figure 4F). To evaluate the role of LINC00624 in ADAR1 ubiquitination, we transfected hemagglutinin (HA)-ubiquitin into WT and LINC00624-KO SK-BR-3 cells. We found that LINC00624-KO promoted the ubiquitination-related degradation of ADAR1 (figure 4G). To verify that the binding of ADAR1 by LINC00624 is critical for ADAR1 ubiquitination inhibition, we coexpressed LINC00624 (isoform 1 or 3), HA-ubiquitin, and FLAG-ADAR1 in 293 T cells. Overexpression of LINC00624, but not the AER region-deleted isoform 3, inhibited the ubiquitination of ADAR1 (figure 4H). The E3 ligase β-TrCP has been demonstrated to be involved in ADAR1 ubiquitination. We found that ADAR1 bound to β-TrCP in BC cells and that overexpression of LINC00624 inhibited the binding of β-TrCP with ADAR1 (figure 4I). These results suggest that LINC00624 stabilizes ADAR1 by inhibiting ADAR1 ubiquitination-related degradation by blocking the interaction of the ubiquitin ligase β-TrCP and ADAR1.
LINC00624 inhibits the immune response and promotes treatment resistance through ADAR1
Although our data confirmed that LINC00624 was A-to-I edited both in vitro and in vivo, we found that overexpression or knockdown of ADAR1 did not affect the RNA expression of LINC00624 (online supplemental figure 5A,B). Next, we questioned whether immune response inhibition by LINC00624 was mediated through ADAR1. To answer this question, we overexpressed LINC00624 with ADAR1 knocked down in SK-BR-3 and BT-474 cells. Antigen presentation-related gene and ISG expression was recovered after ADAR1 knockdown (figure 5A, online supplemental figure 5C), suggesting that ADAR1 was involved in IFN response inhibition by LINC00624. In addition, we found that ADAR1-depleted cells were more sensitive to lapatinib (figure 5B). Overexpression of LINC00624 in ADAR1-KO cells failed to enhance the survival of SK-BR-3 cells treated with lapatinib (figure 5C), indicating that the molecular mechanism of LINC00624 in anti-HER2 treatment resistance depends on ADAR1.

LINC00624 inhibits the immune response and promotes treatment resistance through ADAR1. (A) WT or ADAR1-KO SK-BR-3 cells were overexpressed with pCDH control or LINC00624. After poly(I:C) transfection for 24 hours, RNA levels of ISGs and antigen presentation related genes were analyzed by RT-qPCR. GAPDH were used as reference gene. n=3 biological independent samples. Statistical analysis was performed using two-sided t-test. *p<0.05, **p<0.01, ***p<0.001. Data are shown as mean±SE. (B) Inhibition rate of cells in response to lapatinib. (B) WT compared with ADAR1-KO in SK-BR-3. (C) ADAR1-KO pCDH compared with ADAR1-KO LINC00624 in SK-BR-3. (D) The secondary structure of S3 was predicted by RNAfold web server. The edited base in AER region by ADAR1 was substituted with guanine (G), inosine (I), or cytidine (C) as indicated. Free energy was shown below. (E) Binding of ADAR1 with LINC00624 WT, A-to-C or A-to-G isoforms. Immunoblot of ADAR1 was shown after RNA pull-down. IVT RNA was loaded with same quantity. (F) Recovery assay of LINC00624 in BT474 624-KO cells. pCDH, LINC00624, artificially mutated A-to-C, or A-to-G isoforms were overexpressed. Inhibition rate to lapatinib was plotted. (G) Recovery assay of LINC00624 in SK-BR-3 ADAR1-KO cells. pCDH or A-to-C mutated LINC00624 were overexpressed in ADAR1-KO cells. Inhibition rate to lapatinib was plotted. (B, C, E–G) the experiments were performed twice with similar results. AER, ADAR1 Editing Region; ISGs, IFN stimulated genes; KO, knockout; ns, no significance.
Next, we questioned whether the function of LINC00624 was dependent on editing by ADAR1. When ADAR1 was constitutively expressed, LINC00624 was spontaneously edited in BC cell lines and clinical samples (figure 3H and online supplemental figure 4D). To generate an unedited isoform of LINC00624, we artificially mutated ADAR1-sensitive bases to render them uneditable. RNAfold was used to simulate the secondary structure of LINC00624 with or without edits (figure 5D). When we substituted editable A bases with C bases, the structure and free energy of the mutant S3 region were found to be similar to those of the natural A-to-I edited isoform, while mutating the bases from A to G rendered the mutant S3 similar to that of the unedited wild type (figure 5D). RNA pull-down assays showed that ADAR1 could bind to all three isoforms (figure 5E). Interestingly, ADAR1 bound even more tightly to the spontaneously edited WT or artificially edited A-to-C isoform than to the A-to-G isoform. In addition, we found that in the simulated A-to-C isoform (representing edited LINC00624) and WT isoform, the half-maximal inhibitory concentration (IC50) of lapatinib was higher than that in the A-to-G isoform (representing unedited LINC00624) (figure 5F). These results indicate that LINC00624 relies on ADAR1 A-to-I RNA editing to function. Then, we overexpressed the A-to-C isoform in ADAR1-KO cells. We found that the A-to-C isoform failed to inhibit the lapatinib response (figure 5G), an outcome similar to that of WT LINC00624 in ADAR1-KO cells, indicating that edited LINC00624 cannot function without ADAR1.
LINC00624 inhibits tumor antigen presentation
To further investigate the immune inhibition phenotype of LINC00624 in vivo, mouse cell lines and immunocompetent xenograft mouse model were then used. First, through Pipeline for lncRNA annotation from RNA-seq data (PLAR),37 we did not find orthologs or ‘synteny with sequence conservation’ of LINC00624 in mouse. LINC00624 has orthologs in rhesus and dog only (online supplemental table 5). Therefore, we overexpressed human LINC00624 in mouse cell lines. We found that LINC00624 could inhibit the type I IFN response induced by poly(I:C) in B16-OVA and NF639 cells, which was consistent with the phenotype of human cell lines (figure 6A and online supplemental figure 6A–C). Furthermore, LINC00624 promoted cell proliferation and inhibited the lapatinib response in NF639 cells, a neu-positive cell line derived from MMTV-neu tumors (online supplemental figure 6D-E). Next, we validated that the function of LINC00624 relied on ADAR1 in mouse cells. RNA pull-down confirmed the interaction between ADAR1 and LINC00624 (online supplemental figure 7A). we reconfirmed that LINC00624 could decrease mouse ADAR1 degradation in NF639 cells through ubiquitination inhibition and the blockade of ADAR1-β-TrCP interaction (online supplemental figure 7B–E). Furthermore, KO of ADAR1 in NF639 cells inhibited their proliferation (online supplemental figure 7F). Similar to their human cell counterparts, ADAR1-depleted cells were more sensitive to lapatinib in mouse cells(online supplemental figure 7G). Moreover, overexpression of LINC00624 in WT cells, but not in ADAR1-KO cells, decreased cell sensitivity to lapatinib (online supplemental figure 7G), supporting the idea that the function of LINC00624 was dependent on ADAR1 in mouse cells.

LINC00624 inhibits antitumor immunity and immunotherapy response in vitro and in vivo. (A) pCDH and LINC00624 KO B16-OVA cells were transfected with poly(I:C) for 24 hours with indicated concentration. The levels of the indicated proteins were determined by immunoblot. The experiments were performed twice with similar results. (B, C) Quantitative analysis of SIINFEKL-H-2Kb levels in pCDH and LINC00624 B16-OVA cells with or without (B) IFNα and (C) IFNγ treatment. (B) Left, representative flow cytometry image. Right, statistical analysis. n=3 biologically independent samples. (D) IFNγ released by OT-I CD8+T cells cocultured with isotype IgG or anti-SIINFEKL-H-2Kb pretreated pCDH control or LINC00624 overexpressing B16-OVA cells. n=4 biological independent samples. (E) Tumor growth curve and tumor size of B16-OVA pCDH or LINC00624 cells in C57/B6J. n=8 animals in each group. The experiment was performed twice with similar results. Statistical analysis was performed using two-sided t-test for the tumor volume at the end point. (F) Quantitative analysis of SIINFEKL-H-2Kb levels in pCDH and LINC00624 B16-OVA tumors (n=8 tumors each group). The experiment was performed twice with similar results. (G) RNA expression of ISGs and antigen presentation genes expressed in the B16-OVA tumors quantified by RT-qPCR (n=8 tumors each group). GAPDH were used as reference gene. (H–J) Flow cytometry of immune populations from pCDH control and LINC00624 overexpression B16-OVA tumors (n=8 tumors each group). (H) Percentage of CD45+cells in tumors. (I) The proportion of CD3+, CD8+T, CD4+T, CD49f+, (J) myeloid-derived suppressor cells (MDSCs) and dendritic cells (DCs) in CD45+ cells in tumor. The experiment was performed twice with similar results. Statistical analyses were performed using two-sided t-test. (K), B16-OVA tumor infiltrated CD8+cells were determined by immunohistochemistry. Left, statistical analysis (n=8 tumors each group). Right, representative images. Statistical analyses were performed using two-sided t-test. (L,M) C57/B6J were vaccinated intraperitoneally (I.P.) with poly(I:C) transfected and ultraviolet treated B16-OVA cells as indicated. B16-OVA pCDH or LINC00624 cells were inoculated subcutaneously (S.C.). Mice were treated with anti-PD-1 or IgG isotype control as indicated. (L) Schematic view of treatment protocol. (M) Tumor growth curve of each group. n=6 animals in each group. For (B–D, F–G) statistical analysis was performed using two-sided t-test for the tumor volume at the end point. The experiment was performed twice with similar results. *p<0.05, **p<0.01, ***p<0.001. Data are shown as mean±SE. ISGs, IFN stimulated genes; ns, no significance.
We also evaluated the role of LINC00624 in antigen presentation in mouse model. LINC00624 decreased the levels of major histocompatibility complex (MHC) class I-bound SIINFEKL, an eight-amino-acid peptide derived from OVA, in B16-OVA cells treated with IFNα and IFNγ (figure 6B,C). Coculture of tumor cells overexpressing LINC00624 with CD8+T cells from OT-I mice significantly inhibited IFNγ production (figure 6D), confirming the inhibitory effect of LINC00624 on antigen processing and presentation.
To determine whether LINC00624 can render tumor cells immunotolerant in vivo, we inoculated B16-OVA cells with or without LINC00624 overexpressing vectors into the flanks of immunocompetent C57BL/6J mice. First, LINC00624 overexpression increased B16-OVA xenograft tumor growth, compared with the control group (figure 6E). The MHC class I-bound SIINFEKL level was also lower in LINC00624-overexpressing tumors (figure 6F). Transcription of antigen presentation-related genes and ISGs was inhibited by LINC00624 in vivo, confirming the in vitro results (figure 6G). These results indicate the antigen presentation process of tumor cells were inhibit by LINC00624.
LINC00624 inhibits antitumor immunity and immunotherapy response in vivo
To further analyze whether LINC00624 inhibits antitumor immunity, we first investigated the infiltrated immune cells in mouse tumors. Xenograft tumors from B16-OVA cells with or without LINC00624 were dissected and digested to single cells. Flow cytometry analyses indicated a decrease in CD8+T cells, CD45+immune cells, CD3+T cells, CD4+T cells, CD8+T cells, and CD49f+ monocytes in the immune microenvironment of tumors with high LINC00624 levels, while the population of myeloid-derived suppressor cells was increased significantly (figure 6H–J, online supplemental figure 8A,B). Through immunohistochemical (IHC) staining, we confirmed a significant decrease in the infiltration of CD8+T cells in the immune microenvironment of tumors with high LINC00624 levels (figure 6K).
A previous study showed that loss of ADAR1 sensitized tumors to the innate immune response.14 Type I or type II IFNs led to growth arrest and death of ADAR1-KO B16-OVA cells, indicating that ADAR1 was involved in the modulation of the innate immune response.12 In addition, ADAR1 also promoted the blockade of immune checkpoint inhibitors. Loss of ADAR1 reversed cell resistance to immune therapy. As LINC00624 can inhibit the degradation of ADAR1, we hypothesized that LINC00624 caused resistance to immune checkpoint blockers. To test this, a B16-OVA murine model was used to investigate the role of LINC00624 in immune checkpoint blockade in vivo with a whole tumor cell vaccine loaded with poly(I:C) and anti-PD-1 (figure 6L). We found that LINC00624 significantly inhibited the tumor response to the anti-PD-1 treatment compared with the control group (figure 6M). To investigate whether the inhibition of immune therapy was dependent on ADAR1, ADAR1-KO B16 cells with different LINC0624 expression were used to address this issue. After ADAR1 knocked out, the growth of B16 tumors were significantly reduced (online supplemental figure 9A,B). Overexpression of LINC00624 could not promote tumor growth in ADAR1 null tumors. In addition, tumors were regressed after the treatment of PD-1 in ADAR1 null tumors without the vaccination process, consistent with previous study14 (online supplemental figure 9B). Overexpression of LINC00624 could not further cause treatment resistance of PD-1 (online supplemental figure 9B). Furthermore, we found tumor-infiltrating CD8+cells were significantly increased after ADAR1 KO, while LINC00624 could not inhibit CD8+cells infiltration in ADAR1 null tumors (online supplemental figure 9C), confirming the function of LINC00624 was dependent on ADAR1. All these in vivo data confirmed that LINC00624 inhibited antitumor immunity and promoted immune checkpoint inhibitor blockade.
Translational exploration of LINC00624-targeted treatments with antisense oligonucleotides
To investigate the potential therapeutic target of LINC00624 in BC, we designed five independent antisense oligonucleotides (ASOs) complementary to the LINC00624 S3 region (figure 7A). An ASO with a scrambled sequence in BC was used as the negative control. Transfection with each of the 5 ASOs reduced LINC00624 RNA levels in BT-474 cells, while ASO-2 and ASO-3 showed the highest knockdown efficiency (figure 7B). Consistently, the expression of ADAR1 was reduced, similar to the in vitro results (figure 7C). Next, we synthesized cholesterol-conjugated locked nucleic acid-modified ASOs for in vivo use. Free uptake assay results showed the downregulation of LINC00624 (figure 7D). To determine the potential clinical application of ASOs in treating BC, we generated an orthotopic mammary tumor model with BT-474 WT cells in nude mice, and the mice were treated with either control (ASO-Ctrl) or ASO-2 and ASO-3 mixtures (ASO-2/3) 10 days after inoculation (figure 7E). Although this model did not present with an adaptive immune response, we found that targeting LINC00624 significantly inhibited the proliferation of BT474 tumor cells (figure 7F,G). Indeed, xenograft tumors treated with ASOs exhibited decreased ADAR expression compared with the control, as determined by IHC (figure 7H). Moreover, the expression levels of ISGs and innate immune response genes were significantly increased in the ASO-treated xenograft tumors (figure 7I).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Translational exploration of LINC00624-targeted treatments with ASOs. (A) Schematic view of ASOs designed in the S3 segment of LINC00624. (B) Relative expression of LINC00624 48 hours after ASOs transfection in SK-BR-3 were determined by RT-qPCR, normalized to GAPDH. For each ASO, statistical analysis was performed using two-sided t-test compared with Ctrl, n=3 biological replicates. (C) Immunoblot of ADAR1 expression after ASOs transfection in SK-BR-3 cells. The experiment was performed twice with similar results. (D) LINC00624 expression after cholesterol modified ASOs delivery without transfect reagents. ASOs were added into SK-BR-3 wild type cells at the concentration as indicated. After 48 hours, RNA was extracted, and RT-qPCR was performed. Normalized to GAPDH. For each ASO, statistical analysis was performed using two-sided t-test compared with ASO-Ctrl, n=3 biological replicates. (E–G) BT-474 WT cells were inoculated in nude mice and treated with cholesterol modified ASOs through tail vein injection. (E) Schematic view of treatment plan. (F) Tumor growth curve of each group. (G) Left, mice with tumors after sacrifice were shown. Right, tumors were dissected as shown. n=6 animals in each group. The experiment was performed twice with similar results. Statistical analysis was performed using two-sided t-test for the tumor volume at the end point. ***p<0.001. (H) ADAR1 expression determined by IHC. Representative images were shown. (I) RNA expression of ISGs and antigen presentation genes expressed in the BT474 tumors after ASO treatment quantified by RT-qPCR (n=6 tumors each group), normalized to GAPDH. The experiment was performed twice with similar results. (J) Schematic view of this study. For, (B, C, and I), statistical analyses were performed using two-sided t-test. *p<0.05, **p<0.01, ***p<0.001. Data are shown as mean±SE. ASOs, antisense oligonucleotides; IHC, immunohistochemical; WT, wild type.
Altogether, these data strongly support the supposition that LINC00624 promotes therapy resistance and tumor progression by inhibiting the immune response in BC cells exposed to HER2-targeted treatment. Therefore, LINC00624 can serve as a future therapeutic target in HER2+BC.
Discussion
Tumors can escape elimination by immune cells at the initiation stage. By decreasing the expression of mutated or fusion proteins, reducing antigen presentation of neoantigens, or secreting immune suppressive signals, tumor cells can evade recognition by the immune system.38 The underlying mechanisms that tumors shape the immunosuppressive microenvironment have attracted considerable attention in recent years. Among them, the suppression of the innate immune response that prevents tumors from turning from ‘cold’ to ‘hot’ has been demonstrated.38–40
Type I IFNs recently re-entered the focus of investigation in tumor biology.1 8 Induced by the activation of nucleic sensors through transductors such as TBK1 and IRF3, type I IFNs activate the phosphorylation of STAT1, leading to increased transcription of ISGs.1 41 42 Hundreds of genes have been identified as ISGs under transcriptional regulation, and they are elevated 3- to 100-fold after type I IFN stimulation.42 43 The protein products of ISGs play different roles in antitumor biology, including immune regulation, protein synthesis suppression and apoptosis induction.43–45 Even without the involvement of immune cells, the proliferation of cancer cells were found to be inhibited when the IFN pathway was stimulated,3 consistent with our results showing that proliferation was inhibited and apoptosis was induced in LINC00624-KO cells on IFN signaling activation. In addition, upregulation of the expression of ISGs such as MHC class I proteins enhances antigen presentation to infiltrated T and B cells, eliciting an adaptive immune response.1 As we shown in our work, LINC00624 inhibits antitumor responses both in tumor cells and in the tumor microenvironment.
The tumor cell response to conventional treatments is modulated by the activation of the type I IFN pathway. In addition to cytotoxic drugs, the blockade of growth signaling pathways such as the EGFR and HER2 pathways relies on IFN signaling. Previous studies have shown that PI3K-AKT, a signaling axis downstream of the EGFR and HER2 pathways, can suppress the expression of MHC class I proteins.46 47 HER2 amplification also reduces TBK1/NAK phosphorylation, leading to the inhibition of STING pathway activation, reduction of MHC class I protein expression, and compromise of antitumor immune response.9 18
A previous study has showed that loss of ADAR1 in tumor cells enhances tumor inflammation, increases infiltrated immune cells, and sensitizes tumor cells to the blockade of immune checkpoints.7 13 14 Several researchers are exploring ADAR1 inhibitors.48 49 However, ADAR1 plays immunoregulatory roles in normal cells, inhibition of ADAR1 editase activity may raise concerns about autoimmune reactions. In our study, we found targeting LINC00624 through ASOs can significantly inhibit tumor cell proliferation, suppress ADAR1 activity and promote the type I IFN response. Through the regulation of LINC00624, we can possibly modulate ADAR1 function in tumor cells.
As LINC00624 is evolutionarily new and expressed only in human cells, the experimental models are limited and sometimes artificial models are used, especially in immune research. To tackle this issue, we overexpressed LINC00624 in mammary mouse cell lines and its effectiveness has been proven to be the same as it is in human cells. In addition, B16-OVA contains a model antigen OVA. In our study, it was used to investigate antigen presentation process as many other studies did.14 50 Through this model, we illustrated how LINC00624 regulates antigen presentation, antitumor immunity and immunotherapy response in vivo, confirming the immune suppression role of LINC00624 in immunocompetent model. Bitransgenic mice with LINC00624 overexpression in MMTV/neu mice could be used in further study to confirm how LINC00624 inhibits tumor immunity and the immunotherapy response with anti-HER2 therapy.
In conclusion, our findings demonstrate that LINC00624 plays an important role in inhibiting the IFN response and results in anti-HER2 treatment resistance. Targeting LINC00624 through ASOs shows great therapeutic potential for future clinical use.
Data availability statement
Data relevant to the study are included in the article or uploaded as online supplemental information.
Data availability statement
Original data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
The present study was approved by the Medical Ethics Committee of Fudan University Shanghai Cancer CenterID:2018FUSCC-JS-025. Patients signed the informed consent forms before biosample collection through the central biosample bank of Fudan University Shanghai Cancer Center. The ID number is:050432-4-1212B and 050432-4-1911D. Participants gave informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
QZ, BX, LZ and MC contributed equally.
Contributors QZ and BX designed and performed most of the experiments, analyzed the data, and wrote the manuscript with help from all co-authors. LZ and MC carried out all in vivo experiments and the revision of the manuscript. MC and WC maintained cell culture. LL analyzed clinical data. RG and JX collected and prepared the clinical samples for RNA-seq. XH and BY provided advice on clinical evaluation. SH analyzed the RNA-seq data and AEI index. Z-MS provided assistance in this study. JW, YC and BX initiated the study and provided funding and study supervision. JW is the guarantor for this study.
Funding This work was supported by the National Natural Science Foundation of China (81874115, 82002795, 82072919, and 82173274) and Natural Science Foundation of Shanghai (21ZR1414300).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.