Article Text

Abstract
Objectives To evaluate the morphine-sparing effects of the sequential treatment versus placebo in subjects undergoing total knee arthroplasty (TKA), the effects on pain relief, inflammation control and functional rehabilitation after TKA and safety.
Design Double-blind, pragmatic, randomised, placebo-controlled trial.
Setting Four tertiary hospitals in China.
Participants 246 consecutive patients who underwent elective unilateral TKA because of osteoarthritis (OA).
Interventions Patients were randomised 1:1 to the parecoxib/celecoxib group or the control group. The patients in the parecoxib/celecoxib group were supplied sequential treatment with intravenous parecoxib 40 mg (every 12 hours) for the first 3 days after surgery, followed by oral celecoxib 200 mg (every 12 hours) for up to 6 weeks. The patients in the control group were supplied with the corresponding placebo under the same instructions.
Primary and secondary outcome measures The primary endpoint was the cumulative opioid consumption at 2 weeks post operation (intention-to-treat analysis). Secondary endpoints included the Knee Society Score, patient-reported outcomes and the cumulative opioid consumption.
Results The cumulative opioid consumption at 2 weeks was significantly smaller in the parecoxib/celecoxib group than in the control group (median difference, 57.31 (95% CI 34.66 to 110.33)). The parecoxib/celecoxib group achieving superior Knee Society Scores and EQ-5D scores and greater Visual Analogue Scale score reduction during 6 weeks. Interleukin 6, erythrocyte sedation rate and C-reactive protein levels were reduced at 72 hours, 2 weeks and 4 weeks and prostaglandin E2 levels were reduced at 48 hours and 72 hours in the parecoxib/celecoxib group compared with the placebo group. The occurrence of adverse events (AEs) was significantly lower in the parecoxib/celecoxib group.
Conclusions The sequential intravenous parecoxib followed by oral celecoxib regimen reduces morphine consumption, achieves better pain control and functional recovery and leads to less AEs than placebo after TKA for OA.
Trial registration number ClinicalTrials.gov (ID: NCT02198924).
- celecoxib
- cumulative opioid consumption
- opioid sparing
- parecoxib
- postoperative pain
- total knee arthroplasty
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- celecoxib
- cumulative opioid consumption
- opioid sparing
- parecoxib
- postoperative pain
- total knee arthroplasty
Strengths and limitations of this study
This is the first study to investigate the efficacy and safety of the sequential analgesia regimen of intravenous parecoxib followed by oral celecoxib after total knee arthroplasty surgery.
The study employed a prospective, randomised, multicentre design.
This study explored the benefits of prolonged sequential treatment of parecoxib and celecoxib in medium-term function recovery.
Potential limitations include the need for further validation studies from other institutions outside China, lack of investigation of the long-term (eg, >3 months) effects of the sequential treatment and compromise of the test accuracy of synovial fluid cytokines.
Introduction
Osteoarthritis (OA) is a chronic degenerative joint disorder which frequently occurs in the elderly.1 Total knee arthroplasty (TKA), an effective treatment for end-stage knee OA,2 has been regarded as the most painful orthopaedic surgery due to the weight-bearing characteristics of the knee joint and the high demand of functional exercise post operation.3 Inadequate pain control is correlated with prolonged postoperative bed time, increased incidence of pulmonary infection, deep venous thrombosis, pulmonary embolism and poor functional recovery in some patients after TKA.4 5
Multimodal analgesia is currently recommended for postoperative pain control after TKA.6 As opioid tolerance and related side effects are becoming an increasingly significant problem, and even causing public health emergency, great challenges are faced by pain management post TKA.7 8 Therefore, the value of non-steroidal anti-inflammatory drug (NSAID), especially selective cyclo-oxygenase-2 (COX-2) inhibitors, as an important alternative has become increasingly prominent.9 10
In many Chinese institutions, 40 mg parecoxib is routinely administered intravenously two times per day for the first 3 days after surgery, followed by 200 mg celecoxib administered orally two times per day for 2 weeks or longer. Although this sequential therapeutic strategy has been adopted by most Chinese orthopaedic surgeons for its clinical convenience and satisfactory results during clinical observation, high quality evidence is still lacking to support its use and popularisation.
The PIPFORCE study aimed to investigate the sequential analgesic regimen with intravenous parecoxib followed by oral celecoxib for postsurgical analgesia in OA patients undergoing TKA. The primary objective was to evaluate the morphine-sparing effects of the sequential treatment with parecoxib and celecoxib versus placebo in subjects undergoing TKA. Secondary objectives included comparing the sequential treatment versus placebo for their effects on pain relief, inflammation control and functional rehabilitation after TKA and determining the safety profiles of study and control regimens.
Materials and methods
Study design
This was an investigator-initiated, multicentre, double-blind, randomised, placebo-controlled trial. Details of the trial design have been previously published.11 The project was registered in the ClinicalTrails.gov site. In brief, 246 consecutive patients who underwent elective unilateral TKA because of OA were screened and enrolled in four tertiary care hospitals in China (Peking Union Medical College Hospital as the coordinating centre, West China Hospital of Sichuan University, People’s Hospital of Peking University and Second Affiliated Hospital of Zhejiang University College of Medicine) from 1 December, 2014, to 22 September, 2016. The ethical committees of all participating hospitals approved the study before patient recruitment.
Study participants
Inclusion and exclusion criteria were strictly implemented as stated previously,11 and all patients signed the informed consent form at screening, before any study-specific procedures were conducted. The present study was performed in agreement with the Consolidated Standards of Reporting Trials statement.
Inclusion criteria
Subject eligibility was reviewed and documented by an appropriately qualified member of the investigator’s study team before subject inclusion in the study. In addition, subjects must meet all the following inclusion criteria to be eligible for enrolment:
Planned elective unilateral total knee arthroplasty because of OA, to be performed under a standardised regimen of general anaesthesia, as specified in this protocol.
Evidence of a personally signed and dated informed consent form indicating that the subject (or a legal representative) has been informed of all pertinent aspects of the study.
Age above 18 years (male or female).
Male and female subjects of childbearing potential agreeing to use an effective method of contraception throughout the study and for 42 days after the last dose of the assigned treatment. A subject is of childbearing potential if, in the opinion of the investigators, he/she is biologically capable of having children and sexually active.
Total duration of the surgical procedure of 4 hours or less.
American Society of Anesthesiologists (ASA) grade 1 to 3 cases.
Willingness and ability to comply with scheduled visits, treatment plan, laboratory tests, standardised rehabilitation scheme and other study procedures.
Satisfactory health as determined by the investigators on the basis of medical history and physical exam.
Sufficient psychomotor dexterity and cognitive capacity to use intravenous patient-controlled analgesia.
Subjects residing close to the hospital may be considered in priority for convenient and sufficient follow-up.
Exclusion criteria
Subjects will be excluded with any of the conditions listed below:
Requirement of a revision to a previous knee arthroplasty and/or planned bilateral knee arthroplasties.
Requirement of an emergency knee arthroplasty.
Addiction to any NSAIDs and opioids.
Known hypersensitivity to COX-2 specific inhibitors, sulfonamides, lactose, NSAIDs, opioids or acetaminophen/paracetamol; a history of asthma, urticaria or allergic type reactions after taking aspirin or other NSAIDs.
A history of arthritis (ie, rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis), chronic pain (eg, fibromyalgia), metastasis or Paget’s disease.
Administration of any investigational medication within 30 days prior to the first dose of study medication or plan to receive any investigational drug other than those described in the protocol during the study.
Any known laboratory abnormality, which in the opinion of the investigators, would contraindicate study participation, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen or creatinine ≧1.5 times the upper limits of respective normal reference ranges.
Active malignancy of any type, or a history of malignancy (cases with a history of basal cell carcinoma that has been successfully treated can be entered into the study. Those with a history of other malignancies that have been surgically removed, showing no evidence of recurrence for at least 5 years before study enrolment, were also entered into the study).
Inflammatory bowel disease (eg, Crohn’s disease or ulcerative colitis), chronic or acute renal or hepatic disorder, a significant coagulation defect or any condition which could preclude the use of NSAIDs or COX-2 specific inhibitors.
Active or suspected oesophageal, gastric, pyloric channel or duodenal ulceration.
Treatment with warfarin or other anticoagulants in the 30 days preceding the first dose of study medication (cardioprotective aspirin, ≦ or 325 mg/day permitted, when the dose has been stable for at least a month prior to entering the study; anticoagulation is permitted when related to the surgery, with such medicines as low molecular weight heparin, including Lovenox and Fragmin.
Anticipated or actual requirement of treatment with lithium.
ASA grade 4 to 5 cases.
A history of a psychiatric disorder requiring new or changing treatment (a subject with a stable psychiatric disorder on therapy may enter the study if no therapeutic changes have been required for the 4 weeks prior to study entry and none are anticipated for the 2-week duration of this study).
A history of uncontrolled chronic disease, or a concurrent clinically significant illness or medical condition, which in the investigators’ opinion, would contraindicate study participation or confound data interpretation, including but not limited to: uncontrolled hypertension, uncontrolled ischaemic heart disease, uncontrolled cardiac insufficiency, a history of coronary artery bypass graft surgery, a history of heart valve surgery or coronary stent implantation, a history of peripheral vascular disease or cerebrovascular disease, moderate or severe hepatic impairment, fluid retention, heart failure and abdominal pain of unknown aetiology (or study medication could mask symptoms).
Any cognitive impairment or other characteristics that would in the investigator’s opinion preclude study participation or compliance with protocol mandated procedures.
A history of asthma or bronchospasm, requiring treatment with glucocorticoids.
A history of alcohol, analgesic or narcotic abuse.
Previous randomisation into the study.
Being a staff member of an investigational site or a relative to a site staff member.
Participation in other studies within 3 months before the beginning of the current trial.
Another severe acute or chronic medical or psychiatric condition, or laboratory abnormality that may increase the risk associated with study participation or investigational product administration, may interfere with data interpretation based on investigators’ judgement or would render the subject inappropriate for study enrolment.
Pregnancy or breastfeeding in females, or males and females of childbearing potential not using effective contraceptives or agreeing to continue effective contraception from screening through 42 days after the last dose of investigational product.
Procedures
The study consisted of three phases: an initial screening phase completed within 30 days prior to randomisation, a 6-week double-blind treatment phase and a 6-week follow-up phase.
In the first phase, the investigators initiated the required screening procedures after obtaining written informed consent. All eligible patients after selection by inclusive/exclusive criteria were assigned in the order of enrolment to their allocated treatment groups according to a computer-generated randomisation sequence.
In the second phase, after screening completion, participants who remained eligible entered a 6-week double-blind randomised treatment period. All participants underwent standard TKA on unilateral side under general anaesthesia. The surgical techniques and anaesthetic regimen used in the four centres were the same, and have been described clearly in a previous report.11 Patients in the study group were supplied sequential treatment with parecoxib at 40 mg intravenously two times per day (every 12 hours) for the first 3 days post-surgery followed by celecoxib at 200 mg orally two times per day (every 12 hours) for up to 6 weeks, while control patients were administered the corresponding placebo under the same instructions. Patient-controlled intravenous analgesia with morphine was administrated to all participants starting immediately post-anaesthesia and ending at 24 hours after operation. As long as oral intake is feasible, both groups may receive centrally acting analgesic tramadol hydrochloride in the oral form for rescue analgesia in case of Visual Analogue Scale (VAS) score ≥3. With sufficient pain management, patients were instructed to perform functional exercise according to the standardised post-TKA exercise plan.
In the third phase, a telephone safety follow-up at 12 weeks post-surgery was conducted to record any adverse events that may have occurred. Altogether, there were 10 visits for each participant. Screening was performed at visit 1, and the day of TKA operation was considered day 0. There was a visit 1 day before the operation (visit 2), when patient eligibility was evaluated again, and the visit right after the operation was visit 3. Those on days 1, 2 and 3 post-surgery were regarded as visits 4, 5 and 6, respectively; then there were visits 7, 8 and 9 at 2, 4 and 6 weeks post-surgery, respectively. The last visit, visit 10, was at 12 weeks post-surgery.
Randomisation and blinding
All participants who met the study inclusion and exclusion criteria were randomly assigned in a 1:1 ratio to the parecoxib/celecoxib and placebo groups, respectively. Allocation or randomisation was study site based.
The electronic data capture system automatically generated participant identification numbers in sequence at baseline, which were subsequently linked to treatment assignments at randomisation.
In this trial, a double-blind and imitation design was used to blind patients, treating physicians, investigators and data assessors. All study medications used in the trial were identical in packaging, labelling, usage schedule, appearance, taste and odour.
Ethical review and informed consent
The benefits and risks of patient participation were explained to each patient, legal representative or witness by the investigators or their designees, and signed written informed consent was obtained before the trial. The trial was conducted in accordance with the Declaration of Helsinki.
Outcomes
The primary endpoint was cumulative opioid consumption until 2 weeks post operation, which was calculated as the sum of cumulative morphine consumption over the first postsurgical 24 hours plus opioid consumption until 2 weeks post operation. The conversion equivalent of tramadol to morphine was estimated as 300 mg of tramadol equalling 20 mg of morphine.11
The key secondary endpoint was Knee Society Score (KSS) at 6 weeks post operation. Other secondary endpoints included: (1) Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC),12 KSS,13 VAS score and EQ-5D scores14 prior to operation and at 2, 4 and 6 weeks post operation; (2) cumulative opioid consumption at 24 hours, 48 hours, 72 hours, 4 weeks and 6 weeks post operation.
Exploratory endpoints included: (1) knee circumference (measured 1 cm proximal to the base of the patella); (2) knee skin temperature; (3) peripheral blood, intraoperative intra-articular fluid and postoperative drainage fluid cytokines, including interleukin (IL)-6, IL-8 and prostaglandin E2 (PGE2); (4) erythrocyte sedation rate (ESR) and C-reactive protein (CRP). Safety endpoints included the nature, incidence, duration and severity of adverse events (AEs). AEs occurring during and after trial medication discontinuation and their relationships with study treatment were assessed as well.
Sample size calculation
The primary hypothesis of this trial was that subjects treated with parecoxib/celecoxib would consume less morphine during postoperative 2 weeks. Based on a previous trial,15 we determined that a total of 86 participants per group would have a 90% statistical power in detecting 100 mg or more in the mean difference of cumulative opioid consumption on day 14 between the two groups, assuming a common SD of 200, and a two-sided α of 0.05. This would result in a total of 172 participants. Estimating that 30% of participants would drop out, a sample size of 246 participants was considered to be adequate for this study.
Statistical analysis
The statistician who conducted the analysis was blinded to group allocation. Summary statistics were used to describe the participant characteristics of the trial groups at baseline in the intention-to-treat (ITT) analysis set. The missing data of cumulative opioid consumption was imputed by the multiple imputation method. The results of multiple imputation data were used as a type of sensitivity analysis for comparing cumulative opioid consumption between groups.16
For primary endpoint comparison, the ITT analysis was performed to evaluate differences between groups, and effective analysis population (EAP) and per-protocol (PP) analyses were also performed for sensitivity assessment. The primary endpoint did not follow the Gaussian distribution, and was presented as median (IQR) and tested by the Mann-Whitney U test. Bonferroni correction was used to reduce the significance level as 0.05/6=0.0083. The means (95% CIs) of between-group differences of medians were calculated by the bootstrap method (1000 replications). The generalised linear mixed models (GLMM) were also performed for the primary endpoint, including group, gender, age, height and weight as fixed covariates, and different medical centres as random covariates.
For comparing the secondary and exploratory endpoints, continuous data were presented as means (SDs) or medians (IQRs) as appropriate. The secondary endpoints were analysed by the linear mixed model (LMM), adjusted for gender, age, height, weight and different medical centres. The correlation type of different measurement time points was assumed as the first order autocorrelation. Exploratory endpoints were compared by the Mann-Whitney U test, and the significance level was submitted to Bonferroni correction. For safety endpoints, categorical data were presented as counts and percentages, and tested by the Pearson’s χ2 test or Fisher’s exact test. The 95% CIs of absolute risk differences between groups were calculated by the Newcombe-Wilson Score method.17 All statistical analyses were conducted with the statistical package SPSS, V.18.0 (SPSS Inc) and R 3.4.0 software. Besides Bonferroni correction, statistical significance was defined as p <0.05 with two-sided testing.
Quality control and quality assurance
During the study, the investigators or contracted agents performed periodic monitoring visits to ensure Good Clinical Practices. The monitors reviewed all source documents to confirm that the data recorded on case report forms are accurate. The investigators and institutions allowed monitors to directly access source documents for verification.
Each step was strictly performed according to the trial protocol. Each step of quality control of measured outcomes was performed according to the standard operating and quality control procedures.
Patient and public involvement
No patients were involved in conceiving the research question, setting outcome measures or in any other process of the study design. Nor was any patient involved in trial implementation, data collection, data interpretation or writing of the report. There are no plans to disseminate the results of the study to the subject or the relevant patient communities.
Results
Study patients and follow-up
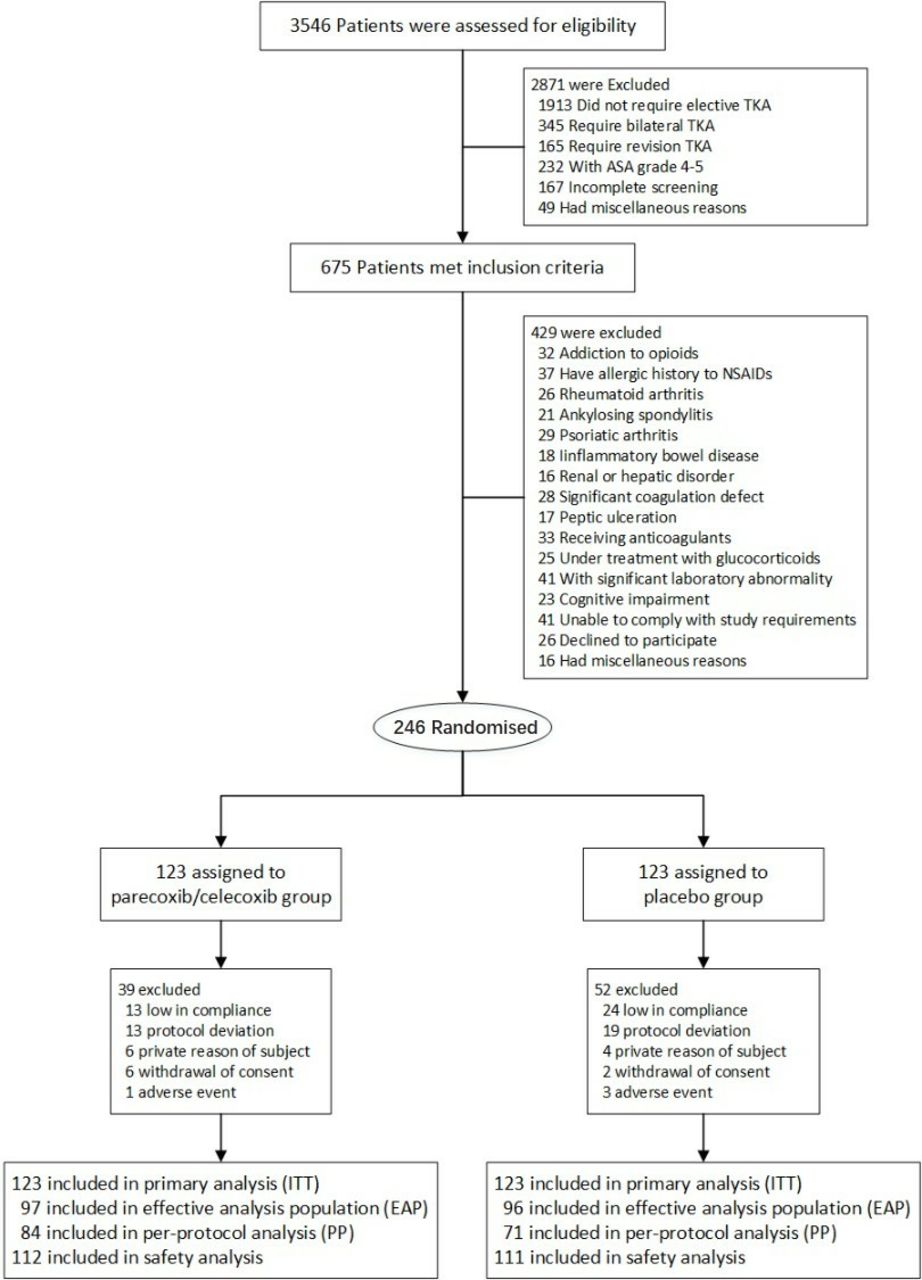
Patient recruitment began on 1 December, 2014, and the study ended on 6 December, 2016. A total of 3546 participants were screened for eligibility, and 246 patients were ultimately enrolled and randomised (figure 1).

Flowchart of the participants through the study. ASA, American Society of Anesthesiologists; ITT, intention-to-treat; NSAID, non-steroidal anti-inflammatory drug; TKA,total knee arthroplasty.
The demographic and baseline characteristics of the randomly assigned patients who received at least one dose of study medication are displayed in table 1 (intention-to-treat set). The baseline characteristics of the two groups were well balanced. There were no statistical differences in age, height and body weight between the placebo and parecoxib/celecoxib groups at baseline.
Demographic and baseline participant characteristics by group (intention-to-treat analysis)
Primary and secondary outcomes
In ITT analysis, cumulative opioid consumption levels until 2 weeks were significantly reduced in the parecoxib/celecoxib group compared with the placebo group (Z=4.849, p<0.001). The bootstrap method showed that the between-group median difference was 57.31 (95% CI 34.66 to 110.33). The results were similar in EAP and PP analyses (Z=6.619, p<0.001; Z=5.992, p<0.001). Meanwhile, longitudinal analysis by the GLMM showed a significant difference between the two groups (p<0.001) in ITT analysis. Besides, significant opioid consumption reductions throughout postoperative 6 weeks were also observed in the parecoxib/celecoxib group compared with the placebo group (p<0.001; table 2). Sensitivity analysis results from the multiple imputation data set also showed that the placebo group had increased opioid consumption compared with the parecoxib/celecoxib group (online supplementary table 1).
Supplemental material
Cumulative opioid consumption of post operation in two groups
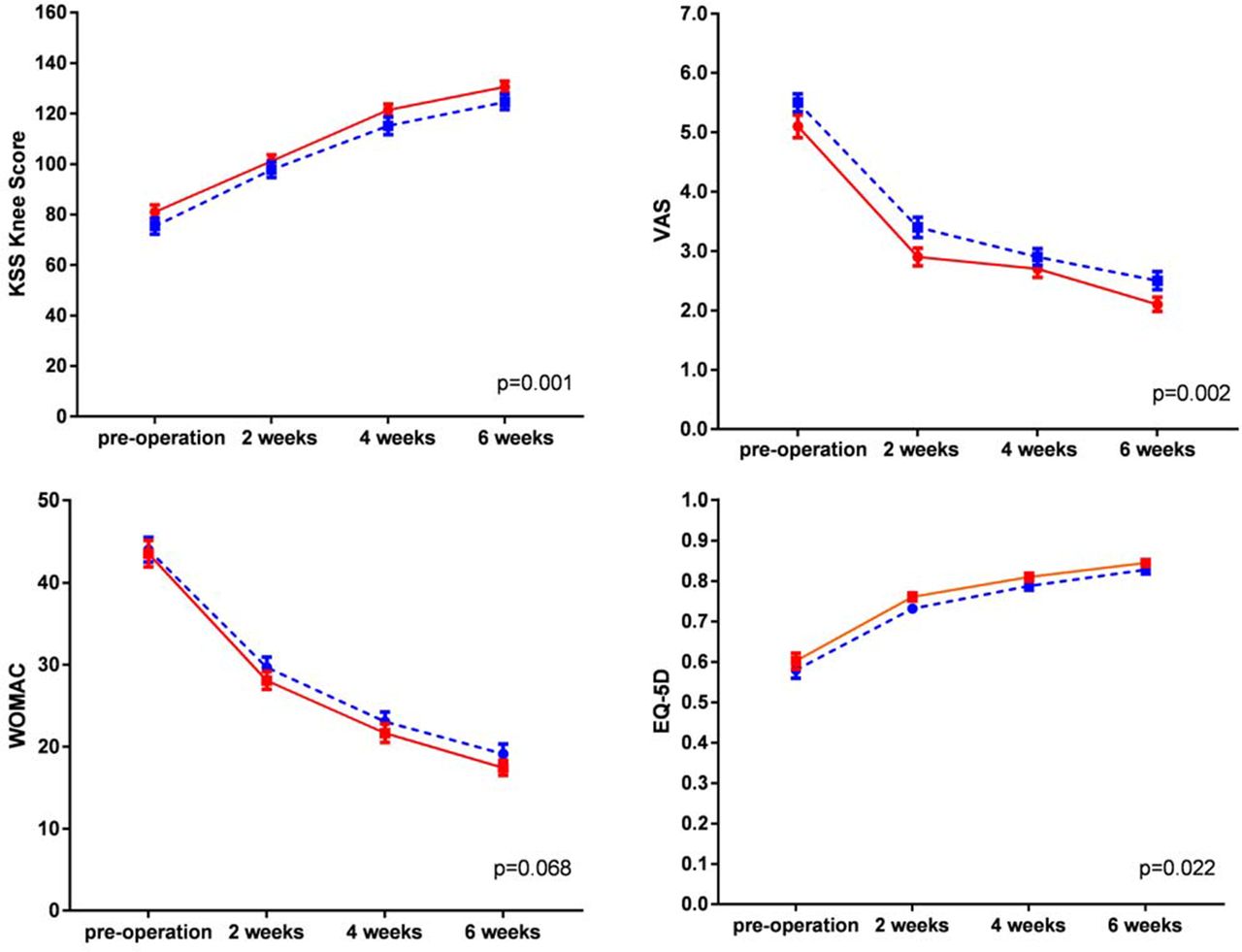
As secondary outcomes, KSS and EQ-5D scores were increased at 2 weeks, 4 weeks and 6 weeks postoperatively in both groups. The LMM showed significant differences between the two groups (p=0.001 and p=0.022, separately), with the parecoxib/celecoxib group achieving superior KSS and EQ-5D scores over the placebo group within 6 postoperative weeks. Similarly, a significant difference between the decreasing tendencies of VAS score was also demonstrated between the two groups (p=0.002). The WOMAC index showed no significant differences between the two groups at the predefined time points (figure 2).

{kind=link}
{kind=link}
KSS, VAS, WOMAC and EQ-5D6 score between the two groups in the effectiveanalysis population set. The red solid lines represent the parecoxib/celecoxib group, the blue dashed lines represent the control group and error bars represent SEs calculated separately for each time point. The differences between groups were tested by linear mixed model adjusted for the gender, age, height, weight and different medical centres. KSS, Knee Society Score; VAS, Visual Analogue Scale; WOMAC, Western Ontarioand McMaster Universities Osteoarthritis Index.
As for the exploratory endpoints, peripheral blood tests revealed that IL-6, ESR and CRP levels were significantly reduced at postoperative 72 hours, 2 weeks and 4 weeks in the parecoxib/celecoxib group compared with the placebo group. PGE2 levels in the intraoperative intra-articular fluid and postoperative drainage fluid, and knee circumference were also significantly reduced at postoperative 48 hours and 72 hours in the treatment group compared with the placebo group. Knee skin temperature was not significantly different between the two groups (table 3).
Cytokine, knee circumference and knee skin temperature of post operation in effectiveanalysis population set
Safety
The incidence rate of AEs was significantly lower in the parecoxib/celecoxib group (22.3%) compared with the placebo group (40.5%), and the absolute rate difference between the two groups was −18.22% (95% CI −30.17% to −6.27%; p=0.003). In addition, there were five serious AEs in the placebo group and zero in the parecoxib/celecoxib group (p=0.029). The five serious AEs included one case of joint stiffness, one case of stenocardia, one case of fever and two cases of pain. All of the serious AEs were resolved timely with no sequel after the proper treatment.
No significant differences were detected in AE durations or expected AEs between the two groups (table 4). Other types of adverse events showed no statistically significant differences between the two groups, except that hyperhidrosis, pain, fever, blood glucose and body temperature were increased (table 4).
Adverse events between groups in the Safety set
Discussion
The enhanced recovery after surgery (ERAS) programme18 has now been recognised and recommended in various elective surgeries.19 20 The ERAS concept aims to adopt standardised multimodal pathways to improve clinical outcomes, specifically in optimising postsurgical pain control and enabling early rehabilitation.19 20 Therefore, effective pain management with minimal systemic opioid use is a key component of the ERAS pathway in TKA patients.21
Pain control
The present study demonstrated better pain control performance and opioid-sparing effects with the sequential analgesia. Patients in the parecoxib/celecoxib group not only required less morphine, but experienced greater pain relief compared with the placebo group at all time points after surgery. Since both pain and opioid-related symptoms can hinder the patient’s mobilisation and may increase the length of recovery,21 our results suggest that the sequential analgesia regimen with parecoxib followed by celecoxib is a potential excellent choice for pain relief and enhanced recovery.
Inflammation control
Our results also showed that peripheral blood IL-6, postoperative drainage fluid PGE2, ESR and CRP were significantly decreased in the parecoxib/celecoxib group. Previous findings22 have suggested that local inflammatory reactions triggered by tissue damage not only increase central and peripheral pain sensitivity but also lead to local intensified pain, oedema and increased bleeding at the knee joint, which is a great challenge in postoperative rehabilitation. Our present findings provide positive evidences that sequential use of COX-2 selective NSAIDs after surgery decreases surgically induced secretion of inflammatory mediators, reduces the incidence of fibrosis and the degree of local oedema, which is beneficial for postoperative exercises.
Opioid consumption
The decreased opioid consumption with the sequential analgesia provides benefits not only in reducing opioid-related adverse effects, but also in reducing overall treatment costs. Opioid drugs are associated with various dose-dependent adverse symptoms.7 8 In the present study, we observed slightly less gastrointestinal adverse events in the parecoxib/celecoxib group (19.64%) compared with the placebo group (20.72%). Athanasakis et al 23 demonstrated that addition of parecoxib to opioid leads to potential savings of €858 per patient compared with opioid use alone. Therefore, reduction in the occurrence of opioid-related adverse events can lead to savings on overall treatment costs.
Adverse events
Our safety data showed that the incidence and severity of adverse events were significantly lower in the parecoxib/celecoxib group compared with the placebo group. The PRECISION trial24 demonstrated that celecoxib at moderate doses is non-inferior to ibuprofen or naproxen with regard to cardiovascular safety. The CONCERN trial25 concluded that in patients at high risk of both cardiovascular and gastrointestinal events, celecoxib plus proton-pump inhibitor is the preferred treatment. All these emerging evidences support the safety of selective NSAID drugs, and therefore the sequential analgesic regimen in this study.
Functional rehabilitation
This study observed significantly higher KSS and function scores of the operated knee, and better EQ-5D scores within 6 weeks post-operation in the parecoxib/celecoxib group. Theoretically, satisfactory pain management and inflammation control are beneficial to rehabilitation effectiveness, quick functional recovery and high patient satisfaction. Malan et al 26 and Desjardins et al 27 demonstrated that both parecoxib and celecoxib result in significantly improved recovery and patient satisfaction. Further well-designed trials with larger sample size and longer treatment period are suggested to elucidate the associations of NSAID use with knee function improvement and patient satisfaction after TKA.
Strengths
This study was, to our knowledge, the first randomised trial to investigate the efficacy and safety of the sequential analgesic regimen of intravenous parecoxib followed by oral celecoxib after TKA, assessing not only morphine consumption, but also pain relief, inflammation control and functional rehabilitation. In addition, compared with previous studies which attempted to observe the short-term effects of single NSAIDs, the present study showed the benefits of prolonged sequential treatment of parecoxib and celecoxib in medium-term recovery. Finally, facing the worldwide problem of opioid tolerance and related side effects, these data of the postoperative sequential regimen of NSAIDs that have been widely accepted as clinical routine in China, may provide important evidence to support the incorporation of this strategy into the standard multimodal analgesic regimen of the ERAS programme in OA patients undergoing TKA.
Limitations
The possible limitations of the PIPFORCE study should be mentioned. First, since the four study centres in this multicentre randomised controlled trial were all in mainland China, the results should be interpreted with caution, and further validation studies of data sets from other institutions outside China are required. Second, the PIPFORCE study did not investigate the long-term (eg, >3 months) effects of the sequential treatment on inflammation control and functional rehabilitation after TKA. Third, although the EAP set reached 193 participants, the PP set consisted of only 155 participants (placebo group 71; parecoxib/celecoxib group 84), which is slightly less than the precalculated 172 participants according to the above sample size estimation. However, these results demonstrated significant differences in the primary outcome between the two groups in ITT, PP and EAP analyses. Furthermore, we used the PASS 14.0 software to calculate the post hoc sample size, when the cumulative opioid consumption levels until 2 weeks between the two groups were 139.3±93.6 and 58.2±44.3, respectively, only 18 participants per group could achieve a 90% power in detecting the difference between the two groups with a significance level of 0.05 (two-sided). Therefore, we believe that the present results have sufficient power to support our conclusion. Fourth, we did not conduct a time-to-event modelling analysis for AE, such as competing risk analysis. Lastly, we used general anaesthesia in this study without combining regional anaesthesia, and it should be noted that our results could not be generalised in every anaesthetic technique.
Summary
In conclusion, the PIPFORCE trial demonstrated that the sequential analgesic regimen with intravenous parecoxib followed by oral celecoxib for postsurgical analgesic treatment requires less morphine in postoperative 2 weeks. Given the increasing recognition of opioid tolerance and related side effects as well as the emerging high quality evidences for cardiovascular and gastrointestinal safety of selective NSAIDs, sequential NSAID use could play a more significant role than currently known in multimodel analgesic regimens. It should be noted that since the PIPFORCE trial was exclusively performed in mainland China, these results still require further validation studies of data sets from other institutions outside China.
Acknowledgments
We would like to thank all study participants and staff who devoted their time and efforts to the study, especially to Dr Shangyi Hui (Peking Union Medical College Hospital, China), Professor Yiming Zhao (Peking University Third Hospital, China) and Miss Zhuang Liu for their critical reading and modification of the manuscript. Thanks to Caddie Qu, Jialing Yang and Math Zhang at Shanghai Bestudy Medical Technology Co, Ltd, for their statistical analysis support.
References
Footnotes
Contributors WXS contribute as the senior author and the principle investigator (PI) of this study. ZQY, as the sub PI, design the protocol, wrote the first draft of the manuscript and contributed to the coordination of the study. TLY, as the medical statistician for the study, contributed to the statistical design, acquisition and analysis of data for the work. JJ, LJ, QWW, BYY, WW, GN, FB, STZ, ZMF recruited patients for the study and participated in coordination. LYL, DYL, PHM, LY, FY were responsible for data input. LJH, YSG, SB, PFX critically revised the report. All authors reviewed the results and approved the final version of the manuscript.
Funding The PIPFORCE study was an investigator-initiated research sponsored by Pfizer (grant number: WI182703). Parecoxib sodium, celecoxib and placebo were provided by Pfizer. The funders had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript and decision to submit the manuscript for publication.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval Approval for this study has been obtained from the Ethics Committee of Peking Union Medical College Hospital, China.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.