
Abstract
Genes specifying long non-coding RNAs (lncRNAs) occupy a large fraction of the genomes of complex organisms. The term ‘lncRNAs’ encompasses RNA polymerase I (Pol I), Pol II and Pol III transcribed RNAs, and RNAs from processed introns. The various functions of lncRNAs and their many isoforms and interleaved relationships with other genes make lncRNA classification and annotation difficult. Most lncRNAs evolve more rapidly than protein-coding sequences, are cell type specific and regulate many aspects of cell differentiation and development and other physiological processes. Many lncRNAs associate with chromatin-modifying complexes, are transcribed from enhancers and nucleate phase separation of nuclear condensates and domains, indicating an intimate link between lncRNA expression and the spatial control of gene expression during development. lncRNAs also have important roles in the cytoplasm and beyond, including in the regulation of translation, metabolism and signalling. lncRNAs often have a modular structure and are rich in repeats, which are increasingly being shown to be relevant to their function. In this Consensus Statement, we address the definition and nomenclature of lncRNAs and their conservation, expression, phenotypic visibility, structure and functions. We also discuss research challenges and provide recommendations to advance the understanding of the roles of lncRNAs in development, cell biology and disease.
Similar content being viewed by others

Introduction
Research on long non-coding RNAs (lncRNAs), a previously unsuspected major output of genomes of complex organisms, has been dogged by uncertainty and controversy from its beginning. lncRNAs have the unfortunate distinction of being named for what they are not, rather than what they are. This loose description has its origins in the belief that the main role of RNA is to act as the intermediate between a gene and a protein, with other ‘housekeeping’ non-coding RNAs such as ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), small nucleolar RNAs (snoRNAs), spliceosomal RNAs and other small nuclear RNAs (snRNAs) being ancillary to this function.
Broad recognition of RNA as a regulatory molecule occurred in the early years of the first decade of the twenty-first century with the unexpected discovery of large numbers of small interfering RNAs (siRNAs), microRNAs (miRNAs) and small PIWI-interacting RNAs (piRNAs) that regulate — through Argonaute family proteins — gene expression at transcriptional, post-transcriptional and translational levels in eukaryotes1, although there were examples of other small regulatory RNAs in the literature, especially in bacteria2. A few long regulatory RNAs, notably meiRNA in the fission yeast Schizosaccharomyces pombe, hsrω, RNA on the X1 (roX1) and roX2 in Drosophila melanogaster, and H19 and X-inactive-specific transcript (XIST) in mammals, had also been reported in the preceding years3,4,5,6,7, but were regarded more as oddities than early examples of a general phenomenon. Moreover, the small regulatory RNAs did not disturb the conceptual framework that most genes encode proteins, but rather fitted comfortably into it. It was later found, however, that while some miRNAs are generated from the introns of pre-mRNAs8, non-coding primary transcripts of miRNAs and of snoRNAs can also have functions9,10 and that rRNAs, tRNAs and snoRNAs are processed to generate small regulatory RNAs, including miRNAs11,12,13,14, in some cases contributing to transgenerational epigenetic inheritance15.
A bigger surprise, and challenge to the reigning understanding of genetic information, came in the early and middle years of the first decade of the twenty-first century, when global transcriptomic analyses, intended to better define the proteome, revealed that most of the genome of animals and plants is dynamically transcribed into longer RNAs that have little or no protein-coding potential16,17,18,19. This surprise was compounded by the associated finding that the number, and to a large extent the repertoire, of protein-coding genes is similar in animals of widely different developmental and cognitive complexity — the nematode worm Caenorhabditis elegans (comprising ~1,000 somatic cells) and humans (~30 × 1012 somatic cells20) both have ~20,000 protein-coding genes — which was termed the ‘g-value paradox’21. By contrast, the extent of non-coding DNA, and consequently the transcription of non-coding RNAs, has increased with increasing developmental complexity22.
Understandably, the common initial reaction of the molecular biology community was to suspect that these unusual RNAs are transcriptional noise, because of their generally low levels of sequence conservation, low levels of expression and low visibility in genetic screens. Since then, however, there has been an explosion in the number of publications reporting the dynamic expression and biological functions of lncRNAs, aided by extensive technology development that has enabled their identification and characterization, although only a minority of lncRNAs have confident annotations and very few have mechanistic information. The realization that the genomes of plants and animals express large numbers of lncRNAs requires a framework for their classification and understanding of their functions and, more profoundly, a reassessment of the amount and type of information required to programme the development of complex organisms.
Purpose of this Consensus Statement
In this Consensus Statement we present a current and coherent picture of the roles of lncRNAs in cell and developmental biology, identify the key issues in understanding their functions and chart the path forward. We address lncRNA definition, nomenclature, conservation, expression, phenotypic visibility, functional assays and molecular mechanisms encompassing lncRNA connections to chromatin architecture, epigenetic processes, enhancer function and biomolecular condensates, as well as the roles of lncRNAs outside the nucleus. We argue that loci expressing lncRNAs should be recognized as bona fide genes and discuss lncRNA structure–function relationships as the means to parse mechanisms and pathways. Finally, we identify the current challenges and offer recommendations for understanding the relationship of lncRNAs to genome architecture, gene regulation and cellular organization.
The authors of this Consensus Statement were suggested by recommendations of colleagues. Consensus was reached by group e-mail and discussion.
Definition and nomenclature of lncRNAs
lncRNAs have been arbitrarily defined as non-coding transcripts of more than 200 nucleotides (200 nt), which is a convenient size cut-off in biochemical and biophysical RNA purification protocols that deplete most infrastructural RNAs, such as 5S rRNAs, tRNAs, snRNAs and snoRNAs, as well as miRNAs, siRNAs and piRNAs23. This definition also excludes some other well-known short RNAs such as the primate-specific snaRs (~80–120 nt), which associate with nuclear factor 90 (ref. 24); Y RNAs (~100 nt), which act as scaffolds for ribonucleoprotein (RNP) complexes25; vault RNAs (88–140 nt), which are involved in transferring extracellular stimuli into intracellular signals26; and promoter-associated RNAs and non-canonical small RNAs produced by post-transcriptional processing27,28,29. Other non-coding RNAs lie close to the 200-nt border, such as 7SK (~330 nt in vertebrates), which controls transcription poising and termination, including at enhancers30,31, and 7SL (~300 nt), which is an integral component of the signal recognition particle that targets proteins to cell membranes32 and the evolutionary ancestor of the widespread primate Alu (~280 nt) and rodent B1 (~135 nt) small interspersed nuclear elements33,34,35. Given this grey zone of sizes, we support the suggestion that non-coding RNAs be divided into three categories36: (1) small RNAs (less than 50 nt); (2) RNA polymerase III (Pol III) transcripts (such as tRNAs, 5S rRNA, 7SK, 7SL, and Alu, vault and Y RNAs37), Pol V transcripts in plants and small Pol II transcripts such as (most) snRNAs and intron-derived snoRNAs38,39 (~50–500 nt); and (3) lncRNAs (more than 500 nt), which are mostly generated by Pol II.
Many lncRNAs are spliced and polyadenylated, which has led to their description as ‘mRNA-like’. However, other lncRNAs are not polyadenylated or 7-methylguanosine capped19,40,41,42, are expressed from Pol I (5.8S, 28S and 18S rRNAs) or Pol III promoters, or are processed from precursors, including from introns and repetitive elements, leading to the more agnostic descriptor ‘transcripts of unknown function’43. With respect to protein-coding genes, lncRNAs can be ‘intergenic’, antisense or intronic. They are also derived from ‘pseudogenes’, which occur commonly in metazoan genomes44, with more than 10,000 pseudogenes identified in the mouse genome45 and almost 15,000 identified in the human genome46, some of which have been shown to be functional44,47. lncRNAs also include circular RNAs generated by back-splicing of coding and non-coding transcripts, also with demonstrated functions48, and trans-acting regulatory RNAs derived from sequences that conventionally act as the 3′ untranslated regions of mRNAs49.
There have been many attempts at nomenclature and classification of lncRNAs, by the HUGO Gene Nomenclature Committee, the GENCODE consortium and others, predominantly based on their genomic position and orientation relative to protein-coding genes46,50,51,52,53. Linking to nearby genes has been useful, as it provides context and has sometimes provided clues to lncRNA function, for example in regulating the expression of these genes, as is often the case with enhancers (see later), although enhancer activity should not be assumed to be directed to the most proximal genes.
Many early studies focused on long intergenic non-coding RNAs (lincRNAs), whose sequences do not trespass on nearby protein-coding loci, owing to the need to distinguish their function from that of proteins. However, many other lncRNAs overlap protein-coding loci or are expressed from enclosed introns. Moreover, the traditional view of genomes as linear arrangements of discrete protein-coding genes fails to accommodate the discovery that eukaryotic transcription, best characterized in human and model organisms, is a fuzzy continuum54, with ‘genes’ within genes, genes interleaved with other genes and non-coding transcripts overlapping or originating within them18,43,55, together posing a growing problem for genome annotations.
In both humans and D. melanogaster, for example, many protein-coding genes have 5′ exons that are incorporated into mRNA in early embryogenesis and lie hundreds of kilobases upstream of the usual first exon, bypassing many other genes in the intervening region56. Indeed, any base may be exonic, intronic or ‘intergenic’, depending on the transcriptional output of the cell at any point in its developmental trajectory or physiological state55. For this reason, unless a lncRNA is antisense to a protein-coding gene, we recommend naming lncRNAs for their own sake with allusion to a discerned characteristic or function (as has been traditional for proteins), such as XIST, antisense IGF2R non-protein-coding RNA57 (AIRN), HOX antisense intergenic RNA58 (HOTAIR), Gomafu (‘spotted pattern’ in Japanese; also known as Miat)59, COOLAIR (referring to plant vernalization)60 and auxin-regulated promoter loop61 (APOLO), for easy recollection, preferably accompanied by complete exon–intron structures and genomic coordinates. If no biological context is available, we recommend naming the lncRNA according to the GENCODE system46.
The wide range of functions of ‘non-coding’ RNAs precludes straightforward classification as specific RNA classes, with some acting locally and some at a distance, or both62. In the absence of more specific categorization, we recommend retention of the general descriptor ‘lncRNA’, noting that most have some type of regulatory or architectural, often related, role in cell and developmental biology, and because there are so many historical articles that use this term or variations thereof. Non-coding RNAs come in all shapes and sizes, and the territory is huge, covering most of the genome and a plethora of functions. Some RNAs have dual functions as coding and regulatory RNAs, and some, perhaps many, cytosolic lncRNAs encode small peptides63,64,65,66. Protein-coding loci also express lncRNAs through alternative splicing67,68,69, and, surprisingly, the major transcript produced by ~17% of human protein-coding loci is non-coding70. Indeed, both lncRNA genes and mRNA genes can produce transcripts that function following different levels of processing. Unspliced transcripts, spliced transcripts, circular RNAs, intronic RNAs and stable small RNAs generated from them can all have a function48,71,72. Any RNA can be regulatory, and any locus can encode both protein-coding and regulatory RNAs.
Well in excess of 100,000 human lncRNAs have been recorded52,73, many of which are specific to the primate lineage74. This is a vastly incomplete list due to the limited analysis of different cells at different developmental stages (see later). There are now hundreds of thousands of catalogued lncRNAs and dozens of databases (and databases of databases) with curated information75,76,77,78,79,80. Over the past decade, there have been ~50,000 publications with ‘long non-coding RNA’ as a key term and more than 2,000 publications reporting validated lncRNA functions81, although most have yet to be followed up in any detail.
From here on, we focus on lncRNAs derived from Pol II primary transcription units (and use the term in that context), as opposed to other non-coding RNAs that are expressed from Pol I or Pol III promoters, processed from introns (which, it should be noted, constitute a major fraction of the non-coding RNA in mammals and other organisms41,82,83,84) or formed by back-splicing, although many of the same considerations apply.
Conservation of lncRNAs
Most lncRNAs are less conserved among species than the mRNA sequences encoding the proteome. Initially, most of the mammalian genome (which included most lncRNA loci) was thought to be evolving neutrally, using the yardstick of the rate of divergence of common ‘ancient repeats’ (derived from transposons) between the human and mouse genomes, on the assumption that these sequences are non-functional and representative of the original distribution in the ancestor85. However, there is increasing evidence that transposable elements are widely co-opted as functional elements of gene expression and structure, forming promoters, regulatory networks, exons and splice junctions in protein-coding genes and lncRNAs86,87,88,89, and therefore cannot be used as indices of neutral evolution.
Regulatory sequences, including promoters and lncRNAs, are known to evolve rapidly due to more relaxed structure–function constraints than protein-coding sequences and due to positive selection during adaptive radiation85,90,91,92. Many lncRNAs are cell lineage specific. Indeed, given their association with developmental enhancers (see later), variation in the complement and sequences of lncRNAs may be a major factor in species diversity.
Loci expressing lncRNAs exhibit many of the characteristics of protein-coding genes, including promoters, multiple exons, alternative splicing, characteristic chromatin signatures, regulation by morphogens and conventional transcription factors, altered expression in cancer and other diseases74,93,94,95,96,97,98, and a range of half-lives similar to those of mRNAs99.
The promoters of lncRNAs exhibit levels of conservation comparable to those of protein-coding genes18,74. lncRNAs also have conserved exon structures, splice junctions and sequence patches18,74,93,97, and they retain orthologous functions despite rapid sequence evolution100,101,102. Indeed, low sequence conservation can be misleading.
The lncRNA telomerase RNA template component (TERC), which is required for telomere maintenance — a vital cellular function — differs widely in size and sequence, but has conserved structural topology from yeast to mammals, albeit with some variation, and a conserved catalytic core103,104,105,106,107,108 (see also later). X chromosome dosage compensation in Drosophila spp. requires the formation of a nuclear domain through phase separation by the lncRNAs roX1 and roX2 interacting with the intrinsically disordered region (IDR) of a specific partner protein, male sex lethal 2 (MSL2). Replacing the IDR of the mammalian orthologue of MSL2 with that of the D. melanogaster protein and expression of roX2 is sufficient to nucleate ectopic X chromosome dosage compensation in mammalian cells, showing that the roX–MSL2 IDR interaction is the primary determinant of compartmentalization of the X chromosome and that such interactions are preserved over vast evolutionary distances109. Similar processes are involved in the regulation of X chromosome dosage compensation in placental mammals by XIST, which performs several functions, including repulsion of euchromatic factors, scaffolding of new heterochromatic factors and reorganization of chromosome structure110,111,112,113.
Expression
Although there are exceptions (such as metastasis-associated lung adenocarcinoma transcript 1 (MALAT1; also known as NEAT2), which is one of the most abundant Pol II transcripts in vertebrate cells114, and nuclear paraspeckle assembly transcript 1 (NEAT1); see later), lncRNAs generally show more restricted expression patterns than mRNAs74,115, and are often highly cell specific116, which is consistent with a role in the definition of cell state and developmental trajectory. They also have specific subcellular locations, often nuclear, although a large fraction is cytoplasmic75. Although it is sometimes asserted that there are a few hundred cell types in a human, broad classifications obscure the fact that each cell occupies a precise place in a developmental ontogeny, illustrated by the differential expression of HOX genes in superficially similar skin cells in different regions of the body117, and by the expression of lncRNAs in various regions of the brain118,119,120,121 and at different stages of development122. lncRNAs are also dynamically expressed during differentiation of mammalian stem, muscle, mammary gland, immune and neural cells, among many others81,116, with a transition during development from broadly expressed and conserved lncRNAs towards an increasing number of lineage-specific and organ-specific lncRNAs123. lncRNA expression can also be strongly influenced by environmental factors, a feature that is especially prominent in plants124,125,126, which include a range of stress responses in animals and drug resistance in cancer127,128,129,130,131,132,133.
The restricted expression of lncRNAs in different cells at different stages of development and their generally low copy number (owing to their regulatory nature) accounts for their sparse representation in bulk-tissue RNA sequencing datasets134, whereas many lncRNAs are relatively easy to detect in particular cells118. The undersampling of lncRNAs is now being rectified by targeted capture98,135, advanced imaging136,137,138, spatial transcriptomics139 and, in some cases, single-cell sequencing120,121,140, which make it clear that, whereas ~20,000 human lncRNA loci have been identified by GENCODE46 and ~30,000 by the FANTOM consortium141, there is likely at least an order of magnitude more.
Due to the high complexity and the variation in transcription initiation and termination sites, expression levels and splicing, comprehensive characterization of transcriptomes is extremely challenging. A recent study showed that the low expression of a lncRNA can be essential for its functional role by ensuring specificity to its regulated targets, suggesting that low abundance levels may be an essential feature of how lncRNAs work142. To fully catalogue the universe of lncRNAs, and properly record their exon–intron organization and splice variants, high-depth sequencing will need to be performed on cells at all stages of differentiation and development, undergoing different neural, immunological and other physiological processes, and in various disease states. This is a huge task, but we recommend that future gene expression profiling should include full transcript analysis not just of mRNAs but also of small RNAs and lncRNAs that are intergenic, antisense and intronic to the annotated genes, and their stoichiometry143.
Phenotypic visibility
Like miRNAs, most lncRNAs have not been identified in genetic screens. There are two reasons for this. First, most genetic screens historically focused on protein-coding mutations, which often have severe consequences that are easy to track; by contrast, regulatory mutations often have subtle consequences that affect quantitative traits. Second, it is difficult to identify causal mutations among the many variations that occur in non-coding sequences. Indeed, most variations that influence human quantitative traits and complex disorders occur in non-coding regions, which are replete with genes expressing lncRNAs144,145 that are transcribed in cell types relevant to the associated trait141,146.
There are exceptions of lncRNAs that have been identified genetically, notably the roX1 and roX2 RNAs involved in X chromosome activation in male fruitflies5, mammalian parentally imprinted H19, Airn and Kcnq1ot1 RNAs in mice6,57,147,148 and others such as Tug1 in mice149, MAENLI (ref. 150) and HELLP (named for ‘haemolysis, elevated liver enzyme levels and low platelet count’; also known as HELLPAR)151, which are associated with disorders or developmental processes. In Arabidopsis thaliana, non-coding intronic single-nucleotide polymorphisms important for flowering-time adaptation were found to alter the splicing of the lncRNA COOLAIR152.
Many lncRNAs have been associated with the cause and progression of cancers, through altered expression of and/or mutations (including translocation breakpoints) in lncRNAs that act as oncogenes or tumour suppressors153,154,155. Other lncRNAs are involved in human genetic disorders81,156,157, including DiGeorge syndrome and other neurodevelopmental and craniofacial defects158,159,160. Phenylketonuria, one of the first documented human genetic disorders, caused mostly by mutations in the enzyme phenylalanine hydroxylase, is caused also by mutations in a lncRNA that can be treated by modified RNA mimics161.
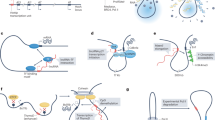
A route to analysing lncRNA biological function is to silence or delete, or (less commonly) ectopically express, lncRNAs that have been identified in RNA sequencing datasets, usually as being differentially expressed. There have been problems with the interpretation of such experiments, however, particularly the difficulty of disentangling the loss of lncRNA expression from the loss of DNA regulatory elements162,163, which has been addressed by strategies such as inserting polyadenylation sites for early transcription termination or transcription repression by CRISPR interference (CRISPRi), replacement of the lncRNA with a reporter gene that leaves the promoter intact or deletion of lncRNA exons (although loss of downstream regulatory elements cannot be ruled out), antisense-mediated blockade of lncRNA splice sites, CRISPR–Cas13 targeting of the lncRNA (rather than its DNA sequence) and transgene rescue163,164. There are now many studies that have demonstrated the biological roles of lncRNAs163, and high-throughput loss-of-function reverse genetic screens are increasing the search speed, identifying, for example, lncRNAs that are required for mammalian cell growth and migration, brain, skeletal, lung, muscle and heart development, immune function, epidermal homeostasis and cancer drug responses or lncRNAs that have fitness effects81,165,166,167,168,169,170 (Fig. 1). CRISPRi-mediated transcription repression of more than 16,000 lncRNAs in seven human cell lines identified almost 500 lncRNAs required for normal cellular proliferation, 89% of which were expressed in only one cell type167.

The following long non-coding RNAs (lncRNAs) are listed in the figure underneath their associated phenotypes: Airn, antisense of IGF2R non-protein-coding RNA147,435; Charme, chromatin architect of muscle expression436; Chaserr, CHD2 adjacent, suppressive regulatory RNA437; Fendrr, FOXF1 adjacent non-coding developmental regulatory RNA165,438; Firre, functional intergenic repeating RNA element316; Gaplinc, gastric adenocarcinoma predictive long intergenic non-coding RNA200; H19, clone pH19 (ref. 439); Handsdown, downstream of the protein-coding gene Hand2 (ref. 440) Kcnq1ot1, Kcnq1 overlapping antisense transcript 1 (ref. 441); linc-Brn1b, long intergenic non-coding RNA (lincRNA) downstream of the Brn1 protein-coding gene165; linc-Epav, endogenous retrovirus-derived lncRNA positively regulates antiviral responses442; lincRNA-Cox2, lincRNA downstream of the inflammation response gene Cox2 (ref. 443); lincRNA-Eps, lincRNA involved in erythroid prosurvival201; lnc-Lsm3b, interferon-inducible non-coding splice variant of the U6 small nuclear RNA-associated Sm-like protein lsm3 gene444; Maenli, master activator of engrailed1 in the limb165; Mdgt, midget165; Meg3, maternally expressed gene 3 (also known as Gtl2)445,446; Norad, non-coding RNA activated by DNA damage447; Peril, perinatal lethal long non-coding RNA165; Pnky, pinky (also known as lnc-Pou3f2)448; Tug1, taurine upregulated gene 1 (refs. 165,166,449) Upperhand, lncRNA upstream of the Hand2 cardiomyocyte transcription factor locus318; Xist, X-inactive-specific transcript450. Figure courtesy of Daniel Andergassen and John Rinn.
Phenotypic consequences of mutations in regulatory RNAs, like some protein-coding mutations, may be context dependent and not evident in laboratory conditions, and may be obscured by the robustness of biological systems171. Loss of Malat1, which localizes in nuclear speckles and associates with splicing factors, has no major phenotypes in mice114,172,173,174; however, it does affect cancer progression and synapse formation, among other physiological and pathophysiological processes175,176. Neat1, which is required for the assembly and function of enigmatic, mammal-specific nuclear organelles called ‘paraspeckles’177,178,179, does not appear to be required for normal development in mice but is important for the differentiation of reproduction-related female tissues such as corpus luteum and mammary gland180. Deletion of brain cytoplasmic RNA 1 (BC1), a highly expressed brain lncRNA, is seemingly harmless in mice but results in behavioural changes that would be lethal in the wild181. So extensive phenotyping is important, especially for cognitive functions. Organoid models may help to identify phenotypes in vitro182,183.
Functional annotation of lncRNAs can also be undertaken by molecular phenotyping184. Analysis of expression patterns, lncRNA–chromatin interactions and other molecular indices following CRISPR–Cas13-mediated depletion of more than 400 lncRNAs in culture indicated that lncRNAs regulate many genes involved in development, cell cycle and cellular adhesion, among other processes185.
Biological functions of lncRNAs
Characterized examples have indicated that RNAs participate in virtually all levels of genome organization, cell structure and gene expression, through RNA–RNA, RNA–DNA and RNA–protein interactions, often involving repeat elements88,186,187, including small interspersed nuclear elements in 3′ untranslated regions188. These interactions are involved in the regulation of chromatin architecture and transcription (see later), splicing (especially by antisense lncRNAs)189,190,191, protein translation and localization188,192,193, and other forms of RNA processing, editing, localization and stability194,195.
Many lncRNAs are involved in the regulation of cell differentiation and development in animals and plants23,81,116,124,196. They also have roles in physiological processes such as (in mammals) the p53-mediated response to DNA damage197, V(D)J recombination and class switch recombination in immune cells198, cytokine expression199, endotoxic shock200, inflammation and neuropathic pain201,202,203, cholesterol biosynthesis and homeostasis204,205, growth hormone and prolactin production206, glucose metabolism207,208, cellular signal transduction and transport pathways209,210,211,212, synapse function213,214 and learning215, and have roles in the response to various biotic and abiotic stresses in plants124,125. There is also an emerging association of lncRNAs with the cell membrane216 and with ribozymes217.
Presently, a growing number of lncRNAs have their own stories, and the literature is becoming replete with them. However, several convergent themes are emerging, which explain lncRNA ubiquity and importance in differentiation and development: the association of lncRNAs with chromatin-modifying proteins; the expression of lncRNAs from developmental ‘enhancers’; and the formation of RNA-nucleated phase-separated coacervates.
Control of chromatin architecture
Epigenetic modifications of chromatin supervise differentiation and development in complex organisms218. DNA methylation is known to be directed by small non-coding RNAs in plants219, and the RNAi pathway is required for heterochromatin formation and epigenetic gene silencing in fungi and animals220. The mammalian de novo DNA (cytosine 5)-methyltransferase 3A (DNMT3A) and DNMT3B, but not the maintenance DNA methylase DNMT1, bind siRNAs with high affinity221. In turn, DNMT1 (which restores methylation at hemimethylated CpG dinucleotides following DNA replication) binds lncRNAs to alter DNA methylation patterns at their cognate loci222,223,224, but this is still largely unexplored territory.
There are more than 100 different histone modifications that are differentially established by enzymes at a myriad of different positions in plant and animal genomes to control gene expression during development. The most studied are Polycomb repressive complex 1 (PRC1) and PRC2, which catalyse monoubiquitylation of histone H2A Lys119 (ref. 225) and dimethylation and trimethylation of histone H3 Lys27 (H3K27), respectively, but in mammals neither complex contains sequence-specific DNA-binding proteins218. Early studies suggested that PRC2 and/or the associated H3K9 methyltransferase G9a are recruited during mouse X chromosome inactivation by Xist186, and the control of parental imprinting in mice by Airn226 and Kcnq1ot1 (ref. 227), although these associations involve complexities and uncertainties228,229.
A subsequent survey of more than 3,300 lncRNAs in human cells showed that ~20% (but only ~2% of mRNAs) interact with PRC2, and that other lncRNAs are associated with other chromatin-modifying complexes230. Moreover, depletion of a selection of these RNAs caused derepression of genes normally silenced by PRC2 (ref. 230). PRC2 associates with many RNAs228,231,232, more than 9,000 in embryonic stem cells233. There are conflicting reports of whether these associations are nonspecific (‘promiscuous’)228,234 or specific high-affinity interactions with different RNAs232,235, although these alternatives are not mutually exclusive229. Some recent studies have shown that RNA is required for PRC2 chromatin occupancy, PRC2 function and cell state definition236, and that the interaction of PRC2 with RNA can regulate transcription elongation232. PRC1 function also appears to be controlled by RNA237,238. However, deconvoluting RNA–protein interactions is complicated by the low affinity of many antibodies used in pulldown assays and the fact that PRC2, for example, has at least two subunits that bind RNA228. The recent development of denaturing crosslinked immunoprecipitation (dCLIP), which is based on high-affinity biotin–streptavidin pulldowns, has indicated that PRC2 interacts with G-rich RNA motifs, including RNA G-quadruplexes, to achieve specificity of RNA-mediated recruitment232,239,240.
Other lncRNAs associate with the gene-activating Trithorax complexes (which methylate H3K4), including enhancer RNAs involved in the maintenance of stem cell fates and lineage specification241,242,243,244,245. H3K9 dimethylation is regulated by lncRNAs during the formation of long-term memory in mice246. lncRNAs also control methylation of a number of non-histone proteins involved in animal cell signalling, gene expression and RNA processing247.
Many other proteins involved in modulating chromatin architecture, including HOX proteins, pioneer transcription factors such as NANOG, OCT4 (also known asPOU5F1), SOX2 and other high mobility group (HMG) proteins, and proteins of SWI/SNF chromatin remodelling complexes, have only vague or promiscuous DNA sequence specificity248,249,250,251, which indicates that other factors are involved in determining their targets at different stages of cell differentiation and development. Moreover, binding-site selection by the zinc-finger transcription factor CTCF, which, together with cohesin complexes, anchors chromosome loops252, was shown to be controlled by the lncRNA just proximal to Xist (Jpx) during early cell differentiation, thereby regulating chromatin topology on a genome-wide scale253. CTCF binds thousands of RNAs, including Xist, Jpx and the lncRNA Xist antisense RNA (Tsix), which targets CTCF to the X inactivation centre254.
There is abundant evidence that RNA may guide chromatin remodelling complexes, although accessibility dictated by DNA and histone modifications (which are also likely directed by regulatory RNAs) may also have a role. The D. melanogaster Hox protein Bicoid (which controls anterior–posterior patterning) binds RNA through its homeodomain255. SOX2 binds RNA with high affinity through its HMG domain256,257, as do other members of the HMGB family257,258,259.
During mouse embryogenesis, the Sox2 locus expresses also an overlapping lncRNA260, and there are well-documented examples of lncRNAs that interact with SOX2 to regulate pluripotency, neurogenesis, neuronal differentiation and brain development257,261,262,263,264. SWI/SNF nucleosome remodelling complexes are directed to specific sites in chromatin or are antagonized by lncRNAs, including XIST and enhancer RNAs, in a wide range of differentiation processes and cancers251,265,266,267,268,269,270.
The lncRNA MaTAR25, which is overexpressed in mammary cancers, acts in trans to regulate the tensin 1 gene through interaction with the transcription co-activator PURB271. The master transcription factor myoblast determination protein (MYOD), which can reprogramme mammalian fibroblasts into muscle cells and is central to muscle differentiation in vivo, is regulated by lncRNAs272,273,274, as are other aspects of muscle gene expression275. The pioneer transcription factor CBP also binds RNAs, including those transcribed from enhancers, to stimulate histone acetylation and consequently transcription276. Some transcription factors (OCT4, NANOG, SOX2 and SOX9) are also regulated by lncRNAs, including pseudogene-derived lncRNAs277,278,279,280,281, and reciprocally regulate the expression of lncRNAs282. Enhancer-derived lncRNAs also regulate the expression of the nuclear hormone receptor ESR1 (ref. 283) and of CCAAT/enhancer-binding protein-α (CEBPA)284.
Enhancer action
Enhancers are non-coding genomic loci that control the spatiotemporal expression of other genes during development. There appear to be ~400,000 (±100,000) enhancers in the mammalian genome285,286,287,288, sometimes clustered into ‘super-enhancers’ or ‘enhancer jungles’288,289,290,291. Enhancers are thought to function by juxtaposing transcription factors bound at the enhancer promoters with the promoters of target genes292,293.
There is no question that enhancer action alters chromatin topology and may be responsible for the formation of chromatin-loop domains that act as local transcription and splicing hubs294,295. Enhancers are transcribed in the cells in which they are active141,289,296,297,298,299, which has led to uncertainty about whether the resulting RNAs are by-products of the binding of transcription factors or have a role in enhancer activity298.
The latter appears to be the case. The epigenetic landscape of and the features of transcription initiation at the promoters of protein-coding genes and enhancers are almost indistinguishable296,297,298,299,300. Enhancers express bidirectional promoter-associated short RNAs301,302,303, termed ‘eRNAs’, although such short RNAs are not specific to enhancers, as similar bidirectional transcripts are produced from the promoters of protein-coding genes304,305. Also analogously to mRNAs produced from protein-coding genes, enhancers express long (non-coding) RNAs (confusingly also referred to as ‘eRNAs’298,306), and transcription is considered the best molecular indicator of enhancer activity in developmental processes296,297,306,307,308 and cancers288. Moreover, enhancer-lncRNA splicing has been shown to modulate enhancer activity309,310.
Although the extent of congruency of combined genetic and high-depth transcriptomic data is uncertain, as their availability is still limited, the data suggest that many if not most lncRNAs are derived from enhancers141,298 and that lncRNAs are required for enhancer activity163,284,311,312,313,314, examples including the lncRNAs Evf2 (also known as Dlx6os1)315, Firre316, Peril317, Upperhand (also known as Hand2os1)318 and Maenli150 in mice. Enhancer RNA function is fertile ground for investigation, but if enhancer loci are considered bona fide ‘genes’, the g-value paradox (the perceived lack of increase in gene number with developmental complexity) is resolved. It also means that a key development in the evolution of complex organisms was the use of RNA to organize developmental trajectories319. It appears that “every cell type expresses precise lncRNA signatures to control lineage-specific regulatory programs”270, and that cell state during ontogeny is likely directed by lncRNAs.
Formation of biomolecular condensates
The past decade has seen the growing appreciation of the role of biomolecular condensates, or phase-separated domains (PSDs), in the organization of cells and chromatin. These condensates are highly dynamic assemblies with high local concentrations of macromolecules, a feature that promotes functional interactions. The condensates usually contain both RNA and proteins320,321,322, the latter having IDRs, which are the major sites of post-translational modifications323. IDRs interact with and are tunable by many partners324. The fraction of the proteome containing IDRs has expanded with cellular and developmental complexity323, and nearly all proteins involved in the regulation of development, including most transcription factors, histones, histone-modifying proteins, other chromatin-binding proteins, RNA-binding proteins, splicing factors, nuclear hormone receptors, cytoskeletal proteins and membrane receptors, contain IDRs323,325,326,327,328,329,330,331,332.
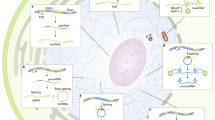
RNA is crucial for the form, composition and function of phase-separated RNA–protein condensates320,321,322. Specific ‘architectural’ lncRNAs333 associate with nuclear condensates of different half-lives and functionalities, including in centrosomes334, nucleoli335 (the lncRNAs SLERT138 and LETN336), nuclear speckles (the lncRNA MALAT1 (refs. 173,337)) rich in RNA-processing factors, speckle-related condensates that contain the lncRNA Gomafu in mice338,339 and paraspeckles (the lncRNA NEAT1 (refs. 340,341)) (Fig. 2), in vertebrates as well as polyadenylation complexes342 and other condensates in plants343. RNP condensates also include cytoplasmic membraneless organelles such as P-granules344,345, subcellular-localized translational messenger RNP assemblies346 and synaptic compartments320,322,347. The mammalian cytoplasmic lncRNA NORAD, which is induced by DNA damage and required for genome stability, prevents aberrant mitosis by sequestering Pumilio proteins (which bind many RNAs to regulate stem cell fate, development and neurological functions) into PSDs through its repeat sequences137,348.

a, 5′ small nucleolar RNA-capped and 3′-polyadenylated long non-coding (lncRNAs) (SPAs)42 and small nucleolar RNA-related lncRNAs (sno-lncRNAs)41 accumulate at their sites of transcription and interact with several splicing factors such as RNA-binding protein FOX-1 homologue 2 (RBFOX2), TAR DNA-binding protein 43 (TDP43) and heterogeneous nuclear ribonucleoprotein M (hnRNPM) to form a microscopically visible nuclear body that is involved in the regulation of alternative splicing42. b, The lncRNA functional intergenic repeating RNA element (Firre) is transcribed from the mouse X chromosome and interacts with the nuclear matrix factor hnRNPU to tether chromosome X (chrX), chr2, chr9, chr15 and chr17 into a nuclear domain451,452. c, The lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) is essential for the formation of paraspeckles178. NEAT1 sequesters numerous paraspeckle proteins to form a highly organized core–shell (dark and light purple, respectively) spheroidal nuclear body453. The middle region of NEAT1 is localized in the centre of paraspeckles, and the 3′-end and 5′-end regions are localized in the periphery453. Different paraspeckle proteins are embedded by NEAT1 into the spheroidal structure in the core region (non-POU domain-containing octamer-binding protein (NONO), fused in sarcoma (FUS) and splicing factor, proline- and glutamine-rich (SFPQ)) or in the shell region (RNA-binding motif protein 14 (RBM14))453. d, The lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is localized at the periphery of nuclear speckles172,454 and is involved in the regulation of pre-mRNA splicing339,455. MALAT1 interacts with the U1 small nuclear RNA (U1 snRNA)428, whereas proteins such as SON DNA- and RNA-binding protein and splicing component 35 kDa (SC35) are localized at the centre of nuclear speckles456. e, The lncRNA CHD2 adjacent, suppressive regulatory RNA (Chaserr) forms a compartment within a region of the mouse chromosome corresponding to a topologically associating domain that includes its own gene as well as the Chd2 gene (encoding chromodomain DNA helicase protein 2 (CHD2))437. Chaserr limits in cis the expression of Chd2, which is important for proper regulation of many genes (not shown). f, The perinucleolar compartment contains the lncRNA pyrimidine-rich non-coding transcript (PNCTR), which sequesters pyrimidine tract-binding protein 1 (PTBP1) and thus suppresses PTPBP1-mediated pre-mRNA splicing elsewhere in the nucleoplasm369. The size of nuclear bodies is indicated where relevant457. Figure adapted from ref. 80, Springer Nature. Part e courtesy of Inna-Marie Strazhnik and Mitch Guttman.
It has been proposed that RNAs have a central role in organizing the genome and gene expression by the formation of spatial compartments and transcriptional condensates349,350,351,352,353. Phase separation appears to drive chromatin long-range interactions and to be required for the action of enhancers and super-enhancers328,351,354,355,356,357 as well as for transcription, transcription factors and polyadenylation complexes342,358,359,360,361, although transcription factor hubs have been reported to operate in the absence of detectable phase separation362. PSDs scaffolded by lncRNAs, including repeat-rich RNAs363,364, mediate the formation of heterochromatin353,365,366, euchromatin367, Polycomb bodies368 and alternative splicing369. lncRNAs are a substantial component of rapidly renaturing, repeat-rich RNA (technically termed ‘CoT-1 RNA’), and high-resolution imaging shows many repeat-containing RNAs bound to chromatin, indicating that the collective presence of thousands of lncRNAs serves to counter chromatin condensation364. High-resolution imaging also shows the localization of many lncRNAs in compartments in the nucleus that resemble PSDs136,353. These data all suggest that there are thousands of low copy number lncRNAs involved in the organization of chromosome territories.
lncRNA structure–function relationships
lncRNAs generally range in size from around 1 kb to longer than 100 kb (refs. 370,371) and have a modular structure372,373,374,375. They are often multi-exonic and highly alternatively spliced (Fig. 3a), a feature that was not obvious before the advent of high-depth sequencing98. They also contain a higher proportion of GC–AG splice sites376 and are therefore less efficiently spliced than protein-coding transcripts377,378, which are properties associated with alternative splicing379. Alternative splicing has, unsurprisingly, been shown to alter the function of lncRNAs42,152,380,381.

a, Targeted RNA sequencing has revealed that human chromosome 21 (chr21) is pervasively transcribed into long non-coding RNAs (lncRNAs) and that lncRNA exons are almost universally (but not randomly) alternatively spliced to form diverse and complex isoforms98. The circle indicates the fraction of non-coding exons across all chr21 transcripts that are alternatively or constitutively spliced. b, Modular structural domains in lncRNAs that fulfil a range of functions372,373,374,375, including targeting DNA, such as in the case of auxin-regulated promoter loop (APOLO)61; binding other RNAs — for example, terminal differentiation-induced non-coding RNA (TINCR)458, potentially involving RNA-binding proteins such as Staufen 1; and recruitment of proteins — for example, pyrimidine-rich non-coding transcript (PNCTR) recruiting of pyrimidine tract-binding protein 1 (PTBP1) through special RNA motifs369 and X-inactive-specific transcript (XIST) recruiting split ends homologue (SPEN) and Polycomb repressive complex 2 (PRC2), perhaps in concert, which is the subject of active exploration and debate142,397,399,423,424,459. Modular functional domains can be repeated within a lncRNA or in multiple different lncRNAs7,87,186,369,388,391,393,394,395,396,397,398,399,400,401. Figure courtesy of Tim R. Mercer.
Some lncRNAs also exhibit common motifs and motif combinations101. At least 18% of the human genome is conserved among mammals at the level of predicted RNA structure382, and similar and potentially paralogous RNA structures occur at many places throughout the genome383,384. Chemical probing has shown that lncRNAs, including Xist, form complex multidomain structures108,385,386,387,388,389, with chemical data matching data predicted by evolutionary conservation of secondary structure389. Moreover, lncRNAs with similar k-base oligonucleotide (short motif) content have related functions despite their lack of general homology, implying that small sequence elements are also key determinants of lncRNA function390.
Many lncRNA exons are derived from transposable elements187,391. The most highly conserved sequences in Xist, which has been intensively studied, are its repeats7, whereas its unique sequences have evolved rapidly392, and many of its biological functions, including recruitment of gene-repressive complexes and gene silencing, are mediated through its modular repeat elements142,186,388,393,394,395,396,397,398,399. Transposable element-derived sequences participate in many RNA–protein interactions369,400,401, which leads to the conclusion that repeat structures are common building blocks of lncRNAs87,391,396 and essential components of their function391.
The molecular mechanisms of lncRNA action are unclear. In most well-characterized cases of RNA regulation, such as RNAi, snoRNAs, CRISPR and telomerase, RNA acts as a guide to target effector protein complexes to complementary RNA or DNA sequences. Data on selected lncRNAs (for example, HOTAIR, roX1, roX2, Meg3, Tug1, PARTICLE (also known as PARTCL), PAPAS and KHPS1) indicates that they form triplex structures with DNA at purine-rich GA stretches to recruit chromatin modifiers to specific loci across the genome402,403,404,405,406,407,408, with evidence that triplex formation by lncRNAs is a widespread phenomenon409,410,411. Others, especially antisense lncRNAs, appear to function through RNA–DNA hybrid formation61,412,413, but detail is presently lacking.
lncRNA RNP structure and function have been well characterized in only one instance, the telomerase complex, which has been studied for decades. Telomerase reverse transcriptase (TERT) catalyses the addition of telomere repeats to chromosome ends, and other proteins in the complex provide nuclear localization, stability or recruitment to telomeres or to Cajal bodies. The lncRNA TERC provides the scaffold for assembly of the RNP and the template for DNA polymerization by TERT, and mutations in TERT and TERC are major contributors to the aetiology of cancer and the cause of hereditary disorders such as dyskeratosis congenita103,104,105,106,107,414,415,416.
By contrast, while we know the phenotypes caused by the loss of some lncRNAs, we know almost nothing about how most of them work, although, considering that as recently as 2010 the very existence of pervasive transcription was still a matter of contention417,418,419 and the sheer number of lncRNAs, substantial progress has been made. It is assumed, in our view reasonably, that generally lncRNAs will engage in multilateral interactions similarly to TERC and the telomerase complex108, and there is some evidence to support this assumption in cases such as XIST (Fig. 3b), but the assumption has not yet been rigorously tested. There are promising discoveries, such as the demonstration that conserved pseudoknots in lncRNA Meg3 are essential for stimulation of the p53 pathway420. There is also growing evidence of discrete structural organization in lncRNAs421. Nonetheless, there is a long journey ahead to understand the structure and function of the many thousands of lncRNAs, and their splice variants, in the context of their associated RNP complexes and biomolecular condensates in both the nucleus and the cytoplasm.
Challenges
If the complex ontogenies of animals and, to a lesser extent plants, require a large number of RNAs to guide the epigenetic decisions at each cell division, then it is not surprising that many lncRNAs have common protein-binding modules and specific targeting sequences that vary between different stages of development. The challenge is to define which lncRNAs and modules within them interact with effector proteins and which convey target (DNA or RNA) specificity. The former is complicated by the multisubunit nature of many RNP complexes, but is being addressed by technologies such as iCLIP422, RAP–MS423, ChIRP-MS388 and iDRiP424. Determining target specificity is even more difficult, as specific targeting requires only short stretches of nucleotide complementarity given the strength of RNA–RNA and RNA–DNA interactions425, but it may be tackled by new methods that analyse RNA–chromatin and RNA–RNA interactions, such as GRID-seq426, RADICL-seq427, RIC-seq428 and RD-SPRITE353. Other lncRNAs are localized in cytoplasmic compartments, whose components also need to be characterized.
Understanding the roles of lncRNAs and how they function in dynamic assemblies with other macromolecules will provide a more comprehensive understanding of cell and developmental biology and of gene–environment interactions. Emerging challenges include understanding the roles of lncRNAs and RNA modifications in functional plasticity, especially in the brain, and the dysregulation of these lncRNA-mediated pathways in neurological disorders, cancer and other diseases.
Recommendations
-
1.
In the absence of more specific categorization, we recommend retention of the general descriptor ‘lncRNA’ for non-coding RNAs greater than 500 nt in length.
-
2.
Unless a lncRNA is antisense to a protein-coding gene (in which case the designation ‘gene name-AS’ should be used), we recommend naming lncRNAs for their own sake with allusion to a discerned characteristic or function (as has been traditional for proteins), preferably accompanied by complete exon–intron structures and genomic coordinates. If no biological context is available, we recommend naming the lncRNA according to the GENCODE system46.
-
3.
We recommend that future gene expression profiling should include full transcript analysis of the isoforms and stoichiometry of mRNAs, lncRNAs and small RNAs in cells at different stages of differentiation, and in various physiological and disease states, learning and stress conditions.
-
4.
These efforts should be complemented by cell-based, organoid-based and in vivo studies using strategies for conditional and tissue-specific or cell type-specific gain-of-function and loss-of-function of lncRNAs.
More broadly, identifying and understanding the roles of lncRNAs and RNA regulatory networks in multicellular development, cell biology and disease will require the following:
-
1.
The determination of the interplay between lncRNAs, chromatin modifications, proteins and the genome in the assembly of the nuclear domains essential for chromatin organization, enhancer function, transcription and splicing. This effort will require the development of antibodies with high specificity for protein–RNA complexes, and of intracellular RNA-tracking methods429.
-
2.
The determination of lncRNA localization, structure–function relationships and interactions using a range of sequencing, chemical probing, imaging methods430,431,432,433 and cryogenic electron microscopy434.
-
3.
The identification and characterization of the many unknown nuclear and cytoplasmic compartments decorated by specific lncRNAs.
-
4.
Harnessing the power of machine learning to interrogate large genomic, epigenomic, transcriptomic, proteomic and phenomic datasets to identify causal links and pathways.
References
Ender, C. & Meister, G. Argonaute proteins at a glance. J. Cell Sci. 123, 1819–1823 (2010).
Wassarman, K. M., Zhang, A. & Storz, G. Small RNAs in Escherichia coli. Trends Microbiol. 7, 37–45 (1999).
Watanabe, Y. & Yamamoto, M. S. pombe mei2+ encodes an RNA-binding protein essential for premeiotic DNA synthesis and meiosis I, which cooperates with a novel RNA species meiRNA. Cell 78, 487–498 (1994).
Lakhotia, S. C. & Sharma, A. The 93D (hsr-omega) locus of Drosophila: non-coding gene with house-keeping functions. Genetica 97, 339–348 (1996).
Kelley, R. L. et al. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98, 513–522 (1999).
Bartolomei, M. S., Zemel, S. & Tilghman, S. M. Parental imprinting of the mouse H19 gene. Nature 351, 153–155 (1991).
Brown, C. J. et al. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71, 527–542 (1992).
Rodriguez, A., Griffiths-Jones, S., Ashurst, J. L. & Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 14, 1902–1910 (2004).
He, D. et al. miRNA-independent function of long noncoding pri-miRNA loci. Proc. Natl Acad. Sci. USA 118, e2017562118 (2021).
Askarian-Amiri, M. E. et al. SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA 17, 878–891 (2011).
Lambert, M., Benmoussa, A. & Provost, P. Small non-coding RNAs derived from eukaryotic ribosomal RNA. Noncoding RNA 5, 16 (2019).
Kawaji, H. et al. Hidden layers of human small RNAs. BMC Genomics 9, 157 (2008).
Krishna, S. et al. Dynamic expression of tRNA-derived small RNAs define cellular states. EMBO Rep. 20, e47789 (2019).
Taft, R. J. et al. Small RNAs derived from snoRNAs. RNA 15, 1233–1240 (2009).
Chen, Q. et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 351, 397–400 (2016).
Kapranov, P. et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296, 916–919 (2002).
Okazaki, Y. et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420, 563–573 (2002).
Carninci, P. et al. The transcriptional landscape of the mammalian genome. Science 309, 1559–1563 (2005).
Cheng, J. et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308, 1149–1154 (2005).
Sender, R., Fuchs, S. & Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, e1002533 (2016).
Hahn, M. W. & Wray, G. A. The g-value paradox. Evol. Dev. 4, 73–75 (2002).
Liu, G., Mattick, J. S. & Taft, R. J. A meta-analysis of the genomic and transcriptomic composition of complex life. Cell Cycle 12, 2061–2072 (2013).
Mercer, T. R., Dinger, M. E. & Mattick, J. S. Long noncoding RNAs: insights into function. Nat. Rev. Genet. 10, 155–159 (2009).
Parrott, A. M. et al. The evolution and expression of the snaR family of small non-coding RNAs. Nucleic Acids Res. 39, 1485–1500 (2011).
Täuber, H., Hüttelmaier, S. & Köhn, M. POLIII-derived non-coding RNAs acting as scaffolds and decoys. J. Mol. Cell Biol. 11, 880–885 (2019).
Hahne, J. C., Lampis, A. & Valeri, N. Vault RNAs: hidden gems in RNA and protein regulation. Cell. Mol. Life Sci. 78, 1487–1499 (2021).
Kapranov, P. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488 (2007).
Fejes-Toth, K. et al. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature 457, 1028–1032 (2009).
Preker, P. et al. PROMoter uPstream Transcripts share characteristics with mRNAs and are produced upstream of all three major types of mammalian promoters. Nucleic Acids Res. 39, 7179–7193 (2011).
Castelo-Branco, G. et al. The non-coding snRNA 7SK controls transcriptional termination, poising, and bidirectionality in embryonic stem cells. Genome Biol. 14, R98 (2013).
Flynn, R. A. et al. 7SK-BAF axis controls pervasive transcription at enhancers. Nat. Struct. Mol. Biol. 23, 231–238 (2016).
Gussakovsky, D. & McKenna, S. A. Alu RNA and their roles in human disease states. RNA Biol. 18, 574–585 (2021).
Ullu, E. & Tschudi, C. Alu sequences are processed 7SL RNA genes. Nature 312, 171–172 (1984).
Tsirigos, A. & Rigoutsos, I. Alu and B1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLoS Comput. Biol. 5, e1000610 (2009).
Zhang, X.-O., Gingeras, T. R. & Weng, Z. Genome-wide analysis of polymerase III–transcribed Alu elements suggests cell-type–specific enhancer function. Genome Res. 29, 1402–1414 (2019).
Deng, W. et al. Organization of the Caenorhabditis elegans small non-coding transcriptome: Genomic features, biogenesis, and expression. Genome Res. 16, 20–29 (2006).
Dieci, G., Conti, A., Pagano, A. & Carnevali, D. Identification of RNA polymerase III-transcribed genes in eukaryotic genomes. Biochim. Biophys. Acta 1829, 296–305 (2013).
Jawdekar, G. W. & Henry, R. W. Transcriptional regulation of human small nuclear RNA genes. Biochim. Biophys. Acta 1779, 295–305 (2008).
Kufel, J. & Grzechnik, P. Small nucleolar RNAs tell a different tale. Trends Genet. 35, 104–117 (2019).
Wilusz, J. E., Freier, S. M. & Spector, D. L. 3’ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135, 919–932 (2008).
Yin, Q.-F. et al. Long noncoding RNAs with snoRNA ends. Mol. Cells 48, 219–230 (2012).
Wu, H. et al. Unusual processing generates SPA lncRNAs that sequester multiple RNA binding proteins. Mol. Cells 64, 534–548 (2016).
Gingeras, T. R. Origin of phenotypes: genes and transcripts. Genome Res. 17, 682–690 (2007).
Cheetham, S. W., Faulkner, G. J. & Dinger, M. E. Overcoming challenges and dogmas to understand the functions of pseudogenes. Nat. Rev. Genet. 21, 191–201 (2020).
Frith, M. C. et al. Pseudo–messenger RNA: phantoms of the transcriptome. PLoS Genet. 2, e23 (2006).
Frankish, A. et al. GENCODE 2021. Nucleic Acids Res. 49, D916–D923 (2021).
Ma, Y. et al. Genome-wide analysis of pseudogenes reveals HBBP1’s human-specific essentiality in erythropoiesis and implication in β-thalassemia. Dev. Cell 56, 478–493 (2021).
Patop, I. L., Wüst, S. & Kadener, S. Past, present, and future of circRNAs. EMBO J. 38, e100836 (2019).
Mercer, T. R. et al. Expression of distinct RNAs from 3’ untranslated regions. Nucleic Acids Res. 39, 2393–2403 (2011).
Wright, M. W. A short guide to long non-coding RNA gene nomenclature. Hum. Genomics 8, 7 (2014).
Mattick, J. S. & Rinn, J. L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 22, 5–7 (2015).
Uszczynska-Ratajczak, B., Lagarde, J., Frankish, A., Guigó, R. & Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 19, 535–548 (2018).
Seal, R. L. et al. A guide to naming human non-coding RNA genes. EMBO J. 39, e103777 (2020).
Mattick, J. S. Challenging the dogma: the hidden layer of non-protein-coding RNAs in complex organisms. Bioessays 25, 930–939 (2003).
Kapranov, P., Willingham, A. T. & Gingeras, T. R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 8, 413–423 (2007).
Willingham, A. T. et al. Transcriptional landscape of the human and fly genomes: nonlinear and multifunctional modular model of transcriptomes. Cold Spring Harb. Symp. Quant. Biol. 71, 101–110 (2006).
Lyle, R. et al. The imprinted antisense RNA at the Igf2r locus overlaps but does not imprint Mas1. Nat. Genet. 25, 19–21 (2000).
Rinn, J. L. et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323 (2007).
Sone, M. et al. The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J. Cell Sci. 120, 2498–2506 (2007).
Ietswaart, R., Wu, Z. & Dean, C. Flowering time control: another window to the connection between antisense RNA and chromatin. Trends Genet. 28, 445–453 (2012).
Ariel, F. et al. R-loop mediated trans action of the APOLO long noncoding RNA. Mol. Cells 77, 1055–1065 (2020).
Kopp, F. & Mendell, J. T. Functional classification and experimental dissection of long noncoding RNAs. Cell 172, 393–407 (2018).
Dinger, M. E., Gascoigne, D. K. & Mattick, J. S. The evolution of RNAs with multiple functions. Biochimie 93, 2013–2018 (2011).
Wu, P. et al. Emerging role of tumor-related functional peptides encoded by lncRNA and circRNA. Mol. Cancer 19, 22 (2020).
Wright, B. W., Yi, Z., Weissman, J. S. & Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends Cell Biol. 32, 243–258 (2022).
Makarewich, C. A. & Olson, E. N. Mining for micropeptides. Trends Cell Biol. 27, 685–696 (2017).
Hube, F. et al. Alternative splicing of the first intron of the steroid receptor RNA activator (SRA) participates in the generation of coding and noncoding RNA isoforms in breast cancer cell lines. DNA Cell Biol. 25, 418–428 (2006).
Williamson, L. et al. UV irradiation induces a non-coding RNA that functionally opposes the protein encoded by the same gene. Cell 168, 843–855 (2017).
Grelet, S. et al. A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nat. Cell Biol. 19, 1105–1115 (2017).
Gonzàlez-Porta, M., Frankish, A., Rung, J., Harrow, J. & Brazma, A. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol. 14, R70 (2013).
Tuck, A. C. & Tollervey, D. RNA in pieces. Trends Genet. 27, 422–432 (2011).
Chan, S. N. & Pek, J. W. Stable intronic sequence RNAs (sisRNAs): an expanding universe. Trends Biochem. Sci. 44, 258–272 (2019).
Fang, S. et al. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 46, D308–D314 (2017).
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012).
Mas-Ponte, D. et al. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 23, 1080–1087 (2017).
Ma, L. et al. LncBook: a curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 47, D128–D134 (2018).
Volders, P.-J. et al. LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 47, D135–D139 (2019).
Seifuddin, F. et al. lncRNAKB, a knowledgebase of tissue-specific functional annotation and trait association of long noncoding RNA. Sci. Data 7, 326 (2020).
Jin, J. et al. PLncDB V2.0: a comprehensive encyclopedia of plant long noncoding RNAs. Nucleic Acids Res. 49, D1489–D1495 (2020).
RNAcentral Consortium. RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res. 49, D212–D220 (2021).
Statello, L., Guo, C.-J., Chen, L.-L. & Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118 (2021).
St Laurent, G. et al. Intronic RNAs constitute the major fraction of the non-coding RNA in mammalian cells. BMC Genomics 13, 504 (2012).
Gardner, E. J., Nizami, Z. F., Talbot, C. C. & Gall, J. G. Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev. 26, 2550–2559 (2012).
Zhang, Y. et al. Circular intronic long noncoding RNAs. Mol. Cells 51, 792–806 (2013).
Pheasant, M. & Mattick, J. S. Raising the estimate of functional human sequences. Genome Res. 17, 1245–1253 (2007).
Faulkner, G. J. et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 41, 563–571 (2009).
Kapusta, A. et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 9, e1003470 (2013).
Kelley, D. & Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 13, R107 (2012).
Fueyo, R., Judd, J., Feschotte, C. & Wysocka, J. Roles of transposable elements in the regulation of mammalian transcription. Nat. Rev. Mol. Cell Biol. 23, 481–497 (2022).
Pang, K. C., Frith, M. C. & Mattick, J. S. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 22, 1–5 (2006).
Kutter, C. et al. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet. 8, e1002841 (2012).
Quinn, J. J. et al. Rapid evolutionary turnover underlies conserved lncRNA-genome interactions. Genes Dev. 30, 191–207 (2016).
Ponjavic, J., Ponting, C. P. & Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 17, 556–565 (2007).
Guttman, M. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227 (2009).
Mattick, J. S. The genetic signatures of noncoding RNAs. PLoS Genet. 5, e1000459 (2009).
Cawley, S. et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116, 499–509 (2004).
Nitsche, A., Rose, D., Fasold, M., Reiche, K. & Stadler, P. F. Comparison of splice sites reveals that long noncoding RNAs are evolutionarily well conserved. RNA 21, 801–812 (2015).
Deveson, I. W. et al. Universal alternative splicing of noncoding exons. Cell Syst. 6, 245–255 (2018).
Clark, M. et al. Genome-wide analysis of long noncoding RNA stability. Genome Res. 21, 885–898 (2012).
Ulitsky, I., Shkumatava, A., Jan, C. H., Sive, H. & Bartel, D. P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147, 1537–1550 (2011).
Ross, C. J. et al. Uncovering deeply conserved motif combinations in rapidly evolving noncoding sequences. Genome Biol. 22, 29 (2021).
Degani, N., Lubelsky, Y., Perry, R. B.-T., Ainbinder, E. & Ulitsky, I. Highly conserved and cis-acting lncRNAs produced from paralogous regions in the center of HOXA and HOXB clusters in the endoderm lineage. PLoS Genet. 17, e1009681 (2021).
Chen, J.-L., Blasco, M. A. & Greider, C. W. Secondary structure of vertebrate telomerase RNA. Cell 100, 503–514 (2000).
Zhang, Q., Kim, N.-K. & Feigon, J. Architecture of human telomerase RNA. Proc. Natl Acad. Sci. USA 108, 20325–20332 (2011).
Wang, Y., Yesselman, J. D., Zhang, Q., Kang, M. & Feigon, J. Structural conservation in the template/pseudoknot domain of vertebrate telomerase RNA from teleost fish to human. Proc. Natl Acad. Sci. USA 113, E5125–E5134 (2016).
Nguyen, T. H. D. et al. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 557, 190–195 (2018).
Mefford, M. A., Hass, E. P. & Zappulla, D. C. A 4-base-pair core-enclosing helix in telomerase RNA is essential for activity and for binding to the telomerase reverse transcriptase catalytic protein subunit. Mol. Cell Biol. 40, e00239-20 (2020).
Zappulla, D. C. Yeast telomerase RNA flexibly scaffolds protein subunits: results and repercussions. Molecules 25, 2750 (2020).
Valsecchi, C. I. K. et al. RNA nucleation by MSL2 induces selective X chromosome compartmentalization. Nature 589, 137–142 (2020).
Galupa, R. & Heard, E. X-chromosome inactivation: a crossroads between chromosome architecture and gene regulation. Annu. Rev. Genet. 52, 535–566 (2018).
van Bemmel, J. G. et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nat. Genet. 51, 1024–1034 (2019).
Pandya-Jones, A. et al. A protein assembly mediates Xist localization and gene silencing. Nature 587, 145–151 (2020).
Jégu, T., Aeby, E. & Lee, J. T. The X chromosome in space. Nat. Rev. Genet. 18, 377–389 (2017).
Eißmann, M. et al. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 9, 1076–1087 (2012).
Gloss, B. S. & Dinger, M. E. The specificity of long noncoding RNA expression. Biochim. Biophys. Acta 1859, 16–22 (2016).
Flynn, R. A. & Chang, H. Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 14, 752–761 (2014).
Rinn, J. L. et al. A dermal HOX transcriptional program regulates site-specific epidermal fate. Genes Dev. 22, 303–307 (2008).
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F. & Mattick, J. S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl Acad. Sci. USA 105, 716–721 (2008).
Goff, L. A. et al. Spatiotemporal expression and transcriptional perturbations by long noncoding RNAs in the mouse brain. Proc. Natl Acad. Sci. USA 112, 6855–6862 (2015).
Liu, S. J. et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 17, 67 (2016).
Bocchi, V. D. et al. The coding and long noncoding single-cell atlas of the developing human fetal striatum. Science 372, eabf5759 (2021).
Kim, D. H. et al. Single-cell transcriptome analysis reveals dynamic changes in lncRNA expression during reprogramming. Cell Stem Cell 16, 88–101 (2015).
Sarropoulos, I., Marin, R., Cardoso-Moreira, M. & Kaessmann, H. Developmental dynamics of lncRNAs across mammalian organs and species. Nature 571, 510–514 (2019).
Chen, L., Zhu, Q.-H. & Kaufmann, K. Long non-coding RNAs in plants: emerging modulators of gene activity in development and stress responses. Planta 252, 92 (2020).
Wierzbicki, A. T., Blevins, T. & Swiezewski, S. Long noncoding RNAs in plants. Annu. Rev. Plant. Biol. 72, 245–271 (2021).
Zhao, Y. et al. Natural temperature fluctuations promote COOLAIR regulation of FLC. Genes. Dev. 35, 888–898 (2021).
Lakhotia, S. C. Long non-coding RNAs coordinate cellular responses to stress. Wiley Interdiscip. Rev. RNA 3, 779–796 (2012).
Kato, M. et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat. Commun. 7, 12864 (2016).
Khan, M. R., Xiang, S., Song, Z. & Wu, M. The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. EMBO J. 36, 3483–3500 (2017).
Barth, D. A. et al. Long-noncoding RNA (lncRNA) in the regulation of hypoxia-inducible factor (HIF) in cancer. Noncoding RNA 6, 27 (2020).
Wang, R. et al. LncRNA GIRGL drives CAPRIN1-mediated phase separation to suppress glutaminase-1 translation under glutamine deprivation. Sci. Adv. 7, eabe5708 (2021).
Connerty, P., Lock, R. B. & de Bock, C. E. Long non-coding RNAs: major regulators of cell stress in cancer. Front. Oncol. 10, 285 (2020).
Liu, K. et al. Long non-coding RNAs regulate drug resistance in cancer. Mol. Cancer 19, 54 (2020).
Deveson, I. W., Hardwick, S. A., Mercer, T. R. & Mattick, J. S. The dimensions, dynamics, and relevance of the mammalian noncoding transcriptome. Trends Genet. 33, 464–478 (2017).
Mercer, T. R. et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 30, 99–104 (2012).
Cabili, M. N. et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 16, 20 (2015).
Elguindy, M. M. & Mendell, J. T. NORAD-induced Pumilio phase separation is required for genome stability. Nature 595, 303–308 (2021).
Wu, M. et al. lncRNA SLERT controls phase separation of FC/DFCs to facilitate Pol I transcription. Science 373, 547–555 (2021).
Asp, M. et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell 179, 1647–1660 (2019).
Ma, Q. & Chang, H. Y. Single-cell profiling of lncRNAs in the developing human brain. Genome Biol. 17, 68 (2016).
Hon, C.-C. et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543, 199–204 (2017).
Jachowicz, J. W. et al. Xist spatially amplifies SHARP/SPEN recruitment to balance chromosome-wide silencing and specificity to the X chromosome. Nat. Struct. Mol. Biol. 29, 239–249 (2022).
Wu, M., Yang, L.-Z. & Chen, L.-L. Long noncoding RNA and protein abundance in lncRNPs. RNA 27, 1427–1440 (2021).
Bartonicek, N. et al. Intergenic disease-associated regions are abundant in novel transcripts. Genome Biol. 18, 241 (2017).
de Goede, O. M. et al. Population-scale tissue transcriptomics maps long non-coding RNAs to complex disease. Cell 184, 2633–2648 (2021).
Nasser, J. et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243 (2021).
Sleutels, F., Zwart, R. & Barlow, D. P. The non-coding air RNA is required for silencing autosomal imprinted genes. Nature 415, 810–813 (2002).
Thakur, N. et al. An antisense RNA regulates the bidirectional silencing property of the Kcnq1 imprinting control region. Mol. Cell Biol. 24, 7855–7862 (2004).
Young, T. L., Matsuda, T. & Cepko, C. L. The noncoding RNA taurine upregulated gene 1 is required for differentiation of the murine retina. Curr. Biol. 15, 501–512 (2005).
Allou, L. et al. Non-coding deletions identify Maenli lncRNA as a limb-specific En1 regulator. Nature 592, 93–98 (2021).
van Dijk, M. et al. HELLP babies link a novel lincRNA to the trophoblast cell cycle. J. Clin. Invest. 122, 4003–4011 (2012).
Li, P., Tao, Z. & Dean, C. Phenotypic evolution through variation in splicing of the noncoding RNA COOLAIR. Genes. Dev. 29, 696–701 (2015).
Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 21, 1253–1261 (2015).
Schmitt, A. M. & Chang, H. Y. Long noncoding RNAs in cancer pathways. Cancer Cell 29, 452–463 (2016).
Carlevaro-Fita, J. et al. Cancer LncRNA census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis. Commun. Biol. 3, 56 (2020).
Sparber, P., Filatova, A., Khantemirova, M. & Skoblov, M. The role of long non-coding RNAs in the pathogenesis of hereditary diseases. BMC Med. Genomics 12, 42 (2019).
Aznaourova, M., Schmerer, N., Schmeck, B. & Schulte, L. N. Disease-causing mutations and rearrangements in long non-coding RNA gene loci. Front. Genet. 11, 527484 (2020).
Sutherland, H. F. et al. Identification of a novel transcript disrupted by a balanced translocation associated with DiGeorge syndrome. Am. J. Hum. Genet. 59, 23–31 (1996).
Ang, C. E. et al. The novel lncRNA lnc-NR2F1 is pro-neurogenic and mutated in human neurodevelopmental disorders. Elife 8, e41770 (2019).
Long, H. K. et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell 27, 765–783 (2020).
Li, Y. et al. A noncoding RNA modulator potentiates phenylalanine metabolism in mice. Science 373, 662–673 (2021).
Gao, F., Cai, Y., Kapranov, P. & Xu, D. Reverse-genetics studies of lncRNAs — what we have learnt and paths forward. Genome Biol. 21, 93 (2020).
Andergassen, D. & Rinn, J. L. From genotype to phenotype: genetics of mammalian long non-coding RNAs in vivo. Nat. Rev. Genet. 23, 229–243 (2021).
Zibitt, M. S., Hartford, C. C. R. & Lal, A. Interrogating lncRNA functions via CRISPR/Cas systems. RNA Biol. 18, 2097–2106 (2021).
Sauvageau, M. et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2, e01749 (2013).
Lai, K.-M. V. et al. Diverse phenotypes and specific transcription patterns in twenty mouse lines with ablated lincRNAs. PLoS ONE 10, e0125522 (2015).
Liu, S. J. et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355, eaah7111 (2017).
Cai, P. et al. A genome-wide long noncoding RNA CRISPRi screen identifies PRANCR as a novel regulator of epidermal homeostasis. Genome Res. 30, 22–34 (2019).
Xu, D. et al. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 10, 1794 (2020).
Horlbeck, M. A., Liu, S. J., Chang, H. Y., Lim, D. A. & Weissman, J. S. Fitness effects of CRISPR/Cas9-targeting of long noncoding RNA genes. Nat. Biotechnol. 38, 573–576 (2020).
Cannavò, E. et al. Shadow enhancers are pervasive features of developmental regulatory networks. Curr. Biol. 26, 38–51 (2016).
Hutchinson, J. N. et al. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8, 39 (2007).
Nakagawa, S. et al. Malat1 is not an essential component of nuclear speckles in mice. RNA 18, 1487–1499 (2012).
Zhang, B. et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2, 111–123 (2012).
Zhang, X., Hamblin, M. H. & Yin, K.-J. The long noncoding RNA Malat1: its physiological and pathophysiological functions. RNA Biol. 14, 1705–1714 (2017).
Arun, G., Aggarwal, D. & Spector, D. L. MALAT1 long non-coding RNA: functional implications. Noncoding RNA 6, 22 (2020).
Sunwoo, H. et al. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 19, 347–359 (2009).
Clemson, C. M. et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cells 33, 717–726 (2009).
Mao, Y. S., Sunwoo, H., Zhang, B. & Spector, D. L. Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat. Cell Biol. 13, 95–101 (2011).
Nakagawa, S. et al. The lncRNA Neat1 is required for corpus luteum formation and the establishment of pregnancy in a subpopulation of mice. Development 141, 4618–4627 (2014).
Lewejohann, L. et al. Role of a neuronal small non-messenger RNA: behavioural alterations in BC1 RNA-deleted mice. Behav. Brain Res. 154, 273–289 (2004).
Field, A. R. et al. Structurally conserved primate lncRNAs are transiently expressed during human cortical differentiation and influence cell-type-specific genes. Stem Cell Rep. 12, 245–257 (2019).
Liu, S. J. et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 21, 83 (2020).
Ramilowski, J. A. et al. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res. 30, 1060–1072 (2020).
Cao, H. et al. Very long intergenic non-coding (vlinc) RNAs directly regulate multiple genes in cis and trans. BMC Biol. 19, 108 (2021).
Zhao, J., Sun, B. K., Erwin, J. A., Song, J. J. & Lee, J. T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756 (2008).
Hacisuleyman, E., Shukla, C. J., Weiner, C. L. & Rinn, J. L. Function and evolution of local repeats in the Firre locus. Nat. Commun. 7, 11021 (2016).
Zucchelli, S. et al. SINEUPs: a new class of natural and synthetic antisense long non-coding RNAs that activate translation. RNA Biol. 12, 771–779 (2015).
Morrissy, A. S., Griffith, M. & Marra, M. A. Extensive relationship between antisense transcription and alternative splicing in the human genome. Genome Res. 21, 1203–1212 (2011).
Romero-Barrios, N., Legascue, M. F., Benhamed, M., Ariel, F. & Crespi, M. Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 46, 2169–2184 (2018).
Pisignano, G. & Ladomery, M. Epigenetic regulation of alternative splicing: how lncRNAs tailor the message. Noncoding RNA 7, 21 (2021).
Carrieri, C. et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491, 454–457 (2012).
Deforges, J. et al. Control of cognate sense mRNA translation by cis-natural antisense RNAs. Plant Physiol. 180, 305–322 (2019).
Peters, N. T., Rohrbach, J. A., Zalewski, B. A., Byrkett, C. M. & Vaughn, J. C. RNA editing and regulation of Drosophila 4f-rnp expression by sas-10 antisense readthrough mRNA transcripts. RNA 9, 698–710 (2003).
Gong, C. & Maquat, L. E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature 470, 284–288 (2011).
Whittaker, C. & Dean, C. The FLC locus: a platform for discoveries in epigenetics and adaptation. Annu. Rev. Cell Dev. Biol. 33, 555–575 (2017).
Huarte, M. et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142, 409–419 (2010).
Rothschild, G. et al. Noncoding RNA transcription alters chromosomal topology to promote isotype-specific class switch recombination. Sci. Immunol. 5, eaay5864 (2020).
Fanucchi, S. et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat. Genet. 51, 138–150 (2019).
Vollmers, A. C. et al. A conserved long noncoding RNA, GAPLINC, modulates the immune response during endotoxic shock. Proc. Natl Acad. Sci. USA 118, e2016648118 (2021).
Atianand, M. K. et al. A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell 165, 1672–1685 (2016).
Hu, G. et al. LincRNA-Cox2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-mediated chromatin remodeling. J. Immunol. 196, 2799–2808 (2016).
Zhao, X. et al. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat. Neurosci. 16, 1024–1031 (2013).
Ruan, X. et al. Identification of human long noncoding RNAs associated with nonalcoholic fatty liver disease and metabolic homeostasis. J. Clin. Invest. 131, e136336 (2021).
Hennessy, E. J. et al. The long noncoding RNA CHROME regulates cholesterol homeostasis in primates. Nat. Metab. 1, 98–110 (2019).
Du, Q. et al. MIR205HG Is a long noncoding RNA that regulates growth hormone and prolactin production in the anterior pituitary. Dev. Cell 49, 618–631 (2019).
Zhang, P., Cao, L., Fan, P., Mei, Y. & Wu, M. LncRNA-MIF, a c-Myc-activated long non-coding RNA, suppresses glycolysis by promoting Fbxw7-mediated c-Myc degradation. EMBO Rep. 17, 1204–1220 (2016).
Zheng, X. et al. LncRNA wires up Hippo and Hedgehog signaling to reprogramme glucose metabolism. EMBO J. 36, 3325–3335 (2017).
McClintock, M. A. et al. RNA-directed activation of cytoplasmic dynein-1 in reconstituted transport RNPs. Elife 7, e36312 (2018).
Lin, A. et al. The LINK-A lncRNA interacts with PtdIns(3,4,5)P3 to hyperactivate AKT and confer resistance to AKT inhibitors. Nat. Cell Biol. 19, 238–251 (2017).
Sang, L. et al. LncRNA CamK-A regulates Ca2+-signaling-mediated tumor microenvironment remodeling. Mol. Cells 72, 71–83 (2018).
Ma, Y., Zhang, J., Wen, L. & Lin, A. Membrane-lipid associated lncRNA: a new regulator in cancer signaling. Cancer Lett. 419, 27–29 (2018).
Wang, F. et al. The long noncoding RNA Synage regulates synapse stability and neuronal function in the cerebellum. Cell Death Differ. 28, 2634–2650 (2021).
Samaddar, S. & Banerjee, S. Far from the nuclear crowd: cytoplasmic lncRNA and their implications in synaptic plasticity and memory. Neurobiol. Learn. Mem. 185, 107522 (2021).
Wei, W. et al. ADRAM is an experience-dependent long noncoding RNA that drives fear extinction through a direct interaction with the chaperone protein 14-3-3. Cell Rep. 38, 110546 (2022).
Wu, E. et al. Discovery of plasma membrane-associated RNAs through APEX-seq. Cell Biochem. Biophys. 79, 905–917 (2021).
Chen, Y. et al. Hovlinc is a recently evolved class of ribozyme found in human lncRNA. Nat. Chem. Biol. 17, 601–607 (2021).
Schuettengruber, B., Chourrout, D., Vervoort, M., Leblanc, B. & Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 128, 735–745 (2007).
Erdmann, R. M. & Picard, C. L. RNA-directed DNA methylation. PLoS Genet. 16, e1009034 (2020).
Djupedal, I. & Ekwall, K. Epigenetics: heterochromatin meets RNAi. Cell Res. 19, 282–295 (2009).
Jeffery, L. & Nakielny, S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 279, 49479–49487 (2004).
Mohammad, F., Mondal, T., Guseva, N., Pandey, G. K. & Kanduri, C. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 137, 2493–2499 (2010).
Di Ruscio, A. et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 503, 371–376 (2013).
Merry, C. R. et al. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 24, 6240–6253 (2015).
Di Croce, L. & Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155 (2013).
Nagano, T. et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720 (2008).
Pandey, R. R. et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cells 32, 232–246 (2008).
Davidovich, C. & Cech, T. R. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA 21, 2007–2022 (2015).
Davidovich, C. et al. Toward a consensus on the binding specificity and promiscuity of PRC2 for RNA. Mol. Cells 57, 552–558 (2015).
Khalil, A. M. et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl Acad. Sci. USA 106, 11667–11672 (2009).
Beltran, M. et al. The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res. 26, 896–907 (2016).
Rosenberg, M. et al. Motif-driven interactions between RNA and PRC2 are rheostats that regulate transcription elongation. Nat. Struct. Mol. Biol. 28, 103–117 (2021).
Zhao, J. et al. Genome-wide identification of Polycomb-associated RNAs by RIP-seq. Mol. Cells 40, 939–953 (2010).
Davidovich, C., Zheng, L., Goodrich, K. J. & Cech, T. R. Promiscuous RNA binding by Polycomb repressive complex 2. Nat. Struct. Mol. Biol. 20, 1250–1257 (2013).
Cifuentes-Rojas, C., Hernandez, A. J., Sarma, K. & Lee, J. T. Regulatory interactions between RNA and Polycomb repressive complex 2. Mol. Cells 55, 171–185 (2014).
Long, Y. et al. RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat. Genet. 52, 931–938 (2020).
Yap, K. L. et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by Polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cells 38, 662–674 (2010).
Rosenberg, M. et al. Denaturing CLIP, dCLIP, pipeline identifies discrete RNA footprints on chromatin-associated proteins and reveals that CBX7 targets 3′ UTRs to regulate mRNA expression. Cell Syst. 5, 368–385 (2017).
Wang, X. et al. Targeting of Polycomb repressive complex 2 to RNA by short repeats of consecutive guanines. Mol. Cells 65, 1056–1067 (2017).
Beltran, M. et al. G-tract RNA removes Polycomb repressive complex 2 from genes. Nat. Struct. Mol. Biol. 26, 899–909 (2019).
Dinger, M. E. et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 18, 1433–1445 (2008).
Wang, K. C. et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124 (2011).
Yang, Y. W. et al. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency. Elife 3, e02046 (2014).
Deng, C. et al. HoxBlinc RNA recruits Set1/MLL complexes to activate Hox gene expression patterns and mesoderm lineage development. Cell Rep. 14, 103–114 (2016).
Subhash, S. et al. H3K4me2 and WDR5 enriched chromatin interacting long non-coding RNAs maintain transcriptionally competent chromatin at divergent transcriptional units. Nucleic Acids Res. 46, 9384–9400 (2018).
Butler, A. A., Johnston, D. R., Kaur, S. & Lubin, F. D. Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci. Signal. 12, eaaw9277 (2019).
Jantrapirom, S. et al. Long noncoding RNA-dependent methylation of nonhistone proteins. WIREs RNA 12, e1661 (2021).
Luo, Z., Rhie, S. K. & Farnham, P. J. The enigmatic HOX genes: can we crack their code? Cancers 11, 323 (2019).
Grosschedl, R., Giese, K. & Pagel, J. HMG domain proteins: architectural elements in the assembly of nucleoprotein structures. Trends Genet. 10, 94–100 (1994).
Tantin, D. Oct transcription factors in development and stem cells: insights and mechanisms. Development 140, 2857–2866 (2013).
Clapier, C. R., Iwasa, J., Cairns, B. R. & Peterson, C. L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 18, 407–422 (2017).
Li, Y. et al. The structural basis for cohesin–CTCF-anchored loops. Nature 578, 472–476 (2020).
Oh, H. J. et al. Jpx RNA regulates CTCF anchor site selection and formation of chromosome loops. Cell 184, P6157–P6173 (2021).
Kung, J. T. et al. Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol. Cells 57, 361–375 (2015).
Rivera-Pomar, R., Niessing, D., Schmidt-Ott, U., Gehring, W. J. & Jacklë, H. RNA binding and translational suppression by bicoid. Nature 379, 746–749 (1996).
Holmes, Z. E. et al. The Sox2 transcription factor binds RNA. Nat. Commun. 11, 1805 (2020).
Cajigas, I. et al. Sox2-Evf2 lncRNA mechanisms of chromosome topological control in developing forebrain. Development 148, dev197202 (2021).
Genzor, P. & Bortvin, A. A unique HMG-box domain of mouse Maelstrom binds structured RNA but not double stranded DNA. PLoS ONE 10, e0120268 (2015).
Zhao, Z., Dammert, M. A., Grummt, I. & Bierhoff, H. lncRNA-Induced nucleosome repositioning reinforces transcriptional repression of RNA genes upon hypotonic stress. Cell Rep. 14, 1876–1882 (2016).
Amaral, P. P. et al. Complex architecture and regulated expression of the Sox2ot locus during vertebrate development. RNA 15, 2013–2027 (2009).
Ng, S.-Y., Johnson, R. & Stanton, L. W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 31, 522–533 (2012).
Ng, S.-Y., Bogu, G. K., Soh, B. S. & Stanton, L. W. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol. Cells 51, 349–359 (2013).
Samudyata et al. Interaction of Sox2 with RNA binding proteins in mouse embryonic stem cells. Exp. Cell Res. 381, 129–138 (2019).
Hou, L. et al. Concurrent binding to DNA and RNA facilitates the pluripotency reprogramming activity of Sox2. Nucleic Acids Res. 48, 3869–3887 (2020).
Tang, Y. et al. Linking long non-coding RNAs and SWI/SNF complexes to chromatin remodeling in cancer. Mol. Cancer 16, 42 (2017).
Jégu, T. et al. Xist RNA antagonizes the SWI/SNF chromatin remodeler BRG1 on the inactive X chromosome. Nat. Struct. Mol. Biol. 26, 96–109 (2019).
Grossi, E. et al. A lncRNA-SWI/SNF complex crosstalk controls transcriptional activation at specific promoter regions. Nat. Commun. 11, 936 (2020).
Schutt, C. et al. Linc-MYH configures INO80 to regulate muscle stem cell numbers and skeletal muscle hypertrophy. EMBO J. 39, e105098 (2020).
Patty, B. J. & Hainer, S. J. Non-coding RNAs and nucleosome remodeling complexes: an intricate regulatory relationship. Biology 9, 213 (2020).
Ducoli, L. et al. LETR1 is a lymphatic endothelial-specific lncRNA governing cell proliferation and migration through KLF4 and SEMA3C. Nat. Commun. 12, 925 (2021).
Chang, K. C. et al. MaTAR25 lncRNA regulates the Tensin1 gene to impact breast cancer progression. Nat. Commun. 11, 6438 (2020).
Caretti, G. et al. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev. Cell 11, 547–560 (2006).
Dong, A. et al. A long noncoding RNA, LncMyoD, modulates chromatin accessibility to regulate muscle stem cell myogenic lineage progression. Proc. Natl Acad. Sci. USA 117, 32464–32475 (2020).
Yu, X. et al. Long non-coding RNA Linc-RAM enhances myogenic differentiation by interacting with MyoD. Nat. Commun. 8, 14016 (2017).
Dou, M. et al. The long noncoding RNA MyHC IIA/X-AS contributes to skeletal muscle myogenesis and maintains the fast fiber phenotype. J. Biol. Chem. 295, 4937–4949 (2020).
Bose, D. A. et al. RNA binding to CBP stimulates histone acetylation and transcription. Cell 168, 135–149 (2017).
Lin, H., Shabbir, A., Molnar, M. & Lee, T. Stem cell regulatory function mediated by expression of a novel mouse Oct4 pseudogene. Biochem. Biophys. Res. Commun. 355, 111–116 (2007).
Hawkins, P. G. & Morris, K. V. Transcriptional regulation of Oct4 by a long non-coding RNA antisense to Oct4-pseudogene 5. Transcription 1, 165–175 (2010).
Wang, Y. et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell 25, 69–80 (2013).
Scarola, M. et al. FUS-dependent loading of SUV39H1 to OCT4 pseudogene-lncRNA programs a silencing complex with OCT4 promoter specificity. Commun. Biol. 3, 632 (2020).
Tariq, A. et al. LncRNA-mediated regulation of SOX9 expression in basal subtype breast cancer cells. RNA 26, 175–185 (2020).
Sheik Mohamed, J., Gaughwin, P. M., Lim, B., Robson, P. & Lipovich, L. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA 16, 324–337 (2010).
Tomita, S. et al. A cluster of noncoding RNAs activates the ESR1 locus during breast cancer adaptation. Nat. Commun. 6, 6966 (2015).
Setten, R. L., Chomchan, P., Epps, E. W., Burnett, J. C. & Rossi, J. J. CRED9: a differentially expressed elncRNA regulates expression of transcription factor CEBPA. RNA 27, 891–906 (2021).
Shen, Y. et al. A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120 (2012).
Thurman, R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012).
Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Chen, H. & Liang, H. A high-resolution map of human enhancer RNA loci characterizes super-enhancer activities in cancer. Cancer Cell 38, 701–715 (2020).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Pott, S. & Lieb, J. D. What are super-enhancers? Nat. Genet. 47, 8–12 (2015).
Li, S. & Ovcharenko, I. Enhancer jungles establish robust tissue-specific regulatory control in the human genome. Genomics 112, 2261–2270 (2020).
Ptashne, M. How eukaryotic transcriptional activators work. Nature 335, 683–689 (1988).
Souaid, C., Bloyer, S. & Noordermeer, D. in Nuclear Architecture and Dynamics Vol. 2 (eds Christophe, L. & Jean-Marc, V.) Ch. 19, 435–456 (Academic Press, 2018).
Lim, B. & Levine, M. S. Enhancer-promoter communication: hubs or loops? Curr. Opin. Genet. Dev. 67, 5–9 (2021).
Zhu, I., Song, W., Ovcharenko, I. & Landsman, D. A model of active transcription hubs that unifies the roles of active promoters and enhancers. Nucleic Acids Res. 49, 4493–4505 (2021).
Kim, T.-K., Hemberg, M. & Gray, J. M. Enhancer RNAs: a class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb. Perspect. Biol. 7, a018622 (2015).
Arner, E. et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014 (2015).
Li, W., Notani, D. & Rosenfeld, M. G. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 17, 207–223 (2016).
Arnold, P. R., Wells, A. D. & Li, X. C. Diversity and emerging roles of enhancer RNA in regulation of gene expression and cell fate. Front. Cell Dev. Biol. 7, 377 (2020).
Core, L. J. et al. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 46, 1311–1320 (2014).
Kim, T.-K. et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187 (2010).
Melgar, M. F., Collins, F. S. & Sethupathy, P. Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol. 12, R113 (2011).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).
Seila, A. C. et al. Divergent transcription from active promoters. Science 322, 1849–1851 (2008).
Young, R. S., Kumar, Y., Bickmore, W. A. & Taylor, M. S. Bidirectional transcription initiation marks accessible chromatin and is not specific to enhancers. Genome Biol. 18, 242 (2017).
Sartorelli, V. & Lauberth, S. M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 27, 521–528 (2020).
Wu, H. et al. Tissue-specific RNA expression marks distant-acting developmental enhancers. PLoS Genet. 10, e1004610 (2014).
Carullo, N. V. N. et al. Enhancer RNAs predict enhancer–gene regulatory links and are critical for enhancer function in neuronal systems. Nucleic Acids Res. 48, 9550–9570 (2020).
Tan, J. Y. & Marques, A. C. The activity of human enhancers is modulated by the splicing of their associated lncRNAs. PLoS Comput. Biol. 18, e1009722 (2022).
Gil, N. & Ulitsky, I. Production of spliced long noncoding RNAs specifies regions with increased enhancer activity. Cell Syst. 7, 537–547 (2018).
Melo, C. A. et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cells 49, 524–535 (2013).
Lam, M. T. Y., Li, W., Rosenfeld, M. G. & Glass, C. K. Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 39, 170–182 (2014).
Yin, Y. et al. Opposing roles for the lncRNA Haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation. Cell Stem Cell 16, 504–516 (2015).
Isoda, T. et al. Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. Cell 171, 103–119 (2017).
Cajigas, I. et al. The Evf2 ultraconserved enhancer lncRNA functionally and spatially organizes megabase distant genes in the developing forebrain. Mol. Cells 71, 956–972 (2018).
Lewandowski, J. P. et al. The Firre locus produces a trans-acting RNA molecule that functions in hematopoiesis. Nat. Commun. 10, 5137 (2019).
Groff, A. F., Barutcu, A. R., Lewandowski, J. P. & Rinn, J. L. Enhancers in the Peril lincRNA locus regulate distant but not local genes. Genome Biol. 19, 219 (2018).
Han, X. et al. The lncRNA Hand2os1/Uph locus orchestrates heart development through regulation of precise expression of Hand2. Development 146, dev176198 (2019).
Mattick, J. S. Deconstructing the dogma: a new view of the evolution and genetic programming of complex organisms. Ann. N. Y. Acad. Sci. 1178, 29–46 (2009).
Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017).
Garcia-Jove Navarro, M. et al. RNA is a critical element for the sizing and the composition of phase-separated RNA–protein condensates. Nat. Commun. 10, 3230 (2019).
Roden, C. & Gladfelter, A. S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 22, 183–195 (2021).
Niklas, K. J., Dunker, A. K. & Yruela, I. The evolutionary origins of cell type diversification and the role of intrinsically disordered proteins. J. Exp. Bot. 69, 1437–1446 (2018).
Macossay-Castillo, M. et al. The balancing act of intrinsically disordered proteins: enabling functional diversity while minimizing promiscuity. J. Mol. Biol. 431, 1650–1670 (2019).
Chen, W. & Moore, M. J. The spliceosome: disorder and dynamics defined. Curr. Opin. Struct. Biol. 24, 141–149 (2014).
Staby, L. et al. Eukaryotic transcription factors: paradigms of protein intrinsic disorder. Biochem. J. 474, 2509–2532 (2017).
Wright, P. E. & Dyson, H. J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 (2015).
Hahn, S. Phase separation, protein disorder, and enhancer function. Cell 175, 1723–1725 (2018).
Peng, Z., Mizianty, M. J., Xue, B., Kurgan, L. & Uversky, V. N. More than just tails: intrinsic disorder in histone proteins. Mol. Biosyst. 8, 1886–1901 (2012).
Watson, M. & Stott, K. Disordered domains in chromatin-binding proteins. Essays Biochem. 63, 147–156 (2019).
Balcerak, A., Trebinska-Stryjewska, A., Konopinski, R., Wakula, M. & Grzybowska, E. A. RNA–protein interactions: disorder, moonlighting and junk contribute to eukaryotic complexity. Open Biol. 9, 190096 (2019).
Musselman, C. A. & Kutateladze, T. G. Characterization of functional disordered regions within chromatin-associated proteins. iScience 24, 102070 (2021).
Takeshi, C. & Tetsuro, H. Nuclear bodies built on architectural long noncoding RNAs: unifying principles of their construction and function. Mol. Cells 40, 889–896 (2017).
Panda, S. et al. Noncoding RNA Ginir functions as an oncogene by associating with centrosomal proteins. PLoS Biol. 16, e2004204 (2018).
Yamazaki, T. & Hirose, T. Control of condensates dictates nucleolar architecture. Science 373, 486–487 (2021).
Wang, X. et al. Mutual dependency between lncRNA LETN and protein NPM1 in controlling the nucleolar structure and functions sustaining cell proliferation. Cell Res. 31, 664–683 (2021).
Spector, D. L. & Lamond, A. I. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 3, a000646 (2011).
Ishizuka, A., Hasegawa, Y., Ishida, K., Yanaka, K. & Nakagawa, S. Formation of nuclear bodies by the lncRNA Gomafu-associating proteins Celf3 and SF1. Genes Cells 19, 704–721 (2014).
Tripathi, V. et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cells 39, 925–938 (2010).
Yamazaki, T. et al. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol. Cells 70, 1038–1053 (2018).
Fox, A. H., Nakagawa, S., Hirose, T. & Bond, C. S. Paraspeckles: where long noncoding RNA meets phase separation. Trends Biochem. Sci. 43, 124–135 (2018).
Fang, X. et al. Arabidopsis FLL2 promotes liquid–liquid phase separation of polyadenylation complexes. Nature 569, 265–269 (2019).
Emenecker, R. J., Holehouse, A. S. & Strader, L. C. Emerging roles for phase separation in plants. Dev. Cell 55, 69–83 (2020).
Brangwynne, C. P. et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 (2009).
Smith, J. et al. Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. Elife 5, e21337 (2016).
Chouaib, R. et al. A dual protein-mRNA localization screen reveals compartmentalized translation and widespread co-translational RNA targeting. Dev. Cell 54, 773–791 (2020).
Chen, X., Wu, X., Wu, H. & Zhang, M. Phase separation at the synapse. Nat. Neurosci. 23, 301–310 (2020).
Tichon, A. et al. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun. 7, 12209 (2016).
Mele, M. & Rinn, J. L. “Cat’s cradling” the 3D genome by the act of lncRNA transcription. Mol. Cells 62, 657–664 (2016).
Bhat, P., Honson, D. & Guttman, M. Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat. Rev. Mol. Cell Biol. 22, 653–670 (2021).
Hnisz, D., Shrinivas, K., Young, R. A., Chakraborty, A. K. & Sharp, P. A. A phase separation model for transcriptional control. Cell 169, 13–23 (2017).
Henninger, J. E. et al. RNA-mediated feedback control of transcriptional condensates. Cell 184, 207–225 (2021).
Quinodoz, S. A. et al. RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790 (2021).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Nair, S. J. et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat. Struct. Mol. Biol. 26, 193–203 (2019).
Shrinivas, K. et al. Enhancer features that drive formation of transcriptional condensates. Mol. Cells 75, 549–561 (2019).
Ahn, J. H. et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 595, 591–595 (2021).
Boehning, M. et al. RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol. 25, 833–840 (2018).
Boija, A. et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855 (2018).
Cho, W.-K. et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018).
Wang, J. et al. Phase separation of OCT4 controls TAD reorganization to promote cell fate transitions. Cell Stem Cell 28, 1868–1883 (2021).
Chong, S. et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555 (2018).
Hall, L. L. et al. Stable C0T-1 repeat RNA Is abundant and is associated with euchromatic interphase chromosomes. Cell 156, 907–919 (2014).
Creamer, K. M., Kolpa, H. J. & Lawrence, J. B. Nascent RNA scaffolds contribute to chromosome territory architecture and counter chromatin compaction. Mol. Cells 81, 3509–3525 (2021).
Strom, A. R. et al. Phase separation drives heterochromatin domain formation. Nature 547, 241–245 (2017).
Lu, J. Y. et al. Homotypic clustering of L1 and B1/Alu repeats compartmentalizes the 3D genome. Cell Res. 31, 613–630 (2021).
Hilbert, L. et al. Transcription organizes euchromatin via microphase separation. Nat. Commun. 12, 1360 (2021).
Plys, A. J. et al. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 33, 1–15 (2019).
Yap, K. et al. A short tandem repeat-enriched RNA assembles a nuclear compartment to control alternative splicing and promote cell survival. Mol. Cells 72, 525–540 (2018).
Furuno, M. et al. Clusters of internally primed transcripts reveal novel long noncoding RNAs. PLoS Genet. 2, e37 (2006).
St Laurent, G. et al. VlincRNAs controlled by retroviral elements are a hallmark of pluripotency and cancer. Genome Biol. 14, R73 (2013).
Guttman, M. & Rinn, J. L. Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346 (2012).
Mercer, T. R. & Mattick, J. S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 20, 300–307 (2013).
Rinn, J. L. & Chang, H. Y. Long noncoding RNAs: molecular modalities to organismal functions. Annu. Rev. Biochem. 89, 283–308 (2020).
Hawkes, E. J. et al. COOLAIR antisense RNAs form evolutionarily conserved elaborate secondary structures. Cell Rep. 16, 3087–3096 (2016).
Abou Alezz, M., Celli, L., Belotti, G., Lisa, A. & Bione, S. GC-AG introns features in long non-coding and protein-coding genes suggest their role in gene expression regulation. Front. Genet. 11, 488 (2020).
Tilgner, H. et al. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 22, 1616–1625 (2012).
Mele, M. et al. Chromatin environment, transcriptional regulation, and splicing distinguish lincRNAs and mRNAs. Genome Res. 27, 27–37 (2017).
Garg, K. & Green, P. Differing patterns of selection in alternative and constitutive splice sites. Genome Res. 17, 1015–1022 (2007).
Guo, C.-J. et al. Distinct processing of lncRNAs contributes to non-conserved functions in stem cells. Cell 181, 621–636 (2020).
Khan, M. R., Wellinger, R. J. & Laurent, B. Exploring the alternative splicing of long noncoding RNAs. Trends Genet. 37, 695–698 (2021).
Smith, M. A., Gesell, T., Stadler, P. F. & Mattick, J. S. Widespread purifying selection on RNA structure in mammals. Nucleic Acids Res. 41, 8220–8236 (2013).
Smith, M. A., Seemann, S. E., Quek, X. C. & Mattick, J. S. DotAligner: identification and clustering of RNA structure motifs. Genome Biol. 18, 244 (2017).
Seemann, S. E. et al. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res. 27, 1371–1383 (2017).
Novikova, I. V., Hennelly, S. P. & Sanbonmatsu, K. Y. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 40, 5034–5051 (2012).
Somarowthu, S. et al. HOTAIR forms an intricate and modular secondary structure. Mol. Cells 58, 353–361 (2015).
Spitale, R. C. et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490 (2015).
Chu, C. et al. Systematic discovery of Xist RNA binding proteins. Cell 161, 404–416 (2015).
Lu, Z. et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 165, 1267–1279 (2016).
Kirk, J. M. et al. Functional classification of long non-coding RNAs by k-mer content. Nat. Genet. 50, 1474–1482 (2018).
Johnson, R. & Guigo, R. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA 20, 959–976 (2014).
Nesterova, T. B. et al. Characterization of the genomic Xist locus in rodents reveals conservation of overall gene structure and tandem repeats but rapid evolution of unique sequence. Genome Res. 11, 833–849 (2001).
Wutz, A., Rasmussen, T. P. & Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 30, 167–174 (2002).
Sunwoo, H., Colognori, D., Froberg, J. E., Jeon, Y. & Lee, J. T. Repeat E anchors Xist RNA to the inactive X chromosomal compartment through CDKN1A-interacting protein (CIZ1). Proc. Natl Acad. Sci. USA 114, 10654–10659 (2017).
Pintacuda, G. et al. hnRNPK recruits PCGF3/5-PRC1 to the Xist RNA B-repeat to establish polycomb-mediated chromosomal silencing. Mol. Cells 68, 955–969 (2017).
Brockdorff, N. Local tandem repeat expansion in Xist RNA as a model for the functionalisation of ncRNA. Noncoding RNA 4, 28 (2018).
Colognori, D., Sunwoo, H., Kriz, A. J., Wang, C.-Y. & Lee, J. T. Xist deletional analysis reveals an interdependency between Xist RNA and Polycomb complexes for spreading along the inactive X. Mol. Cells 74, 101–117 (2019).
Sprague, D. et al. Nonlinear sequence similarity between the Xist and Rsx long noncoding RNAs suggests shared functions of tandem repeat domains. RNA 25, 1004–1019 (2019).
Carter, A. C. et al. Spen links RNA-mediated endogenous retrovirus silencing and X chromosome inactivation. Elife 9, e54508 (2020).
Kelley, D. R., Hendrickson, D. G., Tenen, D. & Rinn, J. L. Transposable elements modulate human RNA abundance and splicing via specific RNA-protein interactions. Genome Biol. 15, 537 (2014).
Percharde, M. et al. A LINE1-nucleolin partnership regulates early development and ESC identity. Cell 174, 391–405 (2018).
Chu, C., Qu, K., Zhong, F. L., Artandi, S. E. & Chang, H. Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cells 44, 667–678 (2011).
Mondal, T. et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 6, 7743 (2015).
Long, J. et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest. 126, 4205–4218 (2016).
O’Leary, V. B. et al. PARTICLE, a triplex-forming long ncRNA, regulates locus-specific methylation in response to low-dose irradiation. Cell Rep. 11, 474–485 (2015).
Zhao, Z., Sentürk, N., Song, C. & Grummt, I. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 32, 836–848 (2018).
Kuo, C.-C. et al. Detection of RNA–DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 47, e32 (2019).
Blank-Giwojna, A., Postepska-Igielska, A. & Grummt, I. lncRNA KHPS1 activates a poised enhancer by triplex-dependent recruitment of epigenomic regulators. Cell Rep. 26, 2904–2915 (2019).
Li, Y., Syed, J. & Sugiyama, H. RNA-DNA triplex formation by long noncoding RNAs. Cell Chem. Biol. 23, 1325–1333 (2016).
Soibam, B. Super-lncRNAs: identification of lncRNAs that target super-enhancers via RNA:DNA:DNA triplex formation. RNA 23, 1729–1742 (2017).
Farabella, I., Di Stefano, M., Soler-Vila, P., Marti-Marimon, M. & Marti-Renom, M. A. Three-dimensional genome organization via triplex-forming RNAs. Nat. Struct. Mol. Biol. 28, 945–954 (2021).
Niehrs, C. & Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 21, 167–178 (2020).
Xu, C. et al. R-loop resolution promotes co-transcriptional chromatin silencing. Nat. Commun. 12, 1790 (2021).
Zappulla, D. C. & Cech, T. R. Yeast telomerase RNA: A flexible scaffold for protein subunits. Proc. Natl Acad. Sci. USA 101, 10024–10029 (2004).
Maciejowski, J. & de Lange, T. Telomeres in cancer: tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 18, 175–186 (2017).
Chen, H. et al. Structural insights into yeast telomerase recruitment to telomeres. Cell 172, 331–343 (2018).
Phillips, M. L. Existence of RNA ‘dark matter’ in doubt. Nature https://doi.org/10.1038/news.2010.1248 (2010).
van Bakel, H., Nislow, C., Blencowe, B. J. & Hughes, T. R. Most “dark matter” transcripts are associated with known genes. PLoS Biol. 8, e1000371 (2010).
Clark, M. B. et al. The reality of pervasive transcription. PLoS Biol. 9, e1000625 (2011).
Uroda, T. et al. Conserved pseudoknots in lncRNA MEG3 are essential for stimulation of the p53 pathway. Mol. Cells 75, 982–995 (2019).
Chillón, I. & Marcia, M. The molecular structure of long non-coding RNAs: emerging patterns and functional implications. Crit. Rev. Biochem. Mol. Biol. 55, 662–690 (2020).
König, J. et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 17, 909–915 (2010).
McHugh, C. A. et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–236 (2015).
Minajigi, A. et al. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349, aab2276 (2015).
Rauzan, B. et al. Kinetics and thermodynamics of DNA, RNA, and hybrid duplex formation. Biochemistry 52, 765–772 (2013).
Zhou, B. et al. GRID-seq for comprehensive analysis of global RNA–chromatin interactions. Nat. Protoc. 14, 2036–2068 (2019).
Bonetti, A. et al. RADICL-seq identifies general and cell type–specific principles of genome-wide RNA-chromatin interactions. Nat. Commun. 11, 1018 (2020).
Cai, Z. et al. RIC-seq for global in situ profiling of RNA–RNA spatial interactions. Nature 582, 432–437 (2020).
George, L., Indig, F. E., Abdelmohsen, K. & Gorospe, M. Intracellular RNA-tracking methods. Open Biol. 8, 180104 (2018).
Fazal, F. M. et al. Atlas of subcellular RNA localization revealed by APEX-seq. Cell 178, 473–490 (2019).
Li, P., Zhou, X., Xu, K. & Zhang, Q. C. RASP: an atlas of transcriptome-wide RNA secondary structure probing data. Nucleic Acids Res. 49, D183–D191 (2020).
Wang, X.-W., Liu, C.-X., Chen, L.-L. & Zhang, Q. C. RNA structure probing uncovers RNA structure-dependent biological functions. Nat. Chem. Biol. 17, 755–766 (2021).
Cao, H. & Kapranov, P. Methods to analyze the non-coding RNA interactome — recent advances and challenges. Front. Genet. 13, 857759 (2022).
Sanbonmatsu, K. Getting to the bottom of lncRNA mechanism: structure–function relationships. Mamm. Genome 33, 343–353 (2021).
Wutz, A. et al. Non-imprinted Igf2r expression decreases growth and rescues the Tme mutation in mice. Development 128, 1881–1887 (2001).
Ballarino, M. et al. Deficiency in the nuclear long noncoding RNA Charme causes myogenic defects and heart remodeling in mice. EMBO J. 37, e99697 (2018).
Rom, A. et al. Regulation of CHD2 expression by the Chaserr long noncoding RNA gene is essential for viability. Nat. Commun. 10, 5092 (2019).
Grote, P. et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 24, 206–214 (2013).
Gabory, A. et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 136, 3413–3421 (2009).
Ritter, N. et al. The lncRNA locus Handsdown regulates cardiac gene programs and is essential for early mouse development. Dev. Cell 50, 644–657 (2019).
Fitzpatrick, G. V., Soloway, P. D. & Higgins, M. J. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat. Genet. 32, 426–431 (2002).
Zhou, B. et al. Endogenous retrovirus-derived long noncoding RNA enhances innate immune responses via derepressing RELA expression. mBio 10, e00937-19 (2019).
Elling, R. et al. Genetic models reveal cis and trans immune-regulatory activities for lincRNA-Cox2. Cell Rep. 25, 1511–1524 (2018).
Jiang, M. et al. Self-recognition of an inducible host lncRNA by RIG-I feedback restricts innate immune response. Cell 173, 906–919 (2018).
Takahashi, N. et al. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum. Mol. Genet. 18, 1879–1888 (2009).
Zhou, Y. et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 137, 2643–2652 (2010).
Kopp, F. et al. PUMILIO hyperactivity drives premature aging of Norad-deficient mice. Elife 8, e42650 (2019).
Andersen, R. E. et al. The long noncoding RNA Pnky is a trans-acting regulator of cortical development in vivo. Dev. Cell 49, 632–642 (2019).
Lewandowski, J. P. et al. The Tug1 lncRNA locus is essential for male fertility. Genome Biol. 21, 237 (2020).
Marahrens, Y., Panning, B., Dausman, J., Strauss, W. & Jaenisch, R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 11, 156–166 (1997).
Hacisuleyman, E. et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 21, 198–206 (2014).
Maass, P. G., Barutcu, A. R., Weiner, C. L. & Rinn, J. L. Inter-chromosomal contact properties in live-cell imaging and in Hi-C. Mol. Cells 69, 1039–1045 (2018).
West, J. A. et al. Structural, super-resolution microscopy analysis of paraspeckle nuclear body organization. J. Cell Biol. 214, 817–830 (2016).
Wilusz, J. E. et al. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 26, 2392–2407 (2012).
Tripathi, V. et al. SRSF1 regulates the assembly of pre-mRNA processing factors in nuclear speckles. Mol. Biol. Cell 23, 3694–3706 (2012).
Fei, J. et al. Quantitative analysis of multilayer organization of proteins and RNA in nuclear speckles at super resolution. J. Cell Sci. 130, 4180–4192 (2017).
Krause, H. M. New and prospective roles for lncRNAs in organelle formation and function. Trends Genet. 34, 736–745 (2018).
Kretz, M. et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493, 231–235 (2013).
Aguilar, R. et al. Targeting Xist with compounds that disrupt RNA structure and X inactivation. Nature 604, 160–166 (2022).
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, made substantial contributions to discussion of the content and reviewed and/or edited the manuscript before submission. J.S.M. drafted the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Molecular Cell Biology and the authors thank Thomas Cech, Xiangting Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
FANTOM: http://fantom.gsc.riken.jp/cat/
GENCODE: https://www.gencodegenes.org
HUGO Gene Nomenclature Committee: https://www.genenames.org/about/guidelines/#!/#tocAnchor-1-4
Glossary
- Cajal bodies
-
Nuclear structures often associated with the nucleolus that have important roles in RNA metabolism and the formation of ribonucleoproteins involved in transcription, splicing, ribosome biogenesis and telomere maintenance.
- Coacervates
-
Condensed liquid-like droplets formed by oppositely charged macromolecules, in vivo involving the interaction of positively charged amino acids in the intrinsically disordered regions of proteins with the negatively charged backbone of RNAs.
- GC–AG splice sites
-
A non-canonical variant of the major U2-type GT–AG splice junctions.
- Intrinsically disordered region
-
A polypeptide segment containing a high proportion of polar or charged residues and insufficient hydrophobic residues to form a stable tertiary structure.
- Modified RNA mimics
-
Chemically synthesized RNA molecules containing chemical modifications to increase their stability or target affinity, facilitate their action and/or bypass detection by innate immunity.
- Nuclear speckles
-
Irregularly shaped nuclear domains enriched in pre-mRNA splicing factors, located in the interchromatin regions of the nucleoplasm of mammalian cells.
- Pol V transcripts
-
Short RNAs transcribed by a specialized plant-specific RNA polymerase (Pol) which maintain the repression of transposons and genomic repeats.
- Polycomb bodies
-
Nuclear foci of Polycomb group proteins, within which Polycomb group protein-bound regions of DNA are localized and contact each other.
- Pioneer transcription factors
-
Proteins that have the unique ability to bind target sites in closed chromatin and open closed chromatin to activate gene expression and implement new cell fates.
- Primary transcription units
-
The long precursor RNAs transcribed from chromosomal regions (‘genes’) before splicing and assembly of the exons into an mRNA or long non-coding RNA and before processing of the intronic RNAs into trans-acting smaller RNAs.
- Quantitative traits
-
Phenotypic characteristics that vary continuously in natural populations, including many aspects of morphology, physiology, behaviour and disease susceptibility.
- Small interspersed nuclear elements
-
Repetitive non-coding sequences of 100–600 bp that are common in animal and plant genomes. They are derived from retrotransposons and are propagated through an RNA intermediate
- Super-enhancers
-
Enhancer-dense genomic regions found near genes that have key roles in determining cellular identity.
- Transcription factor hubs
-
Complex topological assemblies, in which multiple genes and regulatory factors interact with each other.
- X inactivation centre
-
A complex locus in the X chromosome that is required for the near-global inactivation of one of the two X chromosomes in female mammals, a spreading process initiated by the expression of XIST.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mattick, J.S., Amaral, P.P., Carninci, P. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol 24, 430–447 (2023). https://doi.org/10.1038/s41580-022-00566-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41580-022-00566-8
This article is cited by
-
Methylated lncRNAs suppress apoptosis of gastric cancer stem cells via the lncRNA–miRNA/protein axis
Cellular & Molecular Biology Letters (2024)
-
The emerging roles of long non-coding RNA (lncRNA) H19 in gynecologic cancers
BMC Cancer (2024)
-
Noncoding RNAs in skeletal development and disorders
Biological Research (2024)
-
Novel lncRNAs LINC01221, RP11-472G21.2 and CRNDE are markers of differential expression in pediatric patients with T cell acute lymphoblastic leukemia
Cancer Cell International (2024)
-
Tracing vitamins on the long non-coding lane of the transcriptome: vitamin regulation of LncRNAs
Genes & Nutrition (2024)
