

Association between Heavy Metals, Metalloids and Metabolic Syndrome: New Insights and Approaches

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
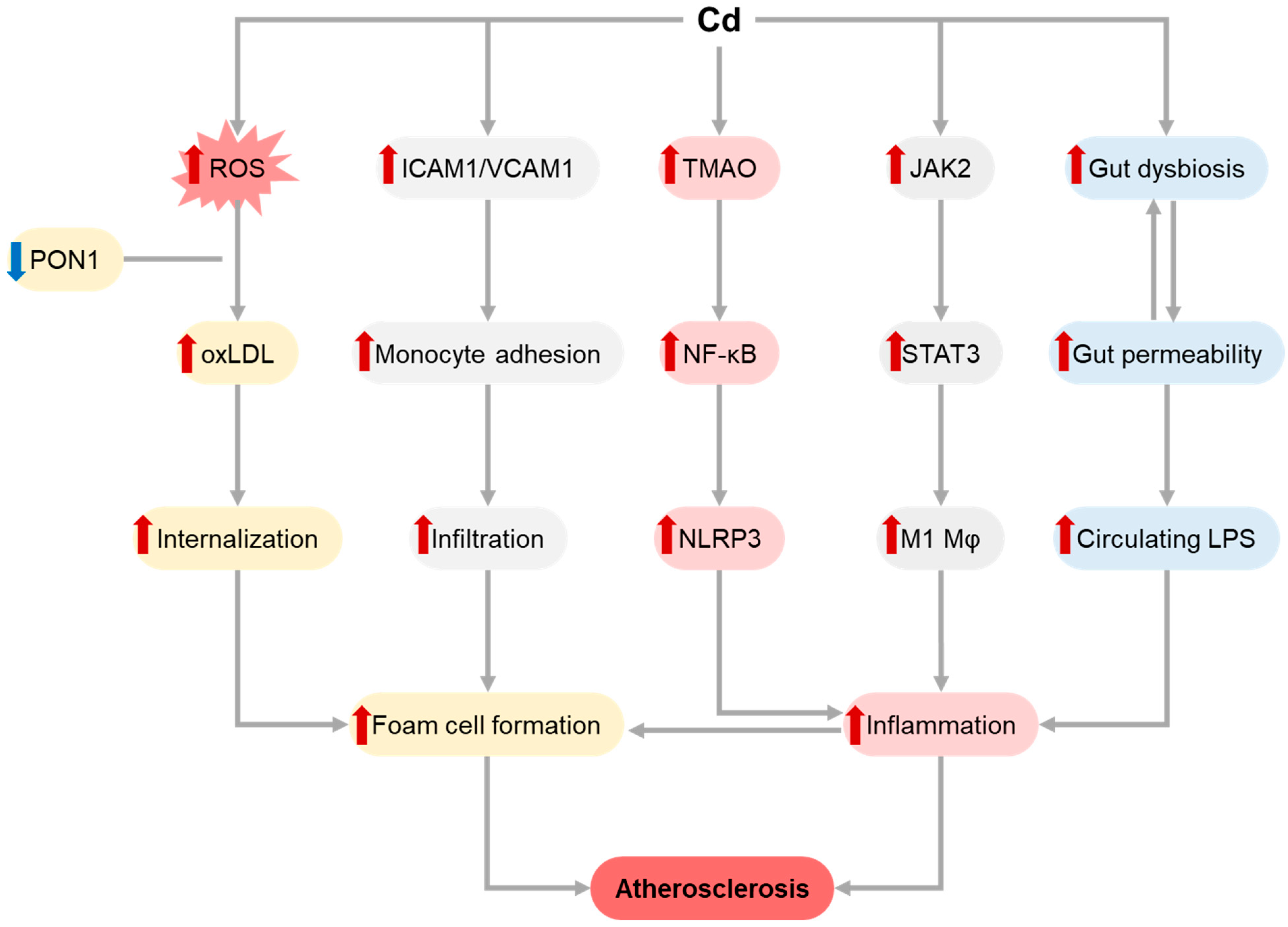
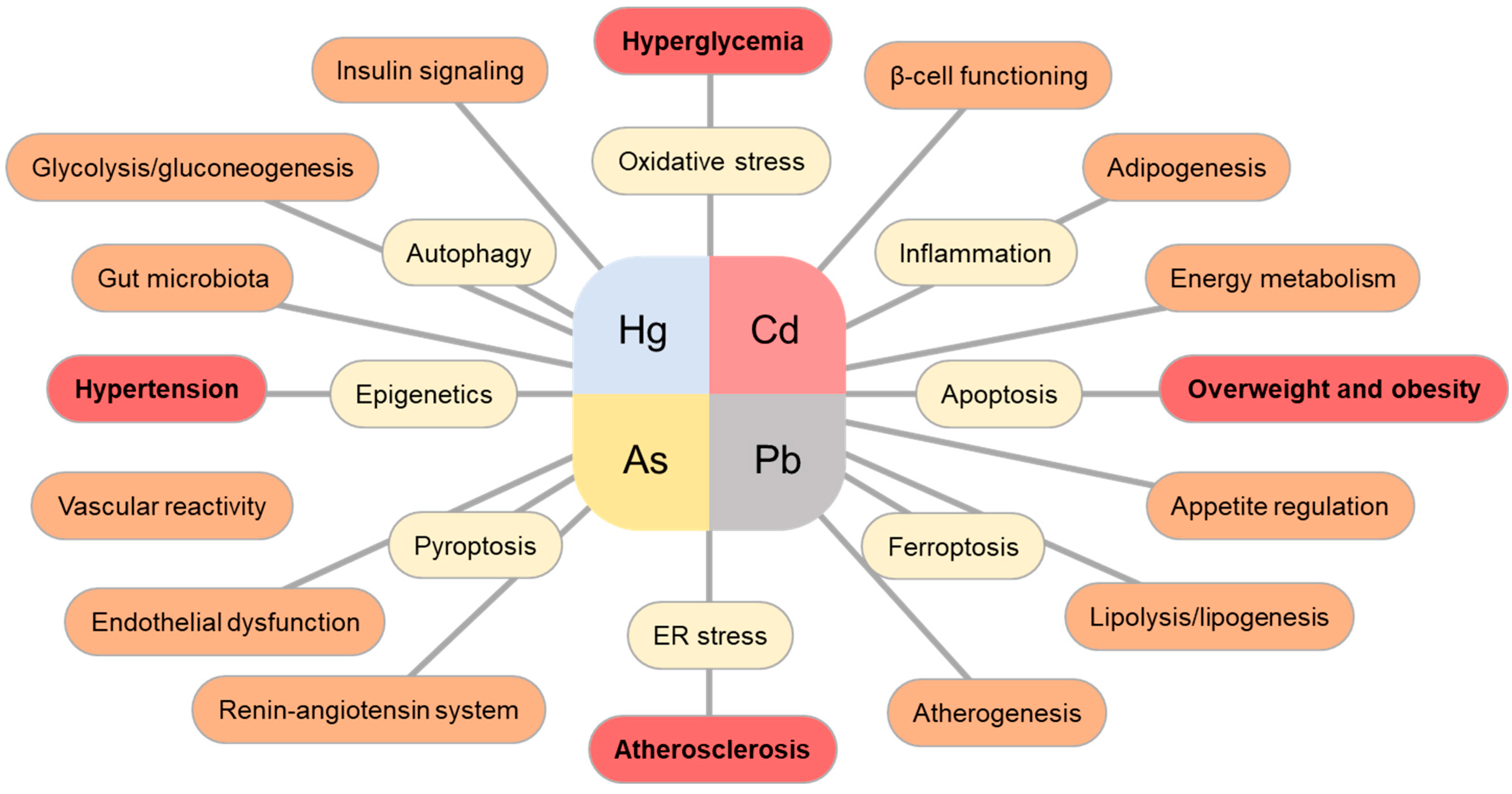
1.1. Cadmium (Cd)
1.2. Arsenic (As)
1.3. Mercury (Hg)
1.4. Lead (Pb)
2. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institutes of Health, National Heart, Lung, and Blood Institute. 2022. Available online: https://www.nhlbi.nih.gov/health/metabolic-syndrome (accessed on 5 May 2023).
- Liang, X.P.; Or, C.Y.; Tsoi, M.F.; Cheung, C.L.; Cheung, B.M.Y. Prevalence of metabolic syndrome in the United States National Health and Nutrition Examination Survey (nhanes) 2011–2018. Eur. Heart J. 2021, 42, 2420. [Google Scholar] [CrossRef]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity among Adults: United States, 2017–2018. NCHS Data Brief 2020, 1–8. [Google Scholar]
- Block, J.P.; He, Y.L.; Zaslavsky, A.M.; Ding, L.; Ayanian, J.Z. Psychosocial Stress and Change in Weight among US Adults. Am. J. Epidemiol. 2009, 170, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, J.E.; Blair, S.N.; Jakicic, J.M.; Manore, M.M.; Rankin, J.W.; Smith, B.K. American College of Sports Medicine Position Stand. Appropriate physical activity intervention strategies for weight loss and prevention of weight regain for adults. Med. Sci. Sports Exerc. 2009, 41, 459–471. [Google Scholar] [CrossRef]
- Goodarzi, M.O. Genetics of obesity: What genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endo. 2018, 6, 223–236. [Google Scholar] [CrossRef]
- Rolls, B.J. The Supersizing of America: Portion Size and the Obesity Epidemic. Nutr. Today 2003, 38, 42–53. [Google Scholar] [CrossRef]
- Taheri, S.; Lin, L.; Austin, D.; Young, T.; Mignot, E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004, 1, 210–217. [Google Scholar] [CrossRef]
- Weiden, P.J.; Mackell, J.A.; McDonnell, D.D. Obesity as a risk factor for antipsychotic noncompliance. Schizophr. Res. 2004, 66, 51–57. [Google Scholar] [CrossRef]
- Duc, H.N.; Oh, H.; Kim, M.S. Effects of Antioxidant Vitamins, Curry Consumption, and Heavy Metal Levels on Metabolic Syndrome with Comorbidities: A Korean Community-Based Cross-Sectional Study. Antioxidants 2021, 10, 808. [Google Scholar] [CrossRef]
- Duc, H.N.; Oh, H.; Kim, M.S. The Effect of Mixture of Heavy Metals on Obesity in Individuals >/=50 Years of Age. Biol. Trace Elem. Res. 2022, 200, 3554–3571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Dong, T.Y.; Hu, W.Y.; Wang, X.; Xu, B.; Lin, Z.N.; Hofer, T.; Stefanoff, P.; Chen, Y.; Wang, X.R.; et al. Association between exposure to a mixture of phenols, pesticides, and phthalates and obesity: Comparison of three statistical models. Environ. Int. 2019, 123, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Stahr, S.; Chiang, T.C.; Bauer, M.A.; Runnells, G.A.; Rogers, L.J.; Vi Do, H.; Kadlubar, S.A.; Joseph Su, L. Low-Level Environmental Heavy Metals are Associated with Obesity among Postmenopausal Women in a Southern State. Expo. Health 2021, 13, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Yang, H.W.; Ayyamperumal, R.; Liu, Y. Pollution, sources, and human health risk assessment of heavy metals in urban areas around industrialization and urbanization-Northwest China. Chemosphere 2022, 308, 136396. [Google Scholar] [CrossRef]
- Pena-Fernandez, A.; Gonzalez-Munoz, M.J.; Lobo-Bedmar, M.C. Establishing the importance of human health risk assessment for metals and metalloids in urban environments. Environ. Int. 2014, 72, 176–185. [Google Scholar] [CrossRef]
- Rehman, K.; Fatima, F.; Waheed, I.; Akash, M.S.H. Prevalence of exposure of heavy metals and their impact on health consequences. J. Cell. Biochem. 2018, 119, 157–184. [Google Scholar] [CrossRef]
- Antunes Dos Santos, A.; Appel Hort, M.; Culbreth, M.; Lopez-Granero, C.; Farina, M.; Rocha, J.B.; Aschner, M. Methylmercury and brain development: A review of recent literature. J. Trace Elem. Med. Biol. 2016, 38, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Sanborn, M.D.; Abelsohn, A.; Campbell, M.; Weir, E. Identifying and managing adverse environmental health effects: 3. Lead exposure. CMAJ Can. Med. Assoc. J. 2002, 166, 1287–1292. [Google Scholar]
- Moon, S.S. Association between Blood Mercury Level and Visceral Adiposity in Adults. Diabetes Metab. J. 2017, 41, 96–98. [Google Scholar] [CrossRef]
- You, C.H.; Kim, B.G.; Kim, J.M.; Yu, S.D.; Kim, Y.M.; Kim, R.B.; Hong, Y.S. Relationship between blood mercury concentration and waist-to-hip ratio in elderly Korean individuals living in coastal areas. J. Prev. Med. Public Health 2011, 44, 218–225. [Google Scholar] [CrossRef]
- Wang, X.; Mukherjee, B.; Park, S.K. Associations of cumulative exposure to heavy metal mixtures with obesity and its comorbidities among U.S. adults in NHANES 2003–2014. Environ. Int. 2018, 121, 683–694. [Google Scholar] [CrossRef]
- Dubuc, P.U.; Reynolds, R.W. Hypothalamic metallic deposition and the production of experimental obesity. Physiol. Behav. 1973, 10, 677–681. [Google Scholar] [CrossRef]
- Hou, Y.Y.; Xue, P.; Woods, C.G.; Wang, X.; Fu, J.Q.; Yarborough, K.; Qu, W.D.; Zhang, Q.; Andersen, M.E.; Pi, J.B. Association between Arsenic Suppression of Adipogenesis and Induction of CHOP10 via the Endoplasmic Reticulum Stress Response. Environ. Health Persp. 2013, 121, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Leasure, J.L.; Giddabasappa, A.; Chaney, S.; Johnson, J.E.; Pothakos, K.; Lau, Y.S.; Fox, D.A. Low-level human equivalent gestational lead exposure produces sex-specific motor and coordination abnormalities and late-onset obesity in year-old mice. Environ. Health Persp. 2008, 116, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Ghaedrahmat, Z.; Cheraghian, B.; Jaafarzadeh, N.; Takdastan, A.; Shahbazian, H.B.; Ahmadi, M. Relationship between urinary heavy metals with metabolic syndrome and its components in population from Hoveyzeh cohort study: A case-control study in Iran. J. Trace Elem. Med. Biol. 2021, 66, 126757. [Google Scholar] [CrossRef]
- Ayoub, N.; Mantash, H.; Dhaini, H.R.; Mourad, A.; Hneino, M.; Daher, Z. Serum Cadmium Levels and Risk of Metabolic Syndrome: A Cross-Sectional Study. Biol. Trace Elem. Res. 2021, 199, 3625–3633. [Google Scholar] [CrossRef]
- Lu, L.; Li, Y.; Chen, C.; Zhang, Y.; Guo, W.; Zhang, S.; Kahe, K. Associations of cadmium exposure with risk of metabolic syndrome and its individual components: A meta-analysis. J. Expo. Sci. Environ. Epidemiol. 2022, 1–9. [Google Scholar] [CrossRef]
- Noor, N.; Zong, G.; Seely, E.W.; Weisskopf, M.; James-Todd, T. Urinary cadmium concentrations and metabolic syndrome in U.S. adults: The National Health and Nutrition Examination Survey 2001–2014. Environ. Int. 2018, 121, 349–356. [Google Scholar] [CrossRef]
- Han, S.J.; Ha, K.H.; Jeon, J.Y.; Kim, H.J.; Lee, K.W.; Kim, D.J. Impact of Cadmium Exposure on the Association between Lipopolysaccharide and Metabolic Syndrome. Int. J. Environ. Res. Public Health 2015, 12, 11396–11409. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Meng, X.; Deng, L.; Liu, N. Non-linear associations between metabolic syndrome and four typical heavy metals: Data from NHANES 2011–2018. Chemosphere 2022, 291, 132953. [Google Scholar] [CrossRef]
- da Cunha Martins, A., Jr.; Carneiro, M.F.H.; Grotto, D.; Adeyemi, J.A.; Barbosa, F., Jr. Arsenic, cadmium, and mercury-induced hypertension: Mechanisms and epidemiological findings. J. Toxicol. Environ. Health B Crit. Rev. 2018, 21, 61–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wei, S. Cadmium affects blood pressure and negatively interacts with obesity: Findings from NHANES 1999–2014. Sci. Total. Environ. 2018, 643, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Gabet, A.; Grave, C.; Blacher, J.; Olie, V. Associations between urinary cadmium levels, blood pressure, and hypertension: The ESTEBAN survey. Environ. Sci. Pollut. Res. Int. 2020, 27, 10748–10756. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhu, X.; Shrubsole, M.J.; Fan, L.; Xia, Z.; Harris, R.C.; Hou, L.; Dai, Q. The modifying effect of kidney function on the association of cadmium exposure with blood pressure and cardiovascular mortality: NHANES 1999–2010. Toxicol. Appl. Pharmacol. 2018, 353, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Oliver-Williams, C.; Howard, A.G.; Navas-Acien, A.; Howard, B.V.; Tellez-Plaza, M.; Franceschini, N. Cadmium body burden, hypertension, and changes in blood pressure over time: Results from a prospective cohort study in American Indians. J. Am. Soc. Hypertens 2018, 12, 426–437.e429. [Google Scholar] [CrossRef]
- Aramjoo, H.; Arab-Zozani, M.; Feyzi, A.; Naghizadeh, A.; Aschner, M.; Naimabadi, A.; Farkhondeh, T.; Samarghandian, S. The association between environmental cadmium exposure, blood pressure, and hypertension: A systematic review and meta-analysis. Environ. Sci. Pollut. Res. Int. 2022, 29, 35682–35706. [Google Scholar] [CrossRef]
- Caciari, T.; Sancini, A.; Fioravanti, M.; Capozzella, A.; Casale, T.; Montuori, L.; Fiaschetti, M.; Schifano, M.P.; Andreozzi, G.; Nardone, N.; et al. Cadmium and hypertension in exposed workers: A meta-analysis. Int. J. Occup. Med. Environ. Health 2013, 26, 440–456. [Google Scholar] [CrossRef]
- Oliveira, T.F.; Batista, P.R.; Leal, M.A.; Campagnaro, B.P.; Nogueira, B.V.; Vassallo, D.V.; Meyrelles, S.S.; Padilha, A.S. Chronic Cadmium Exposure Accelerates the Development of Atherosclerosis and Induces Vascular Dysfunction in the Aorta of ApoE(−/−) Mice. Biol. Trace Elem. Res. 2019, 187, 163–171. [Google Scholar] [CrossRef]
- Pinheiro Junior, J.E.G.; Moraes, P.Z.; Rodriguez, M.D.; Simoes, M.R.; Cibin, F.; Pinton, S.; Barbosa Junior, F.; Pecanha, F.M.; Vassallo, D.V.; Miguel, M.; et al. Cadmium exposure activates NADPH oxidase, renin-angiotensin system and cyclooxygenase 2 pathways in arteries, inducing hypertension and vascular damage. Toxicol. Lett. 2020, 333, 80–89. [Google Scholar] [CrossRef]
- Al-Naemi, H.A.; Das, S.C. Cadmium-induced endothelial dysfunction mediated by asymmetric dimethylarginine. Environ. Sci. Pollut. Res. Int. 2020, 27, 16246–16253. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Li, X.; Nong, Q.; Mao, B.; Pan, X. Metabolic Profiling in Association with Vascular Endothelial Cell Dysfunction Following Non-Toxic Cadmium Exposure. Int. J. Mol. Sci. 2017, 18, 1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukongviriyapan, U.; Pannangpetch, P.; Kukongviriyapan, V.; Donpunha, W.; Sompamit, K.; Surawattanawan, P. Curcumin protects against cadmium-induced vascular dysfunction, hypertension and tissue cadmium accumulation in mice. Nutrients 2014, 6, 1194–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeli, J.K.; Cruz Pereira, C.A.; de Oliveira Faria, T.; Stefanon, I.; Padilha, A.S.; Vassallo, D.V. Cadmium exposure induces vascular injury due to endothelial oxidative stress: The role of local angiotensin II and COX-2. Free Radic. Biol. Med. 2013, 65, 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.M.; Phuagkhaopong, S.; Fang, C.; Wu, J.C.C.; Huang, Y.H.; Vivithanaporn, P.; Lin, H.H.; Tsai, C.Y. Dose-Dependent Acute Circulatory Fates Elicited by Cadmium Are Mediated by Differential Engagements of Cardiovascular Regulatory Mechanisms in Brain. Front. Physiol. 2019, 10, 772. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.C.; Santos, A.A.D.; Lopes, A.; Skalny, A.V.; Aschner, M.; Tinkov, A.A.; Paoliello, M.M.B. Endothelial Dysfunction Induced by Cadmium and Mercury and its Relationship to Hypertension. Curr. Hypertens Rev. 2021, 17, 14–26. [Google Scholar] [CrossRef]
- Liang, H.; Yue, R.; Zhou, C.; Liu, M.; Yu, X.; Lu, S.; Zeng, J.; Yu, Z.; Zhou, Z.; Hu, H. Cadmium exposure induces endothelial dysfunction via disturbing lipid metabolism in human microvascular endothelial cells. J. Appl. Toxicol. 2021, 41, 775–788. [Google Scholar] [CrossRef]
- Almenara, C.C.P.; Oliveira, T.F.; Padilha, A.S. The Role of Antioxidants in the Prevention of Cadmium-Induced Endothelial Dysfunction. Curr. Pharm. Des. 2020, 26, 3667–3675. [Google Scholar] [CrossRef]
- Tang, L.; Su, J.; Liang, P. Modeling cadmium-induced endothelial toxicity using human pluripotent stem cell-derived endothelial cells. Sci. Rep. 2017, 7, 14811. [Google Scholar] [CrossRef] [Green Version]
- Park, S.S.; Skaar, D.A.; Jirtle, R.L.; Hoyo, C. Epigenetics, obesity and early-life cadmium or lead exposure. Epigenomics 2017, 9, 57–75. [Google Scholar] [CrossRef] [Green Version]
- Tinkov, A.A.; Filippini, T.; Ajsuvakova, O.P.; Aaseth, J.; Gluhcheva, Y.G.; Ivanova, J.M.; Bjorklund, G.; Skalnaya, M.G.; Gatiatulina, E.R.; Popova, E.V.; et al. The role of cadmium in obesity and diabetes. Sci. Total. Environ. 2017, 601–602, 741–755. [Google Scholar] [CrossRef]
- Nie, X.; Wang, N.; Chen, Y.; Chen, C.; Han, B.; Zhu, C.; Chen, Y.; Xia, F.; Cang, Z.; Lu, M.; et al. Blood cadmium in Chinese adults and its relationships with diabetes and obesity. Environ. Sci. Pollut. Res. Int. 2016, 23, 18714–18723. [Google Scholar] [CrossRef]
- Salcedo-Bellido, I.; Gomez-Pena, C.; Perez-Carrascosa, F.M.; Vrhovnik, P.; Mustieles, V.; Echeverria, R.; Fiket, Z.; Perez-Diaz, C.; Barrios-Rodriguez, R.; Jimenez-Moleon, J.J.; et al. Adipose tissue cadmium concentrations as a potential risk factor for insulin resistance and future type 2 diabetes mellitus in GraMo adult cohort. Sci. Total. Environ. 2021, 780, 146359. [Google Scholar] [CrossRef]
- Echeverria, R.; Vrhovnik, P.; Salcedo-Bellido, I.; Iribarne-Duran, L.M.; Fiket, Z.; Dolenec, M.; Martin-Olmedo, P.; Olea, N.; Arrebola, J.P. Levels and determinants of adipose tissue cadmium concentrations in an adult cohort from Southern Spain. Sci. Total. Environ. 2019, 670, 1028–1036. [Google Scholar] [CrossRef]
- Green, A.J.; Hoyo, C.; Mattingly, C.J.; Luo, Y.; Tzeng, J.Y.; Murphy, S.K.; Buchwalter, D.B.; Planchart, A. Cadmium exposure increases the risk of juvenile obesity: A human and zebrafish comparative study. Int. J. Obes. 2018, 42, 1285–1295. [Google Scholar] [CrossRef]
- Moynihan, M.; Tellez-Rojo, M.M.; Colacino, J.; Jones, A.; Song, P.X.K.; Cantoral, A.; Mercado-Garcia, A.; Peterson, K.E. Prenatal Cadmium Exposure Is Negatively Associated with Adiposity in Girls Not Boys During Adolescence. Front. Public Health 2019, 7, 61. [Google Scholar] [CrossRef]
- Shao, W.; Liu, Q.; He, X.; Liu, H.; Gu, A.; Jiang, Z. Association between level of urinary trace heavy metals and obesity among children aged 6–19 years: NHANES 1999-2011. Environ. Sci. Pollut. Res. Int. 2017, 24, 11573–11581. [Google Scholar] [CrossRef]
- Attia, S.M.; Varadharajan, K.; Shanmugakonar, M.; Das, S.C.; Al-Naemi, H.A. Cadmium: An Emerging Role in Adipose Tissue Dysfunction. Expos. Health 2022, 14, 171–183. [Google Scholar] [CrossRef]
- Gasser, M.; Lenglet, S.; Bararpour, N.; Sajic, T.; Wiskott, K.; Augsburger, M.; Fracasso, T.; Gilardi, F.; Thomas, A. Cadmium acute exposure induces metabolic and transcriptomic perturbations in human mature adipocytes. Toxicology 2022, 470, 153153. [Google Scholar] [CrossRef] [PubMed]
- Attia, S.M.; Das, S.C.; Varadharajan, K.; Al-Naemi, H.A. White adipose tissue as a target for cadmium toxicity. Front. Pharmacol. 2022, 13, 1010817. [Google Scholar] [CrossRef]
- Lee, E.J.; Moon, J.Y.; Yoo, B.S. Cadmium inhibits the differentiation of 3T3-L1 preadipocyte through the C/EBPalpha and PPARgamma pathways. Drug Chem. Toxicol. 2012, 35, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Sugimoto, H.; Furuichi, R.; Kadota, Y.; Inoue, M.; Setsu, K.; Suzuki, S.; Sato, M. Cadmium reduces adipocyte size and expression levels of adiponectin and Peg1/Mest in adipose tissue. Toxicology 2010, 267, 20–26. [Google Scholar] [CrossRef]
- da Costa, C.S.; Oliveira, T.F.; Freitas-Lima, L.C.; Padilha, A.S.; Krause, M.; Carneiro, M.; Salgado, B.S.; Graceli, J.B. Subacute cadmium exposure disrupts the hypothalamic-pituitary-gonadal axis, leading to polycystic ovarian syndrome and premature ovarian failure features in female rats. Environ. Pollut. 2021, 269, 116154. [Google Scholar] [CrossRef]
- Nguyen, J.; Patel, A.; Gensburg, A.; Bokhari, R.; Lamar, P.; Edwards, J. Diabetogenic and Obesogenic Effects of Cadmium in Db/Db Mice and Rats at a Clinically Relevant Level of Exposure. Toxics 2022, 10, 107. [Google Scholar] [CrossRef]
- Luo, H.; Gu, R.; Ouyang, H.; Wang, L.; Shi, S.; Ji, Y.; Bao, B.; Liao, G.; Xu, B. Cadmium exposure induces osteoporosis through cellular senescence, associated with activation of NF-kappaB pathway and mitochondrial dysfunction. Environ. Pollut. 2021, 290, 118043. [Google Scholar] [CrossRef]
- Knani, L.; Bartolini, D.; Kechiche, S.; Tortoioli, C.; Murdolo, G.; Moretti, M.; Messaoudi, I.; Reiter, R.J.; Galli, F. Melatonin prevents cadmium-induced bone damage: First evidence on an improved osteogenic/adipogenic differentiation balance of mesenchymal stem cells as underlying mechanism. J. Pineal Res. 2019, 67, e12597. [Google Scholar] [CrossRef]
- Ba, Q.; Li, M.; Chen, P.; Huang, C.; Duan, X.; Lu, L.; Li, J.; Chu, R.; Xie, D.; Song, H.; et al. Sex-Dependent Effects of Cadmium Exposure in Early Life on Gut Microbiota and Fat Accumulation in Mice. Environ. Health Perspect. 2017, 125, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.R.; Prozialeck, W.C. Cadmium, diabetes and chronic kidney disease. Toxicol. Appl. Pharmacol. 2009, 238, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Li, W.; Zhu, C.; Yang, S.; Zhou, M.; Wang, B.; Wang, X.; Wang, D.; Ma, J.; Zhou, Y.; et al. Cadmium exposure, fasting blood glucose changes, and type 2 diabetes mellitus: A longitudinal prospective study in China. Environ. Res. 2021, 192, 110259. [Google Scholar] [CrossRef]
- Xiao, L.; Zhou, Y.; Ma, J.; Cao, L.; Zhu, C.; Li, W.; Wang, D.; Fan, L.; Ye, Z.; Chen, W. Roles of C-reactive protein on the association between urinary cadmium and type 2 diabetes. Environ. Pollut. 2019, 255, 113341. [Google Scholar] [CrossRef]
- Jiang, F.; Zhi, X.; Xu, M.; Li, B.; Zhang, Z. Gender-specific differences of interaction between cadmium exposure and obesity on prediabetes in the NHANES 2007–2012 population. Endocrine 2018, 61, 258–266. [Google Scholar] [CrossRef]
- Guo, F.F.; Hu, Z.Y.; Li, B.Y.; Qin, L.Q.; Fu, C.; Yu, H.; Zhang, Z.L. Evaluation of the association between urinary cadmium levels below threshold limits and the risk of diabetes mellitus: A dose-response meta-analysis. Environ. Sci. Pollut. Res. Int. 2019, 26, 19272–19281. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Wang, W.; Wu, Y. Association of urinary cadmium with risk of diabetes: A meta-analysis. Environ. Sci. Pollut. Res. Int. 2017, 24, 10083–10090. [Google Scholar] [CrossRef] [PubMed]
- Filippini, T.; Wise, L.A.; Vinceti, M. Cadmium exposure and risk of diabetes and prediabetes: A systematic review and dose-response meta-analysis. Environ. Int. 2022, 158, 106920. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Song, J.; Zhu, C.; Wang, Y.; Yin, X.; Huang, G.; Zhao, K.; Zhu, J.; Duan, Z.; Su, L. Association between cadmium exposure and diabetes mellitus risk: A prisma-compliant systematic review and meta-analysis. Oncotarget 2017, 8, 113129–113141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buha, A.; Dukic-Cosic, D.; Curcic, M.; Bulat, Z.; Antonijevic, B.; Moulis, J.M.; Goumenou, M.; Wallace, D. Emerging Links between Cadmium Exposure and Insulin Resistance: Human, Animal, and Cell Study Data. Toxics 2020, 8, 63. [Google Scholar] [CrossRef]
- Fitzgerald, R.; Olsen, A.; Nguyen, J.; Wong, W.; El Muayed, M.; Edwards, J. Pancreatic Islets Accumulate Cadmium in a Rodent Model of Cadmium-Induced Hyperglycemia. Int. J. Mol. Sci. 2020, 22, 360. [Google Scholar] [CrossRef]
- Hong, H.; He, H.; Lin, X.; Hayuehashi, T.; Xu, J.; Zhang, J.; Xu, Y.; Tong, T.; Lu, Y.; Zhou, Z. Cadmium exposure suppresses insulin secretion through mtROS-mediated mitochondrial dysfunction and inflammatory response in pancreatic beta cells. J. Trace Elem. Med. Biol. 2022, 71, 126952. [Google Scholar] [CrossRef]
- Aja, P.M.; Izekwe, F.I.; Famurewa, A.C.; Ekpono, E.U.; Nwite, F.E.; Igwenyi, I.O.; Awoke, J.N.; Ani, O.G.; Aloke, C.; Obasi, N.A.; et al. Hesperidin protects against cadmium-induced pancreatitis by modulating insulin secretion, redox imbalance and iNOS/NF-kB signaling in rats. Life Sci. 2020, 259, 118268. [Google Scholar] [CrossRef]
- Hong, H.; Lin, X.; Xu, Y.; Tong, T.; Zhang, J.; He, H.; Yang, L.; Lu, Y.; Zhou, Z. Cadmium induces ferroptosis mediated inflammation by activating Gpx4/Ager/p65 axis in pancreatic beta-cells. Sci. Total. Environ. 2022, 849, 157819. [Google Scholar] [CrossRef]
- Xu, Y.; Wei, X.; Li, X.; Chen, Y.; Mao, X.; Chen, G.; Liu, C. Cadmium inhibits signal transducer and activator of transcription 6 leading to pancreatic beta cell apoptosis. Endocr. J. 2022, 69, 361–371. [Google Scholar] [CrossRef]
- Wu, H.; Zheng, S.; Zhang, J.; Xu, S.; Miao, Z. Cadmium induces endoplasmic reticulum stress-mediated apoptosis in pig pancreas via the increase of Th1 cells. Toxicology 2021, 457, 152790. [Google Scholar] [CrossRef]
- Huang, C.C.; Kuo, C.Y.; Yang, C.Y.; Liu, J.M.; Hsu, R.J.; Lee, K.I.; Su, C.C.; Wu, C.C.; Lin, C.T.; Liu, S.H.; et al. Cadmium exposure induces pancreatic beta-cell death via a Ca(2+)-triggered JNK/CHOP-related apoptotic signaling pathway. Toxicology 2019, 425, 152252. [Google Scholar] [CrossRef]
- Wong, W.P.S.; Wang, J.C.; Schipma, M.J.; Zhang, X.; Edwards, J.R.; El Muayed, M. Cadmium-mediated pancreatic islet transcriptome changes in mice and cultured mouse islets. Toxicol. Appl. Pharmacol. 2021, 433, 115756. [Google Scholar] [CrossRef]
- Hong, H.; Xu, J.; He, H.; Wang, X.; Yang, L.; Deng, P.; Yang, L.; Tan, M.; Zhang, J.; Xu, Y.; et al. Cadmium perturbed metabolomic signature in pancreatic beta cells correlates with disturbed metabolite profile in human urine. Environ. Int. 2022, 161, 107139. [Google Scholar] [CrossRef]
- Sarmiento-Ortega, V.E.; Moroni-Gonzalez, D.; Diaz, A.; Brambila, E.; Trevino, S. ROS and ERK Pathway Mechanistic Approach on Hepatic Insulin Resistance After Chronic Oral Exposure to Cadmium NOAEL Dose. Biol. Trace Elem. Res. 2022, 201, 3903–3918. [Google Scholar] [CrossRef]
- Jacquet, A.; Barbeau, D.; Arnaud, J.; Hijazi, S.; Hazane-Puch, F.; Lamarche, F.; Quiclet, C.; Couturier, K.; Fontaine, E.; Moulis, J.M.; et al. Impact of maternal low-level cadmium exposure on glucose and lipid metabolism of the litter at different ages after weaning. Chemosphere 2019, 219, 109–121. [Google Scholar] [CrossRef]
- Yi, S.J.; Xiong, Y.W.; Zhu, H.L.; Dai, L.M.; Cao, X.L.; Liu, W.B.; Shi, X.T.; Zhou, G.X.; Liu, A.Y.; Zhao, L.L.; et al. Environmental cadmium exposure during pregnancy causes diabetes-like phenotypes in mouse offspring: Association with oxidative stress in the fetal liver. Sci. Total. Environ. 2021, 777, 146006. [Google Scholar] [CrossRef]
- Tinkov, A.A.; Filippini, T.; Ajsuvakova, O.P.; Skalnaya, M.G.; Aaseth, J.; Bjorklund, G.; Gatiatulina, E.R.; Popova, E.V.; Nemereshina, O.N.; Huang, P.T.; et al. Cadmium and atherosclerosis: A review of toxicological mechanisms and a meta-analysis of epidemiologic studies. Environ. Res. 2018, 162, 240–260. [Google Scholar] [CrossRef]
- Kim, D.W.; Ock, J.; Moon, K.W.; Park, C.H. Association between Heavy Metal Exposure and Dyslipidemia among Korean Adults: From the Korean National Environmental Health Survey, 2015–2017. Int. J. Environ. Res. Public Health 2022, 19, 3181. [Google Scholar] [CrossRef]
- Xu, H.; Mao, Y.; Xu, B.; Hu, Y. Low-level environmental lead and cadmium exposures and dyslipidemia in adults: Findings from the NHANES 2005–2016. J. Trace Elem. Med. Biol. 2021, 63, 126651. [Google Scholar] [CrossRef]
- Fagerberg, B.; Barregard, L. Review of cadmium exposure and smoking-independent effects on atherosclerotic cardiovascular disease in the general population. J. Intern. Med. 2021, 290, 1153–1179. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Zhou, D.; Sun, Y.; Zhao, J.; Xu, S.; Zhao, X. Independent association of blood cadmium with subclinical lower extremity atherosclerosis: An observational study based on dose-response analysis. Chemosphere 2023, 313, 137441. [Google Scholar] [CrossRef] [PubMed]
- Barregard, L.; Sallsten, G.; Harari, F.; Andersson, E.M.; Forsgard, N.; Hjelmgren, O.; Angeras, O.; Fagman, E.; Persson, M.; Lundh, T.; et al. Cadmium Exposure and Coronary Artery Atherosclerosis: A Cross-Sectional Population-Based Study of Swedish Middle-Aged Adults. Environ. Health Perspect. 2021, 129, 67007. [Google Scholar] [CrossRef] [PubMed]
- Kumar, W.M.; Ribeiro, M.; Balla, A. Cadmium-induced Macrophage LDL and Oxidized LDL Internalization. FASEB J. 2019, 33, 802–874. [Google Scholar] [CrossRef]
- Kijani, S.; Bergstrom, G.; Lindbom, M.; Levin, M.; Barregard, L.; Fagerberg, B.; Fogelstrand, P.; Boren, J. Non-Toxic Concentrations of Cadmium Accelerate Subendothelial Retention of Atherogenic Lipoproteins in Humanized Atherosclerosis-Susceptible Mice. Atherosclerosis 2017, 263, E1–E2. [Google Scholar] [CrossRef]
- Afolabi, O.K.; Oyewo, E.B.; Adekunle, A.S.; Adedosu, O.T.; Adedeji, A.L. Impaired lipid levels and inflammatory response in rats exposed to cadmium. EXCLI J. 2012, 11, 677–687. [Google Scholar]
- Wan, Y.; Mo, L.; Huang, H.; Mo, L.; Zhu, W.; Li, W.; Yang, G.; Chen, L.; Wu, Y.; Song, J.; et al. Cadmium contributes to atherosclerosis by affecting macrophage polarization. Food Chem. Toxicol. 2023, 173, 113603. [Google Scholar] [CrossRef]
- Zhang, J.; Ou, C.; Chen, M. Curcumin attenuates cadmium-induced atherosclerosis by regulating trimethylamine-N-oxide synthesis and macrophage polarization through remodeling the gut microbiota. Ecotoxicol. Environ. Saf. 2022, 244, 114057. [Google Scholar] [CrossRef]
- Perez Diaz, M.F.F.; Plateo Pignatari, M.G.; Filippa, V.P.; Mohamed, F.H.; Marchevsky, E.J.; Gimenez, M.S.; Ramirez, D.C. A soybean-based diet modulates cadmium-induced vascular apoptosis. J. Trace Elem. Med. Biol. 2019, 52, 239–246. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Sun, R.; Gao, M.; Wang, H. Cadmium exposure enhances VE-cadherin expression in endothelial cells via suppression of ROCK signaling. Exp. Ther. Med. 2022, 23, 355. [Google Scholar] [CrossRef]
- Wang, X.; Dong, F.; Wang, F.; Yan, S.; Chen, X.; Tozawa, H.; Ushijima, T.; Kapron, C.M.; Wada, Y.; Liu, J. Low dose cadmium upregulates the expression of von Willebrand factor in endothelial cells. Toxicol. Lett. 2018, 290, 46–54. [Google Scholar] [CrossRef]
- Yang, J.; Chen, W.; Sun, Y.; Liu, J.; Zhang, W. Effects of cadmium on organ function, gut microbiota and its metabolomics profile in adolescent rats. Ecotoxicol. Environ. Saf. 2021, 222, 112501. [Google Scholar] [CrossRef]
- Panico, P.; Velasco, M.; Salazar, A.M.; Picones, A.; Ortiz-Huidobro, R.I.; Guerrero-Palomo, G.; Salgado-Bernabe, M.E.; Ostrosky-Wegman, P.; Hiriart, M. Is Arsenic Exposure a Risk Factor for Metabolic Syndrome? A Review of the Potential Mechanisms. Front. Endocrinol. 2022, 13, 878280. [Google Scholar] [CrossRef]
- Wang, S.L.; Chang, F.H.; Liou, S.H.; Wang, H.J.; Li, W.F.; Hsieh, D.P. Inorganic arsenic exposure and its relation to metabolic syndrome in an industrial area of Taiwan. Environ. Int. 2007, 33, 805–811. [Google Scholar] [CrossRef]
- Chen, J.W.; Wang, S.L.; Wang, Y.H.; Sun, C.W.; Huang, Y.L.; Chen, C.J.; Li, W.F. Arsenic methylation, GSTO1 polymorphisms, and metabolic syndrome in an arseniasis endemic area of southwestern Taiwan. Chemosphere 2012, 88, 432–438. [Google Scholar] [CrossRef]
- Sarker, M.K.; Tony, S.R.; Siddique, A.; Haque, N.; Islam, M.S.; Hossain, F.; Islam, Z.; Hossain, S.; Hoque, M.A.; Saud, Z.A.; et al. Gender Differences in the Risk of Metabolic Syndrome among Chronic Arsenic-Exposed Individuals in Bangladesh. Expos. Health 2022, 14, 595–608. [Google Scholar] [CrossRef]
- Kazemifar, A.M.; Shafikhani, A.A.; Mozhdehipanah, H.; Khamesi, S.; Arami, M. Evaluation of different types of arsenic methylation and its relationship with metabolic syndrome in an area chronically exposed to arsenic. Environ. Anal. Health Toxicol. 2020, 35, e2020006. [Google Scholar] [CrossRef]
- Wang, X.; Karvonen-Gutierrez, C.A.; Herman, W.H.; Mukherjee, B.; Park, S.K. Metals and risk of incident metabolic syndrome in a prospective cohort of midlife women in the United States. Environ. Res. 2022, 210, 112976. [Google Scholar] [CrossRef]
- Spratlen, M.J.; Grau-Perez, M.; Best, L.G.; Yracheta, J.; Lazo, M.; Vaidya, D.; Balakrishnan, P.; Gamble, M.V.; Francesconi, K.A.; Goessler, W.; et al. The Association of Arsenic Exposure and Arsenic Metabolism with the Metabolic Syndrome and Its Individual Components: Prospective Evidence from the Strong Heart Family Study. Am. J. Epidemiol. 2018, 187, 1598–1612. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Li, Y.; He, Q.; Luo, L.; Zhang, Y.; Gao, Y.; Feng, H.; Zhao, L.; Wei, W.; Fu, S.; et al. Essential hypertension in patients exposed to high-arsenic exposed areas in western China: Genetic susceptibility and urinary arsenic metabolism characteristics. J. Trace Elem. Med. Biol. 2021, 67, 126778. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Samarghandian, S.; Azimi-Nezhad, M. The role of arsenic in obesity and diabetes. J. Cell. Physiol. 2019, 234, 12516–12529. [Google Scholar] [CrossRef] [PubMed]
- Eick, S.M.; Steinmaus, C. Arsenic and Obesity: A Review of Causation and Interaction. Curr. Environ. Health Rep. 2020, 7, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Abuawad, A.; Spratlen, M.J.; Parvez, F.; Slavkovich, V.; Ilievski, V.; Lomax-Luu, A.M.; Saxena, R.; Shahriar, H.; Nasir Uddin, M.; Islam, T.; et al. Association between body mass index and arsenic methylation in three studies of Bangladeshi adults and adolescents. Environ. Int. 2021, 149, 106401. [Google Scholar] [CrossRef] [PubMed]
- Warwick, M.; Marcelo, C.; Marcelo, C.; Shaw, J.; Qayyum, R. The relationship between chronic arsenic exposure and body measures among US adults: National Health and Nutrition Examination Survey 2009–2016. J. Trace Elem. Med. Biol. 2021, 67, 126771. [Google Scholar] [CrossRef]
- Bulka, C.M.; Mabila, S.L.; Lash, J.P.; Turyk, M.E.; Argos, M. Arsenic and Obesity: A Comparison of Urine Dilution Adjustment Methods. Environ. Health Perspect. 2017, 125, 087020. [Google Scholar] [CrossRef] [Green Version]
- Su, C.T.; Lin, H.C.; Choy, C.S.; Huang, Y.K.; Huang, S.R.; Hsueh, Y.M. The relationship between obesity, insulin and arsenic methylation capability in Taiwan adolescents. Sci. Total. Environ. 2012, 414, 152–158. [Google Scholar] [CrossRef]
- Renu, K.; Madhyastha, H.; Madhyastha, R.; Maruyama, M.; Arunachlam, S.; Abilash, V.G. Role of arsenic exposure in adipose tissue dysfunction and its possible implication in diabetes pathophysiology. Toxicol. Lett. 2018, 284, 86–95. [Google Scholar] [CrossRef]
- Srisuporn, P.; Navasumrit, P.; Ngaotepprutaram, T.; Chaisatra, K.; Hunsonti, P.; Ruchirawat, M. Arsenic exposure alters the expression of genes related to metabolic diseases in differentiated adipocytes and in newborns and children. Int. J. Hyg. Environ. Health 2023, 250, 114124. [Google Scholar] [CrossRef]
- Yadav, S.; Anbalagan, M.; Shi, Y.; Wang, F.; Wang, H. Arsenic inhibits the adipogenic differentiation of mesenchymal stem cells by down-regulating peroxisome proliferator-activated receptor gamma and CCAAT enhancer-binding proteins. Toxicol. In Vitro 2013, 27, 211–219. [Google Scholar] [CrossRef]
- Wauson, E.M.; Langan, A.S.; Vorce, R.L. Sodium arsenite inhibits and reverses expression of adipogenic and fat cell-specific genes during in vitro adipogenesis. Toxicol. Sci. 2002, 65, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Beezhold, K.; Klei, L.R.; Barchowsky, A. Regulation of cyclin D1 by arsenic and microRNA inhibits adipogenesis. Toxicol. Lett. 2017, 265, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon-DuPont, D.; Romero-Cordoba, S.L.; Tello, J.K.; Espinosa, A.; Guerrero, B.; Contreras, A.V.; Moran-Ramos, S.; Diaz-Villasenor, A. Impaired white adipose tissue fatty acid metabolism in mice fed a high-fat diet worsened by arsenic exposure, primarily affecting retroperitoneal adipose tissue. Toxicol. Appl. Pharmacol. 2023, 468, 116428. [Google Scholar] [CrossRef] [PubMed]
- Douillet, C.; Huang, M.C.; Saunders, R.J.; Dover, E.N.; Zhang, C.B.; Styblo, M. Knockout of arsenic (+3 oxidation state) methyltransferase is associated with adverse metabolic phenotype in mice: The role of sex and arsenic exposure. Arch. Toxicol. 2017, 91, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Castriota, F.; Zushin, P.J.H.; Sanchez, S.S.; Phillips, R.V.; Hubbard, A.; Stahl, A.; Smith, M.T.; Wang, J.C.; La Merrill, M.A. Chronic arsenic exposure impairs adaptive thermogenesis in male C57BL/6J mice. Am. J. Physiol. Endoc. Metab. 2020, 318, E667–E677. [Google Scholar] [CrossRef]
- Zuo, Z.; Liu, Z.Y.; Gao, T.C.; Yin, Y.Y.; Wang, Z.D.; Hou, Y.Y.; Fu, J.Q.; Liu, S.N.; Wang, H.H.; Xu, Y.Y.; et al. Prolonged inorganic arsenic exposure via drinking water impairs brown adipose tissue function in mice. Sci. Total Environ. 2019, 668, 310–317. [Google Scholar] [CrossRef]
- Bae, J.; Jang, Y.; Kim, H.; Mahato, K.; Schaecher, C.; Kim, I.M.; Kim, E.; Ro, S.H. Arsenite exposure suppresses adipogenesis, mitochondrial biogenesis and thermogenesis via autophagy inhibition in brown adipose tissue. Sci. Rep. 2019, 9, 14464. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.P.; Yadav, S.K.; Patel, K.; Patel, S.; Patil, G.P.; Bijalwan, V.; Singh, G.; Palkhade, R.; Kondepudi, K.K.; Boparai, R.K.; et al. Short-term trivalent arsenic and hexavalent chromium exposures induce gut dysbiosis and transcriptional alteration in adipose tissue of mice. Mol. Biol. Rep. 2023, 50, 1033–1044. [Google Scholar] [CrossRef]
- Gong, Y.Y.; Xue, Y.F.; Li, X.; Zhang, Z.; Zhou, W.J.; Marcolongo, P.; Benedetti, A.; Mao, S.Y.; Han, L.; Ding, G.L.; et al. Inter- and Transgenerational Effects of Paternal Exposure to Inorganic Arsenic. Adv. Sci. 2021, 8, 2002715. [Google Scholar] [CrossRef]
- Ceja-Galicia, Z.A.; Daniel, A.; Salazar, A.M.; Panico, P.; Ostrosky-Wegman, P.; Diaz-Villasenor, A. Effects of arsenic on adipocyte metabolism: Is arsenic an obesogen? Mol. Cell. Endocrinol. 2017, 452, 25–32. [Google Scholar] [CrossRef]
- Tinkov, A.A.; Aschner, M.; Ke, T.; Ferrer, B.; Zhou, J.C.; Chang, J.S.; Santamaria, A.; Chao, J.C.; Aaseth, J.; Skalny, A.V. Adipotropic effects of heavy metals and their potential role in obesity. Fac. Rev. 2021, 10, 32. [Google Scholar] [CrossRef]
- Sung, T.C.; Huang, J.W.; Guo, H.R. Association between Arsenic Exposure and Diabetes: A Meta-Analysis. Biomed Res. Int. 2015, 2015, 368087. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Zhao, E.; Huang, R. Association of urinary arsenic with insulin resistance: Cross-sectional analysis of the National Health and Nutrition Examination Survey, 2015-2016. Ecotoxicol. Environ. Saf. 2022, 231, 113218. [Google Scholar] [CrossRef]
- Grau-Perez, M.; Kuo, C.C.; Gribble, M.O.; Balakrishnan, P.; Jones Spratlen, M.; Vaidya, D.; Francesconi, K.A.; Goessler, W.; Guallar, E.; Silbergeld, E.K.; et al. Association of Low-Moderate Arsenic Exposure and Arsenic Metabolism with Incident Diabetes and Insulin Resistance in the Strong Heart Family Study. Environ. Health. Perspect. 2017, 125, 127004. [Google Scholar] [CrossRef] [Green Version]
- Mondal, V.; Hosen, Z.; Hossen, F.; Siddique, A.E.; Tony, S.R.; Islam, Z.; Islam, M.S.; Hossain, S.; Islam, K.; Sarker, M.K.; et al. Arsenic exposure-related hyperglycemia is linked to insulin resistance with concomitant reduction of skeletal muscle mass. Environ. Int. 2020, 143, 105890. [Google Scholar] [CrossRef]
- Eick, S.M.; Ferreccio, C.; Acevedo, J.; Castriota, F.; Cordero, J.F.; Roh, T.; Smith, A.H.; Smith, M.T.; Steinmaus, C. Socioeconomic status and the association between arsenic exposure and type 2 diabetes. Environ. Res. 2019, 172, 578–585. [Google Scholar] [CrossRef]
- Castriota, F.; Acevedo, J.; Ferreccio, C.; Smith, A.H.; Liaw, J.; Smith, M.T.; Steinmaus, C. Obesity and increased susceptibility to arsenic-related type 2 diabetes in Northern Chile. Environ. Res. 2018, 167, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Grau-Perez, M.; Navas-Acien, A.; Galan-Chilet, I.; Briongos-Figuero, L.S.; Morchon-Simon, D.; Bermudez, J.D.; Crainiceanu, C.M.; de Marco, G.; Rentero-Garrido, P.; Garcia-Barrera, T.; et al. Arsenic exposure, diabetes-related genes and diabetes prevalence in a general population from Spain. Environ. Pollut. 2018, 235, 948–955. [Google Scholar] [CrossRef] [Green Version]
- Castriota, F.; Rieswijk, L.; Dahlberg, S.; La Merrill, M.A.; Steinmaus, C.; Smith, M.T.; Wang, J.C. A State-of-the-Science Review of Arsenic’s Effects on Glucose Homeostasis in Experimental Models. Environ. Health Perspect. 2020, 128, 16001. [Google Scholar] [CrossRef] [Green Version]
- Kirkley, A.G.; Carmean, C.M.; Ruiz, D.; Ye, H.; Regnier, S.M.; Poudel, A.; Hara, M.; Kamau, W.; Johnson, D.N.; Roberts, A.A.; et al. Arsenic exposure induces glucose intolerance and alters global energy metabolism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R294–R303. [Google Scholar] [CrossRef]
- Huang, M.; Douillet, C.; Styblo, M. Arsenite and its trivalent methylated metabolites inhibit glucose-stimulated calcium influx and insulin secretion in murine pancreatic islets. Arch. Toxicol. 2019, 93, 2525–2533. [Google Scholar] [CrossRef]
- Dover, E.N.; Beck, R.; Huang, M.C.; Douillet, C.; Wang, Z.; Klett, E.L.; Styblo, M. Arsenite and methylarsonite inhibit mitochondrial metabolism and glucose-stimulated insulin secretion in INS-1 832/13 beta cells. Arch. Toxicol. 2018, 92, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Douillet, C.; Huang, M.; Beck, R.; Sumner, S.J.; Styblo, M. Exposure to inorganic arsenic and its methylated metabolites alters metabolomics profiles in INS-1 832/13 insulinoma cells and isolated pancreatic islets. Arch. Toxicol. 2020, 94, 1955–1972. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Qiu, T.; Yuan, W.; Shi, Y.; Yao, X.; Jiang, L.; Zhang, J.; Yang, G.; Liu, X.; Bai, J.; et al. Annexin A1 inhibition facilitates NLRP3 inflammasome activation in arsenic-induced insulin resistance in rat liver. Environ. Toxicol. Pharmacol. 2022, 96, 103981. [Google Scholar] [CrossRef]
- Pei, P.; Yao, X.; Jiang, L.; Qiu, T.; Wang, N.; Yang, L.; Gao, N.; Wang, Z.; Yang, G.; Liu, X.; et al. Inorganic arsenic induces pyroptosis and pancreatic beta cells dysfunction through stimulating the IRE1alpha/TNF-alpha pathway and protective effect of taurine. Food Chem. Toxicol. 2019, 125, 392–402. [Google Scholar] [CrossRef]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef]
- Wu, W.; Yao, X.; Jiang, L.; Zhang, Q.; Bai, J.; Qiu, T.; Yang, L.; Gao, N.; Yang, G.; Liu, X.; et al. Pancreatic islet-autonomous effect of arsenic on insulin secretion through endoplasmic reticulum stress-autophagy pathway. Food Chem. Toxicol. 2018, 111, 19–26. [Google Scholar] [CrossRef]
- Khan, F.; Momtaz, S.; Niaz, K.; Hassan, F.I.; Abdollahi, M. Epigenetic mechanisms underlying the toxic effects associated with arsenic exposure and the development of diabetes. Food Chem. Toxicol. 2017, 107, 406–417. [Google Scholar] [CrossRef]
- Khan, F.; Hodjat, M.; Rahimifard, M.; Nigjeh, M.N.; Azizi, M.; Baeeri, M.; Bayrami, Z.; Gholami, M.; Hassani, S.; Abdollahi, M. Assessment of arsenic-induced modifications in the DNA methylation of insulin-related genes in rat pancreatic islets. Ecotoxicol. Environ. Saf. 2020, 201, 110802. [Google Scholar] [CrossRef]
- Ramdas, M.; Sharma, S.; Kaul, D.; Bhatia, A. Possible role of miR-2909 RNomics in arsenic mediated pancreatic beta-cell dysfunction. J. Trace Elem. Med. Biol. 2018, 50, 263–267. [Google Scholar] [CrossRef]
- Sun, Q.; Yang, Q.; Xu, H.; Xue, J.; Chen, C.; Yang, X.; Gao, X.; Liu, Q. miR-149 Negative Regulation of mafA Is Involved in the Arsenite-Induced Dysfunction of Insulin Synthesis and Secretion in Pancreatic Beta Cells. Toxicol. Sci. 2019, 167, 116–125. [Google Scholar] [CrossRef]
- Beck, R.; Chandi, M.; Kanke, M.; Styblo, M.; Sethupathy, P. Arsenic is more potent than cadmium or manganese in disrupting the INS-1 beta cell microRNA landscape. Arch. Toxicol. 2019, 93, 3099–3109. [Google Scholar] [CrossRef]
- Hou, H.; Yu, Y.; Shen, Z.; Liu, S.; Wu, B. Hepatic transcriptomic responses in mice exposed to arsenic and different fat diet. Environ. Sci. Pollut. Res. Int. 2017, 24, 10621–10629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fennel, E.M.J.; Douillet, C.; Styblo, M. Exposures to arsenite and methylarsonite produce insulin resistance and impair insulin-dependent glycogen metabolism in hepatocytes. Arch. Toxicol. 2017, 91, 3811–3821. [Google Scholar] [CrossRef]
- Jia, X.; Qiu, T.; Yao, X.; Jiang, L.; Wang, N.; Wei, S.; Tao, Y.; Pei, P.; Wang, Z.; Zhang, J.; et al. Arsenic induces hepatic insulin resistance via mtROS-NLRP3 inflammasome pathway. J. Hazard. Mater. 2020, 399, 123034. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Wu, C.; Yao, X.; Han, Q.; Wang, N.; Yuan, W.; Zhang, J.; Shi, Y.; Jiang, L.; Liu, X.; et al. AS3MT facilitates NLRP3 inflammasome activation by m(6)A modification during arsenic-induced hepatic insulin resistance. Cell Biol. Toxicol. 2022, 1–17. [Google Scholar] [CrossRef]
- Zhang, J.; Qiu, T.; Jiang, L.; Wang, N.; Zhu, Y.; Yan, R.; Wang, S.; Bai, J.; Shi, X.; Yang, G.; et al. NLRP3 inflammasome blocked the glycolytic pathway via targeting to PKLR in arsenic-induced hepatic insulin resistance. Ecotoxicol. Environ. Saf. 2021, 223, 112590. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, J.; Yao, X.; Qiu, T.; Jiang, L.; Wang, N.; Shi, Y.; Wu, C.; Yuan, W.; Yang, G.; et al. Ubiquitinated gasdermin D mediates arsenic-induced pyroptosis and hepatic insulin resistance in rat liver. Food Chem. Toxicol. 2022, 160, 112771. [Google Scholar] [CrossRef]
- Li, W.; Wu, L.; Sun, Q.; Yang, Q.; Xue, J.; Shi, M.; Tang, H.; Zhang, J.; Liu, Q. MicroRNA-191 blocking the translocation of GLUT4 is involved in arsenite-induced hepatic insulin resistance through inhibiting the IRS1/AKT pathway. Ecotoxicol. Environ. Saf. 2021, 215, 112130. [Google Scholar] [CrossRef]
- Yang, L.; Qiu, T.; Yao, X.; Jiang, L.; Wei, S.; Pei, P.; Wang, Z.; Bai, J.; Liu, X.; Yang, G.; et al. Taurine protects against arsenic trioxide-induced insulin resistance via ROS-Autophagy pathway in skeletal muscle. Int. J. Biochem. Cell. Biol. 2019, 112, 50–60. [Google Scholar] [CrossRef]
- Wisessaowapak, C.; Watcharasit, P.; Satayavivad, J. Arsenic disrupts neuronal insulin signaling through increasing free PI3K-p85 and decreasing PI3K activity. Toxicol. Lett. 2021, 349, 40–50. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.; Tian, X.; Xie, J.; Liu, P.; Ying, X.; Wang, M.; Yuan, J.; Gao, Y.; Tian, F.; et al. Effects of arsenic exposure on lipid metabolism: A systematic review and meta-analysis. Toxicol. Mech. Methods 2021, 31, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Huang, R. Linking the Low-Density Lipoprotein-Cholesterol (LDL) Level to Arsenic Acid, Dimethylarsinic, and Monomethylarsonic: Results from a National Population-Based Study from the NHANES, 2003–2020. Nutrients 2022, 14, 3993. [Google Scholar] [CrossRef]
- Xu, L.; Suman, S.; Sharma, P.; Kumar, R.; Singh, S.K.; Bose, N.; Ghosh, A.; Rahman, M.M.; Polya, D.A.; Mondal, D. Assessment of hypertension association with arsenic exposure from food and drinking water in Bihar, India. Ecotoxicol. Environ. Saf. 2021, 223, 112572. [Google Scholar] [CrossRef]
- Xu, L.; Mondal, D.; Polya, D.A. Positive Association of Cardiovascular Disease (CVD) with Chronic Exposure to Drinking Water Arsenic (As) at Concentrations below the WHO Provisional Guideline Value: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2020, 17, 2536. [Google Scholar] [CrossRef] [Green Version]
- Luo, T.; Chen, S.; Cai, J.; Liu, Q.; Gou, R.; Mo, X.; Tang, X.; He, K.; Xiao, S.; Wei, Y.; et al. Association between combined exposure to plasma heavy metals and dyslipidemia in a chinese population. Lipids Health Dis. 2022, 21, 131. [Google Scholar] [CrossRef]
- Grau-Perez, M.; Caballero-Mateos, M.J.; Domingo-Relloso, A.; Navas-Acien, A.; Gomez-Ariza, J.L.; Garcia-Barrera, T.; Leon-Latre, M.; Soriano-Gil, Z.; Jarauta, E.; Cenarro, A.; et al. Toxic Metals and Subclinical Atherosclerosis in Carotid, Femoral, and Coronary Vascular Territories: The Aragon Workers Health Study. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 87–99. [Google Scholar] [CrossRef]
- Waghe, P.; Sarkar, S.N.; Sarath, T.S.; Kandasamy, K.; Choudhury, S.; Gupta, P.; Harikumar, S.; Mishra, S.K. Subchronic Arsenic Exposure Through Drinking Water Alters Lipid Profile and Electrolyte Status in Rats. Biol. Trace Elem. Res. 2017, 176, 350–354. [Google Scholar] [CrossRef]
- Caglayan, C.; Demir, Y.; Kucukler, S.; Taslimi, P.; Kandemir, F.M.; Gulcin, I. The effects of hesperidin on sodium arsenite-induced different organ toxicity in rats on metabolic enzymes as antidiabetic and anticholinergics potentials: A biochemical approach. J. Food Biochem. 2019, 43, e12720. [Google Scholar] [CrossRef]
- Song, Y.; Zhou, T.; Zong, Y.; Gu, B.; Tan, X.; Yang, L. Arsenic inhibited cholesterol efflux of THP-1 macrophages via ROS-mediated ABCA1 hypermethylation. Toxicology 2019, 424, 152225. [Google Scholar] [CrossRef]
- Ma, Y.; Ma, Z.; Yin, S.; Yan, X.; Wang, J. Arsenic and fluoride induce apoptosis, inflammation and oxidative stress in cultured human umbilical vein endothelial cells. Chemosphere 2017, 167, 454–461. [Google Scholar] [CrossRef]
- Farzan, S.F.; Howe, C.G.; Zens, M.S.; Palys, T.; Channon, J.Y.; Li, Z.; Chen, Y.; Karagas, M.R. Urine Arsenic and Arsenic Metabolites in U.S. Adults and Biomarkers of Inflammation, Oxidative Stress, and Endothelial Dysfunction: A Cross-Sectional Study. Environ. Health Perspect. 2017, 125, 127002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Takahashi, T.; Yamamoto, C.; Yoshida, E.; Kaji, T.; Fujiwara, Y. Arsenite Inhibits Tissue-Type Plasminogen Activator Synthesis through NRF2 Activation in Cultured Human Vascular Endothelial EA.hy926 Cells. Int. J. Mol. Sci. 2021, 22, 739. [Google Scholar] [CrossRef]
- Negro Silva, L.F.; Lemaire, M.; Lemarie, C.A.; Plourde, D.; Bolt, A.M.; Chiavatti, C.; Bohle, D.S.; Slavkovich, V.; Graziano, J.H.; Lehoux, S.; et al. Effects of Inorganic Arsenic, Methylated Arsenicals, and Arsenobetaine on Atherosclerosis in the Mouse Model and the Role of As3mt-Mediated Methylation. Environ. Health Perspect. 2017, 125, 077001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, L.; Lai, Y.; Tu, P.; Liu, C.W.; Xue, J.; Ru, H.; Lu, K. Lipid and Cholesterol Homeostasis after Arsenic Exposure and Antibiotic Treatment in Mice: Potential Role of the Microbiota. Environ. Health Perspect. 2019, 127, 97002. [Google Scholar] [CrossRef] [Green Version]
- Amiri, A.; Mokhayeri, Y.; Mohammadi, R.; Karami, M.A.; Ghaderpoori, M.; Kamarehie, B.; Jafari, A. Dose-response meta-analysis of arsenic exposure in drinking water and hypertension. Heliyon 2021, 7, e06409. [Google Scholar] [CrossRef]
- Zhao, J.; Li, A.; Mei, Y.; Zhou, Q.; Li, Y.; Li, K.; Xu, Q. The association of arsenic exposure with hypertension and blood pressure: A systematic review and dose-response meta-analysis. Environ. Pollut. 2021, 289, 117914. [Google Scholar] [CrossRef]
- Yu, Y.; Guo, Y.; Zhang, J.; Xie, J.; Zhu, Y.; Yan, J.; Wang, B.; Li, Z. A perspective of chronic low exposure of arsenic on non-working women: Risk of hypertension. Sci. Total Environ. 2017, 580, 69–73. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Z.; Yang, L.; Liu, X.; Xing, X.; Zhang, H.; Yun, J.; Ou, X.; Su, X.; Lu, Y.; et al. Arsenic exposure assists ccm3 genetic polymorphism in elevating blood pressure. Oncotarget 2018, 9, 4915–4923. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, F.; Liu, X.; Parvez, F.; LoIacono, N.J.; Gibson, E.A.; Kioumourtzoglou, M.A.; Levy, D.; Shahriar, H.; Uddin, M.N.; et al. Early life and adolescent arsenic exposure from drinking water and blood pressure in adolescence. Environ. Res. 2019, 178, 108681. [Google Scholar] [CrossRef]
- Kaufman, J.A.; Mattison, C.; Fretts, A.M.; Umans, J.G.; Cole, S.A.; Voruganti, V.S.; Goessler, W.; Best, L.G.; Zhang, Y.; Tellez-Plaza, M.; et al. Arsenic, blood pressure, and hypertension in the Strong Heart Family Study. Environ. Res. 2021, 195, 110864. [Google Scholar] [CrossRef]
- Yang, H.T.; Chou, H.J.; Han, B.C.; Huang, S.Y. Lifelong inorganic arsenic compounds consumption affected blood pressure in rats. Food Chem. Toxicol. 2007, 45, 2479–2487. [Google Scholar] [CrossRef]
- Cifuentes, F.; Bravo, J.; Norambuena, M.; Stegen, S.; Ayavire, A.; Palacios, J. Chronic exposure to arsenic in tap water reduces acetylcholine-induced relaxation in the aorta and increases oxidative stress in female rats. Int. J. Toxicol. 2009, 28, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Jyoti, U.; Kansal, S.K.; Kumar, P.; Goyal, S. Possible vasculoprotective role of linagliptin against sodium arsenite-induced vascular endothelial dysfunction. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2016, 389, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Jung, B.I.; Chung, S.M.; Bae, O.N.; Lee, J.Y.; Park, J.D.; Yang, J.S.; Lee, H.; Chung, J.H. Arsenic-induced dysfunction in relaxation of blood vessels. Environ. Health Perspect. 2003, 111, 513–517. [Google Scholar] [CrossRef] [Green Version]
- Sarath, T.S.; Waghe, P.; Gupta, P.; Choudhury, S.; Kannan, K.; Pillai, A.H.; Harikumar, S.K.; Mishra, S.K.; Sarkar, S.N. Atorvastatin ameliorates arsenic-induced hypertension and enhancement of vascular redox signaling in rats. Toxicol. Appl. Pharmacol. 2014, 280, 443–454. [Google Scholar] [CrossRef]
- Osorio-Yanez, C.; Chin-Chan, M.; Sanchez-Pena, L.C.; Atzatzi-Aguilar, O.G.; Olivares-Reyes, J.A.; Segovia, J.; Del Razo, L.M. The ADMA/DDAH/NO pathway in human vein endothelial cells exposed to arsenite. Toxicol. In Vitro 2017, 42, 281–286. [Google Scholar] [CrossRef]
- Waghe, P.; Sarath, T.S.; Gupta, P.; Kandasamy, K.; Choudhury, S.; Kutty, H.S.; Mishra, S.K.; Sarkar, S.N. Arsenic causes aortic dysfunction and systemic hypertension in rats: Augmentation of angiotensin II signaling. Chem. Biol. Interact. 2015, 237, 104–114. [Google Scholar] [CrossRef]
- Rahaman, M.S.; Mise, N.; Ikegami, A.; Zong, C.; Ichihara, G.; Ichihara, S. The mechanism of low-level arsenic exposure-induced hypertension: Inhibition of the activity of the angiotensin-converting enzyme 2. Chemosphere 2023, 318, 137911. [Google Scholar] [CrossRef]
- Balarastaghi, S.; Barangi, S.; Hosseinzadeh, H.; Imenshahidi, M.; Moosavi, Z.; Razavi, B.M.; Karimi, G. Melatonin improves arsenic-induced hypertension through the inactivation of the Sirt1/autophagy pathway in rat. Biomed Pharmacother. 2022, 151, 113135. [Google Scholar] [CrossRef]
- Martins, A.C.; Ke, T.; Bowman, A.B.; Aschner, M. New insights on mechanisms underlying methylmercury-induced and manganese-induced neurotoxicity. Curr. Opin. Toxicol. 2021, 25, 30–35. [Google Scholar] [CrossRef]
- Clarkson, T.W.; Magos, L. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 2006, 36, 609–662. [Google Scholar] [CrossRef] [PubMed]
- Landrigan, P.J.; Sonawane, B.; Butler, R.N.; Trasande, L.; Callan, R.; Droller, D. Early environmental origins of neurodegenerative disease in later life. Environ. Health Perspect. 2005, 113, 1230–1233. [Google Scholar] [CrossRef] [Green Version]
- Berthoud, H.R.; Garman, R.H.; Weiss, B. Food intake, body weight, and brain histopathology in mice following chronic methylmercury treatment. Toxicol. Appl. Pharmacol. 1976, 36, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.A.; Mitchener, M. Life-term effects of mercury, methyl mercury, and nine other trace metals on mice. J. Nutr. 1975, 105, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Magos, L. Neurotoxicity, anorexia and the preferential choice of antidote in methylmercury intoxicated rats. Neurobehav. Toxicol. Teratol. 1982, 4, 643–646. [Google Scholar]
- Ferrer, B.; Peres, T.V.; Dos Santos, A.A.; Bornhorst, J.; Morcillo, P.; Goncalves, C.L.; Aschner, M. Methylmercury Affects the Expression of Hypothalamic Neuropeptides That Control Body Weight in C57BL/6J Mice. Toxicol. Sci. 2018, 163, 557–568. [Google Scholar] [CrossRef]
- Li, T.; Yu, L.; Yang, Z.; Shen, P.; Lin, H.; Shui, L.; Tang, M.; Jin, M.; Chen, K.; Wang, J. Associations of Diet Quality and Heavy Metals with Obesity in Adults: A Cross-Sectional Study from National Health and Nutrition Examination Survey (NHANES). Nutrients 2022, 14, 4038. [Google Scholar] [CrossRef]
- Bulka, C.M.; Persky, V.W.; Daviglus, M.L.; Durazo-Arvizu, R.A.; Argos, M. Multiple metal exposures and metabolic syndrome: A cross-sectional analysis of the National Health and Nutrition Examination Survey 2011–2014. Environ. Res. 2019, 168, 397–405. [Google Scholar] [CrossRef]
- Moon, M.K.; Lee, I.; Lee, A.; Park, H.; Kim, M.J.; Kim, S.; Cho, Y.H.; Hong, S.; Yoo, J.; Cheon, G.J.; et al. Lead, mercury, and cadmium exposures are associated with obesity but not with diabetes mellitus: Korean National Environmental Health Survey (KoNEHS) 2015–2017. Environ. Res. 2022, 204, 111888. [Google Scholar] [CrossRef]
- Lee, K. Blood mercury concentration in relation to metabolic and weight phenotypes using the KNHANES 2011–2013 data. Int. Arch. Occup. Environ. Health 2018, 91, 185–193. [Google Scholar] [CrossRef]
- Lee, S.; Cho, S.R.; Jeong, I.; Park, J.B.; Shin, M.Y.; Kim, S.; Kim, J.H. Mercury Exposure and Associations with Hyperlipidemia and Elevated Liver Enzymes: A Nationwide Cross-Sectional Survey. Toxics 2020, 8, 47. [Google Scholar] [CrossRef]
- Tsai, T.L.; Kuo, C.C.; Pan, W.H.; Wu, T.N.; Lin, P.; Wang, S.L. Type 2 diabetes occurrence and mercury exposure—From the National Nutrition and Health Survey in Taiwan. Environ. Int. 2019, 126, 260–267. [Google Scholar] [CrossRef]
- Zareba, G.; Cernichiari, E.; Goldsmith, L.A.; Clarkson, T.W. Validity of methyl mercury hair analysis: Mercury monitoring in human scalp/nude mouse model. J. Appl. Toxicol. 2008, 28, 535–542. [Google Scholar] [CrossRef]
- Brockman, J.D.; Raymond, L.J.; Ralston, C.R.; Robertson, J.D.; Bodkin, N.; Sharp, N.; Ralston, N.V. The nail as a noninvasive indicator of methylmercury exposures and mercury/selenium molar ratios in brain, kidney, and livers of Long-Evans rats. Biol. Trace Elem. Res. 2011, 144, 812–820. [Google Scholar] [CrossRef]
- Park, K.; Seo, E. Association between Toenail Mercury and Metabolic Syndrome Is Modified by Selenium. Nutrients 2016, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Morris, J.S.; Park, K. Toenail mercury levels positively correlate with obesity and abdominal obesity among Korean adults. J. Trace Elem. Med. Biol. 2021, 64, 126678. [Google Scholar] [CrossRef]
- Skalny, A.V.; Chang, J.S.; Bobrovnitsky, I.P.; Kopylov, P.Y.; Skalnaya, M.G.; Huang, S.Y.; Paoliello, M.M.B.; Ivanova, E.S.; Wang, W.; Tinkov, A.A. Relationship Between Elevated Hair Mercury Levels, Essential Element Status, and Metabolic Profile in Overweight and Obese Adults. Biol. Trace Elem. Res. 2021, 199, 2874–2881. [Google Scholar] [CrossRef]
- Oken, E.; Radesky, J.S.; Wright, R.O.; Bellinger, D.C.; Amarasiriwardena, C.J.; Kleinman, K.P.; Hu, H.; Gillman, M.W. Maternal fish intake during pregnancy, blood mercury levels, and child cognition at age 3 years in a US cohort. Am. J. Epidemiol. 2008, 167, 1171–1181. [Google Scholar] [CrossRef]
- Jia, P. Obesogenic environment and childhood obesity. Obes. Rev. 2021, 22 (Suppl. 1), e13158. [Google Scholar] [CrossRef]
- Leung, A.K.C.; Wong, A.H.C.; Hon, K.L. Childhood Obesity: An Updated Review. Curr. Pediatr. Rev. 2022. [Google Scholar] [CrossRef]
- Smith, A.R.; Lin, P.D.; Rifas-Shiman, S.L.; Wright, R.O.; Coull, B.; Hivert, M.F.; Hubbard, A.; Oken, E.; Cardenas, A. Associations of Prenatal First Trimester Essential and Nonessential Metal Mixtures with Body Size and Adiposity in Childhood. Epidemiology 2023, 34, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Igusa, T.; Wang, G.; Buckley, J.P.; Hong, X.; Bind, E.; Steffens, A.; Mukherjee, J.; Haltmeier, D.; Ji, Y.; et al. In-utero co-exposure to toxic metals and micronutrients on childhood risk of overweight or obesity: New insight on micronutrients counteracting toxic metals. Int. J. Obes. 2022, 46, 1435–1445. [Google Scholar] [CrossRef]
- Karatela, S.; Ward, N.; Paterson, J. Mercury Exposure in Mother-Children Pairs in A Seafood Eating Population: Body Burden and Related Factors. Int. J. Environ. Res. Public Health 2019, 16, 2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; DiBari, J.; Bind, E.; Steffens, A.M.; Mukherjee, J.; Bartell, T.R.; Bellinger, D.C.; Hong, X.; Ji, Y.; Wang, M.C.; et al. In utero exposure to mercury and childhood overweight or obesity: Counteracting effect of maternal folate status. BMC Med. 2019, 17, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.Y.; Ryu, I.K.; Park, M.J.; Kim, S.H. The association of total blood mercury levels and overweight among Korean adolescents: Analysis of the Korean National Health and Nutrition Examination Survey (KNHANES) 2010–2013. Korean J. Pediatr. 2018, 61, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.Y. Association of Blood Mercury Levels with the Risks of Overweight and High Waist-to-Height Ratio in Children and Adolescents: Data from the Korean National Health and Nutrition Examination Survey. Children 2021, 8, 1087. [Google Scholar] [CrossRef]
- Cho, H.W.; Kim, S.H.; Park, M.J. An association of blood mercury levels and hypercholesterolemia among Korean adolescents. Sci. Total. Environ. 2020, 709, 135965. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, C.; Bu, J. Relationship between Selected Serum Metallic Elements and Obesity in Children and Adolescent in the U.S. Nutrients 2017, 9, 104. [Google Scholar] [CrossRef]
- Betanzos-Robledo, L.; Tellez-Rojo, M.M.; Lamadrid-Figueroa, H.; Roldan-Valadez, E.; Peterson, K.E.; Jansen, E.C.; Basu, N.; Cantoral, A. Differential fat accumulation in early adulthood according to adolescent-BMI and heavy metal exposure. New Dir. Child Adolesc. Dev. 2022, 2022, 37–51. [Google Scholar] [CrossRef]
- Caito, S.W.; Jackson, B.P.; Punshon, T.; Scrimale, T.; Grier, A.; Gill, S.R.; Love, T.M.; Watson, G.E.; van Wijngaarden, E.; Rand, M.D. Editor’s Highlight: Variation in Methylmercury Metabolism and Elimination Status in Humans Following Fish Consumption. Toxicol. Sci. 2018, 161, 443–453. [Google Scholar] [CrossRef]
- Sakamoto, M.; Kakita, A.; de Oliveira, R.B.; Sheng Pan, H.; Takahashi, H. Dose-dependent effects of methylmercury administered during neonatal brain spurt in rats. Brain Res. Dev. Brain Res. 2004, 152, 171–176. [Google Scholar] [CrossRef]
- Yamamoto, M.; Motomura, E.; Yanagisawa, R.; Hoang, V.A.T.; Mogi, M.; Mori, T.; Nakamura, M.; Takeya, M.; Eto, K. Evaluation of neurobehavioral impairment in methylmercury-treated KK-Ay mice by dynamic weight-bearing test. J. Appl. Toxicol. 2019, 39, 221–230. [Google Scholar] [CrossRef]
- Ferrer, B.; Prince, L.M.; Tinkov, A.A.; Santamaria, A.; Farina, M.; Rocha, J.B.; Bowman, A.B.; Aschner, M. Chronic exposure to methylmercury enhances the anorexigenic effects of leptin in C57BL/6J male mice. Food Chem. Toxicol. 2021, 147, 111924. [Google Scholar] [CrossRef]
- Dias, L.K.M.; de Medeiros, G.; Silva, A.K.N.; de Araujo Morais, A.H.; da Silva-Maia, J.K. Can polyphenols improve the gut health status in pre-clinical study with diet-induced obesity?: A protocol for systematic review and/or meta-analysis. Medicine 2021, 100, e28162. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yanagisawa, R.; Motomura, E.; Nakamura, M.; Sakamoto, M.; Takeya, M.; Eto, K. Increased methylmercury toxicity related to obesity in diabetic KK-Ay mice. J. Appl. Toxicol. 2014, 34, 914–923. [Google Scholar] [CrossRef]
- Guaraldi, G.; Lonardo, A.; Maia, L.; Palella, F.J., Jr. Metabolic concerns in aging HIV-infected persons: From serum lipid phenotype to fatty liver. AIDS. 2017, 31 (Suppl. 2), S147–S156. [Google Scholar] [CrossRef]
- Nascimento, T.S.; Pinto, D.V.; Dias, R.P.; Raposo, R.S.; Nunes, P.I.G.; Roque, C.R.; Santos, F.A.; Andrade, G.M.; Viana, J.L.; Fostier, A.H.; et al. Chronic Methylmercury Intoxication Induces Systemic Inflammation, Behavioral, and Hippocampal Amino Acid Changes in C57BL6J Adult Mice. Int. J. Mol. Sci. 2022, 23, 13837. [Google Scholar] [CrossRef]
- Silva, J.L.; Leocadio, P.C.L.; Reis, J.M.; Campos, G.P.; Capettini, L.S.A.; Foureaux, G.; Ferreira, A.J.; Windmoller, C.C.; Santos, F.A.; Oria, R.B.; et al. Oral methylmercury intoxication aggravates cardiovascular risk factors and accelerates atherosclerosis lesion development in ApoE knockout and C57BL/6 mice. Toxicol. Res. 2021, 37, 311–321. [Google Scholar] [CrossRef]
- Lacerda Leocadio, P.C.; Dias, R.P.; Pinto, D.V.; Reis, J.M.; Rodrigues Nascimento, J.C.; Anne de Castro Brito, G.; Valenca, J.T., Jr.; Foureaux, G.; Ferreira, A.J.; Windmoller, C.C.; et al. Pollutants and nutrition: Are methylmercury effects on blood pressure and lipoprotein profile comparable to high-fat diet in mice? Ecotoxicol. Environ. Saf. 2020, 204, 111036. [Google Scholar] [CrossRef]
- Alam, R.T.M.; Abu Zeid, E.H.; Khalifa, B.A.; Arisha, A.H.; Reda, R.M. Dietary exposure to methyl mercury chloride induces alterations in hematology, biochemical parameters, and mRNA expression of antioxidant enzymes and metallothionein in Nile tilapia. Environ. Sci. Pollut. Res. Int. 2021, 28, 31391–31402. [Google Scholar] [CrossRef]
- Dutta, H.M.; Haghighi, A.Z. Methylmercuric chloride and serum cholesterol level in the bluegill (Lepomis macrochirus). Bull. Environ. Contam. Toxicol. 1986, 36, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, F.; Bahadar, H.; Niaz, K.; Baeeri, M.; Rahimifard, M.; Navaei-Nigjeh, M.; Ghasemi-Niri, S.F.; Abdollahi, M. Effects of methyl mercury on the activity and gene expression of mouse Langerhans islets and glucose metabolism. Food Chem. Toxicol. 2016, 93, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Dunlap, K.; Duffy, L.K. Effects of Methylmercury and Theaflavin Digallate on Adipokines in Mature 3T3-L1 Adipocytes. Int. J. Mol. Sci. 2019, 20, 2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, D.; Kant, S.; Pandey, S.; Ehtesham, N.Z. Resistin in metabolism, inflammation, and disease. FEBS J. 2020, 287, 3141–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takanezawa, Y.; Kashiwano, Y.; Nakamura, R.; Ohshiro, Y.; Uraguchi, S.; Kiyono, M. Methylmercury drives lipid droplet formation and adipokine expression during the late stages of adipocyte differentiation in 3T3-L1 cells. Toxicology 2023, 486, 153446. [Google Scholar] [CrossRef]
- Tinant, G.; Neefs, I.; Das, K.; Rees, J.F.; Larondelle, Y.; Debier, C. Methylmercury displays pro-adipogenic properties in rainbow trout preadipocytes. Chemosphere 2021, 263, 127917. [Google Scholar] [CrossRef]
- Guo, L.; Li, X.; Tang, Q.Q. Transcriptional regulation of adipocyte differentiation: A central role for CCAAT/enhancer-binding protein (C/EBP) beta. J. Biol. Chem. 2015, 290, 755–761. [Google Scholar] [CrossRef] [Green Version]
- Caito, S.W.; Newell-Caito, J.; Martell, M.; Crawford, N.; Aschner, M. Methylmercury Induces Metabolic Alterations in Caenorhabditis elegans: Role for C/EBP Transcription Factor. Toxicol. Sci. 2020, 174, 112–123. [Google Scholar] [CrossRef]
- Crawford, N.; Martell, M.; Nielsen, T.; Khalil, B.; Imtiaz, F.; Nguidjo, E.; Newell-Caito, J.L.; Bornhorst, J.; Schwerdtle, T.; Caito, S.W. Methylmercury-Induced Metabolic Alterations in Caenorhabditis elegans Are Diet-Dependent. Toxics 2021, 9, 287. [Google Scholar] [CrossRef]
- Iacomino, G.; Siani, A. Role of microRNAs in obesity and obesity-related diseases. Genes Nutr. 2017, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Ibanez-Ventoso, C.; Vora, M.; Driscoll, M. Sequence relationships among C. elegans, D. melanogaster and human microRNAs highlight the extensive conservation of microRNAs in biology. PLoS ONE 2008, 3, e2818. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, T.; Crawford, N.; Martell, M.; Khalil, B.; Imtiaz, F.; Newell-Caito, J.L.; Caito, S. MicroRNA Expression Influences Methylmercury-Induced Lipid Accumulation and Mitochondrial Toxicity in Caenorhabditis elegans. Chem. Res. Toxicol. 2022, 35, 77–88. [Google Scholar] [CrossRef]
- Chen, Y.W.; Huang, C.F.; Tsai, K.S.; Yang, R.S.; Yen, C.C.; Yang, C.Y.; Lin-Shiau, S.Y.; Liu, S.H. Methylmercury induces pancreatic beta-cell apoptosis and dysfunction. Chem. Res. Toxicol. 2006, 19, 1080–1085. [Google Scholar] [CrossRef]
- Yang, C.Y.; Liu, S.H.; Su, C.C.; Fang, K.M.; Yang, T.Y.; Liu, J.M.; Chen, Y.W.; Chang, K.C.; Chuang, H.L.; Wu, C.T.; et al. Methylmercury Induces Mitochondria- and Endoplasmic Reticulum Stress-Dependent Pancreatic beta-Cell Apoptosis via an Oxidative Stress-Mediated JNK Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 2858. [Google Scholar] [CrossRef]
- Rizzetti, D.A.; Corrales, P.; Piagette, J.T.; Uranga-Ocio, J.A.; Medina-Gomez, G.; Pecanha, F.M.; Vassallo, D.V.; Miguel, M.; Wiggers, G.A. Chronic mercury at low doses impairs white adipose tissue plasticity. Toxicology 2019, 418, 41–50. [Google Scholar] [CrossRef]
- Kawakami, T.; Hanao, N.; Nishiyama, K.; Kadota, Y.; Inoue, M.; Sato, M.; Suzuki, S. Differential effects of cobalt and mercury on lipid metabolism in the white adipose tissue of high-fat diet-induced obesity mice. Toxicol. Appl. Pharmacol. 2012, 258, 32–42. [Google Scholar] [CrossRef]
- Barnes, D.M.; Hanlon, P.R.; Kircher, E.A. Effects of inorganic HgCl2 on adipogenesis. Toxicol. Sci. 2003, 75, 368–377. [Google Scholar] [CrossRef] [Green Version]
- da Cunha Martins, A., Jr.; Mazzaron Barcelos, G.R.; Jacob Ferreira, A.L.; de Souza, M.F.; de Syllos Colus, I.M.; Antunes, L.M.; Paoliello, M.M.B.; Adeyemi, J.A.; Barbosa, F., Jr. Effects of Lead Exposure and Genetic Polymorphisms on ALAD and GPx Activities in Brazilian Battery Workers. J. Toxicol. Environ. Health A 2015, 78, 1073–1081. [Google Scholar] [CrossRef]
- Hanna-Attisha, M.; LaChance, J.; Sadler, R.C.; Champney Schnepp, A. Elevated Blood Lead Levels in Children Associated with the Flint Drinking Water Crisis: A Spatial Analysis of Risk and Public Health Response. Am. J. Public Health 2016, 106, 283–290. [Google Scholar] [CrossRef]
- Caito, S.; Aschner, M. Developmental Neurotoxicity of Lead. Adv. Neurobiol. 2017, 18, 3–12. [Google Scholar] [CrossRef]
- Virgolini, M.B.; Aschner, M. Molecular Mechanisms of Lead Neurotoxicity. Adv. Neurotoxicol. 2021, 5, 159–213. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Jung, Y.; Oh, C.U. Relations between the blood lead level and metabolic syndrome risk factors. Public Health Nurs. 2019, 36, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Rotter, I.; Kosik-Bogacka, D.; Dolegowska, B.; Safranow, K.; Lubkowska, A.; Laszczynska, M. Relationship between the concentrations of heavy metals and bioelements in aging men with metabolic syndrome. Int. J. Environ. Res. Public Health 2015, 12, 3944–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scinicariello, F.; Buser, M.C.; Mevissen, M.; Portier, C.J. Blood lead level association with lower body weight in NHANES 1999–2006. Toxicol. Appl. Pharmacol. 2013, 273, 516–523. [Google Scholar] [CrossRef]
- Wang, N.; Sheng, Z.; Zhou, S.; Jiang, F.; Zhang, Z. Chronic lead exposure exacerbates hepatic glucolipid metabolism disorder and gut microbiota dysbiosis in high-fat-diet mice. Food Chem. Toxicol. 2022, 170, 113451. [Google Scholar] [CrossRef]
- Tyrrell, J.B.; Hafida, S.; Stemmer, P.; Adhami, A.; Leff, T. Lead (Pb) exposure promotes diabetes in obese rodents. J. Trace. Elem. Med. Biol. 2017, 39, 221–226. [Google Scholar] [CrossRef]
- Mostafalou, S.; Baeeri, M.; Bahadar, H.; Soltany-Rezaee-Rad, M.; Gholami, M.; Abdollahi, M. Molecular mechanisms involved in lead induced disruption of hepatic and pancreatic glucose metabolism. Environ. Toxicol. Pharmacol. 2015, 39, 16–26. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, A.C.; Ferrer, B.; Tinkov, A.A.; Caito, S.; Deza-Ponzio, R.; Skalny, A.V.; Bowman, A.B.; Aschner, M. Association between Heavy Metals, Metalloids and Metabolic Syndrome: New Insights and Approaches. Toxics 2023, 11, 670. https://doi.org/10.3390/toxics11080670
Martins AC, Ferrer B, Tinkov AA, Caito S, Deza-Ponzio R, Skalny AV, Bowman AB, Aschner M. Association between Heavy Metals, Metalloids and Metabolic Syndrome: New Insights and Approaches. Toxics. 2023; 11(8):670. https://doi.org/10.3390/toxics11080670
Chicago/Turabian StyleMartins, Airton C., Beatriz Ferrer, Alexey A. Tinkov, Samuel Caito, Romina Deza-Ponzio, Anatoly V. Skalny, Aaron B. Bowman, and Michael Aschner. 2023. "Association between Heavy Metals, Metalloids and Metabolic Syndrome: New Insights and Approaches" Toxics 11, no. 8: 670. https://doi.org/10.3390/toxics11080670