
Racemization in Post-Translational Modifications Relevance to Protein Aging, Aggregation and Neurodegeneration: Tip of the Iceberg


1
Virtual Reality Perception Lab (VRPL), The Nathan S. Kline Institute for Psychiatric Research (NKI), Orangeburg, NY 10962, USA
2
Departments of Neurology, Pathology and Psychiatry, Center for Cognitive Neurology, New York University School of Medicine, New York, NY 10016, USA
3
Center for Neurochemistry, The Nathan S. Kline Institute for Psychiatric Research (NKI), Orangeburg, NY 10962, USA
*
Author to whom correspondence should be addressed.
Symmetry 2021, 13(3), 455; https://doi.org/10.3390/sym13030455
Submission received: 7 January 2021
/
Revised: 8 February 2021
/
Accepted: 18 February 2021
/
Published: 11 March 2021
(This article belongs to the Special Issue Bioinformatics and Computational Biology)
{kind=link}
Abstract
:Homochirality of DNA and prevalent chirality of free and protein-bound amino acids in a living organism represents the challenge for modern biochemistry and neuroscience. The idea of an association between age-related disease, neurodegeneration, and racemization originated from the studies of fossils and cataract disease. Under the pressure of new results, this concept has a broader significance linking protein folding, aggregation, and disfunction to an organism’s cognitive and behavioral functions. The integrity of cognitive function is provided by a delicate balance between the evolutionarily imposed molecular homo-chirality and the epigenetic/developmental impact of spontaneous and enzymatic racemization. The chirality of amino acids is the crucial player in the modulation the structure and function of proteins, lipids, and DNA. The collapse of homochirality by racemization is the result of the conformational phase transition. The racemization of protein-bound amino acids (spontaneous and enzymatic) occurs through thermal activation over the energy barrier or by the tunnel transfer effect under the energy barrier. The phase transition is achieved through the intermediate state, where the chirality of alpha carbon vanished. From a thermodynamic consideration, the system in the homo-chiral (single enantiomeric) state is characterized by a decreased level of entropy. The oscillating protein chirality is suggesting its distinct significance in the neurotransmission and flow of perceptual information, adaptive associative learning, and cognitive laterality. The common pathological hallmarks of neurodegenerative disorders include protein misfolding, aging, and the deposition of protease-resistant protein aggregates. Each of the landmarks is influenced by racemization. The brain region, cell type, and age-dependent racemization critically influence the functions of many intracellular, membrane-bound, and extracellular proteins including amyloid precursor protein (APP), TAU, PrP, Huntingtin, α-synuclein, myelin basic protein (MBP), and collagen. The amyloid cascade hypothesis in Alzheimer’s disease (AD) coexists with the failure of amyloid beta (Aβ) targeting drug therapy. According to our view, racemization should be considered as a critical factor of protein conformation with the potential for inducing order, disorder, misfolding, aggregation, toxicity, and malfunctions.
Keywords:
D-amino acids; racemization; post translational modification; protein folding; misfolding; aggregation; neurodegeneration; association; cognitive functions; non-equilibrium phase transitions; intrinsically disordered proteins; protein folding energy landscape; brain information processing; adaptive associative learning; cognitive laterality1. Introduction
Under the pressure of previous and emerging results, the idea of the close association between age-related disease, neurodegeneration, and protein racemization shows a meaningful significance [1,2,3,4,5,6,7]. The diversity of neurodegenerative diseases possesses both specific and common features [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288,289,290,291,292,293,294,295,296,297,298,299,300,301,302,303,304,305,306,307,308,309,310,311,312,313,314,315,316,317,318,319,320,321,322,323,324,325,326,327,328,329,330,331,332,333,334,335,336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353,354,355,356,357,358,359,360,361,362,363,364,365,366,367,368,369,370,371,372,373,374,375,376,377,378,379,380,381,382,383,384,385,386,387,388,389,390,391,392,393,394,395,396,397,398,399,400,401,402,403,404,405,406,407,408,409,410,411,412,413,414,415,416,417,418,419,420,421,422,423,424,425,426,427,428,429,430,431,432,433,434,435,436,437,438,439,440,441,442,443,444,445,446,447,448,449,450,451,452,453,454,455,456,457,458,459,460,461,462,463,464,465,466,467,468,469,470,471,472,473,474,475,476,477,478,479,480,481,482,483,484,485]. In our view, the most common causal mechanism underlying age-related protein misfolding, dysfunction, and aggregation is spontaneous racemization.
The accumulation of misfolded proteins (MPs) is recognized as the most characteristic manifestation of neurodegeneration. The search for the universal mechanism of neurodegenerative diseases eventually considers not only biochemical but also stereochemical processes. Molecular chirality is a critical feature of many biological events in the entire kingdom of life in its normal and pathological forms. Amino acids (AAs) profiles are an effective biomarker (BM) for cardio-genesis [8] and the level of D-amino acids (D-AAs) is increasingly recognized as a novel BM of kidney diseases [9] and neurodegeneration [10]. Chirality is an intrinsic property of peptides and proteins including amyloid beta (Aβ) and microtubule-associated protein TAU, both of which are potent towards the misfolding pathways [11]. It has been shown that dramatic structural perturbations could be triggered by chiral inversions of amino acid chain fragment and any alteration of the physicochemical environment. The effect of chirality perturbations is relevant for the main landmarks of the Alzheimer’s disease (AD): A-β plaques and TAU fibrillary tangles [11]. The racemization of AAs and proteins becomes appreciated as a determinant of most of physiological processes [12,13,14]. D-AAs have been shown to play an adverse role in the physiology of bacteria I [15], (I. D-AAs are found in the cell walls of bacteria, Bacteria are the primary sink for D-AAs contributing to their accumulation in the environments at toxic concentrations [15] and insects [16]).
It is reasonable to expect even more diverse function of D-AAs in animal brain. The biological significance of racemization neuropathogenesis of AD was assessed from as early as year 1994 [17] and remains important [5,18].
1.1. Chiral Phase Transitions
The phase transitions associated with stereo-transformation (racemization and isomerization) of peptides and proteins, are driven by the force of increase in the entropy II. (II. In situations when we can disregard the contribution of other factors such as the enthalpy contribution from the heterochiral interaction [19].)
The first and second order phase transition III in AAs, peptides and proteins, despite being theoretically and experimentally explored, are just at the beginning stages of systematic studies [20,21,22,23]. (III. Phase transitions are classified according to Ehrenfest classification [24]. The order of a phase transition is defined to be the order of the lowest-order derivative, which changes discontinuously at the phase boundary).
The concepts of phase transition and chirality transfer are necessary for understanding the biochemistry of AA signaling and protein folding concerning the cell physiology. The effects of chirality transfer from photons to AAs have been recently reviewed [25]. The understanding, description, and interpretation of experiments of protein folding is based on the physics underlying the electron spin system called “spin-glass” paradigm [26,27,28]. The dynamic behavior of proteins exhibits multiple functional and inactive conformational configurations. The understanding of the structure–function relationship requires the study of kinetic and thermodynamic pathways. At the cellular level, the concept of phase transitions is relevant to the membrane-less organelles (MLO), which represent the coherent structures with the distinct biological functions. Well-known examples include the nucleolus, Cajal bodies, nuclear speckles, cytoplasmic stress granules, P-bodies, and germ granules. MLO molecular structures are environmentally responsive [26,29] and are implicated in the functional protein folding and protein aggregation diseases [30,31]. MLO, peptides, and proteins exhibit various forms of liquid–gel condensations including liquid–liquid and liquid–crystal phase separation, and phase transitions [32,33]. According to [34], the autophagy is considered as a cellular phase transition which maintains the normal cellular functions. The dynamics of the cellular phase transition shown depend on the AAs parameters, however the chirality of AAs is frequently not taken to an account. From the thermodynamic perspective, the existence of a Gibbs potential barrier (energy barrier) between two chiral states is an internal determinant influencing the kinetics of AAs racemization and protein folding. Several external physical and chemical factors influence the rate of stereo-transformation including thermal modulation, photo-stimulation, acoustic-chemical reactions [35], radical reactions, oxidation-reduction sequences, enzyme catalysis, nucleophilic substitutions, and pH of the media IV. (IV. The rate of aspartic acid racemization in the human connective tissue is about 1% per year [36,37,38]).
The widely appreciated “sequence–structure–function” paradigm, postulated by Anfinsen [39], has attracted attention to bio-molecular chirality [23,40]. Three major environmental factors influencing protein conformation are the cytosol, nucleus [30], and cell/organelle membrane. Accordingly, most, if not all, proteins contain segments which have the dual ability to fold into several distinct structures in aqueous and membrane environments [41]. The transport of proteins from the cytosol to the membrane phosphor-lipid or nuclear environment is accompanied by conformational phase transitions. The interplay of the thermodynamic equilibrium and the fundamentally non-equilibrium nature of cellular biochemistry constitutes the basis for the non-equilibrium phase transitions [21,22,33,42].
1.2. Biomolecular Chirality
The major classes V of biomolecules influenced by the phenomena of chirality are: (i) AAs [9,43,44], (ii) peptides, and proteins [45,46,47,48,49], (iii) lipids [50,51,52], (iv) nucleic acids, (v) DNA, and (vi) RNA [53,54,55] group. (V. The most abundant biomolecules belong to four major classes: proteins, lipids, nucleic acids, and carbohydrates. The chirality of carbohydrates (despite its essential role) is beyond the scope of our consideration).
We will focus mainly on the link between AAs chirality and the protein structure–function relationship. Protein function will be considered with regard to the system of post translational modification (PTM-Sys).
1.3. D-Amino Acids in Proteins, Cells, and Neuronal Circuits
The presence of the D-aspartic acid (D-Asp) in myelin and myelin basic protein (MBP) was documented a long time ago [56]. However, the existence of D-AAs in the central nervous system was practically unknown (not discussed) until 2000. Due to the homochirality of biological AAs (L-isoform) in many publications, D-AAs are characterized as “non-biological” [57] or “unnatural” [58]. Presently, diverse D-AAs are found in the body and brain of mammals including, D-serine, D-aspartate, D-alanine, and D-cysteine [59,60,61,62,63,64,65,66,67]. The current finding suggests that the biosynthetic pathway for D-AAs is conserved from bacteria to mammalian [68]. The aspartic acid and serine are among the most studied AAs due to their distinct role in biochemistry and neuroscience. Both are known as the phospho-acceptors. This fact explains the impact of phosphorylation on the structure of corresponding proteins. AAs racemization (along with deamidation, hydrolysis of peptide bonds, breakage of disulfides, and others) is one of the most active mechanisms in the system of PTM associated with plasticity of protein functions [69,70]. The role of AAs in cell biology, to a significant degree, is determined by interplay between two (L and D) isoforms governed by the spontaneous racemization [18], evolutionarily conserved network of PTM [71,72,73,74], and under the environmental factors. Spontaneous, non-enzymatic reactions in proteins are relevant to aging and age-related diseases including AD and cataract. However, the AA-specific mechanisms of spontaneous phase transition are not broadly studied. Recently it was shown that the racemization of the Ser residue occurs preferably in flexible regions of proteins [18]. The translation of peptides/proteins in the eukaryotes utilizes only L-amino acids (L-AAs). The productions of the D-amino acid-containing peptides/proteins through PTM occur via the isomerase enzymes. The isomerization mechanism serves as a yes/no switch of function in the peptide cell- signaling system. The functional significance of racemization is demonstrated by the fact that stereo-transformation modulates the peptide bioactivity in a motor circuit relevant to feeding motor behavior [75].
Growing evidence suggests a vital role of D-AAs not only at the cellular level, but also at the system level; this was shown for immune system [74,75,76,77,78], neuroendocrine system [64,79,80], neurotransmission [81], perception [82], and cognitive functions [83,84]. The ratio of D- AAs to L-AAs increases with the age of the fossil [85] due to the spontaneous racemization VI. (VI. From a physical point of view, racemization is considered because of phase transition between the R and S enantiomers [22,86]).
The half-life of the spontaneous and enzymic PTM racemization can range from several days to 100,000 years [87,88,89].
The racemization has a relevance to the protein/organism aging and age-associated diseases [18], and protein aggregation. Proteins containing D-β- aspartyl (D-Asp) residues were observed in various tissues including cardiac muscle of the heart, blood vessels of the lung, chief cells of the stomach, longitudinal and circular muscle of the stomach, small intestine and large intestine [90]. The presence of free D-Asp in the CNS of rodents and humans was studied [91]. L-Serine (L-Ser) is a major brain metabolite covering functions “from one C-metabolism to transsulfuration, to phospholipid/phosphoprotein function, and to D-serine biosynthesis [92].” The elevated and reduced D-serine level correlates with the progression of many neurological diseases including AD and schizophrenia. D-serine (D-Ser) VII (non-essential AAs are available from the plant-based diet) is abundant in many regions of CNS including forebrain [93,94] cortex, hippocampus, hypothalamus, amygdala, and cerebellum [95]. (VII. Serine (Ser) is a non-essential nucleophilic α-AA, encoded by the codons UCU, UCC, UCA, UCG, AGU, and AGC, used in the biosynthesis of proteins [96,97]).
The distribution of D-Ser and corresponding PLP enzymes suggests an influence on cortico-limbic brain functions [98]. D-Serine (D-Ser) is an endogenous AA implicated in the metabolism of neurons [99], astrocytes [100], oligodendrocytes [101], and microglia cells [102,103] via the variety of signaling pathways. D-Ser and D-Asp were identified as the neurotransmitters. D-Ser is an endogenous co-agonist of the N-methyl-D-aspartate (NMDA) type glutamate receptor at the glycine site [104,105].
Competitive antagonist of AMPA receptor [106] is a key receptor of excitatory neurotransmission in the brain. D-Ser interaction with APP is an essential modulator of the synaptic spine plasticity [107]. In the CNS, D-Ser has a dual (neuronal and glial) origin [105,108]. In addition, D-Ser mediates neurogenesis [109], cellular migration [110], cell proliferation [111], cell death [112], neurotoxicity [113], Neurodegeneration [105], respiratory regulation [114] cardiac activity [115], olfactory perception [116], neuro-endocrine functions [64], immune system [117], learning/memory faculties [118,119] and motor behavior [120]. Proteins and peptides containing D-AAs play an important role in age-related alterations [121,122,123,124,125]. D-glutamic acid (D- Glu) was not believed to be present at any significant level in the brain [84]. However, this was contradicted by the numerous new findings. D-AAs appear to participate in the major biological and neurological mechanisms. D-AAs have been detected in a variety of animal cells’ peptides; these include opiate and antimicrobial peptides from frog skin, neuropeptides from snails, hormones from crustaceans, and venom from spiders. Mammalian hormones and signaling neuropeptides are known as the subject of the functional post-translational racemization (PTM) [126]. However, despite the obvious significance, the role of peptide racemization in cell signaling, aging and neurodegeneration remains the terra-incognito [127]. The presence of D-AAs is detected in brain tissues, cerebrospinal fluid [128], and blood [84,129,130]. Recent measurement suggests that AA levels in brain tissue are typically about 10 to 2000 times higher than in blood [130]. Comparative measurements show that most D-AAs present in the hippocampus are significantly higher in the cortex. Regardless of brain region, the changes in AA chirality cause changes in protein structures (chirality transfer) including forming alpha-helical and beta-sheet structures resulting in changes in metabolic activity and function [19,21,131]. All changes in protein synthesis and degradation are accompanied by sequential spatial (chirality-dependent) transformations. Chirality is also a critical feature of molecular recognition that affects neurotransmission, enzyme activity, and immune functions.
Notably, the processes of protein synthesis and degradation are accompanied by the sequential spatial transformations. D-AAs in organisms are not metabolized by the same pathways as L-AAs and are usually removed by the kidney. In the CNS, an autophagy is known as a pathway for degradation of protein aggregates [132]. It was shown that autophagy, associated with the ubiquitinated aggregates of proteins, was attenuated by a D-Ser in an N-methyl-D-aspartate receptor (NMDAR) pathway [133].
2. Chirality at Protein Level: Role in PTMs
2.1. Protein Racemization, Aging, Folding, Aggregation, and Degradation
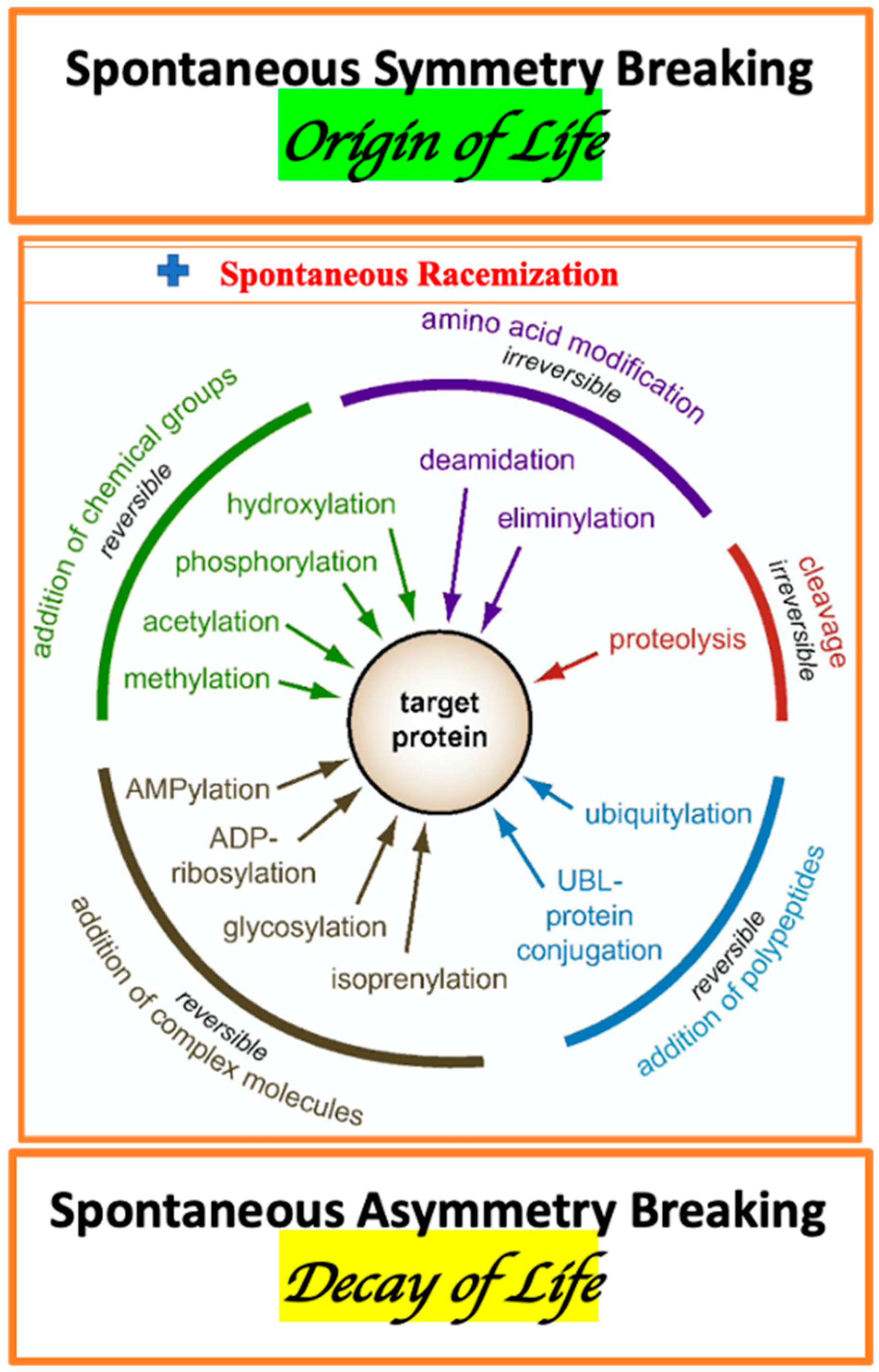
The enormous complexity of a living organism, as the essential elements, includes: AAs metabolism, diversity of membrane- and cytosol-associated proteins, variation of proteins stereo-forms, and multiplicity of enzymes of PTM. The traditional view on PTMs should be complemented by the consideration of the spontaneous, irreversible protein conformations associated with AA racemization (Figure 1) [134].
The essential role of AAs is evident beginning from asymmetric cell division [135]. Biological racemization and isomerization are driven by the interplay of spontaneous and enzymatic mechanisms of PTMs. Enzymatic racemization, to a significant degree, is induced by external factors [136]. Three specific and interconnected forms of PTM such as AA racemization (AAR), isomerization (AAI), and phosphorylation (AAP) are routinely used as biomarkers (BM) of peptide degradation and protein aging [137] and aggregation [10]. The bio-catalysts, which decrease the energy barrier for the phase transition, accelerate racemization rate at least by 104–105 times [37]. A relevant example of such catalysts is serine/threonine phosphatase/kinase [138]. Notably, the majority of kinases act on both serine and threonine residues [139]. L-Ser as a central metabolite in cell biology [90] and phosphorylation is a major mechanism of activating/inactivating enzymes, explaining the role of protein kinases in signaling pathways. Due to the above-mentioned facts, we will focus primarily on the role of Ser residue VIII in the PTM of proteins. (VIII. The role of D-aspartate (D-Asp) in racemization is covered in a recent review [67]).
The metabolism of polar AA D-serine (D-Ser) is highly cell-, organ- and brain region-specific [140].
D-Ser metabolism in the brain is regulated by number of enzymes from which we are targeting to enzyme related to racemization and phosphorylation. IX (IX. Notably, serine is degraded by hydroxymethyltransferase to glycine. Their role in living organisms is determined by the ability to catalyze a wide range of biochemical reactions including deamination and racemization [141]).
Pyridoxal phosphate (PLP) enzymes have multiple evolutionary origins [142]. PLP-dependent enzymes exhibit unique catalytic versatility.
2.1.1. Pyridoxal Phosphate Enzyme
PLP enzymes X are involved in the biosynthesis of protein, glucose and lipid metabolism. We will focus primarily on the Ser racemization. (X. “The functional specialization of most B (6) enzymes seems to have already occurred in the universal ancestor cell before the divergence of eukaryotes, archaebacteria, and eubacteria 1500 million years ago” [142]).
The phosphate ion acts as one of the strongest modulators of biomolecular chirality, including Ser-residue. XI (XI. Phosphoric acid contains a four-coordinated phosphorus atom. Such molecules are tetrahedral. The four s-bonds with sp3 hybridization of the electron orbitals has tetrahedral orientation).
The stereo-configurations of Ser residues are sensitive to the effects of metal ions [143], and di-hydrogen phosphate ion {H2PO4 1-} [18,144]. Under the influence of the variety molecular environments, Ser undergos racemization as internally bound residues of peptides and proteins, providing an opportunity for the normal and pathological protein degradation and for appearance of D-enantiomers in mammalian cells [145]. However, the research devoted to neurodegenerative diseases has not studied the involvement of Ser racemization (along with the other forms of PTM) in the pathological protein misfolding and aggregation. In this review, we illustrate how current studies have examined racemization. The five families of PLP enzymes include: type I—aspartate aminotransferase family, type II—tryptophan synthase family, type III—alanine racemase family (TIM-barrel), type IV—D-amino acid aminotransferase family, type V—glycogen phosphorylase family [146]. The functions of PLP include influence on pi-electron systems and the chemical properties of contiguous sigma bonds [147]. The PLP acts as a coenzyme in all transamination reactions, and in certain decarboxylation, deamination, and racemization reactions of AAs. The aldehyde group of PLP forms a Schiff-base linkage (internal aldimine) with the ε-amino group of a specific lysine group of the aminotransferase enzyme. Trans-amination is involved in the ketamine production [148]. PLP is also involved in various beta-elimination reactions such as the reactions carried out by serine dehydratase [149]. Among the functions relevant to PLP activity are the following: (a) to react with glutamate, which transfers its alpha-amino group to PLP to make pyridoxamine phosphate (PMP) and (b) to provide the catalytic functions for PLP enzymes including serine racemase (SerR).
2.1.2. Serine Racemase
The attention to the significance of racemization in neurodegenerative diseases [17] and its association with proteins’ aggregation emerged a long time ago. Due to the chain of essential facts, we have mainly concentrated on the racemization XII of Ser residues in proteins involved in the neurodegeneration. (XII. The association of serine/threonine phosphorylation with protein disorder is a common landmark of neurodegeneration [150,151,152,153,154]).
Among them are: APP(Aβ) [155,156], TAU [157,158,159], α-Synuclein (α-Syn) [160,161], and prion protein (PrP) [160,162] containing Ser residue in the AAs monomer sequence. An increasing number of experimental findings proves an assumption of the pivotal role of serine racemase (SerR) in the neuronal activity and neurodegeneration. Racemization of protein-bound AAs (including Ser) is important in the aging and pathologies of proteins. Ser undergoes racemization as internally bound residues of functional proteins [163]. Racemization of Ser and Asp residues differently impacts the hydrolysis of proteins. Serine racemase (SerR) is the brain-enriched glial (astro-and micro-glia) cells PLP enzyme [145,164,165,166] which catalyzes racemization of L-Ser to D-Ser. The catalytic mechanism of the SerR is similar to the alanine racemase. The unprotonated PLP-substrate intermediate is stabilized by the interaction of active-site residues with water molecules, contributing to the enzyme’s electrostatic environment. SerR is a homodimeric pyrixidal 5′-phosphate (PLP) dependent enzyme catalyzing beta-elimination of both L- and D-serine to pyruvate and ammonia [167]. The homo-dimer of SerR (each monomer 340 amino acids) consists of two domains (a small and a large) connected by a flexible loop [168,169]. Both mouse and human SR contains functionally active Ser residue (ValSerCys sequence) at their C-terminus [92,169,170,171]. This fact suggests that SR activity itself can be modulated by non-enzymic L-serine racemization. The ValSerCys sequence resembles the (PDZ) domains for binding to PSD95 [170]. SerR is activated by binding to the PDZ6 domain of Grip. This complex of molecular interactions represents the pathway for modulation of synaptic spine activity through PSD95. Full activation of SeR requires binding to the remaining part of the C-terminal region of GRIP [170]. The combination of above-mentioned facts provides the idea of the multiple pathways connecting Ser racemization with synaptic spine function through PSD95 [172,173]. It is notable that substrate of the SerR-protein PSD95 contains multiple Ser residues as active sites of PTM. Two evolutionarily conserved sites of serine phosphorylation (Ser-415 and Ser-418) signify the sensitivity of PDS95 signaling system to Ser racemization. [174]. The association of D-Ser and SeR with PSD-95 maintain an overall stability of glutamatergic synapse [175,176]. Ser-R regulated by many cofactors including phosphorylation [98]. Experimental results indicate that PKC phosphorylates SerR in serine residues and regulates D-Ser availability in the brain [177]. In more general terms, the inherent interaction between racemization and phosphorylation, in our view, is relevant for the regulation of physiological and pathological mechanisms of protein folding. XIII (XIII. The role of glycosylation in protein folding has been considered in a literature review [178]).
This hypothesis is supported by the fact that A-Beta aggregations into filaments become irreversible due to the combined force of several PTMs. The interplay between racemization and phosphorylation promotes incorporated A-β dimers and tetramers into resistant to proteolytic degradation filaments [179].
PLP enzyme serine racemase (SerR) catalyzed D-Ser synthesis [180,181] and D-amino acid oxidase (D-AAO) catalyzed D-Ser degradation [182]. SerR, in addition, degrades L- and D-Ser to pyruvate and ammonia. As a residue prone to racemization and phosphorylation Ser is a primary suspect in protein aggregation. The activation barriers of Ser racemization, estimated in the presence of dihydrogen phosphate ion (H2PO4-), found were consistent with spontaneous rate of reactions occurring at physiological temperature [36,183]. The AAs racemization is driven by spontaneous (non-enzymic) and enzymic process. The rates of AAs racemization in proteins are temperature/protein-dependent, and usually slow under physiological conditions. The progress in the measuring of AAs racemization rate elaborates the concept of protein aging. For aspartic acid (Asp) the rate of racemization occurs over the range from several days to more than 15,000 years [18,38,85,88,184,185,186]. The L-D conversion of aspartic acid in the proteins of human dental enamel (such as dentine) is relatively fast (about 8% conversion in 60 years) and correlates with a chronological age of the organism [187]. The Asp racemization was seen during ageing and cataract formation [188]. Due to a well-known succinimide-mediated mechanism, the Asp residues are the most racemization-prone [189,190,191,192]. Ser is known as one of the main AAs involved in racemization [145,193]. Accordingly, within the lifetime, these AAs residues of long-lived proteins (LLPs) are progressively racemized [18]. In the age-related diseases, this racemization process can be related to protein misfolding and dysfunction [194,195,196]. The idea of conjugality of SerR activity and APP-related AD pathology is supported by the fact that the level of a brain serine racemase expression can be induced by several seine-containing peptides including the APP fragments such as sAPP [197] and pro-inflammatory stimulus including Aβ peptide [198]) and AP1 [197,198,199]. SerR is a component of the complex network PTM. The Ser residues of SerR are the targets of several protein kinases including PKC and PICK1 [200].
2.1.3. Serine Protease
Most of the metabolic enzymes recognize only substrates (proteins and peptides) composed exclusively of L-AAs [10]. Serine proteases (SerPs) are ubiquitous in all organisms. Insights into the atomic level of SerPs structure–function link reveal the significance of the catalytic Ser motions [201,202]. Notably, an evolutionarily conserved catalytic domain Ser–His–Asp contains Ser residue [203,204] providing sensitivity to the AAs racemization. It is in the agreement with a well-known fact that incorporation D-AAs into peptide chain diminishes their susceptibility to proteases [205]. The combination of experimental facts, as mentioned earlier, emphasizes a critical role of racemization on enzyme–substrate interaction. The functions of SerPs are closely associated with the degradative pathway of many PLP enzymes [206,207]. SerPs, representing about one-third of all proteases, serve as essential component of the intracellular and extracellular catalyzing hydrolytic reactions [208]. SerPs participate in many physiological processes including food digestion, embryo development and immune defense [209]. The fact that SerR is degraded through the ubiquitin-proteasomal system [210] and regulated by phosphorylation [180] points to crass-talk between Ser-associated forms of PTM emphasizing the physiologic importance of Ser residues. Quantum calculations reveal the mechanism of SerPs action including four specific residues in a water-containing environment [211,212,213]. SerPs enzymes are involved in the proteolysis of the diverse group of signaling peptides and functional proteins [204] including Aβ [214], APP XIV (XIV. Recent studies have reported that many proteases, besides the canonical α-, β- and γ-secretases, cleave the APP [214], Aβ peptides [215], TAU, and tubulin [216,217,218]).
In humans SerPs comprise several groups: plasmin, acylpeptide, hydrolase, and myelin basic protein (MBP) [215,219]. The group of rhomboid SerPs belongs to the family of intramembranous proteases XV (XV. BACE1 is, known as membrane-associated aspartic protease 2 [220] and plays a key role in major cellular processes [221,222,223]).
SerPs are involved with the degradation of aberrantly folded proteins [222,224]. Notably, the sequential cleavages of APP occur by β- and γ-secretases. Both secretases are members of a new class of intramembrane-cleaving proteases (I-CliPs). These proteases include β-secretase 1 (BACE1) the Rhomboid family of SerPs, and two aspartyl proteases: the signal peptide peptidase (SPP) and γ-secretase. “In sharp contrast to Rhomboid and SPP that function as a single component, γ-secretase is a multi-component protease with complex assembly, maturation and activation processes” [221]. Aβ peptides are subject to proteolytic degradation by a family of peptidases and proteinases known under the common name Aβ-degrading proteases (AβDP) [215]. Among them are SerPs, which are ubiquitous in all organisms. As most proteases SerPs are chiral, meaning they distinguish between L- and D-enantiomers of the substrate. Apparently (based on the summary of experimental facts) that SerPs activity and protein folding and aggregation can be strongly affected by the Ser racemization.
2.1.4. D-Amino Acid Oxidase
The metabolism of D-AAs in a healthy organism is modulated by two stereo-specific enzymes: D-amino acid racemase (in the synthesis), and D-amino acid oxidase (D-AAO) XVI in degradation. (XVI. D-AAO regulates NMDA receptor function through AAs).
As a detoxification enzyme, the D-AAOs (in the presence of molecular oxygen) selectively degrade (by oxidative deamination) only D-enantiomers [28]. DAAO is involved in many aspects of cell physiology. As a detoxification enzyme, the D-AAO (in the presence of molecular oxygen) selectively degrades (by oxidative deamination) only D-enantiomers. D-AAO involved in many aspects of cell physiology [225]. As a D-AAs degrading enzyme, D-AAO is associated with many disease conditions including amyotrophic lateral sclerosis [226] and schizophrenia [227]. In the human brain, DAO expression was found to be both age- and brain region-dependent [140,228].
2.2. Protein Aggregation and Neurodegeneration
The current review focuses on the most common and basic mechanism of protein aggregation-molecular chirality and racemization. Protein aggregation is a prominent feature of many protein misfolding diseases causing neurodegeneration. Among them are Alzheimer’s (AD), Parkinson’s (PD), Huntington’s (HD) diseases, amyotrophic lateral sclerosis (ALS), Lewy Body Dementia (LBD), progressive supranuclear palsy (PSP), spongiform encephalopathies (SE), cataracts, musculo-skeletal disease (MSD) and demyelination diseases (DD) [229,230,231,232,233,234,235]. In our view, racemization should be considered as a common and critical factor of protein conformational stability, potency to aggregation and toxicity [105]. In this review, we focus predominantly on AAs racemization. The first observation of AAs racemization was reported a century ago. The review of the earlier works can be found in [236,237]. AAs undergo spontaneous and catalytic racemization. Two distinct forms of catalytic racemization are base- and acid-catalyzed [19]. In the 1970s–1980s, racemization was used to determine the age of AAs in the biological systems [238,239,240,241]. However, the association of AAs racemization with the pathological protein aggregation and the neurodegeneration, during this period, did not attract attention. Structurally ordered protein aggregates (amyloids) are found in all living organisms including the bacteria [7], plants [242] and animals [243]. Contrary to the common view, in humans they are involved not only in the aggregation-related diseases but also in normal physiological activities associated with cognitive function.
2.2.1. Structurally Ordered Proteins
The comparative studies of amyloids structures (fibrillar, cross beta-sheet quaternary forms) in the bacteria, fungi, insects, invertebrates, and humans reveal two sub-sets of fibrils: pathological and functional [244,245]. New findings suggest that the current knowledge regarding the variety of structural conformations of Aβ is far from complete and probably not enough for the development of an efficient therapeutic strategy. The existence of micelles in the fibrillo-genesis of beta-amyloid peptide was proved by experimental results [246,247,248]. From the bio-physical point of view, it is the spatial distribution of positively and negatively charged domains over surface of protein and spatial orientation of electron spin that tunes the aggregation behavior of proteins [249,250]. Among the broadly studied protein aggregations are inclusion bodies, amyloid fibrils, and other misfolding aggregates. Most protein aggregates contain the secondary structural components such as helical and β sheets elements [251]. The primary hypothesis assumes that aggregation involves the partially folded intermediates and specific intermolecular interactions (molecular chaperones).
2.2.2. Intrinsically Disordered Proteins
The elegant results of such attention to the nature of non-equilibrium phase transitions in proteins is the concept of intrinsically disordered proteins [234]. Intrinsically disordered (ID), intrinsically unstructured protein (IU), or natively unfolded (NA) protein or domain lack a unique three-dimensional structure and exist in a variety of conformations that are in dynamic equilibrium under physiological conditions. It was recognized that some functions of proteins can be associated with the dynamically unstructured conditions. About 40% of eukaryotic proteins have at least one long (>50 AAs) disordered domain [253,254,255]. The mutations within intrinsically disordered regions (IDRs) increase the aggregation propensity, such as those seen in the amyloid β-peptide, α-synuclein, huntingtin, prion protein, and TAU have been directly linked to variety of above previously mentioned IDs. TAU is most studied IDP [256] but unfortunately the experimental design and analysis of experimental results frequently do not involve consideration of D-AAs residues. In this situation, the classification of IDP based on the AAs sequences reveals the roles of L- and D-Ser in the structure and functions relationships [257,258]. Recently the theoretical framework was introduced for the use of D-amino acids as a universal tool to the exploration the aggregation pathways of IDPs [11]. The study of non-equilibrium phase transition [22] in IDP and the role of L- D- AAs substitution is a matter of urgency. The rate of racemization of amino acids (AAs) is temperature dependent and under influence of external physical fields can be altered in the order of 104–105. The balance between physiological protein folding and aggregation relies on the competition between two pathways. The factors promoting aggregation prevent natural folding and vice versa [251]. The chirality of amyloid fibrils is well established [259,260]. The earlier intuitive ideas on the link between the spontaneous phase transition (chirality transfer and chirality inversion) between the polymorphic forms of the amyloid fibrils XVII and protein aggregates have gradually gained objective confirmation [18,260,261,262,263] (XVII. The supramolecular chirality of the amyloid fibrils can be registered by variety of the methods including the microscopy (electron (EM), transmission (TEM), and scanning electron (SEM) microscopy) and vibrational circular dichroism (VCD) [261,262,263,264]).
2.2.3. Racemization Role in Protein Folding, Aggregation and Neurodegeneration
Several authors suggested that the “presence of the isomers may be one of the triggers of abnormal aggregation and may induce the partial unfolding of protein leading to a disease state” [88,90]. Recently, it was shown that amyloid fibrils of different nematic phases, including chiral protein-based systems, undergo liquid-crystalline phase transitions [265]. Spontaneous and enzymatic racemization reactions influence protein misfolding and aggregation associated with aging and age-related diseases [142,180]. The exploration of the AAs racemization [28,266] and protein aggregation phenomena within the bacteria cells opens an evolutionary perspective [267].
The amyloid-like properties of inclusion bodies and protein aggregation in bacterial cells have become the point of attention [268,269]. Spontaneous and enzymatic racemization reactions have relevance to the protein misfolding, aggregation associated with aging, and age-related diseases [267]. Gene mutation and spontaneous racemization of Aβ, TAU, PrP, Huntingtin, and alpha-synuclein proteins are determined as major limiting factors in natural peptide synthesis and incorporation of the peptide into functional proteins, leading to abnormal phosphorylation, aggregation, and deposition [14,270,271,272,273]. The investigation of the biochemistry of mandelic acid-base molecular structures reveals thhe effect of relative chirality of monomers on the aggregation patterns. The structure of dimers and supramolecular aggregates is strongly affected by the relative monomer chirality [274,275,276]. At the molecular orbitals level, the transmission of chirality occurs through the cooperation of hydrogen bonding and π − π stacking interactions [271,275]. The recent studies of L- and D- (Aβ) 42 peptide enantiomers confirm an assumption that the chirality of AAs is the key determinant of the oligomer’s solubility and aggregation [277,278].
3. Hypothesis of Protein Aggregation and Neurodegeneration
The metabolism of Aβ peptides (full length and the truncated forms), despite being a major target of neurodegenerative studies, remains to be elucidated. The various PTMs of Aβ peptides were explored and discussed during recentdecades, including racemization/isomerization, oxidation, nitration, truncated reaction, glycation, and glycosylation. However, “no common perception of the essential foundation of the AD pathology was determinate” [131]. The overall structure of Aβ-40 and Aβ-42 peptides contains the distinct domains characterized by specific physicochemical properties and 3D structures [279]. In particular, Aβ-42 peptide contains hydrophobic regions (such as KLVFF residues 16–20) [280] and α-helical (right-handed) domains (13–26 residues) [281]. The intuitively attractive therapeutic strategy against amyloid-beta aggregation is based on assumption that drugs should exhibit molecular chirality [280]. In other words, the folding pathway of Aβ-42 is valued for its sensitivity to the chirality of the immediate molecular environment. The chirality of molecular environment, in turn, is mediated by the mechanism of PTM. Therefore, the concept of AAs racemization allows the confluence of several hypotheses of protein aggregation and neurodegeneration.
3.1. Amyloid Cascade Hypothesis
The amyloid cascade hypothesis links the misfolding of the Aβ peptide to the cause of AD [282]. The spatial conformations of Aβ peptides are peptide-length specific [283,284]. The aggregation is contributed by multiple pathways directly related to the stereochemistry of the Aβ peptide or indirectly through interaction with aggregated TAU protein [284,285,286] and membrane lipids [287,288]. The Aβ peptide is prone to aggregation through calcium dysregulation [289], oxidative stress [290], phosphorylation [152,291,292], and inflammation [293]. The amyloid cascade hypothesis emphasizes the role of amyloid- β (A-β) peptide aggregation in the pathogenesis of AD [294].
3.2. Glutamate Toxicity Hypothesis
The excitotoxicity of extracellular glutamate was associated with the numerous neurological diseases including ALS, AD, PD, HD, LBD, PSP, and cataracts. We will return to this hypothesis at the consideration of link between D-Ser and functions of glutamate receptors including NMDAR [295], mGluR [296], and AMPAR [106].
3.3. Post Translational Modification Hypothesis
The various PTMs of Aβ peptides were explored and discussed, including oxidation, nitration, truncated reaction, glycation, and glycosylation [297}. However, “no common perception of the essential foundation of the AD pathology was determinate” [130]. The Aβ is the product of the normal proteolytic processing of AβPP, a type 1 trans membrane glycoprotein [298] whose gene is located on chromosome 21 [299,300]. The class of intrinsically disordered (ID) amyloid peptides and proteins includes Aβ, TAU, islet amyloid polypeptide, and α-Synuclein [301,302,303,304,305]. Amyloid consists of linear, unbranched protein or peptide fibrils of approximately 100 Å diameters.
The fibrils are composed of a wide variety of proteins that have no sequence homology and no similarity in three-dimensional structures. However, fibrils share a common secondary structure, the beta-sheet [302,303,304,305,306]. PTM in general and in racemization increases the heterogeneity of protein conformation and, consequently, the diversity of a protein’s physiological functions and pathological pathways. The illustration of protein chirality-related effects is the physiological stereo transformations of the amyloid precursor protein (APP) and TAU. The processing of APP and PTM of amyloid-beta (Aβ) peptides along with the oxidation, phosphorylation, nitration, pyroglutamylation, and glycosylation include racemization and isomerization [307,308]. The racemization of Aβ peptides generates APP fragments with different physiological and pathological properties modulating disease progression. It is important to emphasize the interconnected chain of events.
- (i)
- The accumulation of Aβ and TAU trigger the perturbations in the glutamatergic synapse.
- (ii)
- (iii)
- (iv)
- (v)
- (vi)
- SerR is transcriptionally induced by sAPP [197].
- (vii)
- The NMDA receptor hypofunction is associated with aging, neurodegeneration leading to the impairments of memory, learning and psychosis [316].
- (viii)
- Modified form of TAU in PHFs contains more D-Asp that TAU proteins from normal adult brains (N-TAU) [317]. The chain of the physiological molecular events is inherently linked to the mechanism of racemization. The concept of AAs racemization allows the confluence of three above-mentioned hypotheses of neurodegeneration and protein aggregation. In our view, the racemization is the common relevant factor for the widely circulating hypotheses including the amyloid cascade hypothesis, glutamate toxicity hypothesis [229,231] and hypothesis associated with the functions of PTM network. In support of universal significance of AAs chirality and racemization is speaking the facts that a gradual racemization of peptide and proteins has been observed in aging populations [318], and that mixed chirality proteins evade the known pathway of proteosomal degradation [319]. Notably, the age-related racemization of AAs is critical for function of both the enzymes and their substrates.
4. Racemization Role at Molecular, Cellular, and System (Organ) Levels
4.1. Molecular Level
Aberrant PTM Resulting in Resistance to Proteolytic Degradation
The mechanisms of protein modifications comprise co-translational, post-translational and spontaneous types. The stereo selectivity of the translational machinery of protein synthesis provides reliable defense against the accidental incorporation of D-AAs. It was shown that chirality discrimination occurs at three successive steps (initiation, elongation, and termination) involving tRNA and ribosomal interaction [320,321,322].
The homochirality sustained by the translational machinery provides the platform for the activity of the post-translational modification (PTM).
Following the translation of polypeptide chain, XIX determined by the DNA, most proteins undergo evolutionarily conserved PTM (XIX. Even changing just one AA in a protein’s sequence can affect the protein’s overall structure and function).
For example, phosphoserine is a component of many proteins as the result of post translational modifications [323,324]. Neuronal protein phosphatases in cell signaling pathways are represented by phosphoserine phosphatases XX (PSPs) [325]. (XX. Full activation of SerR requires binding to the remaining part of the C-terminal region of GRIP [170]).
There are several mechanisms of PTM including: covalent modifications (phosphorylation [326,327], methylation [328], glycosylation XXI [329,330]), proteolysis [331], oxidation [332,333,334], deamination [335], cross-linking [336,337], and racemization (enzymic and non-enzymic). The phosphorylation of D-AAs residues is a common way to regulate the activity of proteins. (XXI. “Glycosylation is one of the most common, and the most complex, forms of post-translational modification of proteins” [337]).
The source of phosphate for phosphorylation is ATP. The cross-linking PTM is observed for APP and TAU proteins [337]. The variety of forms of PTM is considered as the mechanism of adaptation to the stereochemistry of the environment. For the purpose of our review, it is essential to note that PTM was linked to abnormal deposition of peptides in the brain tissue [338]. The structural heterogeneity of peptide in the aggregations was associated with structural rearrangements of the L- and D- isoforms of aspartyl residues. The localization of D-AAs in peptide chain (N- and C-terminus, or intermediate position) provides an opportunity for modulation of diverse pathways of PTM. All known mechanisms of PTM directly or indirectly involve racemization. D-AAs containing peptides (characterized by altered 3-D shape and charge distribution) show an increase in resistance to proteolytic degradation of molecular aggregation comprised of insoluble depositions [331,339,340] The stereochemical nature of PTM is most evident in the case of racemization. The modulation of AAs chirality influences the spatial transformation of proteins and distribution of hydrophobic/hydrophilic domains. The increase in “hydrophobicity” results in deposition from aqueous media [341]. This in turn changes the balance of soluble and insoluble components in the cytoplasm and in the intracellular space. The physiology of phosphorylation, oxidation, glycation, and ubiquitination is inevitably influenced by the age-associated, cell-specific racemization [36,100,342,343,344,345,346]. Thus, racemization of AAs could be a common mechanism for many pathogenic pathways. Protein misfolding and aggregation are responsible for the brain neurofibrillary tangles (NFT) and neuritic plaques (NP). Protein aggregates and excitotoxicity, representing the common landmarks of major NDs including ALS, AD, PD, LBD, PSP, HD and cataracts, are inevitably linked to the AAs racemization. For the heat shock proteins in the lens (αA-crystallin (αA) and αB-crystallin (αB)) it was shown that an aggregation and deposition is significantly contributed by several types of PTM. Among them are oxidation, C- and N-terminal truncation, deamidation, phosphorylation, and methylation [333,334]. As we mentioned before, many forms of PTM are directly associated with the protein racemization [194].
4.2. Cellular Level
4.2.1. D-Seine and NMDA-Dependent Neurotransmission
NMDA receptor and corresponding neurotransmitters are one of the best examples of stereoselective interaction. D-AAs (including D-Ser and D-Asp) are involved in many aspects of the brain’s excitatory and inhibitory neurotransmission [67]. For example, in neurons D-Asp serves as a neurotransmitter delivered to NMDA receptor site in synaptic vesicles [67,347,348]. The convincing way to illustrate the role of AAS chirality in the glutamatergic system is to review NMDA receptor ligands.
NMDA receptor agonists and partial agonists include: L-glutamate, D-glutamate, N-methyl-D-aspartate (NMDA), N-methyl-L-aspartate, D-aspartate, L-aspartate, and many others [60,66,67] The co-agonists of NMDA receptors include D-serine, L-serine, D-alanine, and L-alanine [62,63]. In the central excitatory and inhibitory synapses of the mammalian brain, L- and D-isoforms of Ser play a key role in signal transduction [193]. D-Ser participates in the synaptogenesis, synaptic transmission (NMDA and AMPA [106]), synaptic remodeling [349], and spine plasticity [107,350]. In the tripartite synapse, the downregulation of neuronal D-Ser levels under any pathological conditions is naturally associated with an enhanced production and release of D-Ser by astrocytes. The physiology of neuronal-astroglia loop is regulated by the interplay of the enzymes including SerR and D-AAO. The disruption of the natural feedback mechanisms regulating cell–cell and enzyme–enzyme interaction can accelerate the neurodegeneration [351]. As an example, we can point on moto-neuronal death in the mouse model of amyotrophic lateral sclerosis [352]. Astrocytes (as the source of D-Ser) possess the vesicles sequestering and storing D-Ser as gliotransmitter [353,354] The Ser-containing vesicles undergo calcium-dependent exocytosis modulating synaptic NMDA transmission. The activation of opening of the NMDA receptor requires coincidence in occupation of the glutamate and the glycine site. At the post synaptic dendritic spines, an NMDA-dependent endocytosis of GABAB receptors requires the phosphorylation of its intracellular C terminus domain serine 867 residue (Ser867) in the intracellular C terminus [355]. The attenuation of the neuronal nicotinic acetylcholine receptors (nAChRs) by receptor antagonist alters the function and expression of SerR [356] suggesting involvement of cholinergic circuits in modulation of D-serine level. The review of current publications related to the mechanism of Glu-receptors internalization suggests an active role of D-Ser in mediating NMDA and AMPA receptors endocytosis [357]. Pathological roles of free extracellular D-Ser mediating NMDA receptor overactivation are suggested in studies using in vitro culture systems [358]. The internalization of cell membrane receptors involving Ser activity is observed in many cell types. We provide several examples. (1) In liver, parenchymaglucagon-mediated internalization of the serine-phosphorylated glucagon receptor is mediated by serine-phosphorylated residue [359]. (2) Phosphorylation Ser-789 in the C-terminal tail of fibroblast growth factor receptor 1(FGFR1) is required for receptor endocytosis [360]. (3) The low-density lipoprotein receptor-related protein (LRP), which participates in endocytosis, signaling pathways, and phagocytosis of necrotic cells, is mediated by phosphorylation of the serine residues within the LRP receptors cytoplasmic domain by PKCα [361]. (4) The carboxyl-terminal Ser residues (Ser-355, Ser-356, and Ser-364) play a critical role in G protein-coupled receptor kinase (GRK)-mediated phosphorylation and desensitization of β2-adrenergic receptors (β2Ars) [362]. (5) The endocytosis of CD4 (cluster of differentiation antigen is activated by Ser phosphorylation [363,364].
4.2.2. Racemization-Prone Ser Residues
The Ser residues appear more racemization-prone than other residues [5]. The racemization–deracemization dynamics are a natural discriminant between the healthy physiological state, aging, and disease condition [18,267]. For example, D-Ser promotes adult hippocampal neurogenesis enhancing cell proliferation and increase in the survival of new neurons [109]. At the same time, D-Ser is known as a key determinant of glutamate toxicity [352] and D-AAO enzyme-mediated metabolism, results in reduction in the reactive oxygen species (ROS) [365].
SerR/D-Ser/NMDA-receptor pathway is recognized as a regulator of apoptosis and necrosis shift during different forms of excitotoxicity involving microglia activation [102,366,367]. SerR belongs to the class of co-factor-dependent AA racemase enzymes. Accordingly, the activity of SerR is mediated by many co-factors including divalent cations (Mg. Mn, Ca, Fe Ni Cu, Co and Zn) [368,369], nucleotides (ATP, ADP or GTP) [180], and sulfhydryl groups [369,370].
The binding of ATP to serine racemase links the production of D-serine to the energy metabolism [157,371,372,373]. Consequently, racemization, of any origin (spontaneous and induced), will interfere with the cell aerobic metabolism [374]. The presence of D-AAs detected in plants, bacteria, and mammals is associated with the diverse range of biological functions [375]. “The levels of D-Ser in the brain are higher than many L-AAs and account for as much as one-third of L-serine levels” [376]. Free D-aspartic acid and D-alanine are found in the white and gray matter of healthy human brains [377]. d-Ser is known to be involved in glutamate transmission and plays a role in long-term potentiation [378]. D-AAs found in many AD-related proteins including neuronal-specific neurofilament-L [379], MBP [56], and in protein phosphatase PPC1 [380,381]. PPC1 and PPC2 are involved in TAU de-phosphorylation at multiple serine/threonine sites [382,383]. D-Ser was found to be involved in moto-neuron degeneration [384].
4.3. System Level: Morphological and Cognitive Aspects
4.3.1. Aging, Long-Lived Proteins (LLP), and Racemization
“D-amino acids …. play a role in aging-related diseases associated with gradual protein racemization” [318].
Brain laterality is a complex phenomenon widely studied at molecular, cellular, brain morphological and functional levels. Age-related bio-chemical alterations of brain laterality are region, cell-type, and molecular biomarker (type/function) dependent and vary from increase, decrease, and reversal of hemispheric asymmetry [18,385]. Molecular and cellular determinants of an organism aging are evident from asymmetric cell division in embryo [135]. The proteins with a long lifetime have recently become the subject of increasing attention. The nucleoporins [386] and myelin-related proteins of oligodendrocytes [387,388] were identified as the most long-lived proteins in rodent brains. Age-related reduction in D-Ser level with age has been associated with deficiencies in cognitive ability [18,388,389,390]. The intracellular, membrane-bound, and extracellular proteins with long lifetime have been linked to the age-dependent cellular and organism levels of events including fertility and neurodegeneration [387]. Among known LLPs are α-synucleins [391], APP [392], and TAU [393], PrP [394], huntingtin [235], MBP [387] and collagen [87,381]. An accumulation of the altered forms of functional LLPs with aging [18,395] as well as age-dependent protein racemization are considered well-established facts [252]. Aging, at a molecular level is considered a collapse of homochirality of the entire organism including the eye (lens, ciliary body, drusen, and sclera), skin, cardiac muscle, blood vessels of the lung, and heart, stomach (chief cells, longitudinal and circular muscles), small and large intestines, and kidney [396,397]. In Fujii expression, molecular chirality is an “index of aging”. LLPs containing D-AAs are present at many sites in the human body including CNS. However, little is known about the major pathways of PTM that affect protein structure and function in the brain and the studies of link between the racemization, aging and aggregation of proteins are practically absent [194]. The modification and aggregation of functional proteins can be significantly influenced by age-related accumulation of abnormal enzymes [398]. In the prefrontal cortex of mammals (mouse, rat, human) at 1/3 gestation period, more than 50% of the aspartic acid is in D-configuration [399,400]. However, at the time of birth it becomes undetectable. It has been suggested that there is a role for D-Ser in the mechanism of neuronal death in the nervous system [113] that is also associated with pathological protein aggregation. Over the lifetime, a stochastic process leads to alteration of molecular chirality at the DNA and protein levels. The “molecular clock” of aging is influenced by the complex of genetic and epigenetic factors [401,402]. Particularly, a gradual racemization of peptide and protein has been observed in aging populations [318,403] both in humans and animals [379].
Both the proteins relevant to AD (such as TAU and Aβ), have been shown to contain many racemized AAs in brain tissue from elderly human donors [404,405].
The racemization of AAs affects TAU proteolysis and aggregation [317]. Age-related neuro-degradation processes are, to a significant degree, associated with the age-related protein degradation leading to accumulation of misfolding, dysfunctional aggregates. However, it is essential to understand that “protein aggregation is a normal physiological event” with an evolutionarily conserved mechanism balanced by the proteins degradation system [406,407]. It is obvious that distortion of the protein degradation systems will inevitably accelerate neurodegeneration. Autophagy is one of the cell type-specific degradation systems. Autophagy can be up/down-regulated upon many factors including starvation [408] and dietary exposing to environmental toxins [409].
Notably, such toxins can include the environmental/dietary D-AAs. The idea that AAs racemization can modulate proteolytic protein degradation is supported by many facts, some of which are indicated below.
- I.
- II.
- III.
- IV.
4.3.2. Proteolysis vs. Aggregation: APP, Aβ and TAU
In the brain of AD patients, the Aβ peptide’s structural transition is initiated by the monomer to oligomer transition followed by conformation of the oligomers, protofibrils, fibrils, and plaques [3,421]. The detection of D-AAs in the A-β depositions and an affinity of A-β to D-peptides suggest the distinct role of L/D isomerism in the stages of the pathogenesis of neurodegeneration. The stages include a protein aggregation and plaque deposition [422,423]. D-AAs can be localized at the different positions in a peptide chain, including N- and C-termini. The D-AAs-containing peptides are resistant to proteolytic degradation suggesting the possibility of molecular aggregation and creating insoluble depositions [176,339,340]. The racemization of A-β and MBP was observed under different experimental conditions [101,424,425]. The increased level of DAAO was associated with the severity of the cognitive deficits in individuals with mild cognitive impairment and AD [83]. In brains of individuals with AD, D-Alanine (D-Ala) concentration is elevated more than twice [426]. D-Ser levels in the hippocampus and parietal cortex of AD patients are higher than in control subjects [427]. The comparison of physical and biological properties the all-D- and all-L-stereoisomers of Aβ (Aβ25-35) and the full-length peptide (Aβ1-42) reveal practically identical structural and assembly characteristics as well as similar levels of toxicity [428]. The deposition of abnormal protease-resistant proteins is presumably associated with the generation of D-AAs configuration [429]. The distribution of D- and L-aspartic and isoaspartic acids was studied in amyloid β peptides and TAU, designating new potential of the chiral biomarkers [430]. In 2006, Kokkoni et al. showed that ideal inhibitors of Aβ fibril are D-peptides [431] the conclusion is supported by later experiments [280,432]. The discovery of the effect of D-AAs peptides on beta-amyloid aggregation offers an attractive therapeutic strategy against protein misfolding diseases. Replacement of serine 422 with glutamic acid in TAU increases the propensity of TAU aggregation into insoluble fibril deposits of paired helical filaments (PHF) associated with neurodegeneration [433]. If we assume that D-AAs have function, then it is reasonable to link the decline of cognitive function with the changes in the balance between L- and D- AAs.
At present, the multiple isoforms of A-beta [434] and microtubule-associated proteins [435] are useful as biomarkers of neurodegenerative diseases. However, the potential usefulness of examining stereoisomers in protein synthesis and degradation pathways has far been under-appreciated. Recent developments reveal that the morphological and functional hemispheric lateralization and asymmetry originate from spontaneous intracellular symmetry breaking at the molecular and cellular level [436]. As a result, the primary physiological functions of the brain are asymmetrical between the left and right hemispheres. The morphological brain asymmetry correlates with cognitive functions [437,438]. A chain of lateralization is believed to originate from genetic as well as from epigenetic impact [21,439].
4.3.3. Proteolysis
The degradation of proteins is modulated by many PTM pathways including AAs racemization. The specific SerPs family (granzymes) which are expressed exclusively by cytotoxic T-lymphocytes and natural killer (NK) cells play a crucial role in apoptosis [419,420]. It was shown that the racemization results in the accumulation of altered proteins, accompanied by neurodegeneration [413,414]. In AD, the general acceptance of the amyloid cascade hypothesis coexists with the failure of Aβ targeting drug therapy. Resolving this situation requires a broader view on the link between the variety of protein stereo-transformations and the multiplicity of degradation pathways. For example, the heterogeneity of cleavage sites of APP leads to a variety of Aβ peptides forms, of which only a small part of each (such as Aβ1-40 (Aβ40) and Aβ1-42 (Aβ42)) have been currently studied at the stereochemical level [440]. Only close attention to the interaction between the cleavage site of substrate and active site of enzyme will provide an insight to the molecular mechanism of enzyme activity and bring the key for predictive drug therapy [441,442,443]. One of the studies of pathways of APP proteolysis XXII involves the sequential cleavage by two aspartic proteases: β- and γ- secretases. (XXII. Currently about 570 human proteases listed in the human Degradome Database).
However, it has become obvious that the β-/γ- secretases pathway of protein degradation represents only the “tip of the iceberg” complemented by many alternatives. Among them are the proteinase families of hydrolytic enzymes including SerPs, glutamic acid proteases, and metallo-proteinases. All SerPs enzymes (including trypsin, chymotrypsin, elastate, thrombin, subtilisin, plasmin, TPA, and factor D) contain a “catalytic triad” of Ser, His, and Asp. From this perspective, SerPs represent an attractive subject of exploration due to the combination of two facts: (1) their role in the lysosomal-endosomal protein degradation, and (2) expected effect of the racemization. However, SerPs role in the APP and TAU processing is not clearly understood. The SerPs (cathepsin A and G), aspartic proteases (cathepsin D and E) and cysteine proteases (cathepsins (B, C, L, F, H, K, O, S, V, X, and W) belong to proteinase families of hydrolytic enzymes. The proteinase families of hydrolytic enzymes are classified based on the mechanism of catalytic activity as aspartic, metallo, cysteine, serine, or threonine proteases [444,445]. The cathepsins are expressed in the brain in a cell type-specific manner. The activity of serine-cysteine protease was detected within the phagosomes of macrophages [446].
The enzyme activity is strongly influenced by the racemization of active AAs residues such as aspartic acid, threonine, and serine, causing AAs cross-linking and aggregation. It is believed that the deposition of abnormal protease-resistant proteins is associated with the generation of D-AAs [329].
4.3.4. Revision of Aggregation Hypothesis
“Protein aggregation may be exploited by nature to perform specific physiological functions” [406].
‘‘Replacement of serine 422 with glutamic acid in TAU increases the propensity of tau aggregation associated with neurodegeneration” [272].
“TAU Phosphorylation at Ser 422 is observed from the earliest stages of TAU aggregation” [447].
Close attention of researchers to the link between protein aggregation and PTM associated with the Ser residues is evident in the current flow of publications [262,406,447]. We mentioned before that the various PTMs of proteins and peptides (including Aβ) were explored regarding age-related degradation processes [297]. It is not surprising that all hypotheses of protein aggregation in AD are directly or indirectly associated with D-AAs metabolism. Among them are the following: amyloid [448], cholinergic [449,450], proteases [451], N-terminal [452,453], oxidative stress [454,455], branched-chain AAs [456], and amyloid-β crosslinking [457] hypotheses. In these circumstances, the racemization hypothesis of protein aggregation naturally gains its legitimacy. Recent results show that the mutation in D-amino acid oxidase (D-AAO) gene associated with familial ALS impairs D-Ser metabolism and causes protein aggregation [120], suggesting a close association between protein folding and D-AAs metabolism.
It is notable that D-Ser is predominantly released from glia cells (protoplasmic type II astrocytes). These cells enclose nerve terminals and are enriched in specific regions of the gray matter including cerebral cortex, hippocampus, anterior olfactory nucleus, olfactory tubercle, and amygdala [98,228,458,459]. In the brain of AD individuals, the chain of Aβ peptide structural transitions is initiated by the monomer to oligomer transition followed by the protofibrils, fibrils, and plaques formation [3,421]. The studies of the early stage of Aβ42 monomer aggregation reveals the co-existence of two distinct pools of stereo conformation: locally structured “A” and disordered “B” states [460]. The detection of D-AAs in the A-β depositions and an affinity of A-β to D-peptides suggest the distinct role of L/D isomerism in the stages of neurodegeneration. These stages include protein aggregation and plaque deposition [422,423]. D-AAs can be localized at the different position in a peptide chain, including N- and C-termini. The D-AAs-containing peptides are resistant to proteolytic degradation suggesting the possibility of molecular aggregation and insoluble depositions [321,339,340].
The increased level of DAAO was associated with the severity of the cognitive deficits in individuals with mild cognitive impairment and AD [83]. In brains of AD individuals with D-Alanine (D-Ala), the concentration is elevated more than twice [426]. D-Ser levels in the hippocampus and parietal cortex of AD patients are higher than those in control subjects [427]. Comparison of the physical and biological properties of all-D- and all-L-stereoisomers of Aβ (Aβ25-35) and the full-length peptide (Aβ1-42) reveals practically identical structural and assembly characteristics as well as similar levels of toxicity [428].
5. Treatment of Protein Aggregates
The discovery of the effect of D-AAs on Aβ aggregation offers an attractive therapeutic strategy against protein misfolding diseases. Notably, Ser is one of three AA residues (in addition to threonine, and tyrosine) commonly phosphorylated during cell signaling in eukaryotes. Phospho-serine is a component of many proteins as the result of PTM by various types of kinases (more than 50) [461,462]. Hyper phosphorylated TAU is the second major feature of AD. According to contemporary view the network of PTM of TAU protein (monomer) is causal for the assembly of monomers into diverse forms [159,463]. The major forms of aggregation are: oligomers, paired helical filaments (PHFs) and neurofibrillary tangles (NFTs). At a structural level, NFTs consist of PHFs. Among the different TAU isoforms are the neuron-protective and neuron-toxic subsets [464,465,466]. Side-specific phosphorylation of TAU can lead to formation of functional and neuro-protective (inhibits amyloid-β toxicity) iso-forms [465]. Neurodegenerative TAU-pathy is characterized by the hyper-phosphorylation of all TAU isoforms. The PHFs and NFTs do not play a role as the toxic entities leading to disease. The toxicity is ascribed primarily to the TAU oligomer [464]. Hyper-phosphorylated forms of TAU were identified in neuronal somata, neuropil threads, and plaque-like clusters of neuritis [467]. Ser and threonine residues are among the primary targets of phosphorylation. A serine/threonine-proline kinase phosphorylates TAU proteins stereo-transformation forming a paired helical filament [468]. The phosphorylation of TAU is required for hippocampal LTD [469]. TAU protein contains serine 202, 395, and 404 and threonine 205 and 394 residues as targets of differential PTMs [469,470,471]. The replacement of AAs in TAU increases the propensity of TAU aggregation [433].
In summary, the above-mentioned factors play a role in the evolution-supported association between biochemical events, behavioral patterns, and cognitive functions. The aggregation of the β-amyloid (Aβ) peptide into toxic oligomers is a key pathogenic event in the AD. The fact that dietary exposure to the L-Ser containing products reduces the risk of NFT and β-amyloid deposits in the brain suggests the essential role of AAs racemization on neurodegenerative diseases (NDs) [409]. The current strategy for prevention and treatment of existing protein aggregates and their toxicity is aimed at the stereoselective properties of the D-enantiomeric acids and peptides for TAU and Aβ [423,472] associated depositions.
The essential finding is that Aβ42 exhibits an affinity to the D-AAs peptides [472]. The molecules that interfere with aggregation and toxicity potentially may act as therapeutic agents for the treatment of the disease. Many D-AA peptides exhibit an ability to inhibit or promote protein aggregation depending on the binding site [473,474,475]. The studies of the molecular inhibitors of A-beta aggregation successfully used the short peptide fragments homologous to the specific fragment-sequence of full-length wild-type A-beta. It was shown that the effectiveness of the inhibitors is strongly attenuated by replacement of L- to D-AAs or methylation of Aβ fragments [65,431,476].
6. Racemization Hypothesis
Biological evolution has predominantly selected one structural form for AAs (L- levorotary form not the D- dextrorotary form). The consequence of this selection is that proteins being formed will primarily consist of L-AAs. Correspondingly, the enzymes involved in PTM and metabolism of proteins will primarily handle and metabolize (although not completely exclusively) the L- form.
Contemporary studies have revealed that the presence of D-AAs in the organism is not accidental and has fundamental importance for the function of the CNS and adaptation of the single cell and entire organism to the stereochemical environment. The genetic and epigenetic disturbance of the natural balance in the concentration of the free and protein/peptide bound L- and D- AAs in the brain (and or in peptide composition) leads to the misfolding of the D-AAs-containing proteins. This is incompatible with the evolutionary design of protein synthesis, degradation, and repair mechanisms including autophagy [478]. The interference of the spontaneous, enzyme-driven, and environmentally induced racemization can disrupt the functional structure of proteins leading to adverse effects on biological activity [244]. The exploration of the AAs racemization [28,266] and a protein aggregation within the bacterial cells opens an evolutionary perspective on human pathology. The combination of new results suggests specific attention to AAs racemization. It is obvious that along with the functional proteins, most of the enzymes, including Ser-proteases [202] and gamma-secretase [50,479] are involved in the process of AA racemization and APP proteolysis in proteins containing Ser and Asp residues. The fact that both residues are the subject of age dependent racemization allows one to assume an aging effect in enzyme activity. Indeed, it has been found that enzymes undergo age-related modifications which include structural changes and their specific affinity [480,481,482]. The prevalence of proteins receiving non-enzymatic PTM was found to be “increased with aging and is thought to be closely related to age-associated changes” [89]. Since 1975, the racemization of AAs in proteins has been used as a means of assessing the “age” of proteins [89,483]. However, in the current research, the aging of enzymes is not considered usually in respect to the process of racemization and protein aggregation.
7. Conclusions
The internal molecular environment of living systems is characterized by the specific structure–function relationships evident in the activity of signaling proteins, transporters, enzymes, DNA, and RNA. The discovery of a “shape-shifting” molecule (SSM) that is capable of interconversion among thousands of structural isomers has ascertained the dynamic nature of molecular chirality [14]. The stereoselective metabolism of chiral biomolecules emphasizes the significance of the effect of racemization in protein misfolding and aggregation.
We want in this overview to draw attention to the need to further examine the following points:
What is the mechanism linking the residue-specific protein racemization with aggregation?
How does racemization at specific sites contribute to protein aggregation, deposition, and toxicity associated with the major neurodegenerative disorders?
What are the functional consequences of site-specific racemization of A-beta, TAU, Huntingtin, α-synuclein, PrP, and MBP?
Do the residue-specific modulators (inhibitors and enhancers) of racemization have beneficial therapeutic effects?
How does the interplay between the enzymatic and spontaneous PTMs influence protein aggregation in neurodegenerative diseases [484].
For several decades, most treatments for AD have been targeted against the amyloid-β (Aβ) peptide. The frequently asked question is “why this strategy fails” [485,486,487]. The answer lies in the neglect of the link between AAs chirality, the stereochemistry of protein folding, neurodegeneration, and cognitive decline.
We are confident that the review of available information provides the conceptual and experimental background to understand the phenomena of protein aggregation indicating that racemization significantly contributes to brain pathology, and its integrated study would reveal novel therapeutic procedures. The role of racemization in decline of cognitive functions should be studied in conjunction with the major cellular players of protein aggregation pathology. Among them, prior consideration should be given to the trans-membrane receptors (NMDA, AMPA, mGlu5, α7, nAChRs, NGF, and apoE receptors (including receptors in neurons of the olfactory epithelium) [116]), glycoproteins [48], and cell membrane constituents (including cholesterol, and collagen).
The chiral cholesterol was shown to be a mediator of the stereoselective interaction between the cell membrane and proteins [488]. Therefore, the cholesterol of synaptic spine underlines the mechanism of glutamatergic neurotransmission. At the extracellular domain, the molecular mechanisms involved in the collagen aging and aggregation (cross-linking) are also significantly contributed by racemization [87,381]. The dynamic protein chirality, in our view, is a significant determinant of lateral asymmetry of neurotransmitters in the human brain [488].
Author Contributions
V.V.D.: Conceptualization, writing review and editing, supervision. T.M.W.: Data curation, funding acquisition. A.L.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This manuscript is supported by NIH grants AG066512 and AG060882. (Thomas Wisniewski). The payment was initiated 03-19-21.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Amino Acids | AAs |
| Amyloid beta | Aβ |
| Amyloid precursor protein | APP |
| β-secretase 1 | BACE1 |
| Biomarker | BM |
| D-Alanine | D-Ala |
| D-amino acids | D-AAs |
| D-serine | D-Ser |
| Electron microscopy | EM |
| Neurodegenerative diseases | ND |
| Transmission electron microscopy | TEM |
| Scanning electron microscopy | SEM |
| Vibrational circular dichroism | VCD |
| Misfolded proteins | MPs |
| Paired helical filaments | PHFs |
| Post-translational modifications | PTMs |
| System of post translational modification | PTM-Sys |
| Serine racemase | SerR |
| Shape-shifting molecule | SSM |
References
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.L. Beta-amyloid (1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10940. [Google Scholar] [CrossRef] [Green Version]
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Martnez-Rodrguez, C.; Martinez-Gomez, A.L.; Rodrguez-Vico, F.; Clemente-Jimenez, J.M.; Las Heras-Vazquez, J.L. Natural occurrence and industrial applications of d-amino acids: An overview. ChemBioChem 2010, 7, 1531–1548. [Google Scholar] [CrossRef]
- Takahashi, O.; Kobayashi, K.; Oda, A. Computational insight into the mechanism of serine residue racemization. Chem. Biodivers. 2010, 6, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Morinet, F. Prions: A model of conformational disease? Pathol. Biol. 2014, 62, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Ventura, S. Protein solubility and aggregation in bacteria. Front. Microbiol. 2016, 7, 1178. [Google Scholar] [CrossRef] [Green Version]
- Synakiewicz, A.; Sawicka-Zukowska, M.; Adrianowska, N.; Galezowska, G.; Ratajczyk, J.; Owczarzak, A.; Konieczna, L.; Stachowicz-Stencel, T. Amino acid profiles as potential biomarkers for pediatric cancers: A preliminary communication. Biomark. Med. 2017, 11. [Google Scholar] [CrossRef]
- Kimura, T.; Hamase, K.; Miyoshi, Y.; Yamamoto, R.; Yasuda, K.; Mita, M.; Rakugi, H.; Hayashi, T.; Isaka, Y. Chiral amino acid metabolomics for novel biomarker screening in the prognosis of chronic kidney disease. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Ha, S.; Kim, I.; Takata, T.; Kinouchi, T.; Isoyama, M.; Suzuki, M.; Noriko Fuji, N. Identification of d-amino acid-containing peptides in human serum. PLoS ONE 2017, 12, e0189972. [Google Scholar] [CrossRef]
- Raskatov, J.A.; David, B.; Teplow, D.B. Using chirality to probe the conformational dynamics and assembly of intrinsically disordered amyloid proteins. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Fukuda, H.; Murayama, S.; Izumiyama, N.; Shirasawa, T. Isoaspartate formation at position 23 of amyloid beta peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Matsuoka, Y.; Shirasawa, T. Biological significance of isoaspartate and its repair system. Biol. Pharm. Bull. 2005, 28, 1590–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Bode, J.W. Racemization as a stereochemical measure of dynamics and robustness in shape-shifting organic mole-cules. Proc. Natl. Acad. Sci. USA 2011, 108, 4752–14756. [Google Scholar] [CrossRef] [Green Version]
- Radkov, A.D.; Moe, L.A. Amino acid racemization in Pseudomonas putida KT. J. Bacteriol. 2013, 195, 5016–5024. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, J.J.; Srinivasan, N.G. The Occurrence of Certain D-Amino Acids in Insects. Biochemistry 1966, 5, 1185–1190. [Google Scholar] [CrossRef]
- Mori, H.; Ishii, K.; Tomiyama, T.; Furiya, Y.; Sahara, N.; Asano, S.; Endo, N.; Shirasawa, T.; Takio, K. Racemization: Its bio-logical significance on neuropathogenesis of Alzheimer disease. Tohoku J. Exp. Med. 1994, 174, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Racemization of serine residues catalyzed by dihydrogen phosphate ion: A compu-tational study. Catalysts 2017, 7, 363. [Google Scholar] [CrossRef] [Green Version]
- Wolf, C. Dynamic Stereochemistry of Chiral Compounds: Principles and Applications; RSC Publishing: London, UK, 2008. [Google Scholar]
- Wang, W.; Yao, N.; Chen, Y.; Lai, P. Salam hypothesis and the role of phase transition. In Life in the Universe. Cellular Origin and Life in Extreme Habitats and Astrobiology; Seckbach, J., Chela-Flores, J., Owen, T., Raulin, F., Eds.; Springer: Dordrecht, The Netherlands, 2004; pp. 79–82. [Google Scholar]
- Dyakin, V.V.; Lucas, J.; Dyakina-Fagnano, N.V.; Posner, E.V.; Vadasz, C. Chain of chirality transfer as determinant of brain functional laterality. Breaking the chirality silence: Search for new generation of biomarkers. relevance to neurodegenerative diseases, cognitive psychology and nutrition science. Neurol. Neurosci. Res. 2017, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Dyakin, V.V.; Wisniewski, T.M.; Lajtha, A. Chiral Interface of Amyloid Beta (Aβ): Relevance to Protein Aging, Aggregation and Neurodegeneration. Symmetry 2020, 12, 585. [Google Scholar] [CrossRef] [Green Version]
- De Silva, T.N.; Sivised, V. A statistical mechanics perspective for protein folding from q-state Potts model. Cornell Univ. arXiv 2018, arXiv:1709.04813. [Google Scholar]
- Jaeger, G. The ehrenfest classification of phase transitions: Introduction and evolution. Arch. Hist. Exact Sci. 1998, 53, 51–81. [Google Scholar] [CrossRef]
- Sugahara, H.; Meinert, C.; Nahon, L.; Jones, N.C.; Hoffmann, S.V.; Hamase, K.; Takano, Y.; Meierhenrich, U.J. D-Amino acids in molecular evolution in space—Absolute asymmetric photolysis and synthesis of amino acids by circularly polarized light. Biochim. Biophys. Acta 2018, 1866, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Bryngelson, J.D.; Wolynes, P.G. Spin glasses and the statistical mechanics of protein folding (disordered systems/irreversible denaturation/molten-globule state/biomolecular self-assembly). Proc. Nattl. Acad. Sci. USA 1987, 84, 7524–7528. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Kakuichi, T.; Fujii, K.; Kera, Y.; Yamada, R.H. Physiological role of D-aspartate oxidase in the assimilation and detoxification of D-aspartate in the yeast Cryptococcus humicola. Yeast 2005, 22, 1203–1212. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, H.J. Racemization in reverse: Evidence that D-amino acid toxicity on Earth is controlled by bacteria with racemases. PLoS ONE 2014, 9, e92101. [Google Scholar] [CrossRef] [Green Version]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless orga-nelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Brangwynne, C.P. Nuclear bodies: The emerging biophysics of nucleoplasmic phases. Curr. Opin. Cell Biol. 2015, 34, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chakrabartty, A. Phase to phase with TDP-43. Biochemistry 2017, 56, 809–823. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2017, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Kim, J.; Choi, M.Y. Autophagy mediates phase transitions from cell death to life. Heliyon 2015, 1, e00027. [Google Scholar] [CrossRef] [Green Version]
- Gromashevskyi, L.V.; Diakin, V.V.; Salkov, E.A.; Skliarov, S.M.; Khalimonova, N.S.; Dyakin, V.V. Acoustochemical reactions in CdS. Ukr. J. Phys. 1984, 294, 550–554. [Google Scholar]
- Helfman, P.M.; Bada, J.L.; Shou, M.Y. Considerations on the role of aspartic acid racemization in the aging process. Gerontology 1977, 23, 419–425. [Google Scholar] [CrossRef]
- Hassett, A.; Blättler, W.; Knowles, J.R. Pyruvate kinase: Is the mechanism of phospho transfer associative or dissociative? Biochemistry 1982, 21, 6335–6340. [Google Scholar] [CrossRef]
- Schofield, J.D.; Weightman, B. New knowledge of connective tissue ageing. J. Clin. Path. Supl. 1978, 1, 174–190. [Google Scholar] [CrossRef] [Green Version]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Fersht, A.R. From the first protein structures to our current knowledge of protein folding: Delights and scepticisms. Nat. Rev. Mol. Cell. Biol. 2008, 9, 650–654. [Google Scholar] [CrossRef]
- Song, J. Environment-transformable sequence–structure relationship: A general mechanism for proteotoxicity. Biophys. Rev. 2018, 10, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Berry, J.; Brangwynne, C.P.; Haataja, M. Physical principles of intracellular organization via active and passive phase transi-tions. Rep. Prog. Phys. 2018, 81, 046601. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Feng, Y.Y.; Lu, X.; Zhao, S.; Liu, C.W.; Liu, Y.M. D-Amino acids in rat brain measured by liquid chromatog-raphy/tandem mass spectrometry. Neurosci. Lett. 2008, 445, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Hamase, K.; Morikawa, A.; Etoh, S.; Tojo, Y.; Miyoshi, Y.; Zaitsu, K. Analysis of small amounts of D-amino acids and the study of their physiological functions in mammals. Anal. Sci. 2009, 25, 961–968. [Google Scholar] [CrossRef] [Green Version]
- Dedkova, L.M.; Fahmi, N.E.; Golovine, S.Y.; Hecht, S.M. Enhanced D-Amino acid incorporation into protein by modified ribosomes. J. Am. Chem. Soc. 2003, 125, 6616–6617. [Google Scholar] [CrossRef] [PubMed]
- Niemelä, V.; Landtblom, A.M.; Blennow, K.; Sundblom, J. Tau or neurofilament light—Which is the more suitable biomarker for Huntington’s disease? PLoS ONE 2017, 12, e0172762. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.; McColgan, P. Cerebrospinal fluid total tau concentration predicts clinical phenotype in Hunting-ton’s disease. J. Neurochem. 2016, 139, 22–25. [Google Scholar] [CrossRef]
- Yu, N.-N.; Tan, M.-S.; Yu, J.-T.; Xie, A.-M.; Tan, L. The Role of reelin signaling in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 5692–5700. [Google Scholar] [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Scahill, R.I.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: A retrospective cohort analysis. Lancet Neurol. 2017, 16, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Böhm, C.; Möhwald, H.; Leiserowitz, L.; Als-Nielsen, J.; Kjaer, K. Influence of chirality on the structure of phospholipid mon-olayers. Biophys. J. 1993, 64, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Kong, H.; Guan, Y.; Yang, J.; Gu, J.; Yang, S.; Xu, G. Plasma phospholipid metabolic profiling and biomarkers of type 2 diabetes mellitus based on high-performance liquid chromatography/electrospray mass spectrometry and multivariate statistical analysis. Anal. Chem. 2005, 77, 4108–4116. [Google Scholar] [CrossRef]
- Dennis, E.A. Liberating chiral Lipid mediators, inflammatory enzymes, and LIPID MAPS from biological grease. J. Biol. Chem. 2016, 91, 24431–24448. [Google Scholar] [CrossRef] [Green Version]
- Hoover, R.B. Microfossils and biomolecules in carbonaceous meteorites: Possibility of life in water-bearing asteroids and comets. Proc. Nanophotonics Macrophotonics Space Environ. 2014, 9226, 922602. [Google Scholar] [CrossRef]
- Mercier, M.C.; Dontenwill, M.; Choulier, L. Selection of nucleic acid aptamers targeting tumor cell-surface Protein Bi-omarkers. Cancers 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidari, A. Integrative approach to biological networks for emerging roles of proteomics, genomics and transcriptomics in the discovery and validation of human colorectal cancer biomarkers from DNA/RNA sequencing data under synchrotron radiation. Transcriptomics 2017, 5, e117. [Google Scholar] [CrossRef]
- Fisher, H.; Garcia, N.M.; Payan, I.L.; Cadilla-Perezrois, R.; Sheremata, W.A.; Man, E.H. D-aspartate acid in purified myelin and myelin basic protein. Biochem. Biophys. Res. Commun. 1986, 135, 683–687. [Google Scholar] [CrossRef]
- Collins, M.J.; Waite, E.R.; Duin, A.C.T. Predicting protein decomposition: The case of aspartic-acid racemization kinetics. R. Soc. Phil. Trans. R. Soc. Lond. B 1999, 354, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fura, J.M.; Kearns, D.; Pires, M.M. D-amino acid probes for penicillin binding protein-based bacterial surfacel labeling. J. Biological. Chem. 2015, 290, 30540–30550. [Google Scholar] [CrossRef] [Green Version]
- Patneau, D.K.; Mayer, M. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J. Neurosci. 1990, 10, 2385–2399. [Google Scholar] [CrossRef] [PubMed]
- Erreger, K.; Dravid, S.M.; Banke, T.G.; Wyllie, D.I.; Traynelis., S.F. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J. Physiol. 2005, 563 Pt 2, 345–358. [Google Scholar] [CrossRef]
- Erreger, K.; Geballe, M.T.; Kristensen, A.; Chen, P.; Hansen, K.; Lee, C.; Yuan, H.; Le, P.; Lyuboslavsky, P.; Micale, N.; et al. Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate recep-tors. Mol. Pharmacol. 2007, 72, 907–920. [Google Scholar] [CrossRef]
- Chen, P.E.; Geballe, M.T.; Katz, E.; Erreger, K.; Livesey, M.R.; O’Toole, K.K.; Le, P.; Lee, C.J.; Snyder, J.P.; Traynelis, S.F.; et al. Modulation of glycine potency in rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J. Physiol. 2008, 586 Pt 1, 227–245. [Google Scholar] [CrossRef]
- Vyklicky, V.; Korinek, M.; Smejkalova, T.; Balik, A.; Krausova, B.; Kaniakova, M.; Lichnerova, K.; Cerny, J.; Krusek, J.; Dittert, I.; et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol. Res. 2004, 63 (Suppl. S1), S191–S203. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. D-amino acids in the nervous and endocrine systems. Scientifica 2016, 2016, 649462. [Google Scholar] [CrossRef]
- Yoshimura, T.; Nishikawa, T.; Homma, H. D-Amino Acids: Physiology, Metabolism, and Application; Springer: Tokyo, Japan, 2016. [Google Scholar]
- Di Fiore, M.M.; Santillo, A.; Falvo, S.; Longobardi, S.; Baccari, G.C. Molecular mechanisms elicited by d-aspartate in Leydig cells and Spermatogonia. Int. J. Mol. Sci. 2016, 17, 1127. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Han, H.; Yin, J.; Li, T.; Yin, Y. Role of D-aspartate on biosynthesis, racemization, and potential functions: A mini-review. Anim. Nutr. 2018, 4, 311–315. [Google Scholar] [CrossRef]
- Wolosker, H.; Sheth, K.N.; Takahashi, M.; Mothet, J.P.; Brady, R.O.; Ferris, C.D., Jr.; Snyder, S.H. Purification of serine race-mase: Biosynthesis of the neuromodulator D-serine. Proc. Natl. Acad. Sci. USA 1999, 9, 721–725. [Google Scholar] [CrossRef] [Green Version]
- Michels, P.C.; Clark, D.S. Pressure-enhanced activity and stability of a hyperthermophilic protease from a deep-sea Methan-ogen. Appl. Environ. Microbiol. 1997, 63, 3985–3991. [Google Scholar] [CrossRef] [Green Version]
- Le Maux, S.; Nongonierma, A.B.; Lardeux, C.; FitzGerald, R.J. Impact of enzyme inactivation conditions during the generation of whey protein hydrolysates on their physicochemical and bioactive properties. Food Sci. Technol. 2017, 53, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Woodsmith, J.; Stelzl, U. Studying post-translational modifications with protein interaction networks. Curr. Opin. Struct. Biol. 2014, 24, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Loosdregt, J.; Coffer, P.J. Post-translational modification networks regulating FOXP3 function. Trends Immunol. 2014, 35, 368–378. [Google Scholar] [CrossRef]
- Tay, A.P.; Pang, C.N.I.; Winter, D.L.; Wilkins, M.R. PTMOracle: A Cytoscape APP for covisualizing and coanalyzing post-translational modifications in protein interaction networks. J. Proteome Res. 2017, 16, 1988–2003. [Google Scholar] [CrossRef]
- Huang, H.; Arighi, C.N.; Ross, K.E.; Ren, J.; Li, G.; Chen, S.C.; Wang, Q.; Cowart, J.; Vijay-Shanker, K.; Wu, C.H. iPTMnet: An integrated resource for protein post-translational modification network discovery. Nucleic Acids Res. 2018, 46, D542–D550. [Google Scholar] [CrossRef]
- Bai, L.; Livnat, I.; Romanova, E.V.; Alexeeva, V.; Yau, P.M.; Vilim, F.S.; Weiss, K.R.; Jing, J.; Sweedler, J.V. Characterization of GdFFD, a d-amino acid-containing neuropeptide that functions as an extrinsic modulator of the Aplysia feeding circuit. J. Biol. Chem. 2013, 288, 32837–32851. [Google Scholar] [CrossRef] [Green Version]
- Sela, M.; Zisman, E. Different roles of D-amino acids in immune phenomena. FASEB J. 1997, 11, 449–456. [Google Scholar] [CrossRef]
- Tugyi, R.; Uray, K.; Iván, D.; Fellinger, E.; Perkins, A.; Hudecz, F. Partial D-amino acid substitution: Improved enzymatic sta-bility and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Xie, M.; Yin, Y. Amino Acids and Immune Functions. In Nutritional and Physiological Functions of Amino Acids in Pigs; Francois, B., Guoyao, W., Yulong, Y., Eds.; Springer: Wien, Austria, 2013; pp. 175–185. [Google Scholar]
- Soyez, D.; Toullec, J.-Y.; Ollivaux, C.; Géraud, G. L to D Amino Acid Isomerization in a Peptide Hormone Is a Late Post-translational Event Occurring in Specialized Neurosecretory Cells. J. Biol. Chem. 2000, 275, 37870–37875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tom, M.; Manfrin, C.; Mosco, A.; Gerdol, M.; De Moro, G.D.M.; Pallavicini, A.; Giulianini, P.G. Different transcription regula-tion routes are exerted by L- and D-amino acid enantiomers of peptide hormones. J. Exp. Biol. 2014, 17, 4337–4346. [Google Scholar] [CrossRef] [Green Version]
- Billard, J.-M. D-Amino acids in brain neurotransmission and synaptic plasticity. Amino Acids 2012, 43, 1851–1860. [Google Scholar] [CrossRef]
- Laska, M. Olfactory Perception of 6 Amino Acids by Human Subjects. Chem. Senses 2010, 35, 79–87. [Google Scholar] [CrossRef]
- Piubelli, L.; Giulia Murtas, G.; Valentina Rabattoni, V.; Loredano Pollegioni, L. The Role of D-Amino Acids in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Weatherly, C.A.; Du, S.; Parpia, C.; Santos, P.T.; Hartman, A.L.; Armstrong, D.W. D-Amino Acid Levels in Perfused Mouse Brain Tissue and Blood: A Comparative Study. ACS Chem. Neurosci. 2017, 8, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Bada, J.L.; Protsch, R. Racemization Reaction of Aspartic Acid and Its Use in Dating Fossil Bones. Proc. Natl. Acad. Sci. USA 1973, 70, 1331–1334. [Google Scholar] [CrossRef] [Green Version]
- Bonner, W.A.; Lemmon, R.M. Radiolysis, racemization, and the origin of optical activity. Bioorg. Chem. 1978, 7, 175–187. [Google Scholar] [CrossRef]
- Bada, J.L.; Schroeder, R.A.; Protsch, R.; Berger, R. Concordance of Collagen-Based Radiocarbon and Aspartic-Acid Racemi-zation Ages. Proc. Natl. Acad. Sci. USA 1974, 71, 914–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKerrow, J.H. Non-enzymatic, post-translational, amino acid modifications in ageing. A brief review. Mech. Ageing Dev. 1979, 10, 371–377. [Google Scholar] [CrossRef]
- Kaji, Y.; Oshika, T.; Takazawa, Y.; Fukayama, M.; Fujii, N. Pathological Role of D-amino Acid-Containing Proteins and Ad-vanced Glycation End Products in the Development of Age-Related Macular Degeneration. Anti-Aging Med. 2010, 7, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Motoie, R.; Fujii, N.; Tsunoda, S.; Nagata, K.; Shimo-Oka, T.; Kinouchi, T.; Saito, T.; Ono, K. Localization of D-β-Aspartyl Residue-Containing Proteins in Various Tissues. Int. J. Mol. Sci. 2009, 10, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, D.M.; Neidle, A.; McHale, D.M.; Lajtha, A. The presence of free D-aspartic acid in rodents and man. Biochem. Biophys. Res. Commun. 1986, 141, 27–32. [Google Scholar] [CrossRef]
- Nelson, D.L.; Applegate, G.A.; Beio, M.L.; Graham, D.L.; Berkowitz, D.B. Human serine racemase structure/activity relation-ship studies provide mechanistic insight and point to position 84 as a hot spot for β-elimination function. J. Biol. Chem. 2017, 292, 13986–14002. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, M.; Miyoshi, Y.; Ohide, H.; Hamase, K.; Konno, R. D-Amino acids in the brain and mutant rodents lacking d-amino-acid oxidase activity. Amino Acids 2012, 43, 1811–1821. [Google Scholar] [CrossRef]
- Sacchi, S.; Caldinelli, L.; Cappelletti, P.; Pollegioni, L.; Molla, G. Structure–function relationships in human d-amino acid ox-idase. Amino Acids 2012, 43, 1833–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molla, G. Competitive Inhibitors Unveil Structure/Function Relationships in Human D-Amino Acid Oxidase. Front. Mol. Biosci. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature and symbolism for amino acids and peptides. Eur. J. Biochem. 1984, 38, 9–37. [Google Scholar] [CrossRef]
- Griffiths, A.J.F.; Gelbart, W.M.; Miller, J.H.; Lewontin, R.C. Modern Genetic Analysis; W. H. Freeman & Co: New York, NY, USA, 1999. [Google Scholar]
- Schell, M.J.; Brady, R.O.B., Jr.; Molliver, M.E.; Snyder, S.H. D-Serine as a Neuromodulator: Regional and Developmental Lo-calizations in Rat Brain Glia Resemble NMDA Receptors. J. Neurosci. 1997, 17, 1604–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kartvelishvily, E.; Shleper, M.; Balan, L.; Dumin, E.; Wolosker, H. Neuron-derived D-Serine Release Provides a Novel Means to Activate N-Methyl-D-aspartate Receptors. J. Biol. Chem. 2006, 281, 14151–14162. [Google Scholar] [CrossRef] [Green Version]
- Martineau, M.; Parpura, V.; Mothet, J.-P. Cell-type specific mechanisms of D-serine uptake and release in the brain. Front. Synaptic Neurosci. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Shapira, R.; Chou, C. Differential racemization of aspartate and serine in human myelin basic protein. Biochem. Biophys. Res. Commun. 1987, 146, 1342–1349. [Google Scholar] [CrossRef]
- Wu, S.-Z.; Bodles, A.M.; Porter, M.M.; Griffin, W.S.T.; Basile, A.S.; Barger, S.W. Induction of serine racemase expression and D-serine release from microglia by amyloid β-peptide. J. Neuroinflamm. 2004, 1. [Google Scholar] [CrossRef] [Green Version]
- Dhami, K.; Churchward, M.; Baker, G.; Todd, K. Fluoxetine and citalopram decrease microglial release of glutamate and d-serine to promote cortical neuronal viability following ischemic insult. Mol. Cell. Neurosci. 2013, 56, 365–374. [Google Scholar] [CrossRef]
- Wood, P.L.; Hawkinson, J.E.; Goodnough, D.B. Formation of d-Serine from l-Phosphoserine in Brain Synaptosomes. J. Neurochem. 1996, 67, 1485–1490. [Google Scholar] [CrossRef]
- Wolosker, H.; Dumin, E.; Balan, L.; Foltyn, V.D. D-amino acids in the brain: D-serine in neurotransmission and neu-ro-degeneration. FEBS J. 2008, 275, 3514–3526. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.-Q.; Zabek, R.L.; Bai, D. d-Serine inhibits AMPA receptor-mediated current in rat hippocampal neurons. Can. J. Physiol. Pharmacol. 2007, 85, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Montagna, E.; Dorostkar, M.M.; Herms, J. The Role of APP in Structural Spine Plasticity. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Billard, J.-M. D-serine signalling as a prominent determinant of neuronal-glial dialogue in the healthy and diseased brain. J. Cell. Mol. Med. 2008, 12, 1872–1884. [Google Scholar] [CrossRef]
- Sultan, S.; Gebara, E.G.; Moullec, K.; Toni, N. D-serine increases adult hippocampal neurogenesis. Front. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.S.; Aizawa, H.; Huang, A.S.; Wickramasinghe, S.R.; Kashani, A.H.; Barrow, R.K.; Huganir, R.L.; Ghosh, A.; Snyder, S.H. Serine racemase: Activation by glutamate neurotransmission via glutamate receptor interacting protein and mediation of neuronal migration. Proc. Natl. Acad. Sci. USA 2005, 102, 2105–2110. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Kong, H.; Tang, M.; Lu, M.; Ding, J.-H.; Hu, G. d-Serine Regulates Proliferation and Neuronal Differentiation of Neural Stem Cells from Postnatal Mouse Forebrain. CNS Neurosci. Ther. 2012, 18, 4–13. [Google Scholar] [CrossRef]
- Esposito, S.; Pristerà, A.; Maresca, G.; Cavallaro, S.; Felsani, A.; Florenzano, F.; Manni, L.; Ciotti, M.T.; Pollegioni, L.; Borsello, T.; et al. Contribution of serine racemase/d-serine pathway to neuronal apoptosis. Aging Cell 2012, 11, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shleper, M.; Kartvelishvily, E.; Wolosker, H. D-Serine Is the Dominant Endogenous Coagonist for NMDA Receptor Neuro-toxicity in Organotypic Hippocampal Slices. J. Neurosci. 2005, 25, 9413–9417. [Google Scholar] [CrossRef]
- Beltrán-Castillo, S.; Olivares, M.J.; Contreras, R.A.; Zúñiga, G.; Llona, A.; von Bernhardi, R.; Eugenín, J.L. D-serine released by astrocytes in brainstem regulates breathing response to CO2 levels. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Horio, M.; Kohno, M.; Fujita, Y.; Ishima, T.; Inoue, R.; Mori, H.; Hashimoto, K. Levels of d-serine in the brain and peripheral organs of serine racemase (Srr) knock-out mice. Neurochem. Int. 2011, 59, 853–859. [Google Scholar] [CrossRef]
- Zozulya, S.; Echeverri, F.; Nguyen, T. The human olfactory receptor repertoire. Genome Biol. 2011, 2. [Google Scholar] [CrossRef]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 2, 345–357. [Google Scholar] [CrossRef]
- Hashimoto, A.; Nishikawa, T.; Hayashi, T.; Fujii, N.; Harada, K.; Oka, T.; Takahashi, K. The presence of free D-serine in rat brain. FEBS Lett. 1992, 296, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Kakegawa, W.; Miyoshi, Y.; Hamase, K.; Matsuda, S.; Matsuda, K.; Kohda, K.; Emi, K.; Motohashi, J.; Konno, R.; Zaitsu, K.; et al. D-Serine regulates cerebellar LTD and motor coordination through the δ2 glutamate receptor. Nat. Neurosci. 2011, 14, 603–611. [Google Scholar] [CrossRef]
- Kondori, N.R.; Paul, P.; Robbins, J.P.; Liu, K.; Hildyard, J.C.W.; Wells, D.J.; De Belleroche, J.S. Focus on the Role of D-serine and D-amino Acid Oxidase in Amyotrophic Lateral Sclerosis/Motor Neuron Disease (ALS). Front. Mol. Biosci. 2018, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, N.; Awakura, M.; Takemoto, L.; Inomata, M.; Takata, T.; Fujii, N.; Saito, T. Characterization of α A-crystallin from high molecular weight aggregates in the normal human lens. Mol. Vis. 2003, 9, 315–322. [Google Scholar]
- Fujii, N.; Saito, T. Homochirality and life. Chem. Rec. 2004, 4, 267–278. [Google Scholar] [CrossRef]
- Fujii, N. D-Amino Acid in Elderly Tissues. Biol. Pharm. Bull. 2005, 28, 1585–1589. [Google Scholar] [CrossRef] [Green Version]
- Kaji, Y.; Oshika, T.; Takazawa, Y.; Fukayama, M.; Takata, T.; Fujii, N. Localization of D-β-Aspartic Acid–Containing Pro-teins in Human Eyes. Investig. Opthalmol. Vis. Sci. 2007, 48, 3923–3927. [Google Scholar] [CrossRef]
- Fujii, N.; Shimmyo, Y.; Sakai, M.; Sadakane, Y.; Nakamura, T.; Morimoto, Y.; Kinouchi, T.; Goto, Y.; Lampi, K. Age-related changes of alpha-crystallin aggregate in human lens. Amino Acids 2007, 32, 87–94. [Google Scholar] [CrossRef]
- Auvynet, C.; Seddiki, N.; Dunia, I.; Nicolas, P.; Amiche, M.; Lacombe, C. Post-translational amino acid racemization in the frog skin peptide deltorphin I in the secretion granules of cutaneous serous glands. Eur. J. Cell Biol. 1999, 85, 25–34. [Google Scholar] [CrossRef]
- Checco, J.W.; Zhang, G.; Yuan, W.-D.; Yu, K.; Yin, S.-Y.; Roberts-Galbraith, R.H.; Yau, P.M.; Romanova, E.V.; Jing, J.; Sweedler, J.V. Molecular and Physiological Characterization of a Receptor for d-Amino Acid-Containing Neuropeptides. ACS Chem. Biol. 2018, 13, 1343–1352. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.H.; Petrucelli, L.; Gardner, C.; Emory, C.; Frey, W.H., II; Amaducci, L.; Sorbi, S.; Sorrentino, G.; Borghi, M.; D’Aniello, A. Freed-amino acids in human cerebrospinal fluid of alzheimer disease, multiple sclerosis, and healthy control subjects. Mol. Chem. Neuropathol. 1994, 23, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Yang, H.-T.; Chiu, C.-C.; Lane, H.-Y. Blood levels of D-amino acid oxidase vs. D-amino acids in reflecting cognitive aging. Sci. Rep. 2017, 7, 14896. [Google Scholar] [CrossRef]
- Zaragozá, R. Transport of Amino Acids Across the Blood-Brain Barrier. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Zapico, S.C. Mechanisms Linking Aging, Diseases and Biological Age Estimation, 1st ed.; CRC Press: Boca Raton, FL, USA, 2017; pp. 44–45. [Google Scholar]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and Microtubules Are Required for Autophagic Degradation of Aggregated Huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, P.; De Belleroche, J. The role of D-serine and glycine as co-agonists of NMDA receptors in motor neuron degeneration and amyotrophic lateral sclerosis (ALS). Front. Synaptic Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribet, D.; Cossart, P. Post-translational modifications in host cells during bacterial infection. FEBS Lett. 2010, 584, 2748–2758. [Google Scholar] [CrossRef] [Green Version]
- Higuchi-Sanabria, R.; Pernice, W.M.; Vevea, J.D.; Wolken, D.M.A.; Boldogh, I.R.; Pon, L.A. Role of asymmetric cell division in lifespan control inSaccharomyces cerevisiae. FEMS Yeast Res. 2014, 1, 1133–1146. [Google Scholar] [CrossRef] [Green Version]
- Fujii, N.; Harada, K.; Momose, Y.; Ishii, N.; Akaboshi, M. D-Amino Acid Formation Induced by a Chiral Field within a Human Lens Protein during Aging. Biochem. Biophys. Res. Commun. 1999, 263, 322–326. [Google Scholar] [CrossRef]
- Inoue, K.; Hosaka, D.; Mochizuki, N.; Akatsu, H.; Tsutsumiuchi, K.; Hashizume, Y.; Matsukawa, N.; Yamamoto, T.; Toyo’Oka, T. Simultaneous Determination of Post-Translational Racemization and Isomerization ofN-Terminal Amyloid-β in Alzheimer’s Brain Tissues by Covalent Chiral Derivatized Ultraperformance Liquid Chromatography Tandem Mass Spectrometry. Anal. Chem. 2014, 86, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Huang, H.-B.; Kwon, Y.-G.; Greengard, P.; Nairn, A.C.; Kuriyan, J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995, 376, 745–753. [Google Scholar] [CrossRef]
- Dhanasekaran, N.; Reddy, E.P. Signaling by dual specificity kinases. Oncogene 1998, 17, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Jagannath, V.; Brotzakis, Z.F.; Parrinello, M.; Walitza, S.; Grünblatt, E. Controversial Effects of D-Amino Acid Oxidase Ac-tivator (DAOA)/G72 on D-Amino Acid Oxidase (DAO) Activity in Human Neuronal, Astrocyte and Kidney Cell Lines: The N-methyl D-aspartate (NMDA) Receptor Hypofunction Point of View. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Di Salvo, M.L.; Scarsdale, J.N.; Kazanina, G.; Contestabile, R.; Schirch, V.; Wright, H.T. Structure-Based Mechanism for Early PLP-Mediated Steps of Rabbit Cytosolic Serine Hydroxymethyltransferase Reaction. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Christen, P.; Mehta, P.K. From cofactor to enzymes. The molecular evolution of pyridoxal-5’-phosphate-dependent enzymes. Chem. Rec. 2001, 1, 436–447. [Google Scholar] [CrossRef]
- Reddy, G.S.; Reddy, G.V.; Smith, G.G. The effect of Ni(II), Zn(II), Cu(II), Co(II) and Pd(II) ions on racemisation of hydroxy α-amino acids. Inorg. Chim. Acta 1989, 166, 55–58. [Google Scholar] [CrossRef]
- Lyons, B.; Jamie, J.F.; Truscott, R.G.W. Separate mechanisms for age-related truncation and racemisation of peptide-bound serine. Amino Acids 2014, 46, 199–207. [Google Scholar] [CrossRef]
- Demarchi, B.; Collins, M.; Bergström, E.; Dowle, A.; Penkman, K.; Thomas-Oates, J.; Wilson, J. New Experimental Evidence for In-Chain Amino Acid Racemization of Serine in a Model Peptide. Anal. Chem. 2013, 85, 5835–5842. [Google Scholar] [CrossRef]
- Eliot, A.C.; Kirsch, J.F. Pyridoxalphosphateenzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 2004, 73, 383–415. [Google Scholar] [CrossRef] [Green Version]
- Dunathan, H.C. Conformation and reaction specificity in pyridoxal enzymes. Proc. Natl. Acad. Sci. USA 1966, 55, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Toney, M.D. Reaction specificity in pyridoxal phosphate enzymes. Arch. Biochem. Biophys. 2005, 43, 279–287. [Google Scholar] [CrossRef]
- Samuel, G.; Reeves, P. Biosynthesis of O-antigens: Genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr. Res. 2003, 338, 2503–2519. [Google Scholar] [CrossRef]
- Fiske, M.; Valtierra, S.; Solvang, K.; Zorniak, M.; White, M.; Herrera, S.; Konnikova, A.; Brezinsky, R.; DebBurman, S. Contribution of alanine-76 and serine phosphorylation in α-Synuclein membrane association and aggregation in yeasts. Parkinson’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Singh, S.; Hinze, D.; Josten, M.; Sahl, H.G.; Siepmann, M.; Walter, J. Phosphorylation of amyloid-β peptide at serine 8 attenuates its clearance via insulin-degrading and angiotensin-converting enzymes. J. Biol. Chem. 2012, 287, 8641–8651. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wirths, O.; Stüber, K.; Wunderlich, P.; Koch, P.; Theil, S.; Rezaei-Ghaleh, N.; Zweckstetter, M.; Bayer, T.A.; Brüstle, O.; et al. hosphorylation of the amyloid β-peptide at Ser26 stabilizes oligomeric assembly and increases neu-rotoxicity. Acta Neuropathol. 2016, 131, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Zatsepina, O.G.; Kechko, O.I.; Mitkevich, V.A. Amyloid-β with isomerized Asp7 cytotoxicity is coupled to protein phosphor-ylation. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Marchetti, M.; Bruno, S.; Campanini, B.; Bettati, S.; Peracchi, A.; Mozzarelli, A. Regulation of human serine racemase activity and dynamics by halides, ATP and malonate. Amino Acids 2015, 47, 163–173. [Google Scholar] [CrossRef]
- Alsanousi, N.; Sugiki, T.; Furuita, K.; So, M.; Lee, Y.-H.; Fujiwara, T.; Kojima, C. Solution NMR structure and inhibitory effect against amyloid-β fibrillation of Humanin containing a d -isomerized serine residue. Biochem. Biophys. Res. Commun. 2016, 477, 647–653. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Amininasab, M.; Kumar, S.; Walter, J.; Zweckstetter, M. Phosphorylation modifies the molecular stability of β-amyloid deposits. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.B.; Rank, K.B.; Bhattacharya, K.; Thomsen, D.R.; Gurney, M.E.; Sharma, S.K. TAU Phosphorylation at serine 396 and serine 404 by human recombinant TAU protein kinase II inhibits Tau’s Ability to Promote Microtubule Assembly. J. Biol. Chem. 2000, 275, 24977–24983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of TAU protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Cook, C.; Stankowski, J.N.; Carlomagno, Y.; Stetler, C.; Petrucelli, L. Acetylation: A new key to unlock tau’s role in neuro-degeneration. Alzheimer’s Res. Ther. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.-L.; Wang, N.; Sun, F.-R.; Cao, X.-P.; Zhang, W.; Yucorresponding, J.T. TAU in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef]
- Kleinknecht, A.; Popova, B.; Lázaro, D.F.; Pinho, R.; Valerius, O.; Outeiro, T.F.; Braus, G.H. C-Terminal tyrosine residue modifi-cations modulate the protective phosphorylation of Serine 129 of α-Synuclein in a yeast model of Parkinson’s disease. PLoS Genet. 2016, 12, e1006098. [Google Scholar] [CrossRef]
- Agarwal, S.; Döring, K.; Gierusz, L.A.; Iyer, P.; Lane, F.M.; Graham, J.F.; Goldmann, W.; Pinheiro, T.J.T.; Gill, A.C. Complex folding and misfolding effects of deer-specific amino acid substitutions in the β2-α2 loop of murine prion protein. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Pollegioni, L.; Sacchi, S. Metabolism the neuromodulator D-serine. Cell. Mol. Life Sci. 2010, 67, 2387–2404. [Google Scholar] [CrossRef] [PubMed]
- Foltyn, V.N.; Bendikov, I.; De Miranda, J.; Panizzutti, R.; Dumin, E.; Shleper, M.; Li, P.; Toney, M.D.; Kartvelishvily, E.; Wolosker, H. Serine Racemase Modulates Intracellular D-Serine Levels through an α,β-Elimination Activity. J. Biol. Chem. 2005, 280, 1754–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, T.; Goto, M. D-amino acids in the brain: Structure and function of pyridoxal phosphate-dependent amino acid racemases. FEBS J. 2008, 275, 3527–3537. [Google Scholar] [CrossRef]
- Zou, C.; Crux, S.; Marinesco, S.; Montagna, E.; Sgobio, C.; Shi, Y.; Shi, S.; Zhu, K.; Dorostkar, M.M.; Müller, U.C.; et al. Amyloid precursor protein maintains constitutive and adaptive plasticity of dendritic spines in adult brain by regulating D-serine homeostasis. EMBO J. 2016, 35, 2213–2222. [Google Scholar] [CrossRef]
- Goto, M.; Yamauchi, T.; Kamiya, N.; Miyahara, I.; Yoshimura, T.; Mihara, H.; Kurihara, T.; Hirotsu, K.; Esaki, N. Crystal structure of a homolog of mamma-lian serine racemase from Schizosaccharomyces pombe. J. Biol. Chem. 2009, 284, 25944–25952. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Mack, V.; Ebneth, A.; Moraes, I.; Felicetti, B.; Wood, M.; Schonfeld, D.; Mather, O.; Cesura, A.; Barker, J. The structure of mammalian serine racemase: Evi-dence for conformational changes upon inhibitor binding. J. Biol. Chem. 2010, 285, 12873–12881. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, F.; Manchenõ, J.M.; Rodriguez-Crespo, I. Insights into the activation of brain serine racemase by the multi-PDZ domain glutamate receptor interacting protein, divalent cations and ATP. FEBS J. 2007, 274, 4561–4571. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Marchesani, F.; Dellafiora, L.; Margiotta, M.; Faggiano, S.; Campanini, B.; Mozzarelli, A. Human serine racemase is allosterically modulated by NADH and reduced nicotinamide derivatives. Biochem. J. 2016, 473, 3505–3516. [Google Scholar] [CrossRef]
- Steiner, P.; Higley, M.; Xu, W.; Czervionke, B.L.; Malenka, R.C.; Sabatini, B.L. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron 2009, 61, 152. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Bernard, L. Postsynaptic density 95 (PSD-95) serine 561 phosphorylation regulates a conformational switch and bi-directional dendritic spine structural plasticity. J. Biol. Chem. 2017, 292, 16150–16160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Petit, C.M.; King, D.S.; Lee, A.L. Phosphorylation of a PDZ domain extension modulates binding affinity and inter-domain interactions in postsynaptic density-95 (PSD-95) protein, a membrane-associated guanylate kinase (MAGUK). J. Biol. Chem. 2011, 286, 41776–41785. [Google Scholar] [CrossRef] [Green Version]
- Mothet, J.P.; Le Bail, M.; Billard, J.M. Time and space profiling of NMDA receptor co-agonist functions. J. Neurochem. 2015, 135, 210–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Jacobi, A.A.; Anderson, S.A.; Lynch, D.R. D-serine and serine racemase are associated with PSD-95 and glutama-tergic synapse stability. Front. Cell. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Lopes, C.; Madeira, C.; Kahn, S.A.; do Couto, I.A.; Bado, P.; Houzel, J.C.; De Miranda, J.; de Freitas, M.S.; Ferreira, S.; Panizzutti, R. Protein kinase C activity regulates D-serine availability in the brain. J. Neurochem. 2011, 116, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Raboni, S.; Marchetti, M.; Faggiano, S.; Campanini, B.; Bruno, S.; Marchesani, F.; Margiotta, M.; Mozzarelli, A. The Energy Landscape of Human Serine Racemase. Front. Mol. Biosci. 2019, 5. [Google Scholar] [CrossRef]
- Hooi, M.Y.-S.; Raftery, M.J.; Truscott, R.J.W. Racemization of two proteins over our lifespan: Deamidation of asparagine 76 in γS crystallin is greater in cataract than in normal lenses across the age range. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3554–3561. [Google Scholar] [CrossRef]
- Baumgart, F.; Rodríguez-Crespo, I. D-amino acids in the brain: The biochemistry of brain serine racemase. FEBS J. 2008, 27, 3538–3545. [Google Scholar] [CrossRef]
- Foltyn, V.N.; Zehl, M.; Dikopoltsev, E.; Jensen, O.N.; Wolosker, H. Phosphorylation of mouse serine racemase regulates D-serine synthesis. FEBS Lett. 2010, 584, 2937–2941. [Google Scholar] [CrossRef] [Green Version]
- Sacchi, S. D-Serine metabolism: New insights into the modulation of D-amino acid oxidase activity. Biochem. Soc. Trans. 2013, 41, 1551–1556. [Google Scholar] [CrossRef] [Green Version]
- Crow, J.P.; Marecki, J.C.; Thompson, M. 3D-Serine Production, Degradation, and Transport in ALS: Critical Role of Methodology. Neurol. Res. Int. 2012, 2012, 625245. [Google Scholar] [CrossRef] [Green Version]
- Hunter, T. Why nature chose phosphate to modify proteins? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2513–2516. [Google Scholar] [CrossRef]
- Ohtani, E. Rate of aspartic acid racemization in bone. Am. J. Forensic Med. Pathol. 1998, 19, 284–287. [Google Scholar] [CrossRef]
- Arany, S.; Ohtani, S.; Yamamoto, T.; Sugiyama, T. Comparison of aspartic acid racemization between mammoth and human dentinal tissues. Arch. Oral. Biol. 2007, 52, 20–25. [Google Scholar] [CrossRef]
- Buchholz, B.A.; Alkass, K.; Druid, H.; Spalding, K.L. Bomb pulse radiocarbon dating of skeletal tissues. In New Perspectives in Forensic Human Skeletal Identification; Elsevier: Amsterdam, The Netherlands, 2018; pp. 185–196. [Google Scholar] [CrossRef]
- Masters, P.; Bada, J.L.; Zigler, J.S. Amino acid racemization in the human lens during cataract formation. Nature 1977, 268, 71–73. [Google Scholar] [CrossRef]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [CrossRef]
- Stephenson, R.C.; Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar] [CrossRef]
- Radkiewicz, J.L.; Zipse, H.; Clarke, S.; Houk, K.N. Accelerated racemization of aspartic acid and asparagine residues via succinimide intermediates: An ab initio theoretical exploration of mechanism. J. Am. Chem. Soc. 1996, 118, 9148–9155. [Google Scholar] [CrossRef]
- Aki, K.; Norihiko, F.; Noriko, F. Kinetics of isomerization and inversion of aspartate 58 of A-crystallin peptide mimics under physiological conditions. PLoS ONE 2013, 8, e58515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaprakash, N.G.; Surolia, A. Role of glycosylation in nucleating protein folding and stability. Biochem. J. 2017, 474, 333–2347. [Google Scholar] [CrossRef]
- Hooi, M.Y.S.; Raftery, M.J.; Truscott, R.J.W. Age-dependent racemization of serine residues in a human chaperone protein. Protein Sci. 2013, 22, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Truscott, R.J.; Friedrich, M.G. The etiology of human age-related cataract. Proteins don’t last forever. Biochim. Biophys. Acta 2016, 860 Pt B, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K. Isomerization of aspartyl residues in crystallins and its influence upon cataract. Biochim. Biophys. Acta. 2016, 1860 Pt B, 183–191. [Google Scholar] [CrossRef]
- Wu, S.; Basile, A.S.; Barger, S.W. Induction of serine racemase expression and D-serine release from microglia by secreted amyloid precursor protein (sAPP). Curr. Alzheimer Res. 2007, 4, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Barger, S.W. Induction of serine racemase by inflammatory stimuli is dependent on AP-1. Ann. N. Y. Acad. Sci. 2004, 1035, 133–146. [Google Scholar] [CrossRef]
- Mattioli, R.; Francioso, A.; d’Erme, M.; Trovato, M.; Mancini, P.; Piacentini, L.; Casale, A.M.; Wessjohann, L.; Gazzino, R.; Costantino, P.; et al. Anti-inflammatory activity of a polyphenolic extract from Arabidopsis thaliana in in vitro and in vivo models of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 708. [Google Scholar] [CrossRef] [Green Version]
- Miya, K.; Inoue, R.; Takata, Y.; Abe, M.; Natsume, R.; Sakimura, K.; Hongou, K.; Miyawaki, T.; Mori, H. Serine racemase is predominantly localized in neurons in mouse brain. J. Comp. Neurol. 2008, 510, 641–654. [Google Scholar] [CrossRef]
- Radisky, E.S.; Lee, J.M.; Lu, C.-J.; Koshland, D.E., Jr. Insights into the serine protease mechanism from atomic resolution struc-tures of trypsin reaction intermediates. Proc. Natl. Acad. Sci. USA 2006, 103, 6835–6840. [Google Scholar] [CrossRef] [Green Version]
- Zakharova, E.; Horvath, M.P.; Goldenberg, D.P. Structure of a serine protease poised to resynthesize a peptide bond. Proc. Natl. Acad. Sci. USA 2009, 106, 11034–11039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krem, M.M.; Cera, E.D. Molecular markers of serine protease evolution. EMBO J. 2001, 20, 3036–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Zhou, Q.; Qiu, L.; Yao, Q.; Chen, K.; Tang, Q.; Hu, Z. Serine protease Bm-SP142 was differentially expressed in resistant and susceptible Bombyx mori strains, involving in the defence response to viral infection. PLoS ONE 2017, 12, e0175518. [Google Scholar] [CrossRef] [Green Version]
- Eldridge, B.; Cooley, R.N.; Odegrip, R.; McGregor, D.P.; Fitzgerald, K.J.; Ullman, C.G. An in vitro selection strategy for con-ferring protease resistance to ligand binding peptides. Protein Eng. Des. Sel. 2009, 22, 691–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kominami, E.; Kobayashi, K.; Kominami, S.; Katunuma, N. Properties of a Specific Protease for Pyridoxal Enzymes and Its Biological Role. J. Biol. Chem. 1972, 247, 6848–6855. [Google Scholar] [CrossRef]
- Katunuma, N.; Kominami, E.; Kobayashi, K.; Banno, Y.; Suzuki, K.; Chichibu, K.; Hamaguchi, Y.; Katsunuma, T. Studies on New Intracellular Proteases in Various Organs of Rat. Purification and Comparison of Their Properties. Eur. J. Biochem. 1975, 52, 37–50. [Google Scholar] [CrossRef]
- Yang, L.; Mei, Y.; Fang, Q.; Wang, J.; Yan, Z.; Song, Q.; Lin, Z.; Ye, G. Identification and characterization of serine protease in-hibitors in a parasitic wasp, Pteromalus puparum. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Kanost, M.R.; Jiang, H. Clip-domain serine proteases as immune factors in insect hemolymph. Curr. Opin. Insect Sci. 2015, 11, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumin, E.; Bendikov, I.; Foltyn, V.N.; Misumi, Y.; Ikehara, Y.; Kartvelishvily, E.; Wolosker, H. Modulation of D-Serine Levels via Ubiquitin-dependent Proteasomal Degradation of Serine Racemase. J. Biol. Chem. 2006, 281, 20291–20302. [Google Scholar] [CrossRef] [Green Version]
- Voĭtiuk, A.; Vasil’Ev, V.V. Quantum chemical study of the “catalytic triad” of serine proteases. Molekuliarnaia Biologiia 1987, 21, 807–813. [Google Scholar]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. Dec. 2002, 102, 4501–4524. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Analysis of Catalytic Mechanism of Serine Proteases. Viability of the Ring-Flip Hypothesis. J. Phys. Chem. B 2008, 112, 6837–6846. [Google Scholar] [CrossRef]
- Lanchec, E.; Désilets, A.; Béliveau, F.; Flamier, A.; Mahmoud, S.; Bernier, G.; Gris, D.; LeDuc, R.; Lavoie, C. The type II trans-membrane serine protease matriptase cleaves the amyloid precursor protein and reduces its processing to β-amyloid peptide. J. Biol. Chem. 2017, 292, 20669–20682. [Google Scholar] [CrossRef] [Green Version]
- Saido, T.; Leissring, A.M. Proteolytic degradation of amyloid β-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef] [Green Version]
- Tennstaedt, A.; Pöpsel, S.; Truebestein, L.; Hauske, P.; Brockmann, A.; Schmidt, N.; Irle, I.; Sacca, B.; Niemeyer, C.M.; Brandt, R.; et al. Human High Temperature Requirement Serine Protease A1 (HTRA1) Degrades Tau Protein Aggregates. J. Biol. Chem. 2012, 287, 20931–20941. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Garg, S.; Mandelkow, E.-M.; Mandelkow, E. Proteolytic processing of TAU. The Biology of TAU and its Role in Tauopathies. Biochem. Soc. Trans. 2010, 38, 955–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, A.C.; Melo, E.; Roth, D.; Topp, A.; Delobel, F.; Stucki, C.; Chen, C.-Y.; Jakob, P.; Banfai, B.; Dunkley, T.; et al. HtrA1 ac-tivation is driven by an allosteric mechanism of inter-monomer communication. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Leissring, M.A. Aβ-Degrading Proteases: Therapeutic Potential in Alzheimer Disease. CNS Drugs 2016, 30, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Fraering, P.C. Structural and Functional Determinants of γ-Secretase, an Intramembrane Protease Implicated in Alzheimers Disease. Curr. Genom. 2007, 8, 531–549. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Li, X.; Shi, Y. Structural biology of intramembrane proteases: Mechanistic insights from rhomboid and S2P to γ-secretase. Curr. Opin. Struct. Biol. 2016, 37, 97–107. [Google Scholar] [CrossRef]
- Zhou, R.; Shi, Y.; Yang, G. Expression, Purification, and Enzymatic Characterization of Intramembrane Proteases. Methods Enzymol. 2017, 584, 127–155. [Google Scholar] [CrossRef]
- Savopoulos, J.W.; Carter, P.S.; Turconi, S.; Pettman, G.R.; Karran, E.H.; Gray, C.W.; Ward, R.V.; Jenkins, O.; Creasy, C.L. Ex-pression, Purification, and Functional Analysis of the Human Serine Protease HtrA2. Protein Expr. Purif. 2000, 19, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Sikka, P.; Walker, R.; Cockayne, R.; Wood, M.J.; Harrison, P.J.; Burnet, P.W. D-Serine metabolism in C6 glioma cells: Involve-ment of alanine-serine-cysteine transporter (ASCT2) and serine racemase (SRR) but not D-amino acid oxidase (DAO). J. Neurosci. Res. 2010, 88, 1829–1840. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.; Paul, P.; Chen, H.-J.; Morris, A.; Payling, M.; Falchi, M.; Habgood, J.; Panoutsou, S.; Winkler, S.; Tisato, V.; et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc. Natl. Acad. Sci. USA 2010, 10, 7556–7561. [Google Scholar] [CrossRef] [Green Version]
- Verrall, L.; Burnet, P.W.J.; Betts, J.F.; Harrison, P.J. The neurobiology of D-amino acid oxidase and its involvement in schizo-phrenia. Mol. Psychiatry 2010, 15, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, V.; Marinova, Z.; Monoranu, C.-M.; Walitza, S.; Grünblatt, E. Expression of D-Amino Acid Oxidase (DAO/DAAO) and D-Amino Acid Oxidase Activator (DAOA/G72) during Development and Aging in the Human Post-mortem Brain. Front. Neuroanat. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Maragos, W.F.; Greenamyre, J.T.; Penney, J.B., Jr.; Young, A.B. Glutamate dysfunction in Alzheimer’s disease: An hypothesis. Trends Neurosci. 1987, 10, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Price, D.L.; Rockenstein, E.; Ubhi, K.; Phung, V.; MacLean-Lewis, N.; Askay, D.; Cartier, A.; Spencer, B.; Patrick, C.; Desplats, P.; et al. Alterations in mGluR5 Expression and Signaling in Lewy Body Disease and in Transgenic Models of Al-pha-Synucleinopathy—Implications for Excitotoxicity. PLoS ONE 2010, 5, e14020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos-Peña, V.; Meraz-Ríos, M.A. Alzheimer Disease: The role of Aβ in the glutamatergic system. In Neurochemistry; Heinbockel, T., Ed.; IntechOpen: London, UK, 2014. [Google Scholar] [CrossRef] [Green Version]
- Elewerenz, J.; Emaher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases—What is the Evidence? Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Frid, K.; Einstein, O.; Friedman-Levi, Y.; Binyamin, O.; Ben-Hur, T.; Gabizon, R. Aggregation of MBP in chronic demye-lination. Ann. Clin. Transl. Neurol. 2015, 2, 711–721. [Google Scholar] [CrossRef]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [Green Version]
- Juenemann, K.; Jansen, A.H.P.; Van Riel, L.; Merkx, R.; Mulder, M.P.C.; An, H.; Statsyuk, A.; Kirstein, J.; Ovaa, H.; Reits, E.A. Dynamic recruitment of ubiquitin to mutant huntingtin inclusion bodies. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Neuberger, A. Stereochemistry of amino acids. Adv. Protein Chem. 1948, 4, 297–383. [Google Scholar] [CrossRef]
- Berrett, G.C. Chemistry and Biochemistry of the Amino Acids; Springer: Cham, Switzerland, 1985; pp. 6–353. [Google Scholar] [CrossRef]
- Bada, J.L.; Schroeder, R.A. Amino acid racemization reactions and their geochemical implications. Naturwissenschaften 1975, 62, 71–79. [Google Scholar] [CrossRef]
- Williams, K.M.; Smith, G.G. A critical evaluation of the application of amino acid racemization to geochronology and geo-thermometry origins of life. Evol. Biosph. 1977, 8, 91–144. [Google Scholar] [CrossRef]
- Brinton, K.L.; Tsapin, A.I.; Gilichinsky, D.; McDonald, G.D. Aspartic Acid Racemization and Age–Depth Relationships for Organic Carbon in Siberian Permafrost. Astrobiology 2002, 2, 77–82. [Google Scholar] [CrossRef]
- Bada, J.L. Racemization of Amino Acids. Chapter 13 in Chemistry and Biochemistry of the Amino Acids; Berrett, G.C., Ed.; Springer: Cham, Switzerland, 1985; pp. 399–414. [Google Scholar] [CrossRef]
- Ros, R.; Muñoz-Bertomeu, J.; Krueger, S. Serine in plants: Biosynthesis, metabolism, and functions. Trends Plant Sci. 2014, 19, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Furuchi, T.; Sakurako, K.; Katane, M.; Sekine, M.; Homma, H. The Role of Protein L-Isoaspartyl/D-Aspartyl O-Methyltransferase (PIMT) in Intracellular Signal Transduction. Chem. Biodivers. 2010, 7, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Tayeb-Fligelman, E.; Tabachnikov, O.; Moshe, A.; Goldshmidt-Tran, O.; Sawaya, M.R.; Coquelle, N.; Colletier, J.-P.; Landau, M. The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science 2017, 355, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Sabate, R.; Estelrich, J. Evidence of the Existence of Micelles in the Fibrillogenesis of β-Amyloid Peptide. J. Phys. Chem. B 2005, 109, 11027–11132. [Google Scholar] [CrossRef]
- Takahashi, T.; Mihara, H. Peptide and Protein Mimetics Inhibiting Amyloid β-Peptide Aggregation. ACC Chem. Res. 2008, 41, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Vitalis, A.; Caflisch, A. Micelle-Like Architecture of the Monomer Ensemble of Alzheimer’s Amyloid-β Peptide in Aqueous Solution and Its Implications for Aβ Aggregation. J. Mol. Biol. 2010, 403, 148–165. [Google Scholar] [CrossRef]
- Bharti, B. Adsorption, Aggregation and Structure Formation in Systems of Charged Particles; Springer: Berlin, Germany, 2014; pp. 81–102. [Google Scholar]
- Kumar, A.; Capua, E.; Kesharwani, M.K.; Martin, J.M.L.; Sitbon, E.; Waldeck, D.H.; Naaman, R. Chirality-induced spin po-larization places symmetry constraints on biomolecular interactions. Proc. Natl. Acad. Sci. USA 2017, 114, 2474–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Schubert, D.; Sawaya, M.R.; Eisenberg, D.; Riek, R. A multi-dimensional Structure-Activity Relationship of a protein in its aggregated states. Angew. Chem. Int. Ed. Engl. 2010, 49, 3904–3908. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Das, S.; Mukhopadhyay, D. Intrinsically unstructured proteins and neurodegenerative diseases: Conformational promiscuity at its best. IUBMB Life 2011, 63, 478–488. [Google Scholar] [CrossRef]
- Zhu, S.; Shala, A.; Bezginov, A.; Sljoka, A.; Audette, G.; Wilson, D.J. Hyperphosphorylation of Intrinsically Disordered Tau Protein Induces an Amyloidogenic Shift in Its Conformational Ensemble. PLoS ONE 2015, 10, e0120416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.W.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Uversky, V.N. The intrinsic disorder alphabet. III. Dual personality of serine. Proteins 2015, 3, e1027032. [Google Scholar] [CrossRef] [Green Version]
- Rubin, N.; Perugia, E.; Goldschmidt, M.; Fridkin, A.M.; Addadi, L. Chirality of Amyloid Suprastructures. J. Am. Chem. Soc. 2008, 30, 4602–4603. [Google Scholar] [CrossRef]
- Dzwolak, W. Chirality and chiroptical properties of amyloid fibrils. Chirality 2014, 26, 580–587. [Google Scholar] [CrossRef]
- Kurouski, D.; Dukor, R.K.; Lu, X.; Nafie, L.A.; Lednev, I.K. Normal and Reversed Supramolecular Chirality of Insulin Fibrils Probed by Vibrational Circular Dichroism at the Protofilament Level of Fibril Structure. Biophys. J. 2012, 103, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Kurouski, D. Supramolecular organization of amyloid fibrils. In Exploring New Findings on Amyloidosis; Fernan-dez-Escamilla, A.-M., Ed.; IntechOpen: London, UK, 2016; Chapter 2; ISBN 978-953-51-2619-5. [Google Scholar] [CrossRef] [Green Version]
- Volpatti, L.R.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. A Clear View of Polymorphism, Twist, and Chirality in Amy-loid Fibril Formation. ACS Nano 2013, 7, 10443–10448. [Google Scholar] [CrossRef] [PubMed]
- Nyström, G.; Arcari, M.; Mezzenga, R. Confinement-induced liquid crystalline transitions in amyloid fibril cholesteric tac-toids. Nat. Nanotechnol. 2018, 3, 330–336. [Google Scholar] [CrossRef]
- Dunlop, R.A.; Cox, P.A.; Banack, S.A.; Rodgers, K.J. The Non-Protein Amino Acid BMAA Is Misincorporated into Human Proteins in Place of l-Serine Causing Protein Misfolding and Aggregation. PLoS ONE 2013, 8, e75376. [Google Scholar] [CrossRef] [Green Version]
- Desguin, B.; Soumillion, P.; Hausinger, R.P.; Hols, P. Unexpected complexity in the lactate racemization system of lactic acid bacteria. FEMS Microbiol. Rev. 2017, 41, S71–S83. [Google Scholar] [CrossRef]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Racemization of the Succinimide Intermediate Formed in Proteins and Peptides: A Computational Study of the Mechanism Catalyzed by Dihydrogen Phosphate Ion. Int. J. Mol. Sci. 2016, 7, 1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrió, M.; González-Montalbán, N.; Vera, A.; Villaverde, A.; Ventura, S. Amyloid-like Properties of Bacterial Inclusion Bodies. J. Mol. Biol. 2005, 347, 1025–1037. [Google Scholar] [CrossRef]
- Villar-Piqué, A.; Ventura, S. Modeling amyloids in bacteria. Microb. Factories 2012, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wersel, R. Amyloid, prions, and other protein aggregates. In Methods in Enzymology; Academic Press: Cambrigde, MA, USA, 1999; Volume 309, ISBN 9780080496672. [Google Scholar]
- Finder, V.H.; Vodopivec, I.; Nitsch, R.M.; Glockshuber, R. The Recombinant Amyloid-β Peptide Aβ1–42 Aggregates Faster and Is More Neurotoxic than Synthetic Aβ1-41. J. Mol. Biol. 2010, 396, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Asano, S.; Furiya, Y.; Shirasawa, T.; Endo, N.; Mori, H. Racemization of Asp23 residue affects the aggregation properties of Alzheimer amyloid beta protein analogues. J. Biol. Chem. 1994, 269, 10205–10208. [Google Scholar] [CrossRef]
- Sahoo, B.; Singer, D.; Kodali, R.; Zuchner, T.; Wetzel, R. Aggregation Behavior of Chemically Synthesized, Full-Length Hun-tingtin Exon1. Biochemistry 2014, 53, 3897–3907. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, M.; Borba, A.; Le Barbu-Debus, K.; Dittrich, B.; Fausto, R.; Grimme, S.; Mahjoub, A.; Nedić, M.; Schmitt, U.; Schrader, L.; et al. Chirality influence on the aggregation of methyl mandelate. N. J. Chem. 2010, 34, 1266–1285. [Google Scholar] [CrossRef]
- Li, M.; Sun, Q.; Bai, Y.; Duan, C.; Zhang, B.; Meng, Q. Chiral aggregation and spontaneous resolution of thiosemicarbazone metal complexes. Dalton Trans. 2006, 21, 2572–2578. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Gentili, D.; Romeo, A.; Cavallini, M.; Scolaro, L.M. Spatial control of chirality in supramolecular aggre-gates. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.; Namsechi, R.; Sim, V.L. Structure-Based Peptide Design to Modulate Amyloid Beta Aggregation and Reduce Cy-totoxicity. PLoS ONE 2015, 10, e0129087. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Foley, A.R.; Warner, C.J.A.; Zhang, X.; Rolandi, M.; Abrams, B.; Raskatov, J.A. Suppression of Oligomer Formation and Formation of Non-Toxic Fibrils upon Addition of Mirror-Image Aβ42 to the Natural l -Enantiomer. Angew. Chem. Int. Ed. 2017, 56, 11506–11510. [Google Scholar] [CrossRef]
- Vivekanandan, S.; Brender, J.R.; Lee, S.Y.; Ramamoorthy, A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 2011, 411, 312–316. [Google Scholar] [CrossRef]
- Jagota, S.; Rajadas, J. Synthesis of d-amino acid peptides and their effect on beta-amyloid aggregation and toxicity in trans-genic Caenorhabditis elegans. Med. Chem. Res. 2013, 22, 3991–4000. [Google Scholar] [CrossRef]
- Nerelius, C.; Sandegren, A.; Sargsyan, H.; Raunak, R.; Leijonmarck, H.; Chatterjee, U.; Fisahn, A.; Imarisio, S.; Lomas, D.A.; Crowther, D.C.; et al. α-Helix targeting reduces amyloid-β peptide toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9191–9196. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Ha-Duong, T. Exploring the Alzheimer amyloid-β peptide conformational ensemble: A review of molecular dynamics approaches. Peptides 2015, 69, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Fein, J.A.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Yang, F.; Cole, G.M.; Gylys, K.H. Co-Localization of Amyloid Beta and Tau Pathology in Alzheimer’s Disease Synaptosomes. Am. J. Pathol. 2008, 172, 1683–1692. [Google Scholar] [CrossRef] [Green Version]
- Ott, S.; Henkel, A.W.; Henkel, M.K.; Redzic, Z.B.; Kornhuber, J.; Wiltfang, J. Pre-aggregated Aβ1-42 peptide increases tau ag-gregation and hyperphosphorylation after short-term application. Mol. Cell. Biochem. 2011, 349, 169–177. [Google Scholar] [CrossRef]
- Rauk, A. Why is the amyloid beta peptide of Alzheimer’s disease neurotoxic? Dalton Trans. 2008, 1, 1273–1282. [Google Scholar] [CrossRef]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s Disease Amyloid-β Binds Copper and Zinc to Generate an Allosterically Ordered Membrane-penetrating Structure Containing Superoxide Dismutase-like Subunits. J. Biol. Chem. 2001, 276, 20466–20473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium Signaling and Amyloid Toxicity in Alzheimer Disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta 2007, 176, 1976–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Nairn, A.; Gandy, S.; Greengard, P. Phosphorylation of alzheimer amyloid precursor protein by protein kinase C. Neuroscience 1992, 48, 755–761. [Google Scholar] [CrossRef]
- Hung, A.Y.; Selkoe, D.J. Selective ectodomain phosphorylation and regulated cleavage of beta-amyloid precursor protein. EMBO J. 1994, 13, 534–542. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Wolosker, H. NMDA Receptor Regulation by D-serine: New Findings and Perspectives. Mol. Neurobiol. 2007, 36, 152–164. [Google Scholar] [CrossRef]
- Airas, J.M.; Betz, H.; El Far, O. PKC phosphorylation of a conserved serine residue in the C-terminus of group III metabotropic glutamate receptors inhibits calmodulin binding. FEBS Lett. 2001, 494, 60–63. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.L.; Lindner, A.B. Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxidative Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Lemaire, H.-G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.-H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Robakis, N.; Wisniewski, H.; Jenkins, E.; Devine-Gage, E.; Houck, G.; Yao, X.-L.; Ramakrishna, N.; Wolfe, G.; Silverman, W.; Brown, W.T. Chromosome 21q21 sublocalisation of gene encoding beta-amyloid peptide in cerebral vessels and neuritic (se-nile) plaques of people with alzheimer disease and down syndrome. Lancet 1987, 1, 384–385. [Google Scholar] [CrossRef]
- Yoshikai, S.-I.; Sasaki, H.; Doh-Ura, K.; Furuya, H.; Sakaki, Y. Genomic organization of the human-amyloid beta-protein pre-cursor gene. Gene 1991, 102, 291–292. [Google Scholar] [CrossRef]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha -synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Mar, C.; Greenbaum, E.A.; Mayne, L.; Englander, S.W.; Woods, V.L. Structure and properties of alpha-synuclein and other amyloids determined at the amino acid level. Proc. Natl. Acad. Sci. USA 2005, 102, 15477–15482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meredith, S.C. Protein Denaturation and Aggregation: Cellular Responses to Denatured and Aggregated Proteins. Ann. N. Y. Acad. Sci. 2005, 1066, 181–221. [Google Scholar] [CrossRef] [PubMed]
- Tambo, K.; Yamaguchi, T.; Kobayashi, K.; Terauchi, E.; Ichi, I.; Kojo, S. Racemization of the Aspartic Acid Residue of Amy-loid-β Peptide by a Radical Reaction. Biosci. Biotechnol. Biochem. 2013, 77, 416–418. [Google Scholar] [CrossRef]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimer’s Res. Ther. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Panatier, A.; Theodosis, D.T.; Mothet, J.-P.; Touquet, B.; Pollegioni, L.; Poulain, D.A.; Oliet, S.H. Glia-Derived d-Serine Controls NMDA Receptor Activity and Synaptic Memory. Cell 2006, 125, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Rudy, C.C.; Hunsberger, H.C.; Weitzner, D.S.; Reed, M.N. The Role of the Tripartite Glutamatergic Synapse in the Pathophysi-ology of Alzheimer’s Disease. Aging Dis. 2015, 6, 131–148. [Google Scholar] [CrossRef] [Green Version]
- Ying-Luan, Z.; Zhao, Y.L.; Mori, H. Role of D-serine in the mammalian brain. Brain Nerve 2007, 59, 725–730. [Google Scholar]
- Hashimoto, A.; Nishikawa, T.; Oka, T.; Takahashi, K. Endogenous d-Serine in Rat Brain: N-Methyl-d-Aspartate Recep-tor-Related Distribution and Aging. J. Neurochem. 1993, 60, 783–786. [Google Scholar] [CrossRef]
- Matoba, M.; Tomita, U.; Nishikawa, T. Characterization of 5,7-Dichlorokynurenate-Insensitive d-[3H]Serine Binding to Synaptosomal Fraction Isolated from Rat Brain Tissues. J. Neurochem. 1997, 69, 399–405. [Google Scholar] [CrossRef]
- Mothet, J.-P.; Parent, A.T.; Wolosker, H.; Brady, R.O.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef] [Green Version]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-term potentiation depends on release of d-serine from astro-cytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [CrossRef]
- Kenessey, A.; Yen, S.-H.; Liu, W.-K.; Yang, X.-R.; Dunlop, D.S. Detection of d-aspartate in tau proteins associated with Alz-heimer paired helical filaments. Brain Res. 1995, 675, 183–189. [Google Scholar] [CrossRef]
- Towse, C.-L.; Hopping, G.; Vulovic, I.; Daggett, V. Nature versus design: The conformational propensities of d-amino acids and the importance of side chain chirality. Protein Eng. Des. Sel. 2014, 27, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Rabideau, A.E.; Pentelute, B.L. Ad-Amino Acid at the N-Terminus of a Protein Abrogates Its Degradation by the N-End Rule Pathway. ACS Cent. Sci. 2015, 1, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zheng, G.; Peng, X.; Qiang, B.; Yuan, J. D-Amino acids andD-Tyr-tRNATyrdeacylase: Stereospecificity of the trans-lation machine revisited. FEBS Lett. 2003, 552, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Achenbach, J.; Jahnz, M.; Bethge, L.; Paal, K.; Jung, M.; Schuster, M.; Albrecht, R.; Jarosch, F.; Nierhaus, K.H.; Klussmann, S. Outwitting EF-Tu and the ribosome: Translation with d-amino acids. Nucleic Acids Res. 2015, 43, 5687–5698. [Google Scholar] [CrossRef] [Green Version]
- Katoh, T.; Tajima, K.; Suga, H. Consecutive Elongation of D-Amino Acids in Translation. Cell Chem. Biol. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Park, H.-S.; Hohn, M.J.; Umehara, T.; Guo, L.-T.; Osborne, E.M.; Benner, J.; Noren, C.J.; Rinehart, J.; Söll, D. Expanding the Ge-netic Code of Escherichia coli with Phosphoserine. Science 2011, 333, 1151–1154. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphor-ylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Tinette, S.; Feyereisen, R.; Robichon, A. Approach to systematic analysis of serine/threonine phosphoproteome usingBeta elimination and subsequent side effects: Intramolecular linkage and/or racemisation. J. Cell Biochem. 2007, 100, 875–882. [Google Scholar] [CrossRef]
- Silverman, D. PET in the Evaluation of Alzheimer’s Disease and Related Disorders; Springer: New York, NY, USA, 2009. [Google Scholar]
- Belle, R.; Al Temimi, A.H.K.; Kumar, K.; Pieters, B.J.G.E.; Tumber, A.; Dunford, J.E.; Johansson, C.; Oppermann, U.; Brown, T.; Schofield, C.J.; et al. Investigatingd-lysine stereochemistry for epigenetic methylation, demethylation and recognition. Chem. Commun. 2017, 53, 13264–13267. [Google Scholar] [CrossRef]
- Deas, J.; Erecinska, M. Effect of tunicamycin, an inhibitor of protein glycosylation, on the high-affinity transport of acidic amino acid neurotransmitters in C6 glioma cells. Brain Res. 1989, 483, 84–90. [Google Scholar] [CrossRef]
- Janetzko, J.; Walker, S. Aspartate Glycosylation Triggers Isomerization to Isoaspartate. J. Am. Chem. Soc. 2017, 139, 3332–3335. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Li, X.; Xie, Z.; Xie, C.; Zhan, C.; Hu, X.; Shen, Q.; Wei, X.; Su, B.; Wang, J.; et al. D-Peptides as Recognition Molecules and Therapeutic Agents. Chem. Rec. 2016, 16, 1772–1786. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Nissim, A.; Winyard, P.G. Oxidative post-translational modifications and their involvement in the pathogenesis of autoimmune diseases. Redox Biol. 2014, 2, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Tiwary, E.; Hegde, S.; Purushotham, S.; Deivanayagam, C.; Srivastava, O. Deamidation is one of the most common post-translational modifications in crystallins, which results in incorrect interaction and leads to aggregate formation. PLoS ONE 2015, 10, e0144621. [Google Scholar] [CrossRef]
- Thornell, E.; Aquilina, A. Regulation of αA- and αB-crystallins via phosphorylation in cellular homeostasis. Cell. Mol. Life Sci. 2015, 72, 4127–4137. [Google Scholar] [CrossRef]
- Metzler, D.E.; Snell, E.E. Deamination of serine. II. D-Serine dehydrase, a vitamin B6 enzyme from Escherichia coli. J. Biol. Chem. 1952, 198, 363–373. [Google Scholar] [CrossRef]
- Greenberg, C.S.; Birckbichler, P.J.; Rice, R.H. Transglutaminases: Multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991, 5. [Google Scholar] [CrossRef] [PubMed]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281. [Google Scholar] [CrossRef]
- Roher, A.; Lowenson, J.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.; Reardon, I.; Zürcher-Neely, H.; Heinrikson, R.; Ball, M. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Comparison of the proteolytic susceptibilities of homologous L-amino acid, D-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev. Res. 1995, 35, 20–32. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H.; Muller, S. D-peptides as immunogens and diagnostic reagents. Curr. Opin. Biotechnol. 1998, 9, 377–382. [Google Scholar] [CrossRef]
- Munegumi, T. Hydrophobicity of Peptides Containing D-Amino Acids. Chem. Biodivers. 2010, 7, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Helfman, P.M.; Bada, J.L. Aspartic acid racemisation in dentine as a measure of ageing. Nature 1976, 262, 279–281. [Google Scholar] [CrossRef]
- Ingrosso, D.; Perna, A.E. D-amino acids in aging erythrocytes. EXC 1998, 85, 119–141. [Google Scholar] [CrossRef]
- Clarke, S. Aging as war between chemical and biochemical processes: Protein methylation and the recognition of age-damaged proteins for repair. Ageing Res. Rev. 2003, 2, 263–285. [Google Scholar] [CrossRef]
- Meissner, C.; Ritz-Timme, S. Molecular pathology and age estimation. Forensic Sci. Int. 2010, 203, 34–43. [Google Scholar] [CrossRef]
- Lyons, B.; Kwan, A.H.; Jamie, J.; Truscott, R.J.W. Age-dependent modification of proteins: N-terminal racemization. FEBS J. 2013, 280, 1980–1990. [Google Scholar] [CrossRef]
- D’Aniello, A. D-Aspartic acid: An endogenous amino acid with an important neuroendocrine role. Brain Res. Rev. 2007, 53, 215–234. [Google Scholar] [CrossRef]
- D’Aniello, S.; Somorjai, I.M.L.; Garcia-Fernàndez, J.; Topo, E.; D’Aniello, A. D-Aspartic acid is a novel endogenous neuro-transmitter. FASEB J. 2011, 25, 1014–1027. [Google Scholar] [CrossRef] [Green Version]
- Henneberger, C.; Bard, L.; Rusakov, D.A. D-Serine: A key to synaptic plasticity? Int. J. Biochem. Cell Biol. 2012, 44, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gao, Y.; Li, D.; Fleming, J.; Li, H.; Bi, L. Crystal structure of maize serine racemase with pyridoxal 5’-phosphate. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72, 642–645. [Google Scholar] [CrossRef]
- Perez, E.J.; Tapanes, S.A.; Loris, Z.B.; Balu, D.T.; Sick, T.J.; Coyle, J.T.; Liebl, D.J. Enhanced astrocytic d-serine underlies synap-tic damage after traumatic brain injury. J. Clin. Investig. 2017, 127, 3114–3125. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, J.; Chiba, T.; Yamada, M.; Okamoto, K.; Nishimoto, I.; Matsuoka, M.; Aiso, S. D-Serine is a key determinant of gluta-mate toxicity in amyotrophic lateral sclerosis. EMBO J. 2007, 26, 4149–4159. [Google Scholar] [CrossRef] [Green Version]
- Martineau, M.; Galli, T.; Baux, G.; Mothet, J.-P. Confocal imaging and tracking of the exocytotic routes for D-serine-mediated gliotransmission. Glia 2008, 56, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Martineau, M.; Shi, T.; Puyal, J.; Knolhoff, A.M.; Dulong, J.; Gasnier, B.; Klingauf, J.; Sweedler, J.V.; Jahn, R.; Mothet, J.-P. Stor-age and uptake of D-serine into astrocytic synaptic-like vesicles specify gliotransmission. J. Neurosci. 2013, 3, 3413–3423. [Google Scholar] [CrossRef] [Green Version]
- Guetg, N.; Aziz, S.A.; Holbro, N.; Turecek, R.; Rose, T.; Seddik, R.; Gassmann, M.; Moes, S.; Jenoe, P.; Oertner, T.G.; et al. NMDA receptor-dependent GABAB receptor internalization via CaMKII phosphorylation of serine 867 in GABAB. Proc. Natl. Acad. Sci. USA 2010, 107, 13924–13929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.S.; Paul, R.K.; Ramamoorthy, A.; Torjman, M.C.; Moaddel, R.; Bernier, M.; Wainer, I.W. Nicotinic acetylcholine re-ceptor antagonists alter the function and expression of serine racemase in PC-12 and 1321N1 cells. Cell. Signal. 2013, 12, 2634–4265. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, C.; Yang, Q.; Jiao, M.; Qiu, S. Endocytosis of GluN2B-containing NMDA receptor mediates NMDA-induced excitotoxicity. Mol. Pain 2017, 13. [Google Scholar] [CrossRef] [Green Version]
- Inoue, R.; Hashimoto, K.; Harai, T.; Mori, H. NMDA- and β-Amyloid1–42-Induced Neurotoxicity Is Attenuated in Serine Racemase Knock-Out Mice. J. Neurosci. 2008, 28, 14486–14491. [Google Scholar] [CrossRef] [PubMed]
- Merlen, C.; Fabrega, S.; Desbuquois, B.; Unson, C.G.; Authier, F. Glucagon-mediated internalization of serine-phosphorylated glucagon receptor and Gsα in rat liver. FEBS Lett. 2006, 580, 5697–5704. [Google Scholar] [CrossRef]
- Nadratowska-Wesolowska, B.; Haugsten, E.M.; Zakrzewska, M.; Jakimowicz, P.; Zhen, Y.; Pajdzik, D.; Wesche, J.; Wiedlocha, A. RSK2 regulates endocytosis of FGF receptor 1 by phosphorylation on serine 789. Oncogene 2014, 33, 4823–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, S.; Liu, C.-X.; Migliorini, M.M.; Von Arnim, C.A.; Peltan, I.D.; Mikhailenko, I.; Hyman, B.T.; Strickland, D.K. Serine and Threonine Phosphorylation of the Low Density Lipoprotein Receptor-related Protein by Protein Kinase Cα Regu-lates Endocytosis and Association with Adaptor Molecules. J. Biol. Chem. 2004, 279, 40536–40544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, D.J.; Millman, E.E.; Godines, V.; Friedman, J.; Tran, T.M.; Dai, W.; Knoll, B.J.; Clark, R.B.; Moore, R.H. Role of the G Protein-coupled Receptor Kinase Site Serine Cluster in β2-Adrenergic Receptor Internalization, Desensitization, and β-Arrestin Translocation. J. Biol. Chem. 2006, 281, 7684–7692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitcher, C.; Höning, S.; Fingerhut, A.; Bowers, K.; Marsh, M. Cluster of Differentiation Antigen 4 (CD4) Endocytosis and Adaptor Complex Binding Require Activation of the CD4 Endocytosis Signal by Serine Phosphorylation. Mol. Biol. Cell 1999, 10, 677–691. [Google Scholar] [CrossRef]
- Kelly, B.T.; McCoy, A.J.; Späte, K.; Miller, S.E.; Evans, P.R.; Höning, S.; Owen, D.J. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 2008, 456, 976–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, A.W.; Völker, K.; Dantzler, W.H.; Silbernagl, S. Why is d-serine nephrotoxic and α-aminoisobutyric acid protective? Am. J. Physiol. Ren. Physiol. 2007, 293, F382–F390. [Google Scholar] [CrossRef] [Green Version]
- Canu, N.; Ciotti, M.T.; Pollegioni, L. Serine racemase: A key player in apoptosis and necrosis. Front. Synaptic Neurosci. 2014, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, G.; Inoue, R.; Yoshida, T.; Ishimoto, T.; Yaku, K.; Nakagawa, T.; Mori, H. Novel role of serine racemase in an-ti-apoptosis and metabolism. Biochim. Biophys. Acta 2017, 1861, 3378–3387. [Google Scholar] [CrossRef]
- Cook, S.P.; Galve-Roperh, I.; Del Pozo, Á.M.; Rodríguez-Crespo, I. Direct Calcium Binding Results in Activation of Brain Ser-ine Racemase. J. Biol. Chem. 2002, 277, 27782–27792. [Google Scholar] [CrossRef] [Green Version]
- Stříšovský, K.; Jirásková, J.; Bařinka, C.; Majer, P.; Rojas, C.; Slusher, B.S.; Konvalinka, J. Mouse brain serine racemase cata-lyzes specific elimination of L -serine to pyruvate. FEBS Lett. 2003, 535, 44–48. [Google Scholar] [CrossRef] [Green Version]
- Snyder, S.H.; Wolosker, H.; Sheth, K.; Masaaki, T.; Mothet, J.-P.; Brady, R.; Ferris, C.D., Jr. Mammalian serine racemase. US Patent 6,984,484 B1, 10 January 2006. Available online: https://patents.google.com/patent/US6984484 (accessed on 22 February 2021).
- De Miranda, J.; Panizzutti, R.; Foltyn, V.N.; Wolosker, H. Cofactors of serine racemase that physiologically stimulate the synthesis of the N-methyl-D-aspartate (NMDA) receptor coagonist D-serine. Proc. Natl. Acad. Sci. USA 2002, 99, 14542–14547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, M.; Bruno, S.; Campanini, B.; Peracchi, A.; Mai, N.; Mozzarelli, A. ATP binding to human serine racemase is coop-erative and modulated by glycine. FEBS J. 2013, 280, 5853–5863. [Google Scholar] [CrossRef] [PubMed]
- Canosa, A.V.; Faggiano, S.; Marchetti, M.; Armao, S.; Bettati, S.; Bruno, S.; Percudani, R.; Campanini, B.; Mozzarelli, A. Glutamine 89 is a key residue in the allosteric modulation of human serine racemase activity by ATP. Sci Rep. 2018, 8. [Google Scholar] [CrossRef]
- Amery, L.; Fransen, M.; De Nys, K.; Mannaerts, G.P.; Van Veldhoven, P.P. Mitochondrial peroxisomal targeting of 2-methylacyl-CoA racemase in humans. J. Lipid. Res. 2000, 41, 1752–1759. [Google Scholar] [CrossRef]
- Genchi, G. An overview on d-amino acids. Amino Acids 2017, 49, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Wolosker, H.; Mori, H. Serine racemase: An unconventional enzyme for an unconventional transmitter. Amino Acids 2012, 43, 1895–1904. [Google Scholar] [CrossRef]
- Lee, J.M.; Petrucelli, L.; Fisher, G.; Ramdath, S.; Castillo, J.; Di Fiore, M.M.; D’Aniello, A. Evidence for D-Aspartyl-β-Amyloid Secretase Activity in Human Brain. J. Neuropathol. Exp. Neurol. 2002, 61, 125–131. [Google Scholar] [CrossRef]
- Basu, A.C.; Tsai, G.E.; Ma, C.-L.; Ehmsen, J.T.; Mustafa, A.K.; Han, L.; Jiang, Z.I.; Benneyworth, M.A.; Froimowitz, M.P.; Lange, N.; et al. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol. Psychiatry 2009, 14, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Kasai, A. The Increase of D-Amino Acids in Alzheimer′s Disease-Related Proteins Isolated from the Brains of SAMB8 Mice. J. Soc. Jpn. Women Sci. 2012, 11, 57–64. [Google Scholar] [CrossRef]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117 Pt 24, 5721–5729. [Google Scholar] [CrossRef] [Green Version]
- Kasai, A.; Yamashita, N.; Utsunomiya-Tate, N. Collagen Racemization and Deposition in the Lungs of Aged Rats. Biochem. Insights 2010, 2010, 5–8. [Google Scholar] [CrossRef]
- Boxall, S.F.; Kadu, N.; Dever, L.V.; Kneřová, J.; Waller, J.L.; Gould, P.J.D.; Hartwell, J. Kalanchoë PPC1 Is Essential for Crassulacean Acid Metabolism and the Regulation of Core. Circadian Clock and Guard Cell Signaling Genes. Plant Cell 2020, 32, 1136–1160. [Google Scholar] [CrossRef] [Green Version]
- Schweiger, S.; Matthes, F.; Posey, K.; Kickstein, E.; Weber, S.; Hettich, M.M.; Pfurtscheller, S.; Ehninger, D.; Schneider, R.; Krauß, S. Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, J.; Miyoshi, Y.; Suzuki, M.; Mita, M.; Konno, R.; Matsuoka, M.; Hamase, K.; Aiso, S. D-Amino acid oxidase controls motoneuron degeneration through D-serine. Proc. Natl. Acad. Sci. USA 2011, 109, 627–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristofikova, Z.; Ricny, J.; Ort, M.; Řípová, D. Aging and Lateralization of the Rat Brain on a Biochemical Level. Neurochem. Res. 2010, 35, 1138–1146. [Google Scholar] [CrossRef]
- Fichtman, B.; Harel, A. Stress and aging at the nuclear gateway. Mech. Ageing Dev. 2014, 135, 24–32. [Google Scholar] [CrossRef]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R.; Hetzer, M.W. Identification of Long-Lived Proteins Reveals Exceptional Stability of Essential Cellular Structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Thakurela, S.; Garding, A.; Jung, R.B.; Müller, C.; Goebbels, S.; White, R.; Werner, H.B.; Tiwari, V.K. The transcriptome of mouse central nervous system myelin. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Potier, B.; Turpin, F.R.; Sinet, P.-M.; Rouaud, E.; Mothet, J.-P.; Videau, C.; Epelbaum, J.; Dutar, P.; Billard, J.-M. Contribution of the D-Serine-dependent pathway to the cellular mechanisms underlying cognitive aging. Front. Aging Neurosci. 2010, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Panizzutti, R.; Scoriels, L.; Avellar, M. The co-agonist site of NMDA-glutamate receptors: A novel therapeutic target for age-related cognitive decline. Curr. Pharm. Des. 2014, 20, 5160–5168. [Google Scholar] [CrossRef]
- Bourdenx, M.; Bezard, E.; Dehay, B. Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front. Neuroanat. 2014, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, A.; Fujiwara, Y. Abnormal and deficient processing of β-amyloid precursor protein in familial Alzheimer’s disease lymphoblastoid cells. Biochem. Biophys. Res. Commun. 2014, 175, 361–365. [Google Scholar] [CrossRef]
- Fuentes, J.M. Toxicity and Autophagy in Neurodegenerative Disorders; Springer: Berlin, Germany, 2015; p. 132. [Google Scholar]
- Majumdar, A.; Cesario, W.C.; White-Grindley, E.; Jiang, H.; Ren, F.; Khan, M.R.; Li, L.; Choi, E.M.-L.; Kannan, K.; Guo, F.; et al. Critical Role of Amyloid-like Oligomers of Drosophila Orb2 in the Persistence of Memory. Cell 2012, 148, 515–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipkiss, A.R. Accumulation of altered proteins and ageing: Causes and effects. Exp. Gerontol. 2006, 41, 464–473. [Google Scholar] [CrossRef]
- Fujii, N.; Kaji, Y.; Fujii, N.; Nakamura, T.; Motoie, R.; Mori, Y.; Kinouchi, T. Collapse of Homochirality of Amino Acids in Proteins from Various Tissues during Aging. Chem. Biodivers. 2010, 7, 1389–1397. [Google Scholar] [CrossRef]
- Fujii, N.; Takata, T.; Aki, K.; Sakaue, H. D-Amino acids in protein: The mirror of life as a molecular index of aging. Biochim. Biophys. Acta 2018, 1866, 840–847. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein Modification in Aging. J. Gerontol. 1988, 43, B112–B120. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Kumashiro, S.; Nishikawa, T.; Oka, T.; Takahashi, K.; Mito, T.; Takashima, S.; Doi, N.; Mizutani, Y.; Yama-zaki, T.; et al. Embryonic Development and Postnatal Changes in Free d-Aspartate and d-Serine in the Human Prefrontal Cortex. J. Neurochem. 1993, 61, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Konno, R.; Niwa, A. Amino acid levels in d-alanine-administered mutant mice lacking d-amino acid oxidase. Metabolism 1994, 43, 1153–1157. [Google Scholar] [CrossRef]
- Gagneux, P. The Evolution of Developmental Pathways. J. Hered. 2002, 93, 460–461. [Google Scholar] [CrossRef]
- Carmena, A. Signaling networks during development: The case of asymmetric cell division in the Drosophila nervous system. Dev. Biol. 2008, 321, 1–17. [Google Scholar] [CrossRef]
- Truscott, R.J.; Schey, K.L.; Friedrich, M.G. Old Proteins in Man: A Field in its Infancy. Trends Biochem. Sci. 2016, 41, 654–664. [Google Scholar] [CrossRef] [Green Version]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of dis-tinct β-amyloid peptide species, AβN3(pE), in senile plaques. Neuron 1995, 1, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Watanabe, A.; Ogawara, M.; Mori, H.; Shirasawa, T. Isoaspartate Formation and Neurodegeneration in Alz-heimer’s Disease. Arch. Biochem. Biophys. 2000, 381, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M. Protein misfolding and aggregation: New examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 2004, 1739, 5–25. [Google Scholar] [CrossRef] [Green Version]
- Weids, A.J.; Ibstedt, S.; Tamás, M.J.; Grant, C.M. Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Cox, P.A.; Davis, D.A.; Mash, D.C.; Metcalf, J.S.; Banack, S.A. Dietary exposure to an environmental toxin triggers neurofi-brillary tangles and amyloid deposits in the brain. Proc. R. Soc. B 2016, 283. [Google Scholar] [CrossRef] [Green Version]
- Höhn, A.; Jung, T.; Grune, T. Pathophysiological importance of aggregated damaged proteins. Free Radic. Biol. Med. 2014, 71, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Lorin, S.; Blommaart, E.F.; Codogno, P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2015, 47, 2037–2063. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.D.; Mun, S.R.; Ji, C.H.; Sung, K.W.; Kang, K.Y.; Heo, A.J.; Lee, S.H.; An, J.Y.; Hwang, J.; Xie, X.-Q.; et al. N-terminal argi-nylation generates a bimodal degron that modulates autophagic proteolysis. Proc. Natl. Acad. Sci. USA 2018, 115, E2716–E2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Lowenson, J.D.; MacLaren, D.C.; Clarke, S.; Young, S.G. Deficiency of a protein-repair enzyme results in the accumu-lation of altered proteins, retardation of growth, and fatal seizures in mice. Proc. Natl. Acad. Sci. USA 1997, 94, 6132–6137. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Lowenson, J.D.; Clarke, S.; Young, S.G. Phenotypic Analysis of Seizure-prone Mice Lackingl-Isoaspartate (d-Aspartate)O-Methyltransferase. J. Biol. Chem. 1999, 274, 20671–20678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150. [Google Scholar] [CrossRef] [Green Version]
- Young, A.R.J.; Chan, E.Y.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119 Pt 18, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.Y.W.; Kir, S.; Tooze, S.A. siRNA Screening of the Kinome Identifies ULK1 as a Multidomain Modulator of Autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef] [Green Version]
- Kaminskyy, V.; Zhivotovsky, B. Proteases in autophagy. Biochim. Biophys. Acta 2011, 1824, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Trapan, J.A. Granzymes: A family of lymphocyte granule serine proteases. Genome Biol. 2001, 2. [Google Scholar] [CrossRef]
- Moffitt, K.; Martin, S.; Walker, B. The emerging role of serine proteases in apoptosis. Biochem. Soc. Trans. 2007, 35 Pt 3, 559–560. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.; Sim, V. D-amino acid-based peptide inhibitors as early or preventative therapy in Alzheimer disease. Prion 2014, 8, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesehan, K.; Buder, K.; Linke, R.P.; Patt, S.; Stoldt, M.; Unger, E.; Schmitt, B.; Bucci, E.; Willbold, D. Selection of D-Amino-Acid Peptides That Bind to Alzheimer’s Disease Amyloid Peptide Aβ142 by Mirror Image Phage Display. ChemBioChem 2003, 4, 748–753. [Google Scholar] [CrossRef]
- Wiesehan, K.; Stohr, J.; Nagel-Steger, L.; Van Groen, T.; Riesner, D.; Willbold, D. Inhibition of cytotoxicity and amyloid fibril formation by a D-amino acid peptide that specifically binds to Alzheimer’s disease amyloid peptide. Protein Eng. Des. Sel. 2008, 21, 241–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapira, R.; Wilkinson, K.D.; Shapira, G. Racemization of Individual Aspartate Residues in Human Myelin Basic Protein. J. Neurochem. 1988, 50, 649–654. [Google Scholar] [CrossRef]
- Shapira, R.; Austin, G.E.; Mirra, S.S. Neuritic Plaque Amyloid in Alzheimer’s Disease Is Highly Racemized. J. Neurochem. 1988, 50, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.H.; D’Aniello, A.; Vetere, A.; Padula, L.; Cusano, G.P.; Man, E.H. Free D-aspartate and D-alanine in normal and Alzheimer brain. Brain Res. Bull. 1991, 26, 983–985. [Google Scholar] [CrossRef]
- Madeira, C.; Lourenco, M.V.; Vargaslopes, C.; Suemoto, C.; Brandao, C.O.; Reis, T.; Leite, R.E.P.; Laks, J.; Jacobfilho, W.; Pasqualucci, C.A.; et al. D-serine levels in Alzheimer’s disease: Implications for novel biomarker development. Transl. Psychiatry 2015, 5, e561. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, D.H.; Pike, C.J.; Weinstein, S.L.; Velazquez, P.; Cotman, C.W. All-D-Enantiomers of β-Amyloid Exhibit Similar Bio-logical Properties to All-L-β-Amyloids. J. Biol. Chem. 1997, 272, 7431–7436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deloncle, R.; Guillard, O. Is Brain Copper Deficiency in Alzheimer’s, Lewy Body, and Creutzfeldt Jakob Diseases the Common Key for a Free Radical Mechanism and Oxidative Stress-Induced Damage? J. Alzheimer’s Dis. 2015; 43, 1149–1156. [Google Scholar] [CrossRef]
- Zheng, H.-F.; Wang, W.-Q.; Li, X.-M.; Rauw, G.; Baker, G.B. Body fluid levels of neuroactive amino acids in autism spectrum disorders: A review of the literature. Amino Acids 2017, 49, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Kokkoni, N.; Stott, K.; Amijee, H.; Mason, A.J.M.; Doig, A.J. N-Methylated Peptide Inhibitors of β-Amyloid Aggregation and Toxicity. Optimization of the Inhibitor Structure. Biochemistry 2006, 45, 9906–9918. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.A.; Willbold, D. A Survey of Peptides with Effective Therapeutic Potential in Alzheimer’s Disease Rodent Models or in Human Clinical Studies. Med. Chem. 2012, 12, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Haase, C.; Stieler, J.T.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar] [CrossRef]
- Somers, C.; Goossens, J.; Engelborghs, S.; Bjerke, M. Selecting Aβ isoforms for an Alzheimer’s disease cerebrospinal fluid biomarker panel. Biomark. Med. 2017, 11. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Hüsken, U.; Carl, M. The Wnt/beta-catenin signaling pathway establishes neuroanatomical asymmetries and their laterality. Mech. Dev. 2013, 130, 330–335. [Google Scholar] [CrossRef]
- Toga, A.W.; Thompson, P.M. Mapping brain asymmetry. Nat. Rev. Neurosci. 2003, 4, 37–48. [Google Scholar] [CrossRef]
- Chen, C.; Omiya, Y. Brain asymmetry in cortical thickness is correlated with cognitive function. Front. Hum. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J.; Metz, G.A.; Güntürkün, O.; Ocklenburg, S. Beyond the genome—Towards an epigenetic understanding of hand-edness ontogenesis. Prog. Neurobiol. 2017, 159, 69–89. [Google Scholar] [CrossRef]
- Tomaselli, S.; Pagano, K.; D’Arrigo, C.; Molinari, H.; Ragona, L. Evidence of Molecular Interactions of Aβ1–42 with N-Terminal Truncated Beta Amyloids by NMR. ACS Chem. Neurosci. 2017, 8, 759–765. [Google Scholar] [CrossRef]
- Haass, C. Take five—BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef]
- Ben-Shem, A.; Fass, D.; Bibi, E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc. Natl. Acad. Sci. USA 2006, 104, 462–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Bai, X.-C.; Ma, D.; Xie, T.; Yan, C.; Sun, L.; Yang, G.; Zhao, Y.; Zhou, R.; Scheres, S.H.W.; et al. Three-dimensional struc-ture of human γ-secretase. Nature 2014, 512, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Quesada, V.; Ordóñez, G.R.; Sánchez, L.M.; Puente, X.S.; López-Otín, C. The Degradome database: Mammalian proteases and diseases of proteolysis. Nucleic Acids Res. 2009, 37, D239–D243. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Silva, J.G.; Español, Y.; Velasco, G.; Quesada, V. The Degradome database: Expanding roles of mammalian proteases in life and disease. Nucleic Acids Res. 2016, 44, D351–D355. [Google Scholar] [CrossRef] [PubMed]
- Yates, R.M.; Hermetter, A.; Russell, D.G. Recording phagosome maturation through the real-time, spectrofluorometric meas-urement of hydrolytic activities. Methods Mol. Biol. 2009, 531, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 179, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Papouin, T.; Dunphy, J.M.; Tolman, M.; Dineley, K.T.; Haydon, P.G. Septal Cholinergic Neuromodulation Tunes the Astro-cyte-Dependent Gating of Hippocampal NMDA Receptors to Wakefulness. Neuron 2017, 94, 840–854.e7. [Google Scholar] [CrossRef] [Green Version]
- Whalley, K. Neuron-glia interactions: Waking the synapse. Nat. Rev. Neurosci. 2017, 18, 386. [Google Scholar] [CrossRef]
- Bernstein, H.-G. Proteases and Alzheimer’s disease: Present knowledge and emerging concepts of therapy. In Proteases in the Brain; Lendeckel, U., Hooper, N.M., Eds.; Springer: Berlin, Germany, 2005; Chapter 1. [Google Scholar]
- Murray, B.; Sharma, B.; Belfort, G. N-Terminal Hypothesis for Alzheimer’s Disease. ACS Chem. Neurosci. 2017, 8, 432–434. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Jiang, X.; Han, W. Self-Assembly Pathways of β-Sheet-Rich Amyloid-β(1–40) Dimers: Markov State Model Analysis on Millisecond Hybrid-Resolution Simulations. J. Chem. Theory Comput. 2017, 13, 5731–5744. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-J.; Zhang, X.; Chen, W.-W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta pep-tide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Markus, H.S. Branched-chain amino acids and Alzheimer’s disease: A Mendelian randomization analysis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.G.; Morris, K.L.; Sada, A.A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of Amyloid-β in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schell, M.J.; Molliver, M.E.; Snyder, S.H. D-serine, an endogenous synaptic modulator: Localization to astrocytes and gluta-mate-stimulated release. Proc. Natl. Acad. Sci. USA 1995, 92, 3948–3952. [Google Scholar] [CrossRef] [Green Version]
- Wako, K.; Ma, N.; Shiroyama, T.; Semba, R. Glial uptake of intracerebroventricularly injected d-serine in the rat brain: An immunocytochemical study. Neurosci. Lett. 1995, 185, 171–174. [Google Scholar] [CrossRef]
- Zhuang, W.; Sgourakis, N.G.; Li, Z.; Garcia, A.E.; Mukamel, S. Discriminating early stage A 42 monomer structures using chirality-induced 2DIR spectroscopy in a simulation study. Proc. Natl. Acad. Sci. USA 2010, 107, 15687–15692. [Google Scholar] [CrossRef] [Green Version]
- Yaffe, M.B.; Smerdon, S.J. PhosphoSerine/Threonine Binding Domains. Structure 2001, 9, R33–R38. [Google Scholar] [CrossRef] [Green Version]
- Nandi, N. Chirality in Biological Nanospaces; CRC Press: Boca Raton, FL, USA, 2012; pp. 25–37. [Google Scholar]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Farias, G.; Cornejo, A.; Jimenez, J.; Guzman, L.; Maccioni, R.B. Mechanisms of tau self-aggregation and neurotoxicity. Curr. Alzheimer Res. 2011, 8, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; Van Der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; Van Hummel, A.; Ste-vens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Devos, S.L.; Hyman, B.T. Tau at the Crossroads between Neurotoxicity and Neuroprotection. Neuron 2017, 94, 703–704. [Google Scholar] [CrossRef]
- Rosen, R.F.; Farberg, A.S.; Gearing, M.; Dooyema, J.; Long, P.M.; Anderson, D.C.; Davis-Turak, J.; Coppola, G.; Geschwind, D.H.; Paré, J.-F.; et al. Tauopathy with paired helical filaments in an aged chimpanzee. J. Comp. Neurol. 2008, 509, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, K.; Omori, A.; Sato, K.; Tomizawa, K.; Imahori, K.; Uchida, T. A serine/threonine proline kinase activity is included in the tau protein kinase fraction forming a paired helical filament epitope. Neurosci. Lett. 1991, 128, 195–198. [Google Scholar] [CrossRef]
- Regan, P.; Piers, T.; Yi, J.-H.; Kim, D.-H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau Phosphorylation at Serine 396 Residue Is Required for Hippocampal LTD. J. Neurosci. 2015, 3, 4804–4812. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Vanmechelen, E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 1995, 189, 167–169. [Google Scholar] [CrossRef]
- Derkinderen, P.; Scales, T.M.; Hanger, D.P.; Leung, K.Y.; Byers, H.L.; Ward, M.A.; Lenz, C.; Price, C.; Bird, I.N.; Perera, T.; et al. TAU phosphorylated at tyrosine 394 is phosphorylated in Alzheimer’s paired. J. Neurosci. 2005, 25, 6584–6593. [Google Scholar] [CrossRef]
- Van Groen, T.; Kadish, I.; Funke, A.; Bartnik, D.; Willbold, D. Treatment with Abeta42 binding D-amino acid peptides reduce amyloid deposition and inflammation in APP/PS1 double transgenic mice. Adv. Protein Chem. Struct. Biol. 2012, 88, 133–152. [Google Scholar] [CrossRef]
- Griner, S.L.; Seidler, P.; Bowler, J.; Murray, K.A.; Yang, T.P.; Sahay, S.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Philipp, S.; et al. Structure-based inhibitors of amyloid beta core suggest a common interface with TAU. eLife 2019. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, X.; Li, L.; Zheng, J. Inhibition of Amyloid-β Aggregation in Alzheimer’s Disease. Curr. Pharm. Des. 2014, 20, 1223–1243. [Google Scholar] [CrossRef]
- Francioso, A.; Punzi, P.; Boffi, A.; Lori, C.; Martire, S.; Giordano, C.; D’Erme, M.; Mosca, L. β-Sheet interfering molecules act-ing against β-amyloid aggregation and fibrillogenesis. Bioorg. Med. Chem. 2015, 23, 1671–1683. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Ng, V.; McLaurin, J. Small molecule inhibitors of Aβ-aggregation and neurotoxicity. Drug Dev. Res. 2009, 70, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhou, X.-L.; Yang, Q.-G.; Xu, W.-H.; Wang, F.; Chen, Y.-P.; Chen, G.-H. A Peptide That Binds Specifically to the β-Amyloid of Alzheimer’s Disease: Selection and Assessment of Anti-β-Amyloid Neurotoxic Effects. PLoS ONE 2011, 6, e27649. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Gafni, A.; Noy, N. Age-related effects in enzyme catalysis. Mol. Cell. Biochem. 1984, 59, 113–129. [Google Scholar] [CrossRef]
- Comai, S.; Costa, C.V.; Ragazzi, E.; Bertazzo, A.; Allegri, G. The effect of age on the enzyme activities of tryptophan metabo-lism along the kynurenine pathway in rats. Clin. Chim. Acta 2005, 360, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Gavrilova, O.; Brown, A.L.; Soto, J.E.; Bremner, S.; Kim, J.; Xu, X.; Yang, S.; Um, J.-H.; Koch, L.G.; et al. DNA-PK Promotes the Mitochondrial, Metabolic, and Physical Decline that Occurs During Aging. Cell Metab. 2017, 25, 1135–1146.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfman, P.M.; Bada, J.L. Aspartic acid racemization in tooth enamel from living humans. Proc. Natl. Acad. Sci. USA 1975, 72, 2891–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffert, L.-N.; Carter, W.G. Do Post-Translational Modifications Influence Prein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Doig, A.J.; Del Castillo-Frias, M.P.; Berthoumieu, O.; Tarus, B.; Nasica-Labouze, J.; Sterpone, F.; Nguyen, P.H.; Hooper, N.M.; Faller, P.; Derreumaux, P. Why Is Research on Amyloid-β Failing to Give New Drugs for Alzheimer’s Disease? ACS Chem. Neurosci. 2017, 8, 1435–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, P2327–P2329. [Google Scholar] [CrossRef]
- van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, H.; Mizogami, M. Interaction of Local Anesthetics with Biomembranes Consisting of Phospholipids and Cholesterol: Mechanistic and Clinical Implications for Anesthetic and Cardiotoxic Effects. Anesthesiol. Res. Pract. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Glick, S.D.; Ross, D.A.; Hough, L.B. Lateral asymmetry of neurotransmitters in human brain. Brain Res. 1982, 234, 53–63. [Google Scholar] [CrossRef]
Figure 1.
Diversity of post-translational modifications. The spontaneous symmetry breaking in molecular systems resulting in the transfer from the state of thermodynamic equilibrium to the fluctuating non-equilibrium state ( ) is associated with the origin of life. The spontaneous asymmetry breaking in the bio-molecular system resulting in the transfer from the dynamic non-equilibrium state to the state of thermodynamic equilibrium is associated with the decay of life. Part of image is adopted from [134].
Figure 1.
Diversity of post-translational modifications. The spontaneous symmetry breaking in molecular systems resulting in the transfer from the state of thermodynamic equilibrium to the fluctuating non-equilibrium state ( ) is associated with the origin of life. The spontaneous asymmetry breaking in the bio-molecular system resulting in the transfer from the dynamic non-equilibrium state to the state of thermodynamic equilibrium is associated with the decay of life. Part of image is adopted from [134].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dyakin, V.V.; Wisniewski, T.M.; Lajtha, A. Racemization in Post-Translational Modifications Relevance to Protein Aging, Aggregation and Neurodegeneration: Tip of the Iceberg. Symmetry 2021, 13, 455. https://doi.org/10.3390/sym13030455
AMA Style
Dyakin VV, Wisniewski TM, Lajtha A. Racemization in Post-Translational Modifications Relevance to Protein Aging, Aggregation and Neurodegeneration: Tip of the Iceberg. Symmetry. 2021; 13(3):455. https://doi.org/10.3390/sym13030455
Chicago/Turabian StyleDyakin, Victor V., Thomas M. Wisniewski, and Abel Lajtha. 2021. "Racemization in Post-Translational Modifications Relevance to Protein Aging, Aggregation and Neurodegeneration: Tip of the Iceberg" Symmetry 13, no. 3: 455. https://doi.org/10.3390/sym13030455
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.