On the Role of Peripheral Sensory and Gut Mu Opioid Receptors: Peripheral Analgesia and Tolerance

 ,
,  , and
, and
Abstract
:1. Introduction
2. Contribution of Peripheral MORs to Opioid-Induced Analgesia and Analgesic Tolerance
2.1. Peripheral Opioid Analgesia and Tolerance in Animal Models of Acute Thermal Pain
2.2. Peripheral Opioid Analgesia and Tolerance in Animal Acute and Subchronic Inflammatory Pain Models
2.3. Peripheral Opioid Analgesia and Tolerance in Animal Neuropathic Pain Models
3. Drawbacks of Peripheral MORs Activation Related to Opioid Analgesic Tolerance
3.1. The Consequence of MORs Activation on Primary Sensory Neurons
3.2. The Role of MORs in the Gut Microbiota: Dysbiosis, Opioid Tolerance
4. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Holzer, P. Opioid receptors in the gastrointestinal tract. Regul. Pept. 2009, 155, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janson, W.; Stein, C. Peripheral opioid analgesia. Curr. Pharm. Biotechnol. 2003, 4, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.; Fox, C.; Akil, H.; Watson, S. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995, 18, 22–29. [Google Scholar] [CrossRef]
- Kieffer, B.L.; Befort, K.; Gaveriaux-Ruff, C.; Hirth, C.G. The delta-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA 1992, 89, 12048–12052. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Mestek, A.; Liu, J.; Hurley, J.A.; Yu, L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol. Pharmacol. 1993, 44, 8–12. [Google Scholar] [CrossRef]
- Minami, M.; Toya, T.; Katao, Y.; Maekawa, K.; Nakamura, S.; Onogi, T.; Kaneko, S.; Satoh, M. Cloning and expression of a cDNA for the rat kappa-opioid receptor. FEBS Lett. 1993, 329, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Pert, C.B.; Snyder, S.H. Opiate receptor: Demonstration in nervous tissue. Science 1973, 179, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.J.; Hiller, J.M.; Edelman, I. Stereospecific binding of the potent narcotic analgesic (3H) Etorphine to rat-brain homogenate. Proc. Natl. Acad. Sci. USA 1973, 70, 1947–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terenius, L. Characteristics of the “receptor” for narcotic analgesics in synaptic plasma membrane fraction from rat brain. Acta. Pharmacol. Toxicol. 1973, 33, 377–384. [Google Scholar] [CrossRef]
- Law, P.Y.; Wong, Y.H.; Loh, H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 389–430. [Google Scholar] [CrossRef]
- Lord, J.A.; Waterfield, A.A.; Hughes, J.; Kosterlitz, H.W. Endogenous opioid peptides: Multiple agonists and receptors. Nature 1977, 267, 495–499. [Google Scholar] [CrossRef]
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.-L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.-C. ORL1, a novel member of the opioid receptor family. FEBS Lett. 1994, 341, 33–38. [Google Scholar] [CrossRef]
- Wang, J.B.; Johnson, P.S.; Imai, Y.; Persico, A.M.; Ozenberger, B.A.; Eppler, C.M.; Uhl, G.R. cDNA Cloning of an orphan opiate receptor gene family member and its splice variant. FEBS Lett. 1994, 348, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Reinscheid, R.K.; Nothacker, H.P.; Bourson, A.; Ardati, A.; Henningsen, R.A.; Bunzow, J.R.; Grandy, D.K.; Langen, H.; Monsma, F.J.; Civelli, O. Orphanin FQ: A neuropeptide that activates an opioidlike G protein-coupled receptor. Science 1995, 270, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Burford, N.T.; Wang, D.; Sadée, W. G-protein coupling of mu-opioid receptors (OP3): Elevated basal signalling activity. Biochem. J. 2000, 348, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, B.N.; Cesselin, F.; Raghubir, R.; Reisine, T.; Bradley, P.B.; Portoghese, P.S.; Hamon, M. International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol. Rev. 1996, 48, 567–592. [Google Scholar]
- Bian, J.-M.; Wu, N.; Su, R.-B.; Li, J. Opioid receptor trafficking and signaling: What happens after opioid receptor activation? Cell. Mol. Neurobiol. 2012, 32, 167–184. [Google Scholar] [CrossRef]
- Sim, L.J.; Childers, S.R. Anatomical distribution of mu, delta, and kappa opioid- and nociceptin/orphanin FQ-stimulated [35S]guanylyl-5′-O-(gamma-thio)-triphosphate binding in guinea pig brain. J. Comp. Neurol. 1997, 386, 562–572. [Google Scholar] [CrossRef]
- Kaplovitch, E.; Gomes, T.; Camacho, X.; Dhalla, I.A.; Mamdani, M.M.; Juurlink, D.N. Sex differences in dose escalation and overdose death during chronic opioid therapy: A population-based cohort study. PLoS ONE 2015, 10, e0134550. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.M.; Christie, M.J. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br. J. Pharmacol. 2011, 164, 1322–1334. [Google Scholar] [CrossRef] [Green Version]
- Hayhurst, C.J.; Durieux, M.E. Differential opioid tolerance and opioid-induced hyperalgesia. Anesthesiology 2016, 124, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Akbarali, H.I.; Inkisar, A.; Dewey, W.L. Site and mechanism of morphine tolerance in the gastrointestinal tract. Neurogastroenterol. Motil. 2014, 26, 1361–1367. [Google Scholar] [CrossRef]
- Dumas, E.O.; Pollack, G.M. Opioid tolerance development: A pharmacokinetic/pharmacodynamic perspective. AAPS J. 2008, 10, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institute on Drug Abuse. Fentanyl and Other Synthetic Opioids Drug Overdose Deaths. Available online: https://www.drugabuse.gov/related-topics/trends-statistics/infographics/fentanyl-other-synthetic-opioids-drug-overdose-deaths (accessed on 22 May 2020).
- Kanny, D.; Liu, Y.; Brewer, R.D.; Garvin, W.S.; Balluz, L. Vital signs: Binge drinking prevalence, frequency, and intensity among adults-united states, 2010. Morb. Mortal. Wkly. Rep. 2012, 61, 14–19. [Google Scholar]
- Scholl, L.; Seth, P.; Kariisa, M.; Wilson, N.; Baldwin, G. Morbidity and mortality weekly report udrug and opioid-involved overdose deaths-nited States, 2013–2017. MMWR Morb. Mortal. Wkly. Rep. 2019, 67, 1419–1427. [Google Scholar]
- Zoorob, M. Fentanyl shock: The changing geography of overdose in the United States. Int. J. Drug Policy 2019, 70, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.T.; Polomano, R.C.; Berlin, J.A.; Strom, B.L. A comparison of change in the 0-10 numeric rating scale to a pain relief scale and global medication performance scale in a short-term clinical trial of breakthrough pain intensity. Anesthesiology 2010, 112, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Buntin-Mushock, C.; Phillip, L.; Moriyama, K.; Palmer, P.P. Age-dependent opioid escalation in chronic pain patients. Anesth. Analg. 2005, 100, 1740–1745. [Google Scholar] [CrossRef]
- Bowman, W.C.; Rand, M.J. Textbook of Pharmacology, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1980; ISBN 0632099909. [Google Scholar]
- Eisenberg, E.; McNicol, E.D.; Carr, D.B. Efficacy and safety of opioid agonists in the treatment of neuropathic pain of nonmalignant origin: Systematic review and meta-analysis of randomized controlled trials. JAMA 2005, 293, 3043–3052. [Google Scholar] [CrossRef]
- Stein, C.; Machelska, H.; Binder, W.; Schäfer, M. Peripheral opioid analgesia. Curr. Opin. Pharmacol. 2001, 1, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.; Lang, L.J. Peripheral mechanisms of opioid analgesia. Curr. Opin. Pharmacol. 2009, 9, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Fürst, S.; Riba, P.; Friedmann, T.; Tímar, J.; Al-Khrasani, M.; Obara, I.; Makuch, W.; Spetea, M.; Schütz, J.; Przewlocki, R.; et al. Peripheral versus central antinociceptive actions of 6-amino acid-substituted derivatives of 14-O-methyloxymorphone in acute and inflammatory pain in the rat. J. Pharmacol. Exp. Ther. 2005, 312, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bileviciute-Ljungar, I.; Spetea, M.; Guo, Y.; Schutz, J.; Windisch, P.; Schmidhammer, H. Peripherally mediated antinociception of the µ-opioid receptor agonist HS-731 after subcutaneous and oral administration in rats with carrageenan-induced hindpaw inflammation. J. Pharmacol. Exp. Ther. 2005, 317, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Baillie, L.D.; Schmidhammer, H.; Mulligan, S.J. Peripheral μ-opioid receptor mediated inhibition of calcium signaling and action potential-evoked calcium fluorescent transients in primary afferent CGRP nociceptive terminals. Neuropharmacology 2015, 93, 267–273. [Google Scholar] [CrossRef]
- Spetea, M.; Rief, S.B.; Haddou, T.B.; Fink, M.; Kristeva, E.; Mittendorfer, H.; Haas, S.; Hummer, N.; Follia, V.; Guerrieri, E.; et al. Synthesis, biological, and structural explorations of new zwitterionic derivatives of 14-O-methyloxymorphone, as potent μ/δ opioid agonists and peripherally selective antinociceptives. J. Med. Chem. 2019, 62, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Spahn, V.; Del Vecchio, G.; Labuz, D.; Rodriguez-Gaztelumendi, A.; Massaly, N.; Temp, J.; Durmaz, V.; Sabri, P.; Reidelbach, M.; Machelska, H.; et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 2017, 355, 966–969. [Google Scholar] [CrossRef]
- Rodriguez-Gaztelumendi, A.; Spahn, V.; Labuz, D.; Machelska, H.; Stein, C. Analgesic effects of a novel pH-dependent μ-opioid receptor agonist in models of neuropathic and abdominal pain. Pain 2018, 159, 2277–2284. [Google Scholar] [CrossRef]
- He, S.-Q.; Yang, F.; Perez, F.M.; Xu, Q.; Shechter, R.; Cheong, Y.-K.; Carteret, A.F.; Dong, X.; Sweitzer, S.M.; Raja, S.N.; et al. Tolerance develops to the antiallodynic effects of the peripherally acting opioid loperamide hydrochloride in nerve-injured rats. Pain 2013, 154, 2477–2486. [Google Scholar] [CrossRef] [Green Version]
- Akbarali, H.I.; Dewey, W.L. Gastrointestinal motility, dysbiosis and opioid-induced tolerance: Is there a link? Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 323–324. [Google Scholar] [CrossRef]
- He, L.; Kim, J.A.; Whistler, J.L. Biomarkers of morphine tolerance and dependence are prevented by morphine-induced endocytosis of a mutant mu-opioid receptor. FASEB J. 2009, 23, 4327–4334. [Google Scholar] [CrossRef] [Green Version]
- Lackó, E.; Riba, P.; Giricz, Z.; Váradi, A.; Cornic, L.; Balogh, M.; Király, K.; Csekő, K.; Mousa, S.A.; Hosztafi, S.; et al. New morphine analogs produce peripheral antinociception within a certain dose range of their systemic administration. J. Pharmacol. Exp. Ther. 2016, 359, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, M.; Zádori, Z.S.; Lázár, B.; Karádi, D.; László, S.; Mousa, S.A.; Hosztafi, S.; Zádor, F.; Riba, P.; Schäfer, M.; et al. The peripheral versus central antinociception of a novel opioid agonist: Acute inflammatory pain in rats. Neurochem. Res. 2018, 43, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Shannon, H.E.; Lutz, E.A. Comparison of the peripheral and central effects of the opioid agonists loperamide and morphine in the formalin test in rats. Neuropharmacology 2002, 42, 253–261. [Google Scholar] [CrossRef]
- Takasuna, M.; Negus, S.S.; DeCosta, B.R.; Woods, J.H. Opioid pharmacology of the antinociceptive effects of loperamide in mice. Behav. Pharmacol. 1994, 5, 189–195. [Google Scholar] [CrossRef]
- Brown, D.R.; Goldberg, L.I. The use of quaternary narcotic antagonists in opiate research. Neuropharmacology 1985, 24, 181–191. [Google Scholar] [CrossRef]
- Reichert, J.A.; Daughters, R.S.; Rivard, R.; Simone, D.A. Peripheral and preemptive opioid antinociception in a mouse visceral pain model. Pain 2001, 89, 221–227. [Google Scholar] [CrossRef]
- Martínez, V.; Abalo, R. Peripherally acting opioid analgesics and peripherally-induced analgesia. Behav. Pharmacol. 2020, 31, 136–158. [Google Scholar] [CrossRef]
- Labuz, D.; Mousa, S.A.; Schäfer, M.; Stein, C.; Machelska, H. Relative contribution of peripheral versus central opioid receptors to antinociception. Brain Res. 2007, 1160, 30–38. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Spetea, M.; Friedmann, T.; Riba, P.; Király, K.; Schmidhammer, H.; Furst, S. DAMGO and 6β-glycine substituted 14-O-methyloxymorphone but not morphine show peripheral, preemptive antinociception after systemic administration in a mouse visceral pain model and high intrinsic efficacy in the isolated rat vas deferens. Brain Res. Bull. 2007, 74, 369–375. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Lackó, E.; Riba, P.; Király, K.; Sobor, M.; Timár, J.; Mousa, S.; Schäfer, M.; Fürst, S. The central versus peripheral antinociceptive effects of μ-opioid receptor agonists in the new model of rat visceral pain. Brain Res. Bull. 2012, 87, 238–243. [Google Scholar] [CrossRef]
- Khalefa, B.I.; Shaqura, M.; Al-Khrasani, M.; Fürst, S.; Mousa, S.A.; Schäfer, M. Relative contributions of peripheral versus supraspinal or spinal opioid receptors to the antinociception of systemic opioids. Eur. J. Pain 2012, 16, 690–705. [Google Scholar] [CrossRef] [PubMed]
- Obara, I.; Przewlocki, R.; Przewlocka, B. Local peripheral effects of μ-opioid receptor agonists in neuropathic pain in rats. Neurosci. Lett. 2004, 360, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Obara, I.; Makuch, W.; Spetea, M.; Schütz, J.; Schmidhammer, H.; Przewlocki, R.; Przewlocka, B. Local peripheral antinociceptive effects of 14-O-methyloxymorphone derivatives in inflammatory and neuropathic pain in the rat. Eur. J. Pharmacol. 2007, 558, 60–67. [Google Scholar] [CrossRef]
- Balogh, M.; Zádor, F.; Zádori, Z.S.; Shaqura, M.; Király, K.; Mohammadzadeh, A.; Varga, B.; Lázár, B.; Mousa, S.A.; Hosztafi, S.; et al. Efficacy-based perspective to overcome reduced opioid analgesia of advanced painful diabetic neuropathy in rats. Front. Pharmacol. 2019, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Truong, W.; Cheng, C.; Xu, Q.-G.; Li, X.-Q.; Zochodne, D.W. Mu Opioid receptors and analgesia at the site of a peripheral nerve injury. Ann. Neurol. 2003, 53, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Johanek, L.M.; Hartke, T.V.; Shim, B.; Tao, Y.-X.; Ringkamp, M.; Meyer, R.A.; Raja, S.N. Peripherally acting mu-opioid receptor agonist attenuates neuropathic pain in rats after L5 spinal nerve injury. Pain 2008, 138, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Shinoda, K.; Hruby, V.J.; Porreca, F. Antihyperalgesic effects of loperamide in a model of rat neuropathic pain are mediated by peripheral δ-opioid receptors. Neurosci. Lett. 2007, 411, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Zuckerman, A.; Bolan, E.; de Paulis, T.; Schmidt, D.; Spector, S.; Pasternak, G.W. Pharmacological characterization of morphine-6-sulfate and codeine-6-sulfate. Brain Res. 1999, 842, 1–5. [Google Scholar] [CrossRef]
- Botros, S.; Lipkowski, A.W.; Larson, D.L.; Stark, P.A.; Takemori, A.E.; Portoghese, P.S. Opioid agonist and antagonist activities of peripherally selective derivatives of naltrexamine and oxymorphamine. J. Med. Chem. 1989, 32, 2068–2071. [Google Scholar] [CrossRef] [PubMed]
- Lacko, E.; Varadi, A.; Rapavi, R.; Zador, F.; Riba, P.; Benyhe, S.; Borsodi, A.; Hosztafi, S.; Timar, J.; Noszal, B.; et al. A novel µ-opioid receptor ligand with high in vitro and in vivo agonist efficacy. Curr. Med. Chem. 2012, 19, 4699–4707. [Google Scholar] [CrossRef]
- Smith, T.W.; Buchan, P.; Parsons, D.N.; Wilkinson, S. Peripheral antinociceptive effects of N-methyl morphine. Life Sci. 1982, 31, 1205–1208. [Google Scholar] [CrossRef]
- Spetea, M.; Schmidhammer, H. Recent advances in the development of 14-alkoxy substituted morphinans as potent and safer opioid analgesics. Curr. Med. Chem. 2012, 19, 2442–2457. [Google Scholar] [CrossRef]
- Zádor, F.; Mohammadzadeh, A.; Balogh, M.; Zádori, Z.S.; Király, K.; Barsi, S.; Galambos, A.R.; László, S.B.; Hutka, B.; Váradi, A.; et al. Comparisons of In Vivo and In Vitro Opioid Effects of Newly Synthesized 14-Methoxycodeine-6-O-sulfate and Codeine-6-O-sulfate. Molecules 2020, 25, 1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadivelu, N.; Mitra, S.; Hines, R.L. Peripheral opioid receptor agonists for analgesia: A comprehensive review. J. Opioid Manag. 2011, 7, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, N.; Smith, H.S.; Manchikanti, L. Peripherally acting opioids and clinical implications for pain control. Pain Physician 2011, 14, 249–258. [Google Scholar] [PubMed]
- Zöllner, C.; Mousa, S.A.; Fischer, O.; Rittner, H.L.; Shaqura, M.; Brack, A.; Shakibaei, M.; Binder, W.; Urban, F.; Stein, C.; et al. Chronic morphine use does not induce peripheral tolerance in a rat model of inflammatory pain. J. Clin. Invest. 2008, 118, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, C.; Pflüger, M.; Yassouridis, A.; Hoelzl, J.; Lehrberger, K.; Welte, C.; Hassan, A.H. No tolerance to peripheral morphine analgesia in presence of opioid expression in inflamed synovia. J. Clin. Invest. 1996, 98, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Allouche, S.; Noble, F.; Marie, N. Opioid receptor desensitization: Mechanisms and its link to tolerance. Front. Pharmacol. 2014, 5, 280. [Google Scholar] [CrossRef] [Green Version]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [Green Version]
- Uniyal, A.; Gadepalli, A.; Akhilesh; Tiwari, V. Underpinning the neurobiological intricacies associated with opioid tolerance. ACS Chem. Neurosci. 2020, 11, 830–839. [Google Scholar] [CrossRef]
- Mori, M.; Oguri, K.; Yoshimura, H.; Shimomura, K.; Kamata, O.; Ueki, S. Chemical synthesis and analgesic effect of morphine ethereal sulfates. Life Sci. I. 1972, 11, 525–533. [Google Scholar] [CrossRef]
- Kolesnikov, Y.A.; Jain, S.; Wilson, R.; Pasternak, G.W. Peripheral morphine analgesia: Synergy with central sites and a target of morphine tolerance. J. Pharmacol. Exp. Ther. 1996, 279, 502–506. [Google Scholar] [PubMed]
- Kolesnikov, Y.; Pasternak, G.W. Topical opioids in mice: Analgesia and reversal of tolerance by a topical N-methyl-D-aspartate antagonist. J. Pharmacol. Exp. Ther. 1999, 290, 247–252. [Google Scholar] [PubMed]
- Aley, K.O.; Levine, J.D. Dissociation of tolerance and dependence for opioid peripheral antinociception in rats. J. Neurosci. 1997, 17, 3907–3912. [Google Scholar] [CrossRef]
- Danysz, W.; Kozela, E.; Parsons, C.G.; Sladek, M.; Bauer, T.; Popik, P. Peripherally acting NMDA receptor/glycineB site receptor antagonists inhibit morphine tolerance. Neuropharmacology 2005, 48, 360–371. [Google Scholar] [CrossRef]
- Ben-Eliyahu, S.; Marek, P.; Vaccarino, A.L.; Mogil, J.S.; Sternberg, W.F.; Liebeskind, J.C. The NMDA receptor antagonist MK-801 prevents long-lasting non-associative morphine tolerance in the rat. Brain Res. 1992, 575, 304–308. [Google Scholar] [CrossRef]
- Trujillo, K.A. Are NMDA receptors involved in opiate-induced neural and behavioral plasticity? Psychopharmacology (Berl.) 2000, 151, 121–141. [Google Scholar] [CrossRef]
- Carlton, S.M.; Hargett, G.L.; Coggeshall, R.E. Localization and activation of glutamate receptors in unmyelinated axons of rat glabrous skin. Neurosci. Lett. 1995, 197, 25–28. [Google Scholar] [CrossRef]
- Zhou, S.; Bonasera, L.; Carlton, S.M. Peripheral administration of NMDA, AMPA or KA results in pain behaviors in rats. Neuroreport 1996, 7, 895–900. [Google Scholar] [CrossRef]
- Davidson, E.M.; Coggeshall, R.E.; Carlton, S.M. Peripheral NMDA and non-NMDA glutamate receptors contribute to nociceptive behaviors in the rat formalin test. Neuroreport 1997, 8, 941–946. [Google Scholar] [CrossRef]
- Meuser, T.; Giesecke, T.; Gabriel, A.; Horsch, M.; Sabatowski, R.; Hescheler, J.; Grond, S.; Palmer, P.P. Mu-opioid receptor mRNA regulation during morphine tolerance in the rat peripheral nervous system. Anesth. Analg. 2003, 97, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, S.; Chen, H.; Pan, H. μ-Opioid receptors in primary sensory neurons are essential for opioid analgesic effect on acute and inflammatory pain and opioid-induced hyperalgesia. J. Physiol. 2019, 597, 1661–1675. [Google Scholar] [CrossRef] [Green Version]
- Kiraly, K.; Caputi, F.F.; Hanuska, A.; Kató, E.; Balogh, M.; Köles, L.; Palmisano, M.; Riba, P.; Hosztafi, S.; Romualdi, P.; et al. A new potent analgesic agent with reduced liability to produce morphine tolerance. Brain Res. Bull. 2015, 117, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Aley, K.O.; Green, P.G.; Levine, J.D. Opioid and adenosine peripheral antinociception are subject to tolerance and withdrawal. J. Neurosci. 1995, 15, 8031–8038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honoré, P.; Catheline, G.; Le Guen, S.; Besson, J.M. Chronic treatment with systemic morphine induced tolerance to the systemic and peripheral antinociceptive effects of morphine on both carrageenin induced mechanical hyperalgesia and spinal c-Fos expression in awake rats. Pain 1997, 71, 99–108. [Google Scholar] [CrossRef]
- Pethő, G.; Reeh, P.W. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol. Rev. 2012, 92, 1699–1775. [Google Scholar] [CrossRef]
- Tokuyama, S.; Inoue, M.; Fuchigami, T.; Ueda, H. Lack of tolerance in peripheral opioid analgesia in mice. Life Sci. 1998, 62, 1677–1681. [Google Scholar] [CrossRef]
- Inoue, M.; Shimohira, I.; Yoshida, A.; Zimmer, A.; Takeshima, H.; Sakurada, T.; Ueda, H. Dose-related opposite modulation by nociceptin/orphanin FQ of substance P nociception in the nociceptors and spinal cord. J. Pharmacol. Exp. Ther. 1999, 291, 308–313. [Google Scholar]
- Inoue, M.; Ueda, H. Protein kinase C-mediated acute tolerance to peripheral mu-opioid analgesia in the bradykinin-nociception test in mice. J. Pharmacol. Exp. Ther. 2000, 293, 662–669. [Google Scholar]
- Shaqura, M.A.; Zöllner, C.; Mousa, S.A.; Stein, C.; Schäfer, M. Characterization of μ opioid receptor binding and g protein coupling in rat hypothalamus, spinal cord, and primary afferent neurons during inflammatory pain. J. Pharmacol. Exp. Ther. 2004, 308, 712–718. [Google Scholar] [CrossRef]
- Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Schäfer, M.; Machelska, H. Attacking pain at its source: New perspectives on opioids. Nat. Med. 2003, 9, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Straub, R.H.; Schäfer, M.; Stein, C. Beta-endorphin, Met-enkephalin and corresponding opioid receptors within synovium of patients with joint trauma, osteoarthritis and rheumatoid arthritis. Ann. Rheum. Dis. 2007, 66, 871–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, C.; Zöllner, C. Opioids and Sensory Nerves. In Sensory Nerves; Canning, B., Ed.; Springer: Berlin, Germany, 2009; pp. 495–518. ISBN 978-3-540-79089-1. [Google Scholar]
- Del Vecchio, G.; Spahn, V.; Stein, C. Novel opioid analgesics and side effects. ACS Chem. Neurosci. 2017, 8, 1638–1640. [Google Scholar] [CrossRef] [PubMed]
- Ninković, J.; Roy, S. Role of the mu-opioid receptor in opioid modulation of immune function. Amino Acids 2013, 45, 9–24. [Google Scholar] [CrossRef]
- Fernández-Dueñas, V.; Pol, O.; García-Nogales, P.; Hernández, L.; Planas, E.; Puig, M.M. Tolerance to the antinociceptive and antiexudative effects of morphine in a murine model of peripheral inflammation. J. Pharmacol. Exp. Ther. 2007, 322, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Hernández, L.; Romero, A.; Almela, P.; García-Nogales, P.; Laorden, M.L.; Puig, M.M. Tolerance to the antinociceptive effects of peripherally administered opioids: Expression of β-arrestins. Brain Res. 2009, 1248, 31–39. [Google Scholar] [CrossRef]
- Eidson, L.N.; Murphy, A.Z. Persistent peripheral inflammation attenuates morphine-induced periaqueductal gray glial cell activation and analgesic tolerance in the male rat. J. Pain 2013, 14, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Bruce, D.J.; Peterson, C.D.; Kitto, K.F.; Akgün, E.; Lazzaroni, S.; Portoghese, P.S.; Fairbanks, C.A.; Wilcox, G.L. Combination of a δ-opioid receptor agonist and loperamide produces peripherally-mediated analgesic synergy in mice. Anesthesiology 2019, 131, 649–663. [Google Scholar] [CrossRef]
- Hoffman, E.M.; Watson, J.C.; St Sauver, J.; Staff, N.P.; Klein, C.J. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol. 2017, 74, 773–779. [Google Scholar] [CrossRef]
- Furlan, A.D.; Sandoval, J.A.; Mailis-Gagnon, A.; Tunks, E. Opioids for chronic noncancer pain: A meta-analysis of effectiveness and side effects. CMAJ 2006, 174, 1589–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khrasani, M.; Mohammadzadeh, A.; Balogh, M.; Király, K.; Barsi, S.; Hajnal, B.; Köles, L.; Zádori, Z.S.; Harsing, L.G. Glycine transporter inhibitors: A new avenue for managing neuropathic pain. Brain Res. Bull. 2019, 152, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Porreca, F.; Tang, Q.B.; Bian, D.; Riedl, M.; Elde, R.; Lai, J. Spinal opioid mu receptor expression in lumbar spinal cord of rats following nerve injury. Brain Res. 1998, 795, 197–203. [Google Scholar] [CrossRef]
- Kohno, T.; Ji, R.-R.; Ito, N.; Allchorne, A.J.; Befort, K.; Karchewski, L.A.; Woolf, C.J. Peripheral axonal injury results in reduced mu opioid receptor pre- and post-synaptic action in the spinal cord. Pain 2005, 117, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Mansikka, H.; Zhao, C.; Sheth, R.N.; Sora, I.; Uhl, G.; Raja, S.N. Nerve injury induces a tonic bilateral mu-opioid receptor-mediated inhibitory effect on mechanical allodynia in mice. Anesthesiology 2004, 100, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.H.; Mohammad, H.K.; Ali, R.; Peper, B.; Wilson, S.P.; Raja, S.N.; Ringkamp, M.; Sweitzer, S. Overexpression of µ-opioid receptors in peripheral afferents, but not in combination with enkephalin, decreases neuropathic pain behavior and enhances opioid analgesia in mouse. Anesthesiology 2018, 128, 967–983. [Google Scholar] [CrossRef]
- Kayser, V.; Lee, S.H.; Guilbaud, G. Evidence for a peripheral component in the enhanced antinociceptive effect of a low dose of systemic morphine in rats with peripheral mononeuropathy. Neuroscience 1995, 64, 537–545. [Google Scholar] [CrossRef]
- Idänpään-Heikkilä, J.J.; Guilbaud, G.; Kayser, V. Prevention of tolerance to the antinociceptive effects of systemic morphine by a selective cholecystokinin-B receptor antagonist in a rat model of peripheral neuropathy. J. Pharmacol. Exp. Ther. 1997, 282, 1366–1372. [Google Scholar]
- Chu, L.F.; Angst, M.S.; Clark, D. Opioid-induced hyperalgesia in humans. Clin. J. Pain 2008, 24, 479–496. [Google Scholar] [CrossRef]
- Busserolles, J.; Lolignier, S.; Kerckhove, N.; Bertin, C.; Authier, N.; Eschalier, A. Replacement of current opioid drugs focusing on MOR-related strategies. Pharmacol. Ther. 2020, 210, 107519. [Google Scholar] [CrossRef]
- Corder, G.; Tawfik, V.L.; Wang, D.; Sypek, E.I.; Low, S.A.; Dickinson, J.R.; Sotoudeh, C.; Clark, J.D.; Barres, B.A.; Bohlen, C.J.; et al. Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med. 2017, 23, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.; Catheline, G.; Guilbaud, G.; Kayser, V. Lack of cross-tolerance between the antinociceptive effects of systemic morphine and asimadoline, a peripherally-selective κ-opioid agonist, in CCI- neuropathic rats. Pain 1999, 83, 509–516. [Google Scholar] [CrossRef]
- Albert-Vartanian, A.; Boyd, M.R.; Hall, A.L.; Morgado, S.J.; Nguyen, E.; Nguyen, V.P.H.; Patel, S.P.; Russo, L.J.; Shao, A.J.; Raffa, R.B. Will peripherally restricted kappa-opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential? J. Clin. Pharm. Ther. 2016, 41, 371–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo, K.A.; Akil, H. Inhibition of opiate tolerance by non-competitive N-methyl-D-aspartate receptor antagonists. Brain Res. 1994, 633, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, V.; Nogueira, L.; Aponte, Y.; Vanegas, H. Involvement of cholecystokinin in the opioid tolerance induced by dipyrone (metamizol) microinjections into the periaqueductal gray matter of rats. Pain 2004, 112, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Zhou, L.; Stein, C. Cholecystokinin inhibits peripheral opioid analgesia in inflamed tissue. Neuroscience 1997, 82, 603–611. [Google Scholar] [CrossRef]
- Ebert, B.; Thorkildsen, C.; Andersen, S.; Christrup, L.L.; Hjeds, H. Opioid analgesics as noncompetitive N-methyl-D-aspartate (NMDA) antagonists. Biochem. Pharmacol. 1998, 56, 553–559. [Google Scholar] [CrossRef]
- Zhang, X.; de Araujo Lucas, G.; Elde, R.; Wiesenfeld-Hallin, Z.; Hökfelt, T. Effect of morphine on cholecystokinin and μ-opioid receptor-like immunoreactivities in rat spinal dorsal horn neurons after peripheral axotomy and inflammation. Neuroscience 1999, 95, 197–207. [Google Scholar] [CrossRef]
- Kohno, T.; Kumamoto, E.; Higashi, H.; Shimoji, K.; Yoshimura, M. Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J. Physiol. 1999, 518, 803–813. [Google Scholar] [CrossRef]
- Shaqura, M.; Khalefa, B.I.; Shakibaei, M.; Winkler, J.; Al-Khrasani, M.; Fürst, S.; Mousa, S.A.; Schäfer, M. Reduced number, G protein coupling, and antinociceptive efficacy of spinal mu-opioid receptors in diabetic rats are reversed by nerve growth factor. J. Pain 2013, 14, 720–730. [Google Scholar] [CrossRef]
- Fisher, C.; Johnson, K.; Okerman, T.; Jurgenson, T.; Nickell, A.; Salo, E.; Moore, M.; Doucette, A.; Bjork, J.; Klein, A.H. Morphine efficacy, tolerance, and hypersensitivity are altered after modulation of SUR1 subtype KATP channel activity in mice. Front. Neurosci. 2019, 13, 1122. [Google Scholar] [CrossRef]
- DuPont, A.W.; DuPont, H.L. The intestinal microbiota and chronic disorders of the gut. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The central nervous system and the gut microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meng, J.; Ban, Y.; Jalodia, R.; Chupikova, I.; Fernandez, I.; Brito, N.; Sharma, U.; Abreu, M.T.; Ramakrishnan, S.; et al. Morphine tolerance is attenuated in germfree mice and reversed by probiotics, implicating the role of gut microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 13523–13532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Meng, J.; Zhang, L.; Johnson, T.; Chen, C.; Roy, S. Morphine induces changes in the gut microbiome and metabolome in a morphine dependence model. Sci. Rep. 2018, 8, 3596. [Google Scholar] [CrossRef] [Green Version]
- Mischel, R.A.; Dewey, W.L.; Akbarali, H.I. Tolerance to morphine-induced inhibition of TTX-R sodium channels in dorsal root ganglia neurons is modulated by gut-derived mediators. iScience 2018, 2, 193–209. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Mischel, R.A.; Bhave, S.; Komla, E.; Cho, A.; Huang, C.; Dewey, W.L.; Akbarali, H.I. The effect of gut microbiome on tolerance to morphine mediated antinociception in mice. Sci. Rep. 2017, 7, 42658. [Google Scholar] [CrossRef] [Green Version]
- Mischel, R.A.; Muchhala, K.H.; Dewey, W.L.; Akbarali, H.I. The “culture” of pain control: A review of opioid-induced dysbiosis (OID) in antinociceptive tolerance. J. Pain 2019. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Banerjee, S.; Li, D.; Sindberg, G.M.; Wang, F.; Ma, J.; Roy, S. Opioid exacerbation of Gram-positive sepsis, induced by gut microbial modulation, is rescued by IL-17A neutralization. Sci. Rep. 2015, 5, 10918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Sindberg, G.; Wang, F.; Meng, J.; Sharma, U.; Zhang, L.; Dauer, P.; Chen, C.; Dalluge, J.; Johnson, T.; et al. Opioid-induced gut microbial disruption and bile dysregulation leads to gut barrier compromise and sustained systemic inflammation. Mucosal Immunol. 2016, 9, 1418–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Vuong, H.E.; Nusbaum, D.J.; Hsiao, E.Y.; Evans, C.J.; Taylor, A.M.W. The gut microbiota mediates reward and sensory responses associated with regimen-selective morphine dependence. Neuropsychopharmacology 2018, 43, 2606–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyap, P.C.; Marcobal, A.; Ursell, L.K.; Larauche, M.; Duboc, H.; Earle, K.A.; Sonnenburg, E.D.; Ferreyra, J.A.; Higginbottom, S.K.; Million, M.; et al. Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology 2013, 144, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touw, K.; Ringus, D.L.; Hubert, N.; Wang, Y.; Leone, V.A.; Nadimpalli, A.; Theriault, B.R.; Huang, Y.E.; Tune, J.D.; Herring, P.B.; et al. Mutual reinforcement of pathophysiological host-microbe interactions in intestinal stasis models. Physiol. Rep. 2017, 5, e13182. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Olson, R.K.; Erhart, F.N.; Zhang, L.; Meng, J.; Segura, B.; Banerjee, S.; Sharma, M.; Saluja, A.K.; Ramakrishnan, S.; et al. Prescription opioid induce gut dysbiosis and exacerbate colitis in a murine model of Inflammatory Bowel Disease. J. Crohns. Colitis 2019. [Google Scholar] [CrossRef] [PubMed]
- Sindberg, G.M.; Callen, S.E.; Banerjee, S.; Meng, J.; Hale, V.L.; Hegde, R.; Cheney, P.D.; Villinger, F.; Roy, S.; Buch, S. Morphine potentiates dysbiotic microbial and metabolic shifts in acute SIV infection. J. Neuroimmune Pharmacol. 2019, 14, 200–214. [Google Scholar] [CrossRef]
- Acharya, C.; Betrapally, N.S.; Gillevet, P.M.; Sterling, R.K.; Akbarali, H.; White, M.B.; Ganapathy, D.; Fagan, A.; Sikaroodi, M.; Bajaj, J.S. Chronic opioid use is associated with altered gut microbiota and predicts readmissions in patients with cirrhosis. Aliment. Pharmacol. Ther. 2017, 45, 319–331. [Google Scholar] [CrossRef]
- Xu, Y.; Xie, Z.; Wang, H.; Shen, Z.; Guo, Y.; Gao, Y.; Chen, X.; Wu, Q.; Li, X.; Wang, K. Bacterial diversity of intestinal microbiota in patients with substance use disorders revealed by 16S rRNA gene deep sequencing. Sci. Rep. 2017, 7, 3628. [Google Scholar] [CrossRef]
- Kim, S.C.; Tonkonogy, S.L.; Albright, C.A.; Tsang, J.; Balish, E.J.; Braun, J.; Huycke, M.M.; Sartor, R.B. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 2005, 128, 891–906. [Google Scholar] [CrossRef]
- Moon, C.; Baldridge, M.T.; Wallace, M.A.; Burnham, C.-A.D.; Virgin, H.W.; Stappenbeck, T.S. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 2015, 521, 90–93. [Google Scholar] [CrossRef]
- Lane, E.R.; Zisman, T.L.; Suskind, D.L. The microbiota in inflammatory bowel disease: Current and therapeutic insights. J. Inflamm. Res. 2017, 10, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geirnaert, A.; Calatayud, M.; Grootaert, C.; Laukens, D.; Devriese, S.; Smagghe, G.; De Vos, M.; Boon, N.; Van de Wiele, T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep. 2017, 7, 11450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.C.; Hart, A.L.; Kamm, M.A.; Stagg, A.J.; Knight, S.C. Mechanisms of action of probiotics: Recent advances. Inflamm. Bowel Dis. 2009, 15, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.A.; Stewart, T.M.R.; Hill, C.; Vanner, S.J. TNBS ileitis evokes hyperexcitability and changes in ionic membrane properties of nociceptive DRG neurons. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, 1045–1051. [Google Scholar] [CrossRef]
- Komla, E.; Stevens, D.L.; Zheng, Y.; Zhang, Y.; Dewey, W.L.; Akbarali, H.I. Experimental colitis enhances the rate of antinociceptive tolerance to morphine via peripheral opioid receptors. J. Pharmacol. Exp. Ther. 2019, 370, 504–513. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Shavit, Y.; Grace, P.M.; Rice, K.C.; Maier, S.F.; Watkins, L.R. Exploring the neuroimmunopharmacology of opioids: An integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol. Rev. 2011, 63, 772–810. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, S.D.; Aalinkeel, R.; Sykes, D.E.; Reynolds, J.L.; Bindukumar, B.; Fernandez, S.F.; Chawda, R.; Shanahan, T.C.; Schwartz, S.A. Tight junction regulation by morphine and HIV-1 tat modulates blood-brain barrier permeability. J. Clin. Immunol. 2008, 28, 528–541. [Google Scholar] [CrossRef]
- Plein, L.M.; Rittner, H.L. Opioids and the immune system - friend or foe. Br. J. Pharmacol. 2018, 175, 2717–2725. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Barbara, G.; Stanghellini, V.; Brandi, G.; Cremon, C.; Di Nardo, G.; De Giorgio, R.; Corinaldesi, R. Interactions between commensal bacteria and gut sensorimotor function in health and disease. Am. J. Gastroenterol. 2005, 100, 2560–2568. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
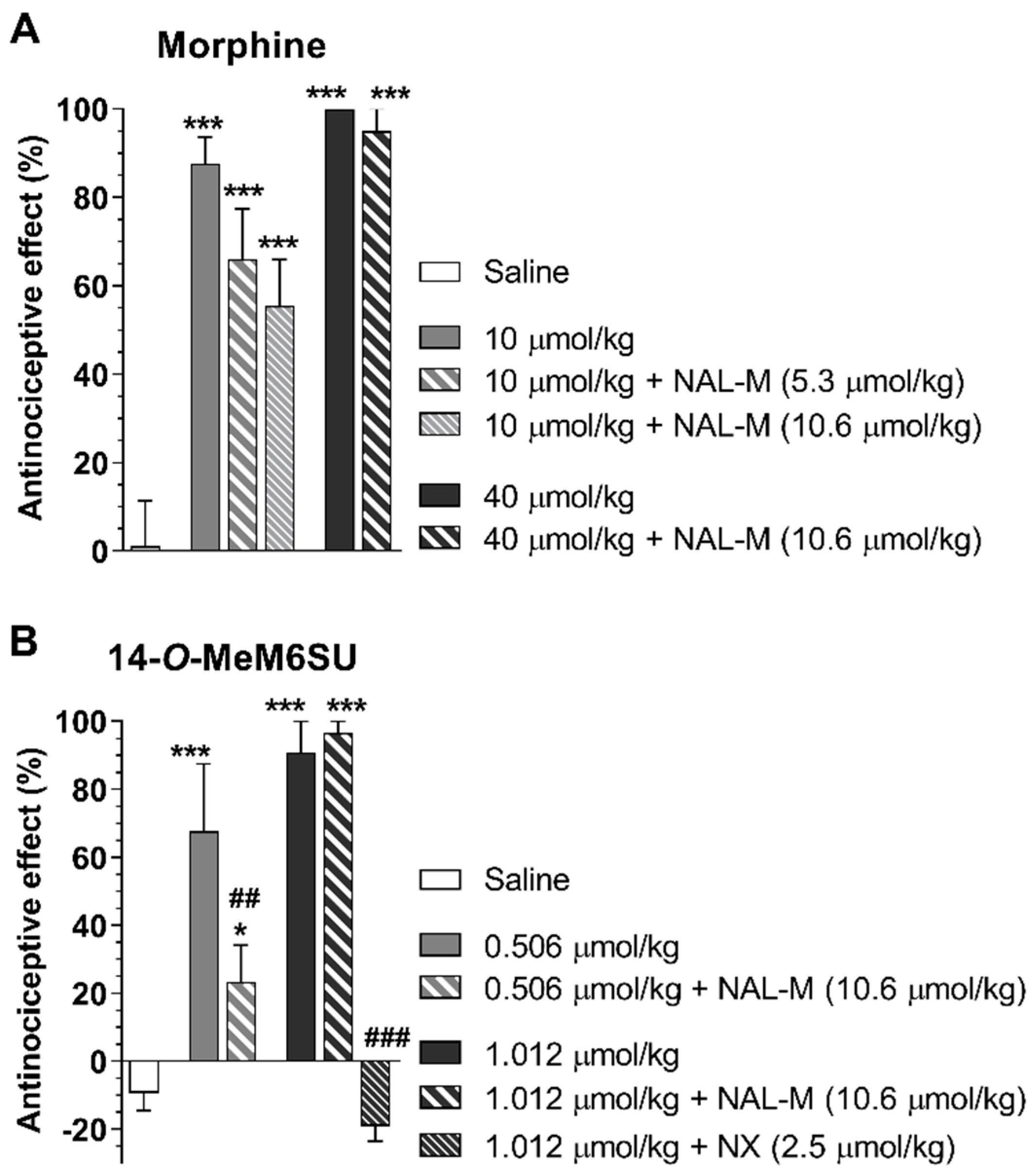{kind=link}
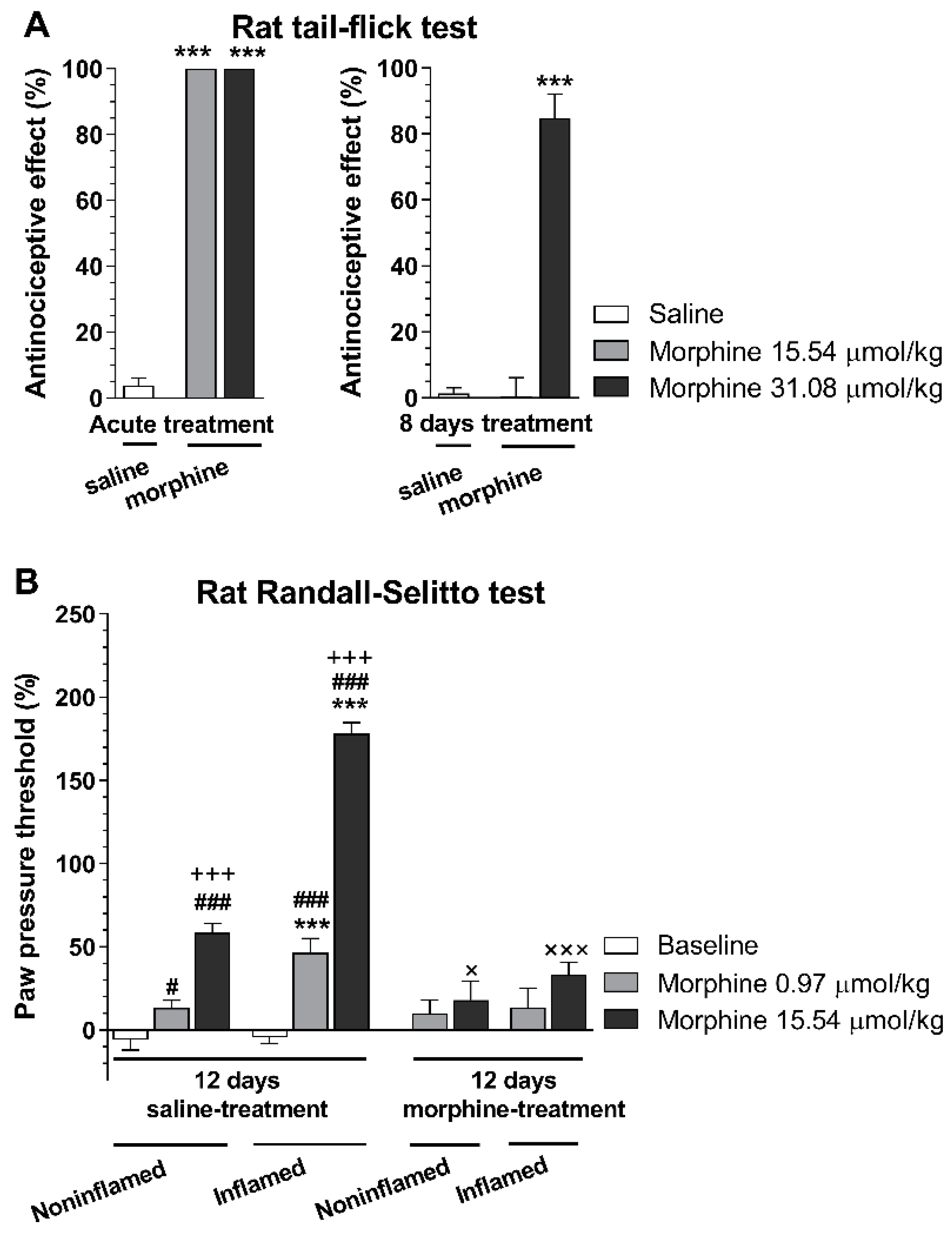{kind=link}
| Structure | R1 | R2 | R3 | Compound | Ref. |
|---|---|---|---|---|---|
 | H | OSO3− | H | Morphine-6-O-sulfate | [73] |
| OCH3 | OSO3− | H | 14-O-methylmorphine-6-O-sulfate | [62] | |
| H | OSO3− | CH3 | Codeine-6-O-sulfate | [60] | |
| OCH3 | OSO3− | CH3 | 14-O-methylcodeine-6-O-sulfate | [65] | |
 | |||||
| OCH3 | HNCH2COOH | H | 14-O-methyloxymorphone-6β-glycine (HS-731) | [34,35] | |
| Pain Model | Assay and Species | Test Compound | Route of Administration | Tolerance Induction | Main Findings | Ref. |
|---|---|---|---|---|---|---|
| Acute thermal | MTF | Morphine M6G | Tail injection | s.c. morphine | High grade tolerance | [74] |
| Acute thermal | MTF | Morphine M6G DAMGO | DMSO solution immersion | topical morphine | High grade tolerance | [75] |
| PGE2-induced pain | RS in rat | DAMGO | i.pl. injection | i.pl. injection DAMGO | Tolerance, withdrawal hyperalgesia | [76,86] |
| Bradykinin-induced flexion reflex | mouse | DAMGO U69,593 DESLET morphine | i.pl. infusion | s.c. morphine | No tolerance to local effect | [89,90] |
| Bradykinin-induced flexion reflex | mouse | Morphine | i.pl. infusion | i.pl. morphine | Tolerance to local effect | [91] |
| Carrageenan-induced inflammation | RS in rat, spinal c-Fos | Morphine | i.pl. (and i.v.) injection | s.c. morphine 3 days 2x | Tolerance to i.pl. or i.v. | [87] |
| CFA-induced inflammation | Plantar, RS, von Frey in mouse | Morphine | s.c. | morphine pellet 3 days | Higher grade tolerance on CFA treated paw than noninflamed | [99] |
| CFA-induced inflammation | RS in mouse | Morphine Fentanyl Bup. DPDPE U50488H CRF | i.pl. | morphine pellet 3 days | Emax↓ in all case (exc. buprenorphine) and rightward shift (exc. buprenorphine and fentanyl) | [100] |
| CFA-induced inflammation | RS in rat | Fentanyl | i.pl. | s.c. morphine (4 days 2x) | Tolerance developed only in absence of inflammation | [68] |
| SNL | von Frey in mouse | Loperamide | i.pl./s.c. | i.pl./s.c. loperamide | i.pl. loperamide caused tolerance only on i.pl., caused both for i.pl. and s.c.; s.c. morphine remained effective | [40] |
| CFA-induced inflammation | Hargreaves assay in rat | Loperamide–oxymorphindole | topical (left hind paw) | twice a daily for 3 days | No tolerance to local effect | [102] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fürst, S.; Zádori, Z.S.; Zádor, F.; Király, K.; Balogh, M.; László, S.B.; Hutka, B.; Mohammadzadeh, A.; Calabrese, C.; Galambos, A.R.; et al. On the Role of Peripheral Sensory and Gut Mu Opioid Receptors: Peripheral Analgesia and Tolerance. Molecules 2020, 25, 2473. https://doi.org/10.3390/molecules25112473
Fürst S, Zádori ZS, Zádor F, Király K, Balogh M, László SB, Hutka B, Mohammadzadeh A, Calabrese C, Galambos AR, et al. On the Role of Peripheral Sensory and Gut Mu Opioid Receptors: Peripheral Analgesia and Tolerance. Molecules. 2020; 25(11):2473. https://doi.org/10.3390/molecules25112473
Chicago/Turabian StyleFürst, Susanna, Zoltán S. Zádori, Ferenc Zádor, Kornél Király, Mihály Balogh, Szilvia B. László, Barbara Hutka, Amir Mohammadzadeh, Chiara Calabrese, Anna Rita Galambos, and et al. 2020. "On the Role of Peripheral Sensory and Gut Mu Opioid Receptors: Peripheral Analgesia and Tolerance" Molecules 25, no. 11: 2473. https://doi.org/10.3390/molecules25112473