
Insights into the Genomic and Phenotypic Landscape of the Oleaginous Yeast Yarrowia lipolytica

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains
2.2. DNA Extraction
2.3. Genome Sequencing
2.4. Bioinformatics
2.5. Growth Conditions and Phenotyping
2.6. Fluorescence Microscopy
2.7. Lipid Content Analysis
2.8. Statistical Analysis
3. Results
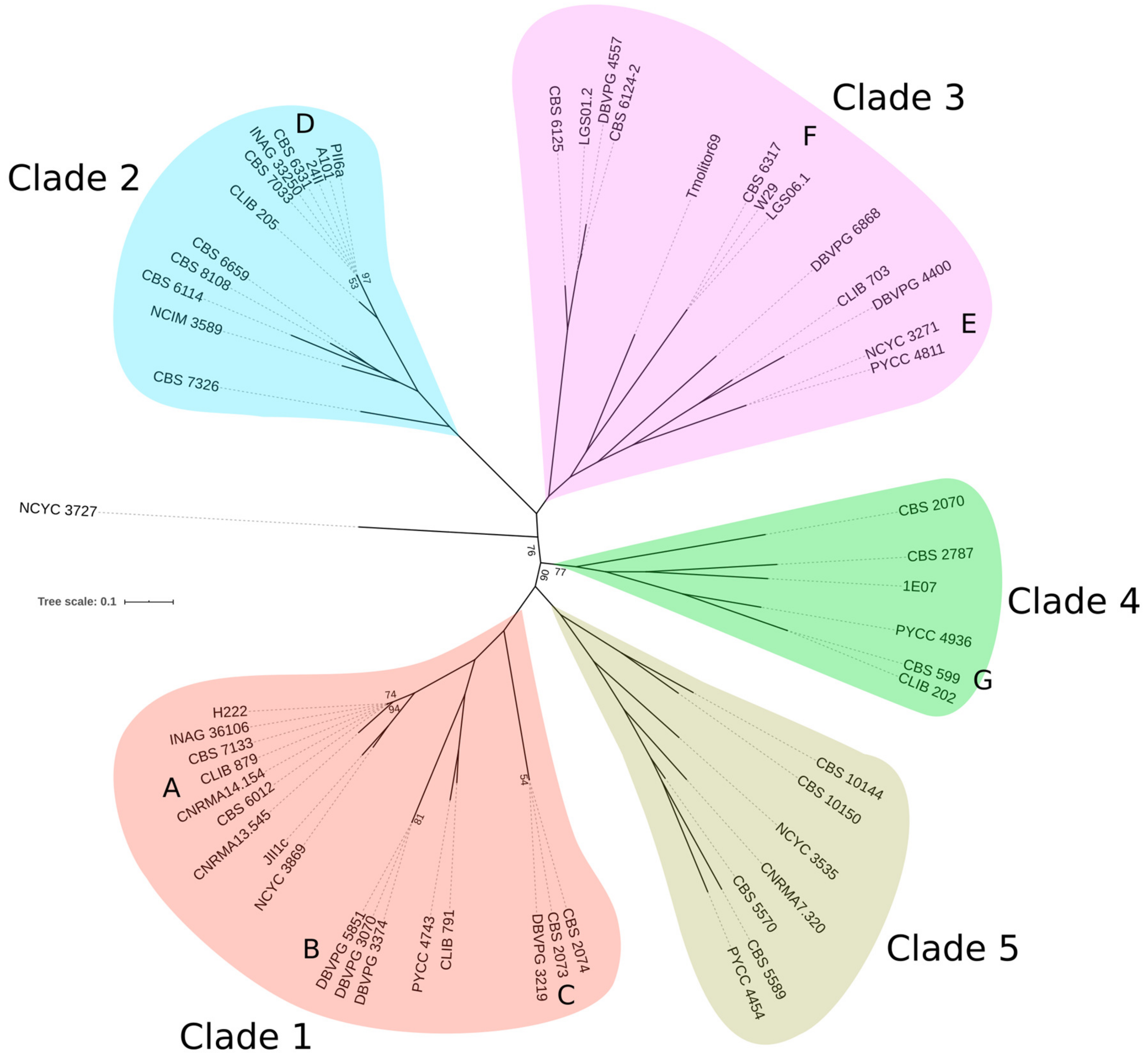
3.1. Nucleotide Diversity, Phylogeny and Population Structure
3.2. Genome Variation: Segmental Duplication, Copy Number Variation of Genes
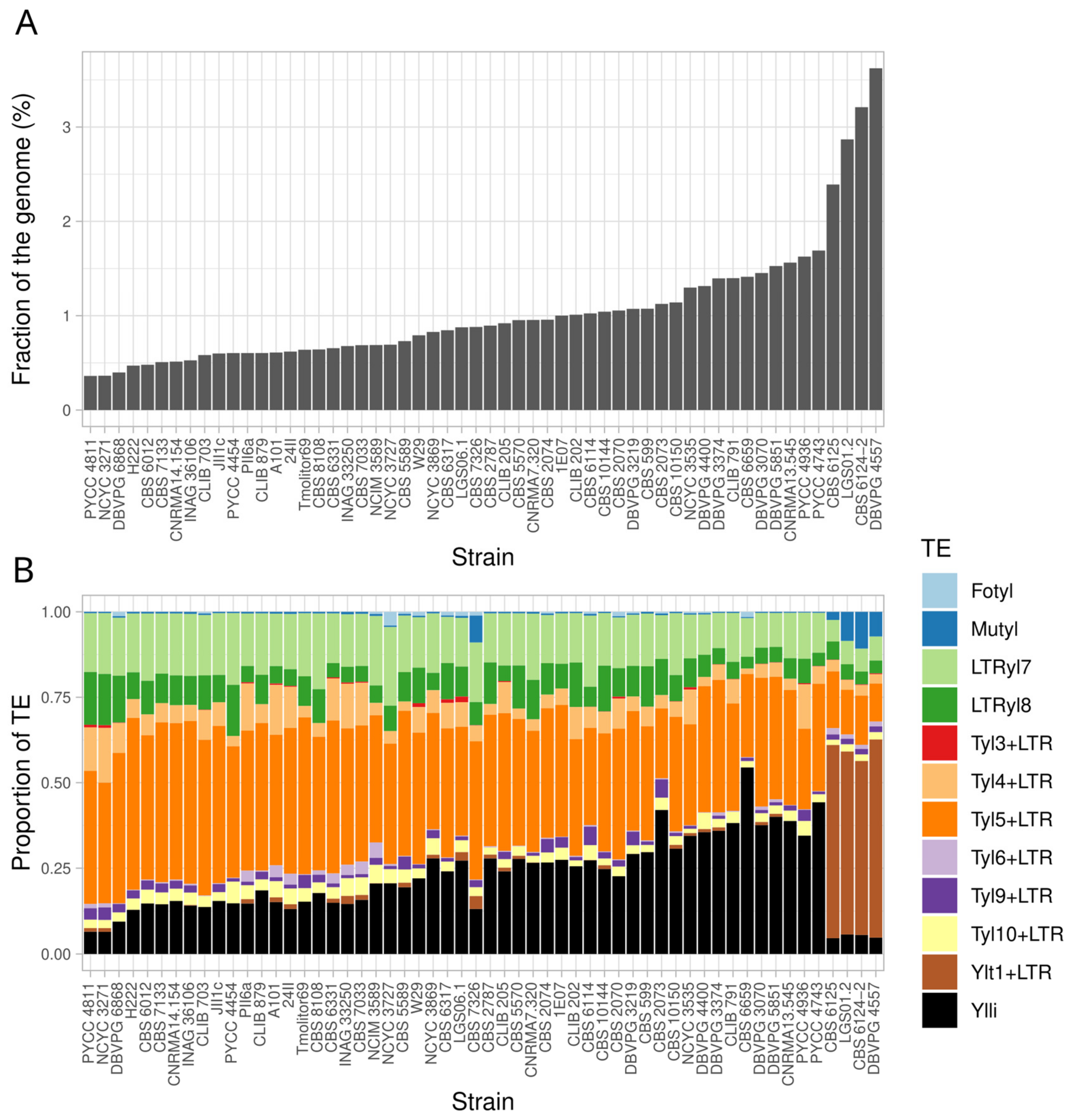
3.3. Transposable Elements
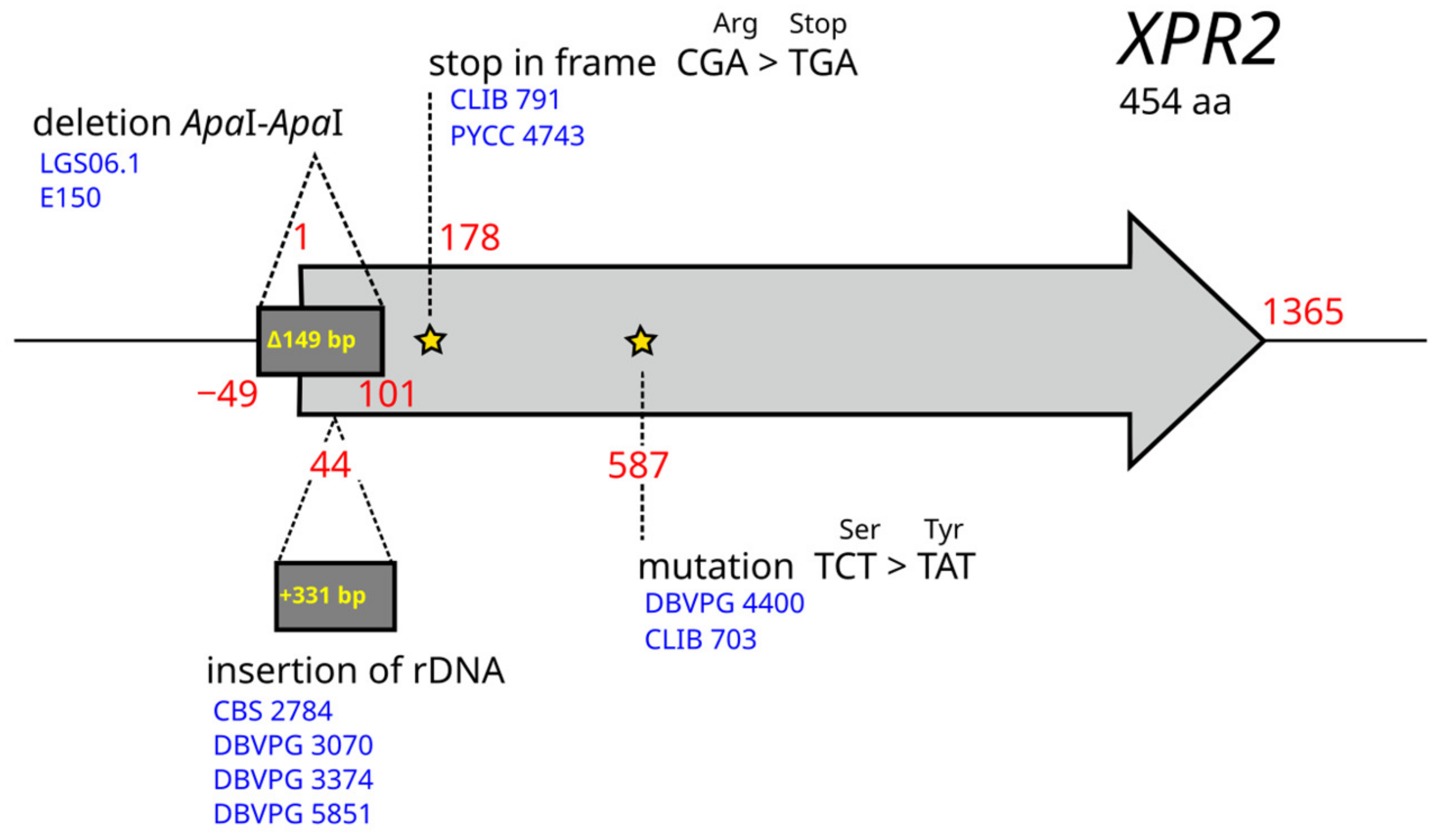
3.4. Lipid Metabolism
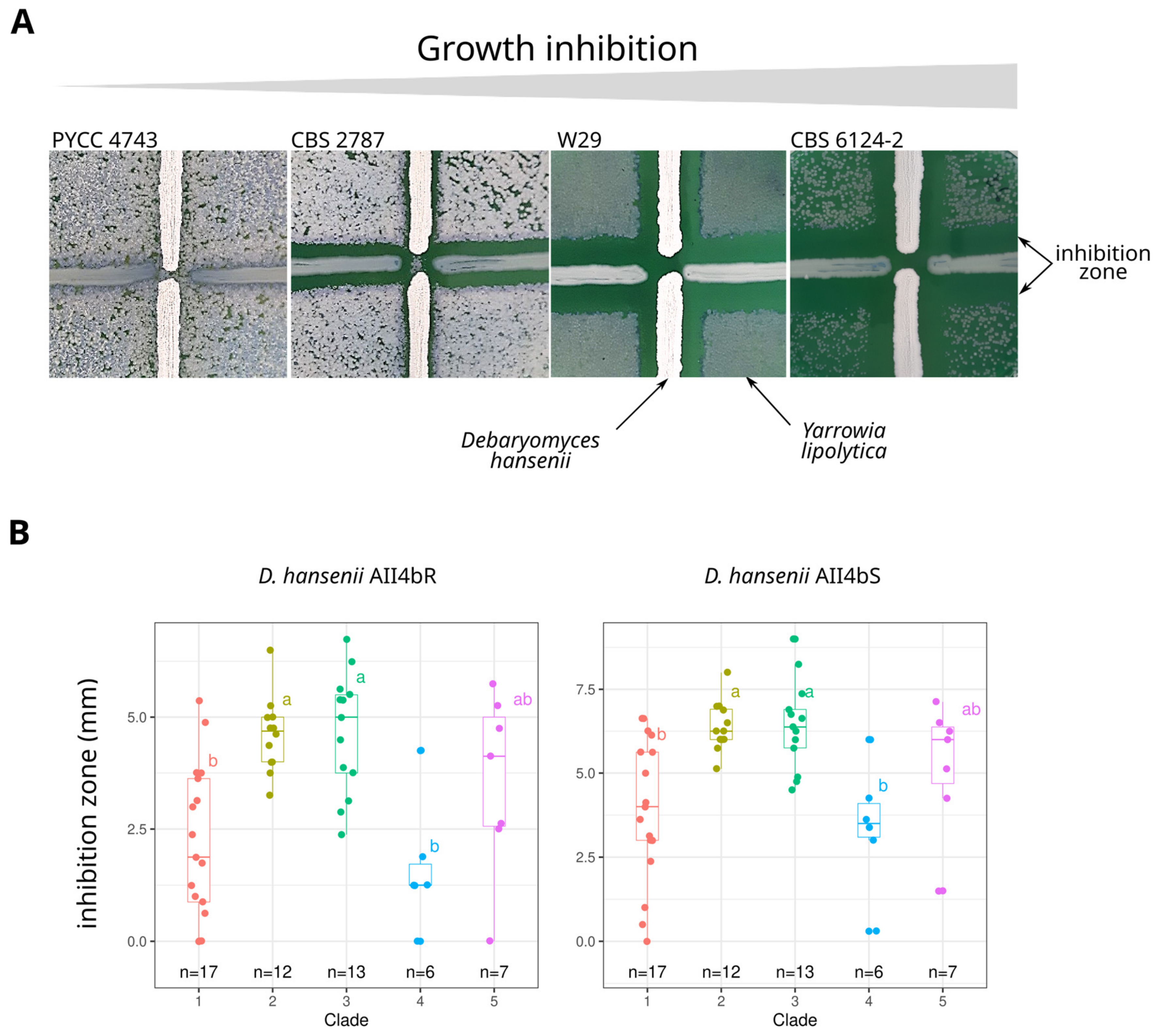
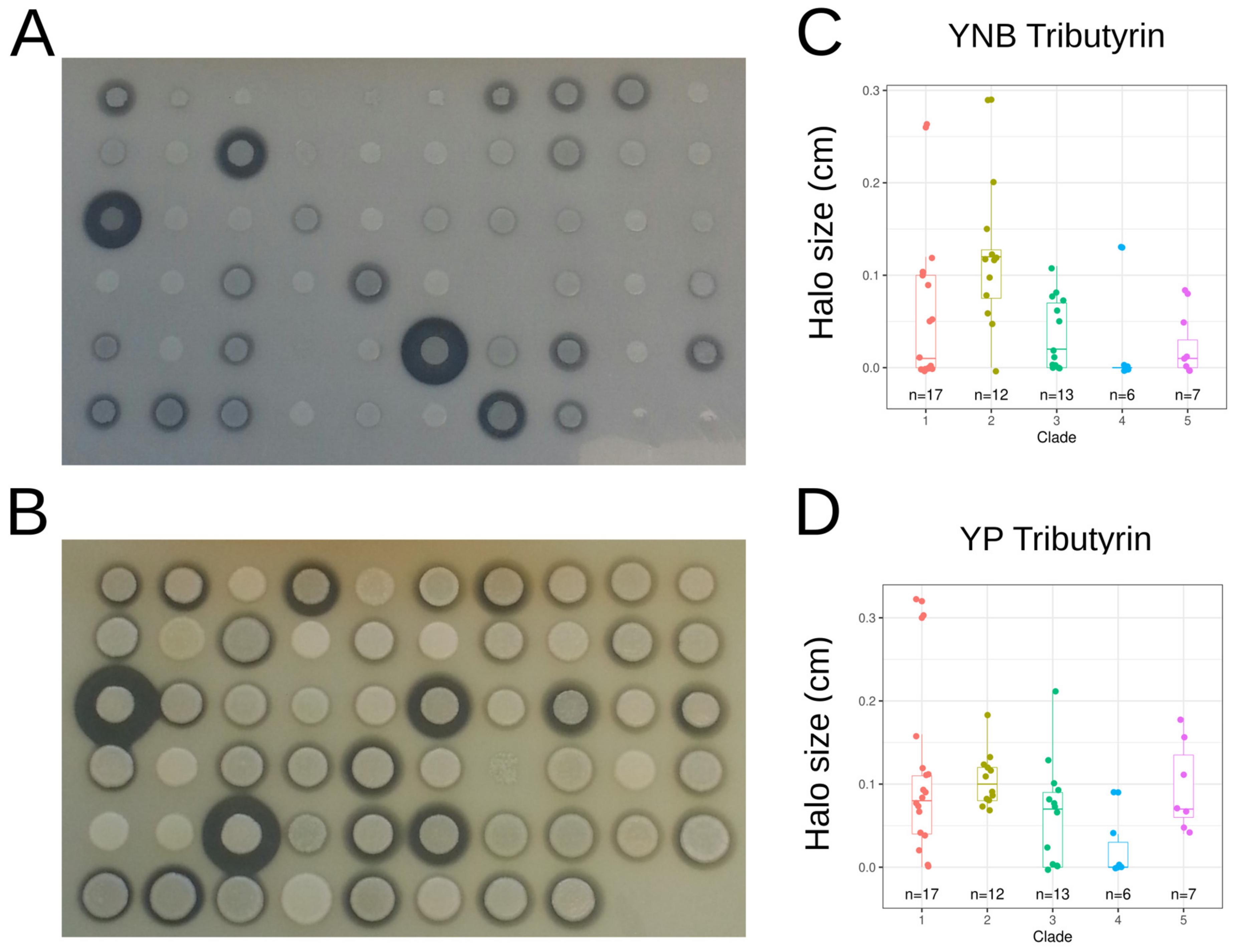
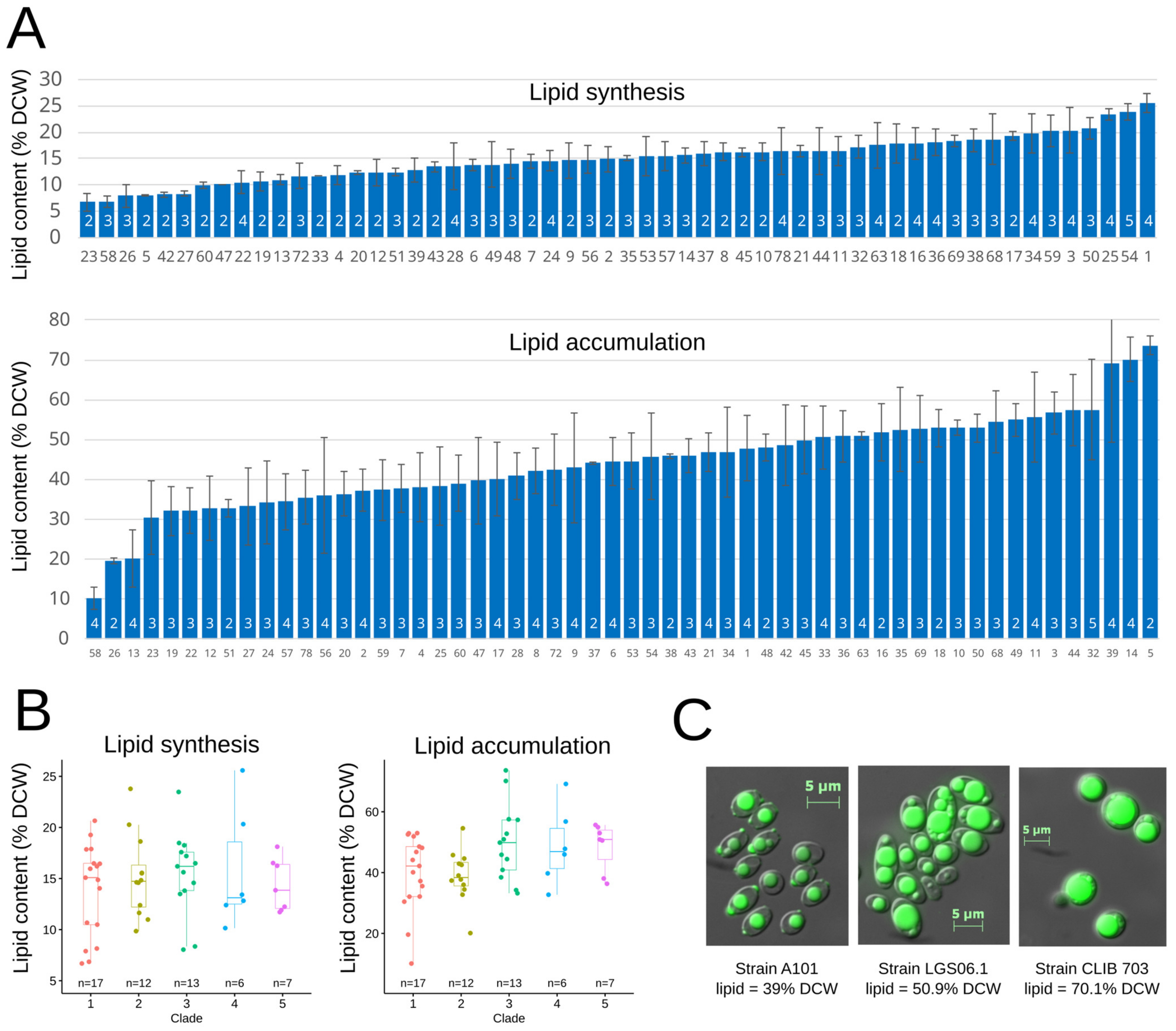
3.5. Phenotypic Variation
3.6. Links between Genotypes and Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mamaev, D.; Zvyagilskaya, R. Yarrowia lipolytica: A multitalented yeast species of ecological significance. FEMS Yeast Res. 2021, 21, foab008. [Google Scholar] [CrossRef] [PubMed]
- Salvador Lopez, J.M.; Vandeputte, M.; Van Bogaert, I.N.A. Oleaginous yeasts: Time to rethink the definition? Yeast 2022, 39, 553–606. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Cao, L.; Wang, K.; Ledesma-Amaro, R.; Ji, X.J. Engineering Yarrowia lipolytica to produce advanced biofuels: Current status and perspectives. Bioresour. Technol. 2021, 341, 125877. [Google Scholar] [CrossRef] [PubMed]
- Poli, J.S.; da Silva, M.A.; Siqueira, E.P.; Pasa, V.M.; Rosa, C.A.; Valente, P. Microbial lipid produced by Yarrowia lipolytica QU21 using industrial waste: A potential feedstock for biodiesel production. Bioresour. Technol. 2014, 161, 320–326. [Google Scholar] [CrossRef]
- Rong, L.; Miao, L.; Wang, S.; Wang, Y.; Liu, S.; Lu, Z.; Zhao, B.; Zhang, C.; Xiao, D.; Pushpanathan, K.; et al. Engineering Yarrowia lipolytica to Produce Itaconic Acid From Waste Cooking Oil. Front. Bioeng. Biotechnol. 2022, 10, 888869. [Google Scholar] [CrossRef]
- Park, Y.K.; Ledesma-Amaro, R. What makes Yarrowia lipolytica well suited for industry? Trends Biotechnol. 2022. [Google Scholar] [CrossRef]
- Madzak, C. Yarrowia lipolytica Strains and Their Biotechnological Applications: How Natural Biodiversity and Metabolic Engineering Could Contribute to Cell Factories Improvement. J. Fungi 2021, 7, 548. [Google Scholar] [CrossRef]
- Barth, G.; Gaillardin, C. Physiology and genetics of the dimorphic fungus Yarrowia lipolytica. FEMS Microbiol. Rev. 1997, 19, 219–237. [Google Scholar] [CrossRef]
- Dujon, B.; Sherman, D.; Fischer, G.; Durrens, P.; Casaregola, S.; Lafontaine, I.; De Montigny, J.; Marck, C.; Neuveglise, C.; Talla, E.; et al. Genome evolution in yeasts. Nature 2004, 430, 35–44. [Google Scholar] [CrossRef]
- Mekouar, M.; Blanc-Lenfle, I.; Ozanne, C.; Da Silva, C.; Cruaud, C.; Wincker, P.; Gaillardin, C.; Neuveglise, C. Detection and analysis of alternative splicing in Yarrowia lipolytica reveal structural constraints facilitating nonsense-mediated decay of intron-retaining transcripts. Genome Biol. 2010, 11, R65. [Google Scholar] [CrossRef]
- Liu, L.; Alper, H.S. Draft Genome Sequence of the Oleaginous Yeast Yarrowia lipolytica PO1f, a Commonly Used Metabolic Engineering Host. Genome Announc. 2014, 2, e00652-14. [Google Scholar] [CrossRef] [Green Version]
- Magnan, C.; Yu, J.; Chang, I.; Jahn, E.; Kanomata, Y.; Wu, J.; Zeller, M.; Oakes, M.; Baldi, P.; Sandmeyer, S. Sequence Assembly of Yarrowia lipolytica Strain W29/CLIB89 Shows Transposable Element Diversity. PLoS ONE 2016, 11, e0162363. [Google Scholar] [CrossRef] [Green Version]
- Devillers, H.; Brunel, F.; Polomska, X.; Sarilar, V.; Lazar, Z.; Robak, M.; Neuveglise, C. Draft Genome Sequence of Yarrowia lipolytica Strain A-101 Isolated from Polluted Soil in Poland. Genome Announc. 2016, 4, e01094-16. [Google Scholar] [CrossRef] [Green Version]
- Devillers, H.; Neuveglise, C. Genome Sequence of the Oleaginous Yeast Yarrowia lipolytica H222. Microbiol. Resour. Announc. 2019, 8, e01547-18. [Google Scholar] [CrossRef] [Green Version]
- Luttermann, T.; Ruckert, C.; Wibberg, D.; Busche, T.; Schwarzhans, J.P.; Friehs, K.; Kalinowski, J. Establishment of a near-contiguous genome sequence of the citric acid producing yeast Yarrowia lipolytica DSM 3286 with resolution of rDNA clusters and telomeres. NAR Genom. Bioinform. 2021, 3, lqab085. [Google Scholar] [CrossRef]
- Casaregola, S.; Feynerol, C.; Diez, M.; Fournier, P.; Gaillardin, C. Genomic organization of the yeast Yarrowia lipolytica. Chromosoma 1997, 106, 380–390. [Google Scholar]
- Schmid-Berger, N.; Schmid, B.; Barth, G. Ylt1, a highly repetitive retrotransposon in the genome of the dimorphic fungus Yarrowia lipolytica. J. Bacteriol. 1994, 176, 2477–2482. [Google Scholar] [CrossRef] [Green Version]
- Casaregola, S.; Neuveglise, C.; Bon, E.; Gaillardin, C. Ylli, a non-LTR retrotransposon L1 family in the dimorphic yeast Yarrowia lipolytica. Mol. Biol. Evol. 2002, 19, 664–677. [Google Scholar] [CrossRef] [Green Version]
- Kovalchuk, A.; Senam, S.; Mauersberger, S.; Barth, G. Tyl6, a novel Ty3/gypsy-like retrotransposon in the genome of the dimorphic fungus Yarrowia lipolytica. Yeast 2005, 22, 979–991. [Google Scholar] [CrossRef]
- Neuveglise, C.; Chalvet, F.; Wincker, P.; Gaillardin, C.; Casaregola, S. Mutator-like element in the yeast Yarrowia lipolytica displays multiple alternative splicings. Eukaryot. Cell 2005, 4, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Fickers, P.; Benetti, P.H.; Wache, Y.; Marty, A.; Mauersberger, S.; Smit, M.S.; Nicaud, J.M. Hydrophobic substrate utilisation by the yeast Yarrowia lipolytica, and its potential applications. FEMS Yeast Res. 2005, 5, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Meunchan, M.; Michely, S.; Devillers, H.; Nicaud, J.M.; Marty, A.; Neuveglise, C. Comprehensive Analysis of a Yeast Lipase Family in the Yarrowia Clade. PLoS ONE 2015, 10, e0143096. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Iwama, R.; Kobayashi, S.; Horiuchi, H.; Fukuda, R.; Ohta, A. Construction and characterization of a Yarrowia lipolytica mutant lacking genes encoding cytochromes P450 subfamily 52. Fungal Genet. Biol. 2012, 49, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Haddouche, R.; Delessert, S.; Sabirova, J.; Neuveglise, C.; Poirier, Y.; Nicaud, J.M. Roles of multiple acyl-CoA oxidases in the routing of carbon flow towards beta-oxidation and polyhydroxyalkanoate biosynthesis in Yarrowia lipolytica. FEMS Yeast Res. 2010, 10, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, C.G.P.; Fitzpatrick, D.A. Pangloss: A Tool for Pan-Genome Analysis of Microbial Eukaryotes. Genes 2019, 10, 521. [Google Scholar] [CrossRef] [Green Version]
- Carsanba, E.; Papanikolaou, S.; Fickers, P.; Erten, H. Lipids by Yarrowia lipolytica Strains Cultivated on Glucose in Batch Cultures. Microorganisms 2020, 8, 10154. [Google Scholar] [CrossRef]
- Carsanba, E.; Papanikolaou, S.; Fickers, P.; Erten, H. Screening various Yarrowia lipolytica strains for citric acid production. Yeast 2019, 36, 319–327. [Google Scholar] [CrossRef]
- Hackenschmidt, S.; Bracharz, F.; Daniel, R.; Thurmer, A.; Bruder, S.; Kabisch, J. Effects of a high-cultivation temperature on the physiology of three different Yarrowia lipolytica strains. FEMS Yeast Res. 2019, 19, foz068. [Google Scholar] [CrossRef]
- Walker, C.; Ryu, S.; Na, H.; Zane, M.; LaButti, K.; Lipzen, A.; Haridas, S.; Barry, K.; Grigoriev, I.V.; Quarterman, J.; et al. Draft Genome Assemblies of Five Robust Yarrowia lipolytica Strains Exhibiting High Lipid Production, Pentose Sugar Utilization, and Sugar Alcohol Secretion from Undetoxified Lignocellulosic Biomass Hydrolysates. Microbiol. Resour. Announc. 2018, 7, e01040-18. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Pfeifer, B.; Wittelsburger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Casaregola, S.; Neuveglise, C.; Lepingle, A.; Bon, E.; Feynerol, C.; Artiguenave, F.; Wincker, P.; Gaillardin, C. Genomic exploration of the hemiascomycetous yeasts: 17. Yarrowia lipolytica. FEBS Lett. 2000, 487, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Abdelmoteleb, A.; Troncoso-Rojas, R.; Gonzalez-Soto, T.; González-Mendoza, D. Antifungical Activity of Autochthonous Bacillus subtilis Isolated from Prosopis juliflora against Phytopathogenic Fungi. Mycobiology 2017, 45, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Wagih, O.; Parts, L. gitter: A robust and accurate method for quantification of colony sizes from plate images. G3 2014, 4, 547–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, D.R.; Bevan, E.A. Studies on the nature of the killer factor produced by Saccharomyces cerevisiae. J. Gen. Microbiol. 1968, 51, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarowska, B.; Wojtatowicz, M.; Polomska, X.; Juszczyk, P.; Chrzanowska, J. Factors affecting killer activity of some yeast species occurring in Rokpol cheese. Folia Microbiol. 2004, 49, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Browse, J.; McCourt, P.J.; Somerville, C.R. Fatty acid composition of leaf lipids determined after combined digestion and fatty acid methyl ester formation from fresh tissue. Anal. Biochem. 1986, 152, 141–145. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- van Rossum, B.; Kruijer, W. statgenGWAS: Genome Wide Association Studies. Available online: https://biometris.github.io/statgenGWAS/index.html; https://github.com/Biometris/statgenGWAS/ (accessed on 14 October 2022).
- Neuveglise, C.; Feldmann, H.; Bon, E.; Gaillardin, C.; Casaregola, S. Genomic evolution of the long terminal repeat retrotransposons in hemiascomycetous yeasts. Genome Res. 2002, 12, 930–943. [Google Scholar] [CrossRef] [Green Version]
- Strijbis, K.; van Roermund, C.W.; Hardy, G.P.; van den Burg, J.; Bloem, K.; de Haan, J.; van Vlies, N.; Wanders, R.J.; Vaz, F.M.; Distel, B. Identification and characterization of a complete carnitine biosynthesis pathway in Candida albicans. FASEB J. 2009, 23, 2349–2359. [Google Scholar] [CrossRef]
- Fukuda, R. Metabolism of hydrophobic carbon sources and regulation of it in n-alkane-assimilating yeast Yarrowia lipolytica. Biosci. Biotechnol. Biochem. 2013, 77, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Endoh-Yamagami, S.; Hirakawa, K.; Morioka, D.; Fukuda, R.; Ohta, A. Basic helix-loop-helix transcription factor heterocomplex of Yas1p and Yas2p regulates cytochrome P450 expression in response to alkanes in the yeast Yarrowia lipolytica. Eukaryot. Cell 2007, 6, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, K.; Kobayashi, S.; Inoue, T.; Endoh-Yamagami, S.; Fukuda, R.; Ohta, A. Yas3p, an Opi1 family transcription factor, regulates cytochrome P450 expression in response to n-alkanes in Yarrowia lipolytica. J. Biol. Chem. 2009, 284, 7126–7137. [Google Scholar] [CrossRef]
- Michely, S.; Gaillardin, C.; Nicaud, J.M.; Neuveglise, C. Comparative physiology of oleaginous species from the Yarrowia clade. PLoS ONE 2013, 8, e63356. [Google Scholar] [CrossRef]
- Polomska, X.; Neuveglise, C.; Zyzak, J.; Zarowska, B.; Casaregola, S.; Lazar, Z. New Cytoplasmic Virus-Like Elements (VLEs) in the Yeast Debaryomyces hansenii. Toxins 2021, 13, 615. [Google Scholar] [CrossRef]
- Barth, G.; Gaillardin, C. Yarrowia lipolytica. In Genetics, Biochemistry and Molecular Biology of Non Conventional Yeasts in Biotechnology; Wolf, W.K., Ed.; Springer: Berlin, Germany, 1996; Volume 1, pp. 313–388. [Google Scholar]
- Ogrydziak, D.M.; Mortimer, R.K. Genetics of Extracellular Protease Production in Saccharomycopsis lipolytica. Genetics 1977, 87, 621–632. [Google Scholar] [CrossRef]
- Gounot, J.S.; Neuveglise, C.; Freel, K.C.; Devillers, H.; Piskur, J.; Friedrich, A.; Schacherer, J. High Complexity and Degree of Genetic Variation in Brettanomyces bruxellensis Population. Genome Biol. Evol. 2020, 12, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, A.; Jung, P.; Reisser, C.; Fischer, G.; Schacherer, J. Population genomics reveals chromosome-scale heterogeneous evolution in a protoploid yeast. Mol. Biol. Evol. 2015, 32, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.X.; Pflieger, D.; Bergstrom, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Jeffares, D.C.; Rallis, C.; Rieux, A.; Speed, D.; Prevorovsky, M.; Mourier, T.; Marsellach, F.X.; Iqbal, Z.; Lau, W.; Cheng, T.M.; et al. The genomic and phenotypic diversity of Schizosaccharomyces pombe. Nat. Genet. 2015, 47, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, C.A.; Giamberardino, C.; Sykes, S.M.; Yu, C.H.; Tenor, J.L.; Chen, Y.; Yang, T.; Jones, A.M.; Sun, S.; Haverkamp, M.R.; et al. Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans. Genome Res. 2017, 27, 1207–1219. [Google Scholar] [CrossRef] [Green Version]
- Naumova, E.; Naumov, G.; Fournier, P.; Nguyen, H.V.; Gaillardin, C. Chromosomal polymorphism of the yeast Yarrowia lipolytica and related species: Electrophoretic karyotyping and hybridization with cloned genes. Curr. Genet. 1993, 23, 450–454. [Google Scholar] [CrossRef]
- Bleykasten-Grosshans, C.; Neuveglise, C. Transposable elements in yeasts. Comptes Rendus Biol. 2011, 334, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Dean, A.C.R.; Hinshelwood, C.N. The formation of papillae on bacterial colonies. Proc. R. Soc. B 1957, 147, 1–9. [Google Scholar]
- Liu, L.; Pan, A.; Spofford, C.; Zhou, N.; Alper, H.S. An evolutionary metabolic engineering approach for enhancing lipogenesis in Yarrowia lipolytica. Metab. Eng. 2015, 29, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Daskalaki, A.; Perdikouli, N.; Aggeli, D.; Aggelis, G. Laboratory evolution strategies for improving lipid accumulation in Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 2019, 103, 8585–8596. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, H.; Li, C.; Lin, C.S.K. Restoring of Glucose Metabolism of Engineered Yarrowia lipolytica for Succinic Acid Production via a Simple and Efficient Adaptive Evolution Strategy. J. Agric. Food Chem. 2017, 65, 4133–4139. [Google Scholar] [CrossRef]
- Walker, C.; Ryu, S.; Trinh, C.T. Exceptional solvent tolerance in Yarrowia lipolytica is enhanced by sterols. Metab. Eng. 2019, 54, 83–95. [Google Scholar] [CrossRef]
- Li, J.; Zhu, K.; Miao, L.; Rong, L.; Zhao, Y.; Li, S.; Ma, L.; Li, J.; Zhang, C.; Xiao, D.; et al. Simultaneous Improvement of Limonene Production and Tolerance in Yarrowia lipolytica through Tolerance Engineering and Evolutionary Engineering. ACS Synth. Biol. 2021, 10, 884–896. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, L.; Lu, M.; Zhang, Y.; Perveen, S.; Zhou, H.; Wen, Z.; Xu, Z.; Jin, M. Adaptive laboratory evolution of Yarrowia lipolytica improves ferulic acid tolerance. Appl. Microbiol. Biotechnol. 2021, 105, 1745–1758. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, Z.; Wen, Z.; Lu, M.; Wang, Z.; Zhang, Y.; Zhou, H.; Jin, M. Combined adaptive evolution and transcriptomic profiles reveal aromatic aldehydes tolerance mechanisms in Yarrowia lipolytica. Bioresour. Technol. 2021, 329, 124910. [Google Scholar] [CrossRef]
- Narisetty, V.; Prabhu, A.A.; Bommareddy, R.R.; Cox, R.; Agrawal, D.; Misra, A.; Haider, M.A.; Bhatnagar, A.; Pandey, A.; Kumar, V. Development of Hypertolerant Strain of Yarrowia lipolytica Accumulating Succinic Acid Using High Levels of Acetate. ACS Sustain. Chem. Eng. 2022, 10, 10858–10869. [Google Scholar] [CrossRef]
- Qiu, X.; Gu, Y.; Du, G.; Zhang, J.; Xu, P.; Li, J. Conferring thermotolerant phenotype to wild-type Yarrowia lipolytica improves cell growth and erythritol production. Biotechnol. Bioeng. 2021, 118, 3117–3127. [Google Scholar] [CrossRef]
- Papanikolaou, S.; Galiotou-Panayotou, M.; Chevalot, I.; Komaitis, M.; Marc, I.; Aggelis, G. Influence of glucose and saturated free-fatty acid mixtures on citric acid and lipid production by Yarrowia lipolytica. Curr. Microbiol. 2006, 52, 134–142. [Google Scholar] [CrossRef]
- Bati, N.; Hammond, E.G.; Glatz, B.A. Biomodification of fats and oils: Trials with Candida lipolytica. J. Am. Oil Chem. Soc. 1984, 61, 1743–1746. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bigey, F.; Pasteur, E.; Połomska, X.; Thomas, S.; Crutz-Le Coq, A.-M.; Devillers, H.; Neuvéglise, C. Insights into the Genomic and Phenotypic Landscape of the Oleaginous Yeast Yarrowia lipolytica. J. Fungi 2023, 9, 76. https://doi.org/10.3390/jof9010076
Bigey F, Pasteur E, Połomska X, Thomas S, Crutz-Le Coq A-M, Devillers H, Neuvéglise C. Insights into the Genomic and Phenotypic Landscape of the Oleaginous Yeast Yarrowia lipolytica. Journal of Fungi. 2023; 9(1):76. https://doi.org/10.3390/jof9010076
Chicago/Turabian StyleBigey, Frédéric, Emilie Pasteur, Xymena Połomska, Stéphane Thomas, Anne-Marie Crutz-Le Coq, Hugo Devillers, and Cécile Neuvéglise. 2023. "Insights into the Genomic and Phenotypic Landscape of the Oleaginous Yeast Yarrowia lipolytica" Journal of Fungi 9, no. 1: 76. https://doi.org/10.3390/jof9010076