
Deficiency of Acute-Phase Serum Amyloid A Exacerbates Sepsis-Induced Mortality and Lung Injury in Mice
 and
and 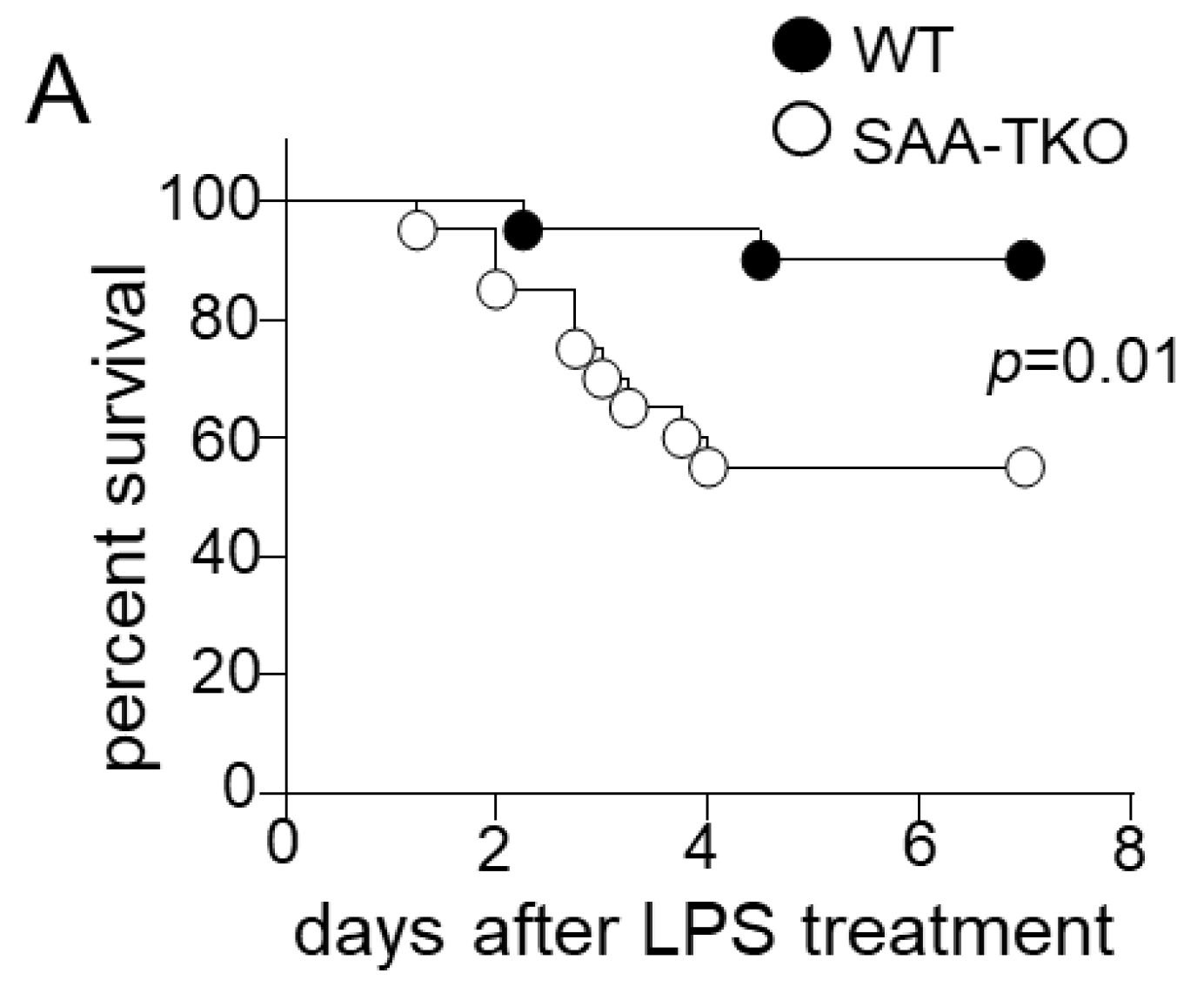{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Whole-Body SAA Deficiency Aggravates Sepsis-Induced Mortality
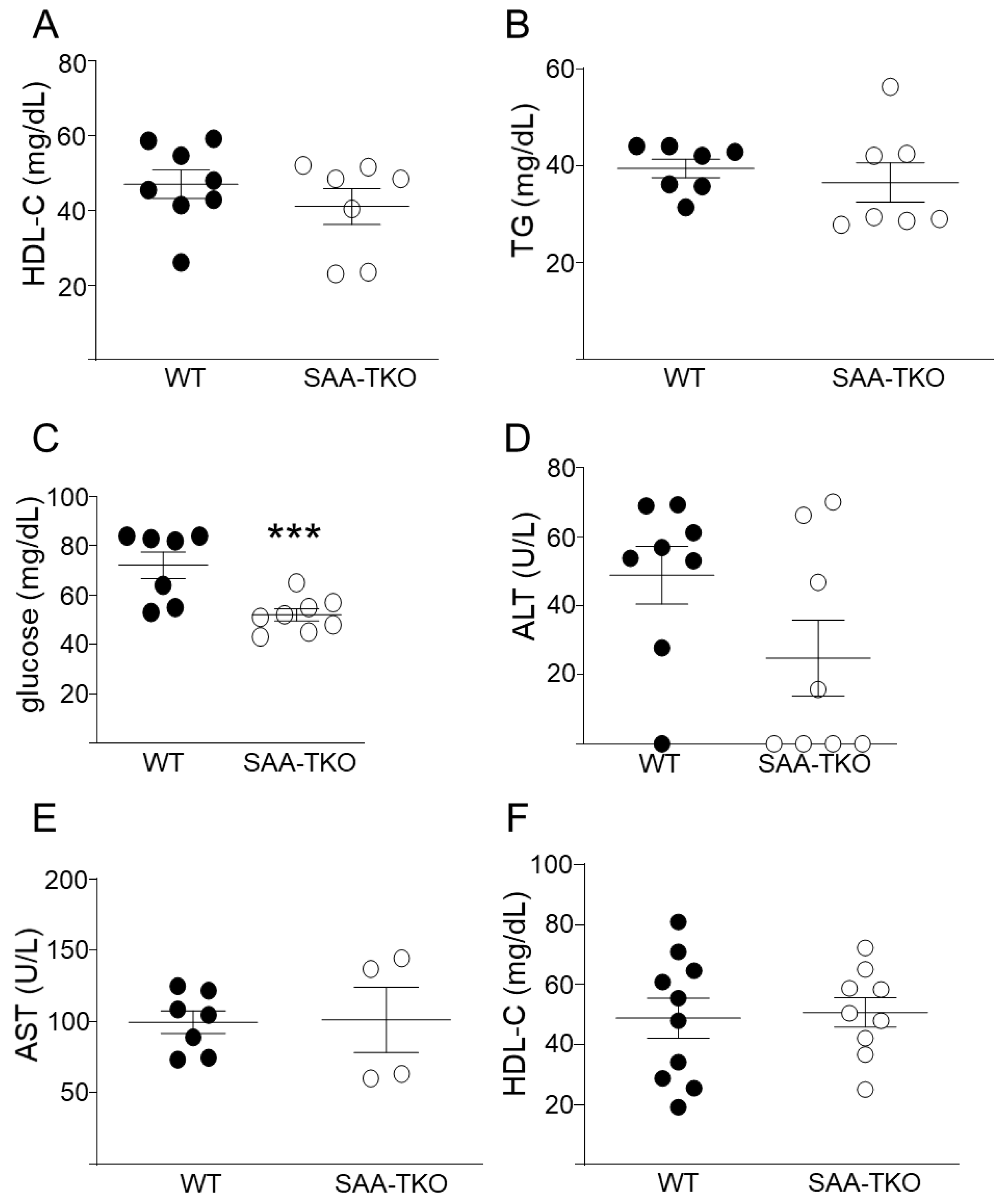
2.2. SAA Is Dispensable for Endotoxin and Bacterial Clearance or Systemic Proinflammatory Responses
2.3. Deficiency of SAA Does Not Change Plasma HDL-Cholesterol (HDL-C) Levels in Mice during Sepsis
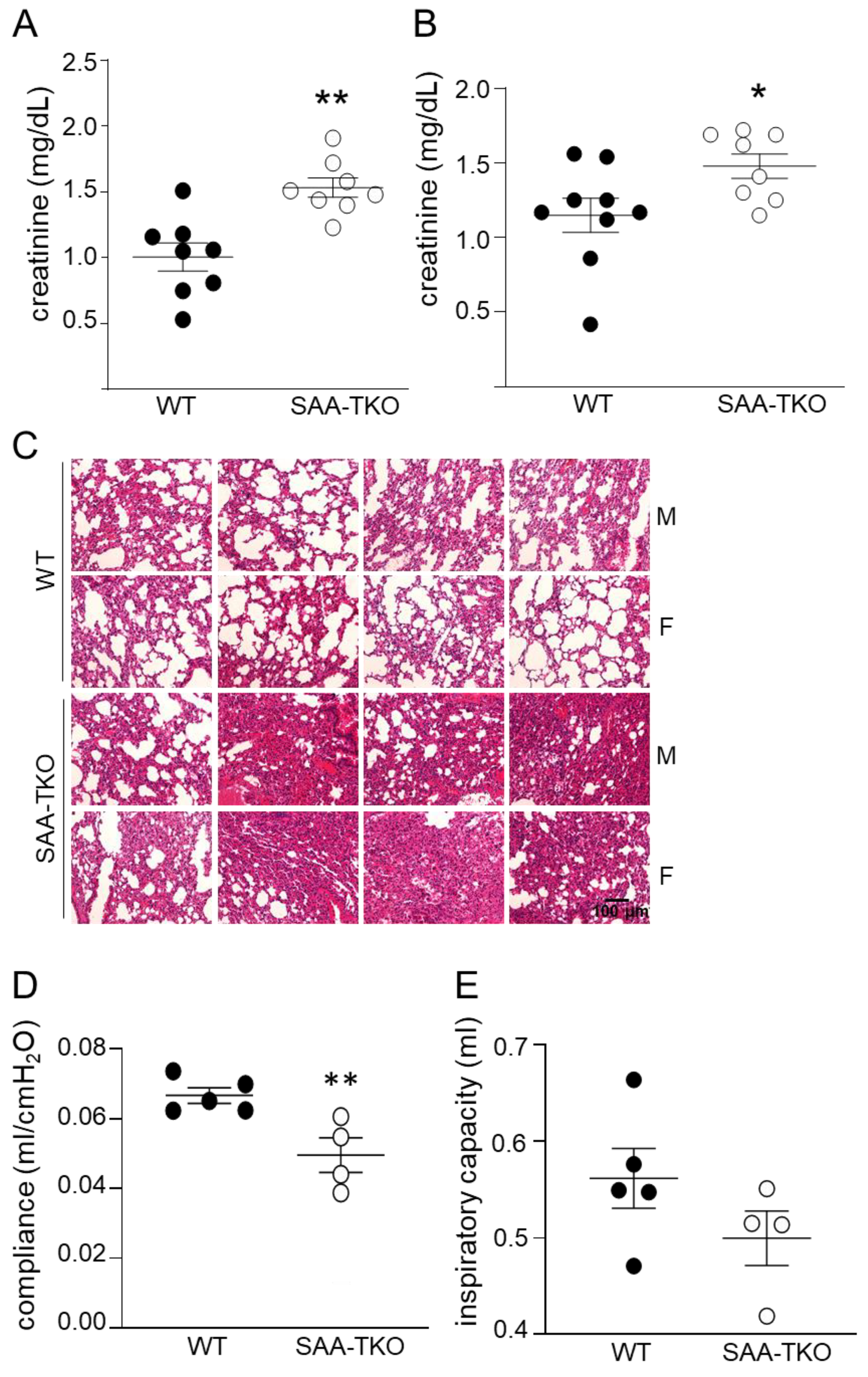
2.4. Deficiency of SAA Exacerbates Sepsis-Induced Kidney and Lung Injury in Mice
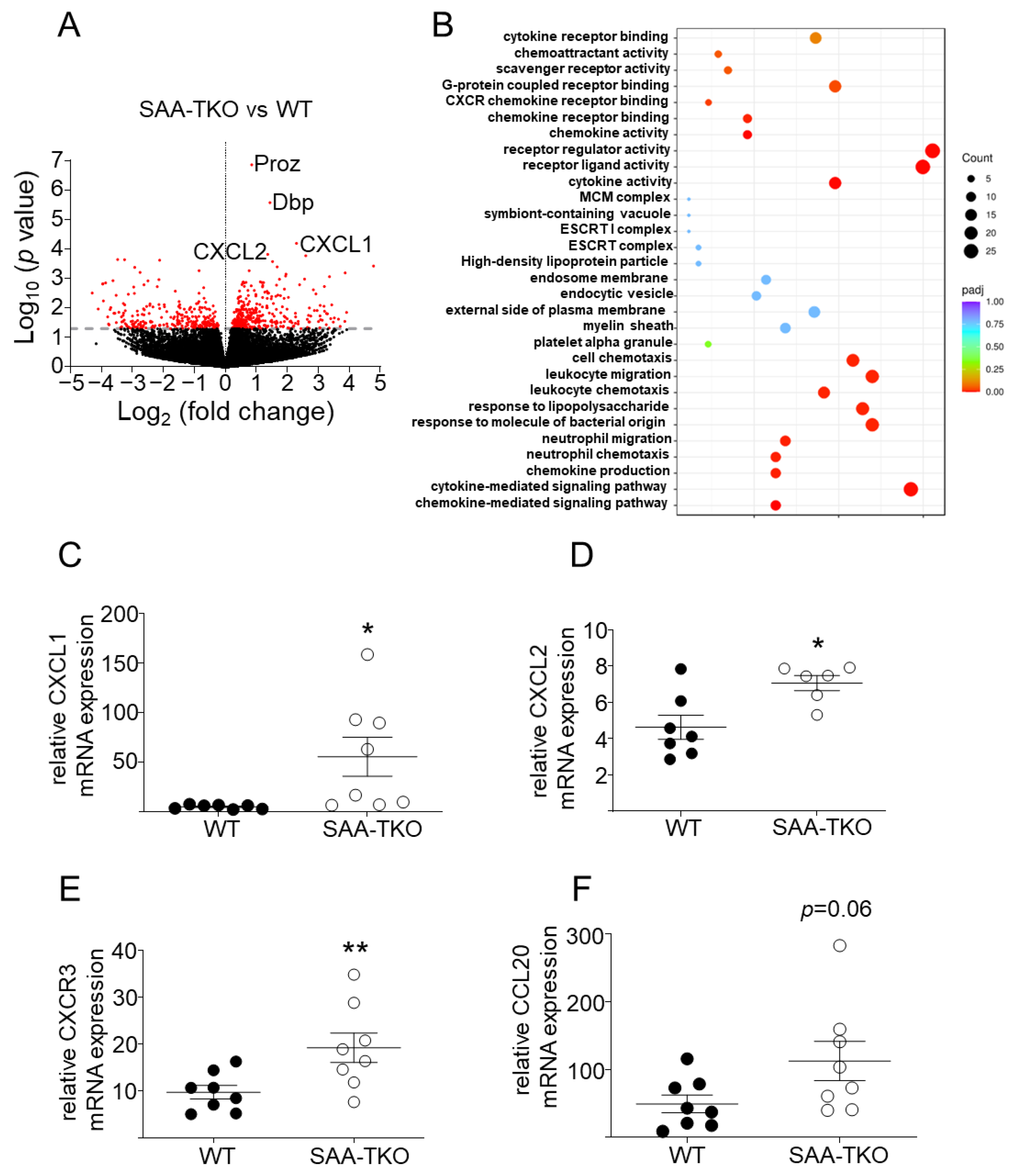
2.5. Deficiency of SAA Increases the Expression of Genes Involved in Leukocyte Chemotaxis in Lung Tissues during Sepsis
2.6. Deficiency of SAA Increases the Accumulation of Neutrophils in the Lungs of Septic Mice and SAA Attenuates In Vitro Neutrophil Transmigration
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Mouse Models of Sepsis
4.3. Cytokine, Glucose, Endotoxin, and Tissue Injury Marker Analysis
4.4. Analysis of Bacterial Burden
4.5. Plasma SAA, Total Cholesterol, and Triglyceride Measurements
4.6. RNA Isolation and Quantitative RT-PCR
4.7. Histology
4.8. Myeloperoxidase (MPO) Activity
4.9. Neutrophil Transmigration Assay
4.10. Lung Function Measurements
4.11. RNA Sequencing and Analysis
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.C. Why have clinical trials in sepsis failed? Trends Mol. Med. 2014, 20, 195–203. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Vandenbroucke, R.E.; Libert, C. An acute phase protein ready to go therapeutic for sepsis. EMBO Mol. Med. 2014, 6, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Kluve-Beckerman, B.; Drumm, M.L.; Benson, M.D. Nonexpression of the Human Serum Amyloid A Three (SAA3) Gene. DNA Cell Biol. 1991, 10, 651–661. [Google Scholar] [CrossRef]
- Sack, G.H. Serum amyloid A—A review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R. High-Density Lipoproteins and Serum Amyloid A (SAA). Curr. Atheroscler. Rep. 2021, 23, 7. [Google Scholar] [CrossRef]
- Benditt, E.P.; Eriksen, N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc. Natl. Acad. Sci. USA 1977, 74, 4025–4028. [Google Scholar] [CrossRef]
- Guo, L.; Song, Z.; Li, M.; Wu, Q.; Wang, D.; Feng, H.; Bernard, P.; Daugherty, A.; Huang, B.; Li, X.-A. Scavenger Receptor BI Protects against Septic Death through Its Role in Modulating Inflammatory Response. J. Biol. Chem. 2009, 284, 19826–19834. [Google Scholar] [CrossRef]
- Steele, A.M.; Starr, M.E.; Saito, H. Late Therapeutic Intervention with Antibiotics and Fluid Resuscitation Allows for a Prolonged Disease Course with High Survival in a Severe Murine Model of Sepsis. Shock 2017, 47, 726–734. [Google Scholar] [CrossRef]
- Cheng, N.; Liang, Y.; Du, X.; Ye, R.D. Serum amyloid A promotes LPS clearance and suppresses LPS -induced inflammation and tissue injury. EMBO Rep. 2018, 19, e45517. [Google Scholar] [CrossRef]
- Zimetti, F.; De Vuono, S.; Gomaraschi, M.; Adorni, M.P.; Favari, E.; Ronda, N.; Ricci, M.A.; Veglia, F.; Calabresi, L.; Lupattelli, G. Plasma cholesterol homeostasis, HDL remodeling and function during the acute phase reaction. J. Lipid Res. 2017, 58, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Diallo, D.; Delbosc, S.; Genève, C.; Zappella, N.; Yong-Sang, J.; Patche, J.; Harrois, A.; Hamada, S.; Denamur, E.; et al. High-density lipoprotein (HDL) particle size and concentration changes in septic shock patients. Ann. Intensiv. Care 2019, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- de Beer, M.C.; Webb, N.R.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; Ji, A.; van der Westhuyzen, D.R.; de Beer, F.C. Impact of serum amyloid A on high density lipoprotein composition and levels. J. Lipid Res. 2010, 51, 3117–3125. [Google Scholar] [CrossRef]
- Hosoai, H.; Webb, N.R.; Glick, J.M.; Tietge, U.J.; Purdom, M.S.; de Beer, F.C.; Rader, D.J. Expression of serum amyloid A protein in the absence of the acute phase response does not reduce HDL cholesterol or apoA-I levels in human apoA-I transgenic mice. J. Lipid Res. 1999, 40, 648–653. [Google Scholar] [CrossRef]
- Preciado-Patt, L.; Levartowsky, D.; Prass, M.; Hershkoviz, R.; Lider, O.; Fridkin, M. Inhibition of cell adhesion to glycoproteins of the extracellular matrix by peptides corresponding to serum amyloid A. Toward understanding the physiological role of an enigmatic protein. JBIC J. Biol. Inorg. Chem. 1994, 223, 35–42. [Google Scholar] [CrossRef]
- Urieli-Shoval, S.; Shubinsky, G.; Linke, R.P.; Fridkin, M.; Tabi, I.; Matzner, Y. Adhesion of human platelets to serum amyloid A. Blood 2002, 99, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- McEneny, J.; Daniels, J.-A.; McGowan, A.; Gunness, A.; Moore, K.; Stevenson, M.; Young, I.S.; Gibney, J. A Cross-Sectional Study Demonstrating Increased Serum Amyloid A Related Inflammation in High-Density Lipoproteins from Subjects with Type 1 Diabetes Mellitus and How This Association Was Augmented by Poor Glycaemic Control. J. Diabetes Res. 2015, 2015, 351601. [Google Scholar] [CrossRef]
- Shridas, P.; Patrick, A.C.; Tannock, L.R. Role of Serum Amyloid A in Abdominal Aortic Aneurysm and Related Cardiovascular Diseases. Biomolecules 2021, 11, 1883. [Google Scholar] [CrossRef]
- Zhou, J.; Sheng, J.; Fan, Y.; Zhu, X.; Tao, Q.; He, Y.; Wang, S. Association between serum amyloid A levels and cancers: A systematic review and meta-analysis. Postgrad. Med. J. 2018, 94, 499–507. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, G.; Vong, C.T.; Ye, R.D. Serum amyloid A3 confers protection against acute lung injury in Pseudomonas aeruginosa-infected mice. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 318, L314–L322. [Google Scholar] [CrossRef]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Arnon, S.; Litmanovitz, I.; Regev, R.H.; Bauer, S.; Shainkin-Kestenbaum, R.; Dolfin, T. Serum amyloid A: An early and accurate marker of neonatal early-onset sepsis. J. Perinatol. 2007, 27, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Lee, S.Y.; Lee, H.; Cho, K.-H.; Yun, J.; Bae, Y.-S. Serum Amyloid A Induces CCL2 Production via Formyl Peptide Receptor-Like 1-Mediated Signaling in Human Monocytes. J. Immunol. 2008, 181, 4332–4339. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Fellowes, R.; Coade, S.; Woo, P. Human Serum Amyloid A has Cytokine-Like Properties. Scand. J. Immunol. 1998, 48, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; Johnston, J.A.; Wang, J.M.; McVicar, D.; Xu, L.L.; Oppenheim, J.J.; Kelvin, D.J. Serum amyloid A induces calcium mobilization and chemotaxis of human monocytes by activating a pertussis toxin-sensitive signaling pathway. J. Immunol. 1995, 155, 4004–4010. [Google Scholar] [CrossRef]
- Baranova, I.N.; Vishnyakova, T.G.; Bocharov, A.V.; Kurlander, R.; Chen, Z.; Kimelman, M.L.; Remaley, A.T.; Csako, G.; Thomas, F.; Eggerman, T.L.; et al. Serum Amyloid A Binding to CLA-1 (CD36 and LIMPII Analogous-1) Mediates Serum Amyloid A Protein-induced Activation of ERK1/2 and p38 Mitogen-activated Protein Kinases. J. Biol. Chem. 2005, 280, 8031–8040. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Hsu, K.; Yamen, E.; Yan, W.; Fock, J.; Witting, P.K.; Geczy, C.L.; Ben Freedman, S. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis 2009, 207, 374–383. [Google Scholar] [CrossRef]
- Badolato, R.; Wang, J.M.; Murphy, W.J.; Lloyd, A.R.; Michiel, D.F.; Bausserman, L.L.; Kelvin, D.J.; Oppenheim, J.J.; Fava, R.A.; Olsen, N.J.; et al. Serum amyloid A is a chemoattractant: Induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J. Exp. Med. 1994, 180, 203–209. [Google Scholar] [CrossRef]
- Franco, A.G.; Sandri, S.; Campa, A. High-density lipoprotein prevents SAA-induced production of TNF-α in THP-1 monocytic cells and peripheral blood mononuclear cells. Memórias Inst. Oswaldo Cruz 2011, 106, 986–992. [Google Scholar] [CrossRef]
- Witting, P.K.; Song, C.; Hsu, K.; Hua, S.; Parry, S.N.; Aran, R.; Geczy, C.; Freedman, S.B. The acute-phase protein serum amyloid A induces endothelial dysfunction that is inhibited by high-density lipoprotein. Free Radic. Biol. Med. 2011, 51, 1390–1398. [Google Scholar] [CrossRef]
- Simons, J.P.; Al-Shawi, R.; Ellmerich, S.; Speck, I.; Aslam, S.; Hutchinson, W.L.; Mangione, P.P.; Disterer, P.; Gilbertson, J.A.; Hunt, T.; et al. Pathogenetic mechanisms of amyloid A amyloidosis. Proc. Natl. Acad. Sci. USA 2013, 110, 16115–16120. [Google Scholar] [CrossRef] [PubMed]
- Bannenberg, G.L.; Chiang, N.; Ariel, A.; Arita, M.; Tjonahen, E.; Gotlinger, K.H.; Hong, S.; Serhan, C.N. Molecular Circuits of Resolution: Formation and Actions of Resolvins and Protectins. J. Immunol. 2005, 174, 4345–4355. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-H. Human Serum Amyloid a Impaired Structural Stability of High-Density Lipoproteins (HDL) and Apolipoprotein (Apo) A-I and Exacerbated Glycation Susceptibility of ApoA-I and HDL. Molecules 2022, 27, 4255. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Ather, J.L.; Dienz, O.; Boyson, J.E.; Anathy, V.; Amiel, E.; Poynter, M.E. Serum Amyloid A3 is required for normal lung development and survival following influenza infection. Sci. Rep. 2018, 8, 16571. [Google Scholar] [CrossRef] [PubMed]
- Renckens, R.; Roelofs, J.J.T.H.; Knapp, S.; de Vos, A.F.; Florquin, S.; van der Poll, T. The Acute-Phase Response and Serum Amyloid A Inhibit the Inflammatory Response to Acinetobacter baumannii Pneumonia. J. Infect. Dis. 2006, 193, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.F.; Cao, K.; Jiang, J.P.; Guan, W.X.; Du, J.F. Neutrophil dysregulation during sepsis: An overview and update. J. Cell. Mol. Med. 2017, 21, 1687–1697. [Google Scholar] [CrossRef]
- Spite, M.; Serhan, C.N. Novel Lipid Mediators Promote Resolution of Acute Inflammation. Circ. Res. 2010, 107, 1170–1184. [Google Scholar] [CrossRef]
- Jarczak, D.; Kluge, S.; Nierhaus, A. Sepsis—Pathophysiology and Therapeutic Concepts. Front. Med. 2021, 8, 628302. [Google Scholar] [CrossRef]
- Preciado-Patt, L.; Pras, M.; Fridkin, M. Binding of human serum amyloid A (hSAA) and its high-density Iipoprotein3 complex (hSAA-HDL3) to human neutrophils. Possible implication to the function of a protein of an unknown physiological role. Int. J. Pept. Protein Res. 1996, 48, 503–513. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, J.; Wu, L.; Fan, Y.; Sun, L.; Qian, F.; Chen, D.; Ye, R.D. Elevated Expression of Serum Amyloid A 3 Protects Colon Epithelium Against Acute Injury Through TLR2-Dependent Induction of Neutrophil IL-22 Expression in a Mouse Model of Colitis. Front. Immunol. 2018, 9, 1503. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Hall, J.A.; Kroehling, L.; Wu, L.; Najar, T.; Nguyen, H.H.; Lin, W.-Y.; Yeung, S.T.; Silva, H.M.; Li, D.; et al. Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell 2019, 180, 79–91.e16. [Google Scholar] [CrossRef] [PubMed]
- Starr, M.E.; Steele, A.M.; Saito, M.; Hacker, B.J.; Evers, B.M.; Saito, H. A New Cecal Slurry Preparation Protocol with Improved Long-Term Reproducibility for Animal Models of Sepsis. PLoS ONE 2014, 9, e115705. [Google Scholar] [CrossRef] [PubMed]
- Speer, E.M.; Diago-Navarro, E.; Ozog, L.S.; Raheel, M.; Levy, O.; Fries, B.C. A Neonatal Murine Escherichia coli Sepsis Model Demonstrates That Adjunctive Pentoxifylline Enhances the Ratio of Anti- vs. Pro-inflammatory Cytokines in Blood and Organ Tissues. Front. Immunol. 2020, 11, 577878. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M. An Official American Thoracic Society Workshop Report: Features and Measurements of Experimental Acute Lung Injury in Animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef]
- Li, S.-C.; Tsai, K.-W.; Huang, L.-H.; Weng, K.-P.; Chien, K.-J.; Lin, Y.; Tu, C.-Y.; Lin, P.-H. Serum proteins may facilitate the identification of Kawasaki disease and promote in vitro neutrophil infiltration. Sci. Rep. 2020, 10, 15645. [Google Scholar] [CrossRef]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A–mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef]
- Valenca, S.S.; Dong, B.E.; Gordon, E.M.; Sun, R.C.; Waters, C.M. ASK1 Regulates Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 484–496. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, A.; Trumbauer, A.C.; Noffsinger, V.P.; Meredith, L.W.; Dong, B.; Wang, Q.; Guo, L.; Li, X.; De Beer, F.C.; Webb, N.R.; et al. Deficiency of Acute-Phase Serum Amyloid A Exacerbates Sepsis-Induced Mortality and Lung Injury in Mice. Int. J. Mol. Sci. 2023, 24, 17501. https://doi.org/10.3390/ijms242417501
Ji A, Trumbauer AC, Noffsinger VP, Meredith LW, Dong B, Wang Q, Guo L, Li X, De Beer FC, Webb NR, et al. Deficiency of Acute-Phase Serum Amyloid A Exacerbates Sepsis-Induced Mortality and Lung Injury in Mice. International Journal of Molecular Sciences. 2023; 24(24):17501. https://doi.org/10.3390/ijms242417501
Chicago/Turabian StyleJi, Ailing, Andrea C. Trumbauer, Victoria P. Noffsinger, Luke W. Meredith, Brittany Dong, Qian Wang, Ling Guo, Xiangan Li, Frederick C. De Beer, Nancy R. Webb, and et al. 2023. "Deficiency of Acute-Phase Serum Amyloid A Exacerbates Sepsis-Induced Mortality and Lung Injury in Mice" International Journal of Molecular Sciences 24, no. 24: 17501. https://doi.org/10.3390/ijms242417501