
Compromised Mitotic Fidelity in Human Pluripotent Stem Cells


1
Católica Biomedical Research Centre, Católica Medical School, Universidade Católica Portuguesa, 1649-023 Lisbon, Portugal
2
Instituto Gulbenkian de Ciência, 2780-156 Oeiras, Portugal
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(15), 11933; https://doi.org/10.3390/ijms241511933
Submission received: 12 June 2023
/
Revised: 19 July 2023
/
Accepted: 21 July 2023
/
Published: 25 July 2023
(This article belongs to the Special Issue Scientific Discoveries Supporting Theories in Science: From Thinking to Practice 2.0)
Abstract
:Human pluripotent stem cells (PSCs), which include both embryonic and induced pluripotent stem cells, are widely used in fundamental and applied biomedical research. They have been instrumental for better understanding development and cell differentiation processes, disease origin and progression and can aid in the discovery of new drugs. PSCs also hold great potential in regenerative medicine to treat or diminish the effects of certain debilitating diseases, such as degenerative disorders. However, some concerns have recently been raised over their safety for use in regenerative medicine. One of the major concerns is the fact that PSCs are prone to errors in passing the correct number of chromosomes to daughter cells, resulting in aneuploid cells. Aneuploidy, characterised by an imbalance in chromosome number, elicits the upregulation of different stress pathways that are deleterious to cell homeostasis, impair proper embryo development and potentiate cancer development. In this review, we will summarize known molecular mechanisms recently revealed to impair mitotic fidelity in human PSCs and the consequences of the decreased mitotic fidelity of these cells. We will finish with speculative views on how the physiological characteristics of PSCs can affect the mitotic machinery and how their suboptimal mitotic fidelity may be circumvented.
1. Human Pluripotent Stem Cells Present Frequent Karyotypic Abnormalities
Pluripotent stem cells (PSCs) can give rise to all cell types of the organism [1]. Their potential for self-renewal and differentiation makes them very appealing for use in basic research as well as drug screening and discovery, toxicity tests and ultimately in cellular therapy [2]. Human embryonic stem cells (ESCs), a type of PSC, were first derived from human embryos in 1998 [3]. These cells have the capacity for self-renewal, can be cultured in vitro indefinitely and have the ability to differentiate into cells of the three germ layers: endoderm, ectoderm and mesoderm [3]. In 2006, the revolutionary work by Takahashi and Yamanaka demonstrated that it was possible to reprogram adult and embryonic fibroblasts to obtain “embryonic stem cell-like” cells, which they termed induced pluripotent stem cells (iPSCs) [1], another type of PSC. By induced expression of only four transcription factors (Oct4, Sox2, Klf4 and c-Myc), mouse fibroblasts were reprogrammed into iPSCs. Only a year later, Yamanaka and others showed it was also possible to use the same minimal cocktail of proteins to obtain iPSCs from several human cell types [4,5,6]. Already numerous clinical trials are ongoing using either ESCs or iPSCs, aiming to cure and/or ameliorate symptoms of a plethora of human diseases, including many degenerative disorders and ageing-related complications, such as Parkinson’s disease, macular degeneration and heart failure [7]. It is therefore essential that these cells are safe and robust while maintaining a stable karyotype (the species’ complete set of chromosomes), since karyotypic abnormalities hinder the safe use of PSCs in regenerative medicine and in translational and basic research [7,8]. Aneuploidy—an abnormal chromosome number—can impair proper embryo development, potentiate cancer development and trigger different types of stresses in cells [9]. Yet, PSCs are sometimes aneuploid, frequently having partial or whole chromosome gains or losses [10,11,12,13] and/or recurrent copy number variations [14,15,16].
Aneuploidy occurs in a wide range of biological settings. For example, mammalian female meiosis is highly erroneous, resulting in aneuploid embryos [17]. Additionally, early embryos accumulate aneuploid cells due to mitotic errors [18]. Aneuploidy is also a key aspect of tumour biology, found in around 90% of solid tumours and 75% of hematopoietic cancers [19,20,21]. Likewise, culture conditions, such as medium acidification, may affect the genomic stability of PSCs [22]: up to 30% of PSC lines used in laboratories all over the world present karyotypic abnormalities, including whole chromosome aneuploidies [10,11]. Yet, we are only now starting to characterize and understand the molecular mechanisms underlying this chromosomal imbalance, its link to pluripotency and the consequences for PSC maintenance. Recently, several papers have begun to clarify the specific mitotic pathways that are malfunctioning in human PSCs. In this review we will focus on the known molecular pathways and physiological properties that can contribute to whole chromosome gains or losses in human PSCs. We will start by summarising recent findings that have identified specific mitotic molecular pathways that are malfunctioning or suboptimal in PSCs. Next, we will discuss how certain properties of these cells may underlie their reduced mitotic fidelity and how particular differences in the cellular responses to abnormal chromosome numbers may contribute to a differential survival of aneuploid PSCs. We will finalize with speculative views on how basic knowledge on the properties that render PSCs more prone to mitotic defects can be used to improve their mitotic fidelity and, thus, their safe applicability.
2. Molecular Mechanisms Underlying the Compromised Mitotic Fidelity in Pluripotent Stem Cells
PSCs have been described as having karyotypic abnormalities for more than a decade [10,11,12,13]; however, the molecular mechanisms that account for the problems in mitosis have only recently been explored (see Figure 1 and Table 1).
Proper chromosome segregation is central to genome stability and the maintenance of the correct karyotype relies on the mitotic machinery that segregates chromosomes with high fidelity [32,33,34,35,36,37]. PSCs have an atypical cell cycle structure with truncated gap phases and proliferate at an unusually rapid rate [38]. Additionally, the chromatin of PSCs is characterised by being open, decondensed and more dynamic than that of differentiated cells [39]. It is possible that the intrinsic characteristics of the cell cycle and chromatin state impose differences in mitotic machinery function and regulation in PSCs, resulting in compromised mitotic fidelity.
2.1. DNA Replication
Several studies have shown that PSCs undergo much higher replication stress during S phase [23,27,28,29,30] than differentiated cells, although the duration of S phase is comparable in both cell types. Replication stress, such as incomplete, under-replicated or unresolved chromosomes from S phase, if not corrected, can persist into mitosis and hinder proper chromosome segregation [40]. Replication stress in PSCs arises mainly due to slower rates of DNA synthesis, activation of latent origins of replication and stalling of replication forks, which results in abortive and abnormal mitosis [23,27]. The high levels of replication stress that these cells undergo result in DNA damage and double strand breaks [27]. It is likely that these problems during replication are a consequence of the truncated cell cycle, characterised by a very fast G1 [38], which would preclude cells from properly preparing for the subsequent replication of DNA in the S phase. In accordance with this, the addition of exogenous nucleosides to the culture medium of PSCs results in a partial rescue of the replication dynamics, reducing the levels of DNA damage [27,41], decreasing the incidence of abnormal mitosis and increasing survival of these cells [27].
The response to replication stress of PSCs is distinct from that of differentiated cells and may result in the increased aneuploidy of these cells. In response to this type of stress, PSCs tend to fail to activate DNA repair pathways and favour instead the activation of apoptosis [42], which may help control the deleterious effects of replication stress. One exception to this is when telomere shortening is induced in these cells, which, combined with p53 deletion, leads to an accumulation of mitotic errors [43]. Although apoptosis is PSCs’ preferred response to DNA damage [42], they can proceed to mitosis after replication stress [23,30]. This can result in either abortive or erroneous mitosis, which can then result in daughter cells that are aneuploid. Aneuploid PSCs with an extra copy of chromosome 12, chromosome 17 or a combination of both, suffer even higher levels of replication stress and make more mistakes in mitosis, when compared to euploid PSCs [23], which could lead to further chromosomal instability.
Replication stress in PSCs has been well established [23,27,28,29,30] and has recently been shown to affect downstream mitosis [23,27,30]. However, recent papers have demonstrated that there are mitotic-specific processes that are also affected and may work in a sub-optimal way in these cells. Below we will focus on the recent advances in the mitotic processes of pluripotent stem cells and how they differ from those of differentiated cells.
2.2. Chromosome Condensation
In interphase, PSC chromatin is more open, mobile and accessible [39,44] than that of differentiated cells and extensive remodelling of the chromatin during iPSC reprogramming is necessary to induce pluripotency [45]. It is conceivable that these differences in mitotic chromatin properties affect how the mitotic machinery functions and is regulated in these cells, which can impact efficient chromosome segregation. During mitosis, euploid PSCs have problems in chromosome condensation, and this is exacerbated in PSCs with extra copies of chromosome 12, 17 or both [23]. These mitotic defects, as well as the increased replication stress described in the previous section of these aneuploid PSCs, seem to be due to the downregulation of a transcription factor involved in the regulation of several actin–cytoskeleton genes—the serum response factor (SRF) [23]. Overexpression of this factor in aneuploid cells resulted in a decrease in replication stress and a partial rescue of the errors and condensation defects during mitosis. Consistently, silencing SRF in euploid pluripotent stem cells led to an increase in replication stress and condensation defects [23]. However, it is unclear how known key players important for mitotic chromosome condensation in differentiated cells are regulated in either euploid or aneuploid PSCs. These include specific histone post-translational modifications [46] and key proteins, such as condensins and DNA topoisomerase II alpha [36]. Equally unclear is if differences in the regulation of these structural factors affect chromosome condensation and, therefore, the genomic integrity of pluripotent stem cells.
2.3. The Centromere
At the heart of faithful genome distribution is a specialised chromosomal locus—the centromere—that is present exactly once in each chromosome to ensure the binding and/or regulation of various components of the mitotic machinery. The centromere forms a scaffold for the assembly of the kinetochore, a multi-subunit protein complex that acts as a platform for microtubule attachment during mitosis to drive chromosome segregation [47]. The histone H3 variant CENP-A was identified as a crucial component in the epigenetic specification of the centromere [48]. Our own recent findings show that centromere strength is compromised in PSCs, which present reduced levels of this histone variant [24]. While PSCs maintain abundant cytoplasmatic pools of CENP-A, CENP-C and CENP-T, these essential centromere components are strongly reduced at stem cell centromeres [24]. And CENP-B, another important centromeric protein, is expressed at low levels and almost undetected at the centromeres of these cells.
The weak centromere affects the recruitment of kinetochore proteins in mitosis, at least partially, resulting in reduced levels of key proteins in kinetochore–microtubule attachment and chromosome motility [24]. Assembly of CENP-A chromatin occurs in the G1 phase of the stem cell cycle [24], as is the case in human differentiated and cancer cell lines [49,50]. The reduction in centromeric chromatin size is induced early during iPSC reprogramming [24], coincident with the time of increased proliferation rates. Additionally, chromosomes that lack CENP-B at the centromere, which is not essential for cell viability, have been shown to mis-segregate more frequently and have slightly lower levels of CENP-A and CENP-C [51]. Low CENP-A, -B and -C levels may be sufficient to impact on chromosome segregation efficiency in PSCs. Accordingly, lagging chromosomes are the major mitotic defect in these cells [25,26]. Other mitotic errors, such as anaphase bridges, micronuclei and multipolar divisions, were also detected [25,26] but did not differ greatly in frequency from those detected in differentiated cells [26]. Hence, sub-optimal CENP-A levels may be one of the most critical abnormalities in PSCs that underlies their mitotic defects.
2.4. Kinetochore–Microtubule Dynamics
Improper kinetochore–microtubule attachments are corrected by a dedicated error correction machinery primarily mediated by the Aurora B kinase [52]. This pathway ensures dynamic detachment and reattachment of microtubules to allow that each sister chromatid is attached to microtubules coming from one spindle pole and that the other sister is attached only to the opposite pole [53].
If error correction pathways are not working properly, cells can accumulate erroneous kinetochore–microtubule attachments at anaphase onset [54]. It has been shown that Aurora B is maintained at normal levels at the inner centromere of PSCs [24], but it is unknown if its activity is altered in these cells, which may interfere with proper error correction. Indeed, merotelic attachments (when the kinetochore of one sister chromatid is attached to one pole and the other to both poles) were observed in more than 70% of all lagging chromosomes in PSCs [26]. This high frequency is possibly the result of hyperstabilisation of microtubule attachments [26,31] concomitantly with impaired error correction. Accordingly, artificial de-stabilization of kinetochore–microtubule attachments using small molecules restores mitotic fidelity in PSCs [26]. Moreover, impairing the correct localisation of Aurora B to the inner centromere exacerbates errors in mitosis observed in PSCs [55]. These results point to altered error-correction efficiency in PSCs, which impacts on correct chromosome segregation.
2.5. Spindle Assembly Checkpoint
The outer kinetochore, which assembles at the centromere in mitosis, is also the site for the accumulation of proteins from the spindle assembly checkpoint (SAC). This checkpoint senses unattached or improperly attached kinetochores, preventing mitotic exit until all chromosomes are properly attached to the spindle [56]. PSCs can activate the SAC in response to spindle poisons [57], delaying anaphase onset. However, it remains unknown if SAC activation is as robust as that of differentiated cells. Interestingly, prolonging mitosis using APC/C inhibitors can decrease the amount of mitotic defects in PSCs [26], suggesting that PSCs may not halt mitosis until all attachments are properly established.
3. Physiological Properties of PSCs Which Can Affect Mitotic Fidelity
Pluripotent stem cells possess distinct physiological characteristics that set them apart from differentiated cells, giving them unique properties that can influence the accuracy of cell division. These include differences in cell cycle length and checkpoint function, chromatin marks and organisation, mitotic machinery regulation and function and potential divergences in their tolerance and response to aneuploidy. In this part of the review, we will focus on these differences in physiology, consider how these physiological traits of PSCs can affect mitosis (Figure 2) and discuss potential strategies to overcome their suboptimal mitotic fidelity.
3.1. Cell Cycle
PSCs have atypical or compromised cell cycle checkpoints [58]. The cell cycle is comprised of discrete phases—mitosis, gap phase 1 (G1 phase), synthesis phase (S phase) and gap phase 2 (G2 phase). Each of these phases needs to be completed before cells enter the next phase, and to ensure this, checkpoint signalling pathways are activated if problems are detected. These checkpoints give cells the opportunity to correct any mistakes before progressing in the cell cycle [59]. For example, cells with entangled sister chromatids delay entry into mitosis to allow DNA topoisomerase II alpha to correct them. However, mouse ESCs complete cell division even in the presence of catenated sister chromatids, which results in aneuploid daughter cells [60]. Moreover, checkpoint activation can sometimes be uncoupled from the canonical response seen in differentiated cells, where, for instance, prolonged SAC activation leads to apoptosis during or after mitosis [61]. In contrast, SAC activation in PSCs is uncoupled from apoptosis [57], which may result in mitotic slippage in cells with improper attachments.
Pluripotent stem cells proliferate very fast, which may reflect the need for fast cell division in the embryo. They do this by shortening the G1 phase to approximately 2–3 h [38] (Figure 2). The short G1 phase is intrinsically linked to cell fate decisions, and G1 length increases upon differentiation [62,63]. Reciprocally, commitment of pluripotent stem cells to differentiation occurs during the G1 phase, and cells in the S and G2 phases are more impervious to exit the pluripotent state [64,65]. It is also during G1 that cells prepare for DNA replication by synthesising RNA, ATP nucleotides, amino acids and proteins and when components of centromeres and centrosomes—key structures for proper chromosome segregation—are replenished. CENP-A loading onto the centromeric chromatin occurs slowly during the long G1 of differentiated cells, for approximately 10 h [66]. It is therefore tempting to speculate that PSCs are not able to fully assemble CENP-A [24] in this short time window. The truncated pluripotent stem cell cycle can therefore lead to errors in mitosis by affecting the proper loading of CENP-A at the centromere or by not producing enough proteins and nucleotides for the next round of DNA replication.
3.2. Histone Post-Translational Modifications
Inducing pluripotency in terminally differentiated cells requires reprogramming, i.e., a genome-wide remodelling of the epigenome, such as histone modifications [67] and DNA methylation [68,69]. This remodelling allows for two cells with the same genome (iPSCs and the differentiated cells they were reprogrammed from) to have completely different cell identities and confers iPSCs with the characteristic plastic chromatin associated with the pluripotent state (Figure 2). This results in the repression of somatic genes and activation of self-renewal and pluripotency associated genes [70]. One of the earliest events in reprogramming is the rapid genome-wide re-distribution of H3K4me2 observed both in mouse and human iPSC reprogramming [71,72]. H3K4me2, together with H3K9me3, are considered barriers to reprogramming, as failure to remove or re-distribute these marks results in the inability of cells to reach pluripotency [67]. Moreover, PSCs have much lower levels of H4K20me1/2 when compared to differentiated cells [73]. Because chromatin structure and histone modifications also influence the recruitment of key mitotic players to chromosomes [74], it is possible that the lower levels and/or distinct distribution of specific histone modifications, such as H3K9me3, H3K4me2 and H4K20me1/2 in PSCs, influences the correct recruitment of pivotal mitotic players to mitotic chromosomes. Moreover, impaired recruitment of centromeric [75,76] as well as structural maintenance of chromosomes [77,78], kinases [79] and other proteins [78], to chromosomes, lead to defects in recruiting the downstream effectors of mitosis [80]. This characteristic chromatin of PSCs may be disrupting the fine-tuning of mitosis in these cells. Since this characteristically fluid chromatin is inherently linked to the pluripotent state, it is plausible that a compromised mitotic fidelity is an intrinsic characteristic of PSCs.
3.3. Mitotic Machinery of PSCs
Studies of the mitotic machinery in PSCs have, until recently, been mainly focused on the maintenance of stem cell identity [81]. In particular, cohesin has mainly been studied in the context of higher order chromatin organisation and the maintenance of stem cell identity [82], but it remains unknown how it functions in mitosis in these cells. Additionally, although condensation defects have been proposed as a cause for genomic instability in human PSCs [23], the underlying role of condensins, DNA topoisomerases as well as histone post-translational modifications important for chromosome condensation remain unknown. In addition to the structural components, critical regulators of chromosome attachments/checkpoint signalling have also been hypothesised to be altered in PSCs. The chromosomal passenger complex (CPC) component survivin was shown to be upregulated in ESCs [83]. Moreover, chemical inhibition of the critical CPC kinase Aurora B led to an increase in aberrant mitosis and apoptosis in ESCs [25]. How CPC is regulated in PSCs and how it affects mitotic fidelity is still unknown. The impact of the different regulation of all these proteins in mitosis and in the correct chromosome segregation of PSCs has not been investigated. Understanding which are the major differences in the composition, recruitment and regulation of the mitotic machinery of PSCs represents a clear knowledge gap. It is important to assess why these pathways are at least partially compromised and what are the mechanisms that become rate limiting during the extensive chromatin remodelling or upon the acquisition of pluripotency.
4. Consequences of Decreased Mitotic Fidelity in Pluripotent Stem Cells
The effects of aneuploidy have been studied in yeast, Drosophila and mammalian cancer and differentiated cells [9]. Aneuploidy can arise due to errors in mitosis, which can then impact the physiology of cells that inherit an abnormal chromosome number. Having an extra copy of a chromosome can lead to an imbalance in gene expression. Dosage compensation effects can overcome this problem, which is what happens in triple X syndrome (47, XXX). This is achieved by silencing the supernumerary X chromosomes by the dosage compensation mechanism of X chromosome inactivation [84]. Although in some species some autosomal dosage compensation similarly occurs [85], this has not been shown to naturally happen in mammalian systems so far. It has recently been shown that cancer aneuploid cells do not scale their protein expression to the DNA content [86,87,88], suggesting that some dosage compensation does occur (the mechanisms of which remain unknown). Even if DNA content and protein expression do not linearly scale, having extra copies of chromosomes results in increased expression of the genes of the trisomic chromosome, which results in extra protein production [89]. This can lead to several problems, since chaperones may become rate-limiting for correct protein folding and degradation, causing proteotoxic stress. Moreover, proper metabolism and replication need precise control of the exact stoichiometry of specific proteins/enzymes [85]. When cells produce extra amounts of proteins, the fine balance in these tightly regulated processes can be disrupted, resulting in metabolic and replication stress. Furthermore, having extra copies of chromosomes can in itself affect mitosis, resulting in chromosomal instability and in further mitotic stress [85,90]. Also, specific aneuploidies differentially impair the post-implantation developmental potential of human embryos, resulting in diverse developmental fates [91].
Karyotype analysis of several PSCs revealed that recurrent aneuploidies are commonly observed and proposed to directly impact on their biology (Figure 3). PSCs with chromosome 12 trisomy present higher tumorigenicity [15,92] and proliferate better than euploid PSCs in culture by increasing their replication potential [93]. Human PSCs trisomic for chromosome 12, 17 or both chromosomes 12 and 17, present higher incidence of mitotic errors and suffer increased replication stress than euploid PSCs [23]. Mouse stem cells trisomic for specific chromosomes were shown to have decreased differentiation potential and increased neoplastic properties [94]. Very recently, it was also shown that human PSCs trisomic for all chromosomes (triploid cells) have a decreased differentiation potential [95]. Some specific aneuploidies, such as those for chromosomes 1, 8, 12, 17 and X, are recurrent in PSCs [11]. The fact that there are recurrent aneuploidies also demonstrates that these cells are tolerant to specific aneuploidies, which may reflect that these chromosome gains confer advantages for cells that can then overtake euploid cells in culture. In differentiated cells, the stresses imposed by the aneuploid state ultimately lead to cell cycle arrest or cell death (Figure 3). In contrast, the high frequency of karyotypic abnormalities found in PSCs, including whole chromosome aneuploidies [10,11], implies continued proliferation despite the aneuploid state. This conundrum suggests that checkpoints are less functioning in PSCs. The intrinsic biological characteristics of pluripotency, such as increased proteasome activity, higher replicative stress and glycolytic-based metabolism, may affect the way they maintain cell homeostasis and tolerate aneuploidy. Studies in Drosophila have shown that aneuploid neuronal stem cells have delayed p53 activation, which confers tolerance to aneuploidy [96] and may impact on karyotypic evolution. It is possible that this also plays a role in human PSC tolerance to aneuploidy, as these cells frequently acquire and expand p53-dominant negative mutations [97]. However, the physiological pathways that render human PSCs tolerant to aneuploidy remain largely unknown. Uncovering this unique response to aneuploidy is crucial not only to understand critical aspects of stem cell physiology but also to allow for the elimination of these cells from PSCs cultures, similarly to what has recently been accomplished for cancer aneuploid cells [98].
5. Can We Increase the Mitotic Fidelity of PSCs?
Here we have summarized the recent evidence on the sub-optimal mitotic pathways of PSCs, which render these cells more prone to segregation defects and account for the elevated rates of aneuploidy. Characterising the mitotic machinery of PSCs opens the possibility of rescuing mitotic fidelity in these cells. Is it possible to modulate these pathways to obtain safer and more robust PSCs and accomplish their full potential?
It is possible that pluripotency cell identity is intrinsically linked with changes in mitotic machinery composition. For example, the intricate connection between cell cycle stages and cell fate [64] may impose an additional challenge on the attempts to overcome mitotic infidelity. It has been previously proposed that the acquisition of a unique pluripotent stem cell cycle organization during iPSC reprogramming is functionally linked to the acquisition of pluripotency. Consequently, modulating these mitotic pathways can prime cells to differentiate and consequently make them no longer pluripotent. Therefore, it also remains to be answered if restoration of mitotic fidelity is linked to loss of cell identity. This raises the hypothesis that aneuploidy is an intrinsic feature of pluripotency, implying that there is an intricate trade-off regarding sub-optimal mitosis and pluripotency.
On the other hand, attempts have been successful at decreasing mitotic errors in PSCs (e.g., prolonging mitotic duration or de-stabilization of kinetochore–microtubule attachments) [26]. Unfortunately, these revealed not to be a long term solution [26]. In contrast to what has been shown using similar strategies in fibroblasts [99], PSCs adapt to the drugs and stop responding to them, thus continuing to display errors in chromosome segregation [26]. This fast adaptation is similar to what was observed in cancer cells [100]. Nevertheless, it is possible that other strategies, manipulating other pathways and/or using other drugs may reveal more successful in the future.
6. Concluding Remarks
ESCs are derived from the inner cell mass of early embryos and can give rise to almost all the cell types of the organism. Induced pluripotent stem cell technology is a breakthrough methodology that relies on the ectopic expression of only four transcription factors to generate iPSCs [1]. PSCs, which include both ESCs and iPSCs, share many biological features and have an immeasurable potential to be used in research and in regenerative medicine. Yet, PSCs often display abnormal chromosome numbers (aneuploidy) which hinders their safe use. Mitosis is a carefully orchestrated and highly regulated process, which results in the equal segregation of the duplicated genome to the daughter cells. Understanding the mechanisms that support proper chromosome segregation in PSCs is an important step toward unravelling the underlying causes of compromised mitotic fidelity in these cells. Although recent studies are trying to delve into the mitotic machinery and mitotic fidelity of PSCs, we have still only unveiled the tip of the iceberg and several questions remain to be answered. By characterising the mechanisms regulating mitotic pathways in PSCs and/or conferring them increased tolerance to aneuploidy, we can design strategies to increase mitotic fidelity by modulating the key players of mitosis or selectively eliminating aneuploid cells from our cultures. Only then can we use these cells to their full potential.
Author Contributions
Conceptualization, I.M., C.P. and R.A.O.; writing—original draft preparation, I.M.; writing—review and editing, C.P. and R.A.O.; visualization, I.M. and C.P.; funding acquisition, I.M. and R.A.O. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funding from Fundação para a Ciência e Tecnologia: project n° LISBOA-01-0145-FEDER-031502, co-funded by the Programa Operacional Regional de Lisboa (Lisboa 2020), through Portugal 2020 and the European Regional Development Fund (FEDER) and by the FCT/MCTES through national funds (PIDDAC) (to I.M.) and PhD fellowship 2020.04801.BD (to C.P.), as well as the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 101002391) (to R.A.O.).
Acknowledgments
We thank all members of the CHR lab and Paulo Navarro-Costa for helpful comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced Pluripotent Stem Cells: The New Patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In Vitro Reprogramming of Fibroblasts into a Pluripotent ES-Cell-like State. Nature 2007, 448, 318–324. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Andrews, P.W.; Ben-David, U.; Benvenisty, N.; Coffey, P.; Eggan, K.; Knowles, B.B.; Nagy, A.; Pera, M.; Reubinoff, B.; Rugg-Gunn, P.J.; et al. Assessing the Safety of Human Pluripotent Stem Cells and Their Derivatives for Clinical Applications. Stem Cell Rep. 2017, 9, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Santaguida, S.; Amon, A. Short- and Long-Term Effects of Chromosome Mis-Segregation and Aneuploidy. Nat. Rev. Mol. Cell Biol. 2015, 16, 473–485. [Google Scholar] [CrossRef] [Green Version]
- The International Stem Cell Initiative. Screening Ethnically Diverse Human Embryonic Stem Cells Identifies a Chromosome 20 Minimal Amplicon Conferring Growth Advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karyotypic Abnormalities in Human Induced Pluripotent Stem Cells and Embryonic Stem Cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.-C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and Classification of Chromosomal Aberrations in Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draper, J.S.; Smith, K.; Gokhale, P.; Moore, H.D.; Maltby, E.; Johnson, J.; Meisner, L.; Zwaka, T.P.; Thomson, J.A.; Andrews, P.W. Recurrent Gain of Chromosomes 17q and 12 in Cultured Human Embryonic Stem Cells. Nat. Biotechnol. 2004, 22, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Martins-Taylor, K.; Nisler, B.S.; Taapken, S.M.; Compton, T.; Crandall, L.; Montgomery, K.D.; Lalande, M.; Xu, R.-H. Recurrent Copy Number Variations in Human Induced Pluripotent Stem Cells. Nat. Biotechnol. 2011, 29, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.E.C.; Harrison, N.J.; Maltby, E.; Smith, K.; Moore, H.D.; Shaw, P.J.; Heath, P.R.; Holden, H.; Andrews, P.W. Adaptation to Culture of Human Embryonic Stem Cells and Oncogenesis in Vivo. Nat. Biotechnol. 2007, 25, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic Changes in the Copy Number of Pluripotency and Cell Proliferation Genes in Human ESCs and IPSCs during Reprogramming and Time in Culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Cavazza, T.; Schuh, M. Aneuploidy in Human Eggs: Contributions of the Meiotic Spindle. Biochem. Soc. Trans. 2021, 49, 107–118. [Google Scholar] [CrossRef]
- Chunduri, N.K.; Storchová, Z. The Diverse Consequences of Aneuploidy. Nat. Cell Biol. 2019, 21, 54–62. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Does Aneuploidy Cause Cancer? Curr. Opin. Cell Biol. 2006, 18, 658–667. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. The Aneuploidy Paradox in Cell Growth and Tumorigenesis. Cancer Cell 2008, 14, 431–433. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.J.; Cleveland, D.W. Losing Balance: The Origin and Impact of Aneuploidy in Cancer: ‘Exploring Aneuploidy: The Significance of Chromosomal Imbalance’ Review Series. EMBO Rep. 2012, 13, 501–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, K.; Zambelli, F.; Mertzanidou, A.; Smolders, I.; Geens, M.; Nguyen, H.T.; Barbé, L.; Sermon, K.; Spits, C. Higher-Density Culture in Human Embryonic Stem Cells Results in DNA Damage and Genome Instability. Stem Cell Rep. 2016, 6, 330–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and Consequences of Aneuploidy in Cancer. Nat. Rev. Genet. 2012, 13, 189–203. [Google Scholar] [CrossRef]
- Compton, D.A. Mechanisms of Aneuploidy. Curr. Opin. Cell Biol. 2011, 23, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.; Salmon, E.D. The Spindle-Assembly Checkpoint in Space and Time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Mirkovic, M.; Oliveira, R.A. Centromeric Cohesin: Molecular Glue and Much More. Prog. Mol. Subcell. Biol. 2017, 56, 485–513. [Google Scholar] [CrossRef]
- Piskadlo, E.; Oliveira, R.A. A Topology-Centric View on Mitotic Chromosome Architecture. IJMS 2017, 18, 2751. [Google Scholar] [CrossRef] [Green Version]
- Soto, M.; Raaijmakers, J.A.; Medema, R.H. Consequences of Genomic Diversification Induced by Segregation Errors. Trends Genet. 2019, 35, 279–291. [Google Scholar] [CrossRef]
- Becker, K.A.; Ghule, P.N.; Therrien, J.A.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Self-Renewal of Human Embryonic Stem Cells Is Supported by a Shortened G1 Cell Cycle Phase. J. Cell. Physiol. 2006, 209, 883–893. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kikyo, N. Epigenetic Regulation of Open Chromatin in Pluripotent Stem Cells. Transl. Res. 2015, 165, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Lamm, N.; Ben-David, U.; Golan-Lev, T.; Storchová, Z.; Benvenisty, N.; Kerem, B. Genomic Instability in Human Pluripotent Stem Cells Arises from Replicative Stress and Chromosome Condensation Defects. Cell Stem Cell 2016, 18, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, J.A.; Frith, T.J.R.; Laing, O.; Price, C.J.; Bower, O.J.; Stavish, D.; Gokhale, P.J.; Hewitt, Z.; El-Khamisy, S.F.; Barbaric, I.; et al. Nucleosides Rescue Replication-Mediated Genome Instability of Human Pluripotent Stem Cells. Stem Cell Rep. 2020, 14, 1009–1017. [Google Scholar] [CrossRef]
- Simara, P.; Tesarova, L.; Rehakova, D.; Matula, P.; Stejskal, S.; Hampl, A.; Koutna, I. DNA Double-Strand Breaks in Human Induced Pluripotent Stem Cell Reprogramming and Long-Term in Vitro Culturing. Stem Cell Res. Ther. 2017, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhaneni, H.; Lynch, P.J.; Chen, G.; Park, K.; Liu, Y.; Goehe, R.; Mallon, B.S.; Boehm, M.; Hursh, D.A. High Basal Levels of ΓH2AX in Human Induced Pluripotent Stem Cells Are Linked to Replication-Associated DNA Damage and Repair. Stem Cells 2018, 36, 1501–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kislova, A.V.; Zheglo, D.; Pozhitnova, V.O.; Sviridov, P.S.; Gadzhieva, E.P.; Voronina, E.S. Replication Stress Causes Delayed Mitotic Entry and Chromosome 12 Fragility at the ANKS1B Large Neuronal Gene in Human Induced Pluripotent Stem Cells. preprint 2023. [Google Scholar] [CrossRef]
- Mankouri, H.W.; Huttner, D.; Hickson, I.D. How Unfinished Business from S-Phase Affects Mitosis and Beyond. EMBO J. 2013, 32, 2661–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, S.; Lopez-Contreras, A.J.; Gabut, M.; Marion, R.M.; Gutierrez-Martinez, P.; Bua, S.; Ramirez, O.; Olalde, I.; Rodrigo-Perez, S.; Li, H.; et al. Limiting Replication Stress during Somatic Cell Reprogramming Reduces Genomic Instability in Induced Pluripotent Stem Cells. Nat. Commun. 2015, 6, 8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmarais, J.A.; Hoffmann, M.J.; Bingham, G.; Gagou, M.E.; Meuth, M.; Andrews, P.W. Human Embryonic Stem Cells Fail to Activate CHK1 and Commit to Apoptosis in Response to DNA Replication Stress. Stem Cells 2012, 30, 1385–1393. [Google Scholar] [CrossRef]
- Vessoni, A.T.; Zhang, T.; Quinet, A.; Jeong, H.-C.; Munroe, M.; Wood, M.; Tedone, E.; Vindigni, A.; Shay, J.W.; Greenberg, R.A.; et al. Telomere Erosion in Human Pluripotent Stem Cells Leads to ATR-Mediated Mitotic Catastrophe. J. Cell Biol. 2021, 220, e202011014. [Google Scholar] [CrossRef]
- Delgado-Olguin, P.; Recillas-Targa, F. Chromatin Structure of Pluripotent Stem Cells and Induced Pluripotent Stem Cells. Brief. Funct. Genom. 2011, 10, 37–49. [Google Scholar] [CrossRef]
- Li, D.; Shu, X.; Zhu, P.; Pei, D. Chromatin Accessibility Dynamics during Cell Fate Reprogramming. EMBO Rep. 2021, 22, e51644. [Google Scholar] [CrossRef]
- Wilkins, B.J.; Rall, N.A.; Ostwal, Y.; Kruitwagen, T.; Hiragami-Hamada, K.; Winkler, M.; Barral, Y.; Fischle, W.; Neumann, H. A Cascade of Histone Modifications Induces Chromatin Condensation in Mitosis. Science 2014, 343, 77–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, J.R.; Faesen, A.C.; Klare, K.; Petrovic, A.; Basilico, F.; Fischböck, J.; Pentakota, S.; Keller, J.; Pesenti, M.E.; Pan, D.; et al. Insights from Biochemical Reconstitution into the Architecture of Human Kinetochores. Nature 2016, 537, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, B.E.; Jansen, L.E.T.; Foltz, D.R.; Cleveland, D.W. Centromere Identity, Function, and Epigenetic Propagation across Cell Divisions. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 403–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milagre, I.; Pereira, C.; Oliveira, R.A.; Jansen, L.E.T. Reprogramming of Human Cells to Pluripotency Induces CENP-A Chromatin Depletion. Open Biol. 2020, 10, 200227. [Google Scholar] [CrossRef]
- Bodor, D.L.; Valente, L.P.; Mata, J.F.; Black, B.E.; Jansen, L.E.T. Assembly in G1 Phase and Long-Term Stability Are Unique Intrinsic Features of CENP-A Nucleosomes. MBoC 2013, 24, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Jansen, L.E.T.; Black, B.E.; Foltz, D.R.; Cleveland, D.W. Propagation of Centromeric Chromatin Requires Exit from Mitosis. J. Cell Biol. 2007, 176, 795–805. [Google Scholar] [CrossRef]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA Sequence-Specific Binding of CENP-B Enhances the Fidelity of Human Centromere Function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hirst, A.J.; Duan, F.; Qiu, H.; Huang, R.; Ji, Y.; Bai, L.; Zhang, F.; Robinson, D.; Jones, M.; et al. Anti-Apoptotic Mutations Desensitize Human Pluripotent Stem Cells to Mitotic Stress and Enable Aneuploid Cell Survival. Stem Cell Rep. 2019, 12, 557–571. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Ya, A.; Compton, D.A.; Godek, K.M. A Pluripotent Developmental State Confers a Low Fidelity of Chromosome Segregation. Stem Cell Rep. 2023, 18, 475–488. [Google Scholar] [CrossRef]
- Gregan, J.; Polakova, S.; Zhang, L.; Tolić-Nørrelykke, I.M.; Cimini, D. Merotelic Kinetochore Attachment: Causes and Effects. Trends Cell Biol. 2011, 21, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, M.; Lee, S.H. The Chromosomal Passenger Complex (CPC) as a Key Orchestrator of Orderly Mitotic Exit and Cytokinesis. Front. Cell Dev. Biol. 2015, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Vukušić, K.; Tolić, I.M. Polar Chromosomes—Challenges of a Risky Path. Cells 2022, 11, 1531. [Google Scholar] [CrossRef]
- Lyu, R.; Wu, X.; Ma, N.; Wang, D.; Sun, S.; Luo, Y.; Zhou, J.; Lu, X.; Liu, M.; Li, D. The Specialized Mitotic Behavior of Human Embryonic Stem Cells. Cell Tissue Res. 2022, 387, 85–93. [Google Scholar] [CrossRef]
- Francois, L.; Boskovic, P.; Knerr, J.; He, W.; Sigismondo, G.; Schwan, C.; More, T.H.; Schlotter, M.; Krijgsveld, J.; Hiller, K.; et al. BCAT1 Redox Function Maintains Mitotic Fidelity. Cell Rep. 2022, 41, 111524. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Prifti, D.; Gui, P.; Liu, X.; Elowe, S.; Yao, X. Recent Progress on the Localization of the Spindle Assembly Checkpoint Machinery to Kinetochores. Cells 2019, 8, 278. [Google Scholar] [CrossRef] [Green Version]
- Mantel, C.; Guo, Y.; Lee, M.R.; Kim, M.-K.; Han, M.-K.; Shibayama, H.; Fukuda, S.; Yoder, M.C.; Pelus, L.M.; Kim, K.-S.; et al. Checkpoint-Apoptosis Uncoupling in Human and Mouse Embryonic Stem Cells: A Source of Karyotpic Instability. Blood 2007, 109, 4518–4527. [Google Scholar] [CrossRef] [Green Version]
- Padgett, J.; Santos, S.D.M. From Clocks to Dominoes: Lessons on Cell Cycle Remodelling from Embryonic Stem Cells. FEBS Lett. 2020, 594, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. In Cell Cycle Control; Noguchi, E., Gadaleta, M.C., Eds.; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2014; Volume 1170, pp. 29–40. ISBN 978-1-4939-0887-5. [Google Scholar]
- Damelin, M.; Sun, Y.E.; Sodja, V.B.; Bestor, T.H. Decatenation Checkpoint Deficiency in Stem and Progenitor Cells. Cancer Cell 2005, 8, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Rieder, C.L.; Maiato, H. Stuck in Division or Passing Through. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.A.; Stein, J.L.; Lian, J.B.; van Wijnen, A.J.; Stein, G.S. Human Embryonic Stem Cells Are Pre-Mitotically Committed to Self-Renewal and Acquire a Lengthened G1 Phase upon Lineage Programming. J. Cell Physiol. 2010, 222, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipczyk, A.A.; Laslett, A.L.; Mummery, C.; Pera, M.F. Differentiation Is Coupled to Changes in the Cell Cycle Regulatory Apparatus of Human Embryonic Stem Cells. Stem Cell Res. 2007, 1, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauklin, S.; Vallier, L. The Cell-Cycle State of Stem Cells Determines Cell Fate Propensity. Cell 2013, 155, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, K.A.U.; Liang, H.; Lim, Y.-S.; Chan, Y.-S.; Yeo, J.-C.; Tan, C.-P.; Gao, B.; Le, B.; Tan, Z.-Y.; Low, K.-Y.; et al. Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. Cell 2015, 162, 564–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagana, A.; Dorn, J.F.; De Rop, V.; Ladouceur, A.-M.; Maddox, A.S.; Maddox, P.S. A Small GTPase Molecular Switch Regulates Epigenetic Centromere Maintenance by Stabilizing Newly Incorporated CENP-A. Nat. Cell Biol. 2010, 12, 1186–1193. [Google Scholar] [CrossRef]
- Nashun, B.; Hill, P.W.; Hajkova, P. Reprogramming of Cell Fate: Epigenetic Memory and the Erasure of Memories Past. EMBO J. 2015, 34, 1296–1308. [Google Scholar] [CrossRef]
- Milagre, I.; Stubbs, T.M.; King, M.R.; Spindel, J.; Santos, F.; Krueger, F.; Bachman, M.; Segonds-Pichon, A.; Balasubramanian, S.; Andrews, S.R.; et al. Gender Differences in Global but Not Targeted Demethylation in IPSC Reprogramming. Cell Rep. 2017, 18, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-S.; Shin, J.-Y.; Tonge, P.D.; Puri, M.C.; Lee, S.; Park, H.; Lee, W.-C.; Hussein, S.M.I.; Bleazard, T.; Yun, J.-Y.; et al. An Epigenomic Roadmap to Induced Pluripotency Reveals DNA Methylation as a Reprogramming Modulator. Nat. Commun. 2014, 5, 5619. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and Cellular Reprogramming: Facts, Hypotheses, Unresolved Issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [Green Version]
- Cacchiarelli, D.; Trapnell, C.; Ziller, M.J.; Soumillon, M.; Cesana, M.; Karnik, R.; Donaghey, J.; Smith, Z.D.; Ratanasirintrawoot, S.; Zhang, X.; et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Koche, R.P.; Smith, Z.D.; Adli, M.; Gu, H.; Ku, M.; Gnirke, A.; Bernstein, B.E.; Meissner, A. Reprogramming Factor Expression Initiates Widespread Targeted Chromatin Remodeling. Cell Stem Cell 2011, 8, 96–105. [Google Scholar] [CrossRef] [Green Version]
- van Nuland, R.; Gozani, O. Histone H4 Lysine 20 (H4K20) Methylation, Expanding the Signaling Potential of the Proteome One Methyl Moiety at a Time. Mol. Cell Proteom. 2016, 15, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andonegui-Elguera, M.A.; Cáceres-Gutiérrez, R.E.; López-Saavedra, A.; Cisneros-Soberanis, F.; Justo-Garrido, M.; Díaz-Chávez, J.; Herrera, L.A. The Roles of Histone Post-Translational Modifications in the Formation and Function of a Mitotic Chromosome. IJMS 2022, 23, 8704. [Google Scholar] [CrossRef]
- Bergmann, J.H.; Rodríguez, M.G.; Martins, N.M.C.; Kimura, H.; Kelly, D.A.; Masumoto, H.; Larionov, V.; Jansen, L.E.T.; Earnshaw, W.C. Epigenetic Engineering Shows H3K4me2 Is Required for HJURP Targeting and CENP-A Assembly on a Synthetic Human Kinetochore: H3K4me2 and Kinetochore Maintenance. EMBO J. 2011, 30, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, O.; Vargiu, G.; Abad, M.A.; Zhiteneva, A.; Jeyaprakash, A.A.; Masumoto, H.; Kouprina, N.; Larionov, V.; Earnshaw, W.C. Epigenetic Engineering Reveals a Balance between Histone Modifications and Transcription in Kinetochore Maintenance. Nat. Commun. 2016, 7, 13334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Tanasa, B.; Tyurina, O.V.; Zhou, T.Y.; Gassmann, R.; Liu, W.T.; Ohgi, K.A.; Benner, C.; Garcia-Bassets, I.; Aggarwal, A.K.; et al. PHF8 Mediates Histone H4 Lysine 20 Demethylation Events Involved in Cell Cycle Progression. Nature 2010, 466, 508–512. [Google Scholar] [CrossRef] [Green Version]
- Schalch, T.; Steiner, F.A. Structure of Centromere Chromatin: From Nucleosome to Chromosomal Architecture. Chromosoma 2017, 126, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Sako, K.; Takagaki, K.; Hirayama, Y.; Uchida, K.S.K.; Herman, J.A.; DeLuca, J.G.; Hirota, T. HP1-Assisted Aurora B Kinase Activity Prevents Chromosome Segregation Errors. Dev. Cell 2016, 36, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Higgins, J.M.G. Histone Modifications and Mitosis: Countermarks, Landmarks, and Bookmarks. Trends Cell Biol. 2013, 23, 175–184. [Google Scholar] [CrossRef]
- Soufi, A.; Dalton, S. Cycling through Developmental Decisions: How Cell Cycle Dynamics Control Pluripotency, Differentiation and Reprogramming. Development 2016, 143, 4301–4311. [Google Scholar] [CrossRef] [Green Version]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and Cohesin Connect Gene Expression and Chromatin Architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Blum, B.; Bar-Nur, O.; Golan-Lev, T.; Benvenisty, N. The Anti-Apoptotic Gene Survivin Contributes to Teratoma Formation by Human Embryonic Stem Cells. Nat. Biotechnol. 2009, 27, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Payer, B.; Lee, J.T. X Chromosome Dosage Compensation: How Mammals Keep the Balance. Annu. Rev. Genet. 2008, 42, 733–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Tsai, H.-J.; Gordon, M.R.; Li, R. Cellular Stress Associated with Aneuploidy. Dev. Cell 2018, 44, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schukken, K.M.; Sheltzer, J.M. Extensive Protein Dosage Compensation in Aneuploid Human Cancers. Genome Res. 2022, 32, 1254–1270. [Google Scholar] [CrossRef] [PubMed]
- Senger, G.; Santaguida, S.; Schaefer, M.H. Regulation of Protein Complex Partners as a Compensatory Mechanism in Aneuploid Tumors. eLife 2022, 11, e75526. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Zhao, X.; Katsnelson, L.; Camacho-Hernandez, E.M.; Mermerian, A.; Mays, J.C.; Lippman, S.M.; Rosales-Alvarez, R.E.; Moya, R.; Shwetar, J.; et al. Proteogenomic Analysis of Cancer Aneuploidy and Normal Tissues Reveals Divergent Modes of Gene Regulation across Cellular Pathways. eLife 2022, 11, e75227. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C.A.; Housman, D.E.; Amon, A. Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science 2008, 322, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, J.M.; Macedo, J.C.; Mattingly, A.J.; Wangsa, D.; Camps, J.; Lima, V.; Gomes, A.M.; Dória, S.; Ried, T.; Logarinho, E.; et al. Chromosome Mis-Segregation and Cytokinesis Failure in Trisomic Human Cells. eLife 2015, 4, e05068. [Google Scholar] [CrossRef]
- Shahbazi, M.N.; Wang, T.; Tao, X.; Weatherbee, B.A.T.; Sun, L.; Zhan, Y.; Keller, L.; Smith, G.D.; Pellicer, A.; Scott, R.T.; et al. Developmental Potential of Aneuploid Human Embryos Cultured beyond Implantation. Nat. Commun. 2020, 11, 3987. [Google Scholar] [CrossRef]
- Yang, S.; Lin, G.; Tan, Y.-Q.; Zhou, D.; Deng, L.-Y.; Cheng, D.-H.; Luo, S.-W.; Liu, T.-C.; Zhou, X.-Y.; Sun, Z.; et al. Tumor Progression of Culture-Adapted Human Embryonic Stem Cells during Long-Term Culture. Genes Chromosomes Cancer 2008, 47, 665–679. [Google Scholar] [CrossRef]
- Ben-David, U.; Arad, G.; Weissbein, U.; Mandefro, B.; Maimon, A.; Golan-Lev, T.; Narwani, K.; Clark, A.T.; Andrews, P.W.; Benvenisty, N.; et al. Aneuploidy Induces Profound Changes in Gene Expression, Proliferation and Tumorigenicity of Human Pluripotent Stem Cells. Nat. Commun. 2014, 5, 4825. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Cheng, L.; Jia, Y.; Liu, G.; Li, C.; Song, S.; Bradley, A.; Huang, Y. Aneuploid Embryonic Stem Cells Exhibit Impaired Differentiation and Increased Neoplastic Potential. EMBO J. 2016, 35, 2285–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haim-Abadi, G.; Golan-Lev, T.; Koren, A.; Benvenisty, N. Generation, Genomic Characterization, and Differentiation of Triploid Human Embryonic Stem Cells. Stem Cell Rep. 2023, 18, 1049–1060. [Google Scholar] [CrossRef]
- Mirkovic, M.; Guilgur, L.G.; Tavares, A.; Passagem-Santos, D.; Oliveira, R.A. Induced Aneuploidy in Neural Stem Cells Triggers a Delayed Stress Response and Impairs Adult Life Span in Flies. PLoS Biol. 2019, 17, e3000016. [Google Scholar] [CrossRef] [Green Version]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human Pluripotent Stem Cells Recurrently Acquire and Expand Dominant Negative P53 Mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Sharir, Y.; McFarland, J.M.; Abdusamad, M.; Marquis, C.; Bernhard, S.V.; Kazachkova, M.; Tang, H.; Ippolito, M.R.; Laue, K.; Zerbib, J.; et al. Aneuploidy Renders Cancer Cells Vulnerable to Mitotic Checkpoint Inhibition. Nature 2021, 590, 486–491. [Google Scholar] [CrossRef]
- Barroso-Vilares, M.; Macedo, J.C.; Reis, M.; Warren, J.D.; Compton, D.; Logarinho, E. Small-molecule Inhibition of Aging-associated Chromosomal Instability Delays Cellular Senescence. EMBO Rep. 2020, 21, e49248. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Talje, L.; Liu, Z.; Kwok, B.H.; Compton, D.A. Adaptive Resistance to an Inhibitor of Chromosomal Instability in Human Cancer Cells. Cell Rep. 2016, 17, 1755–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
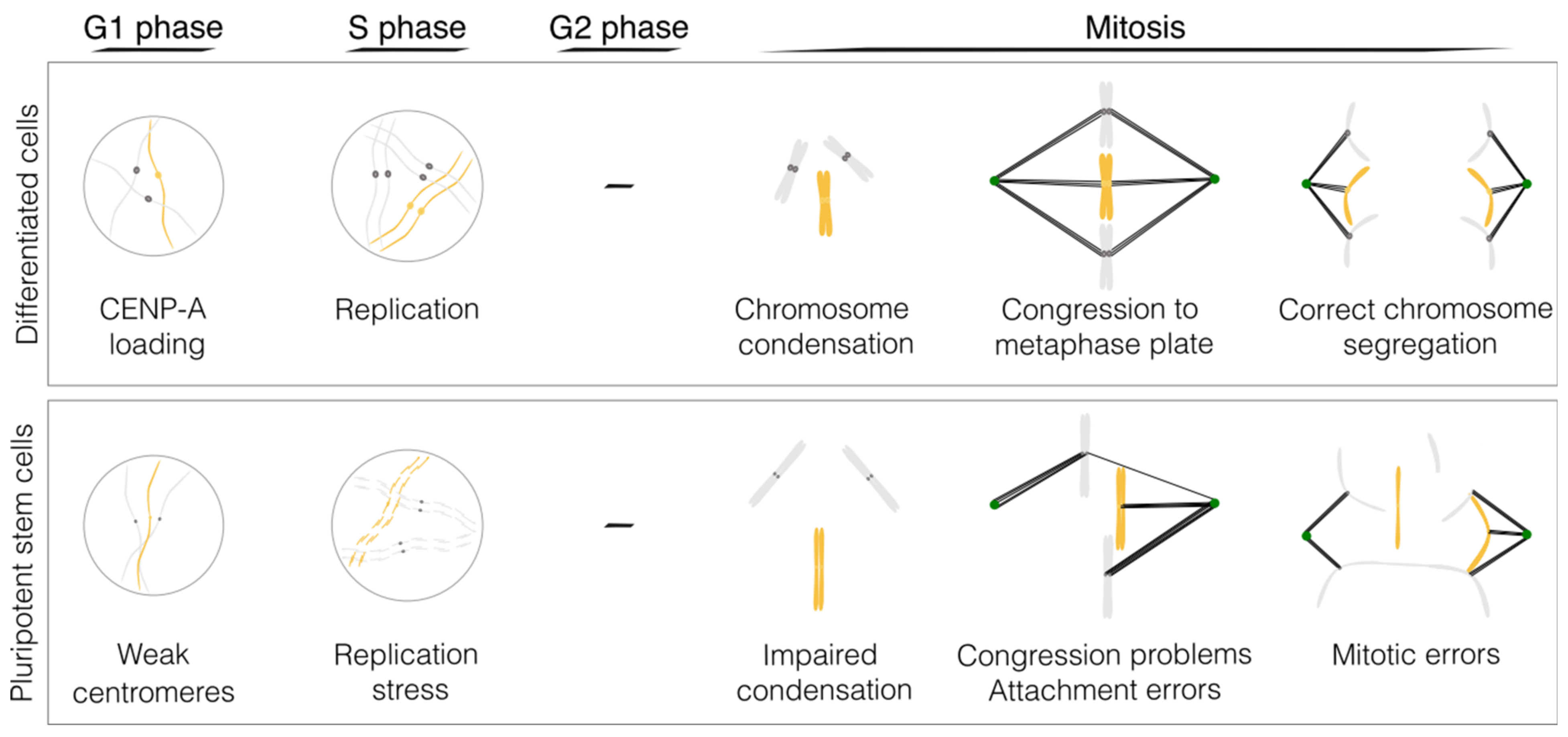
Figure 1.
Molecular mechanisms underlying decreased mitotic fidelity in pluripotent stem cells. Schematic of the major mitosis problems in PSCs (please refer to Table 1 for the references and the main text for a detailed explanation). Figure designed in Inkscape v1.2.2.
Figure 1.
Molecular mechanisms underlying decreased mitotic fidelity in pluripotent stem cells. Schematic of the major mitosis problems in PSCs (please refer to Table 1 for the references and the main text for a detailed explanation). Figure designed in Inkscape v1.2.2.

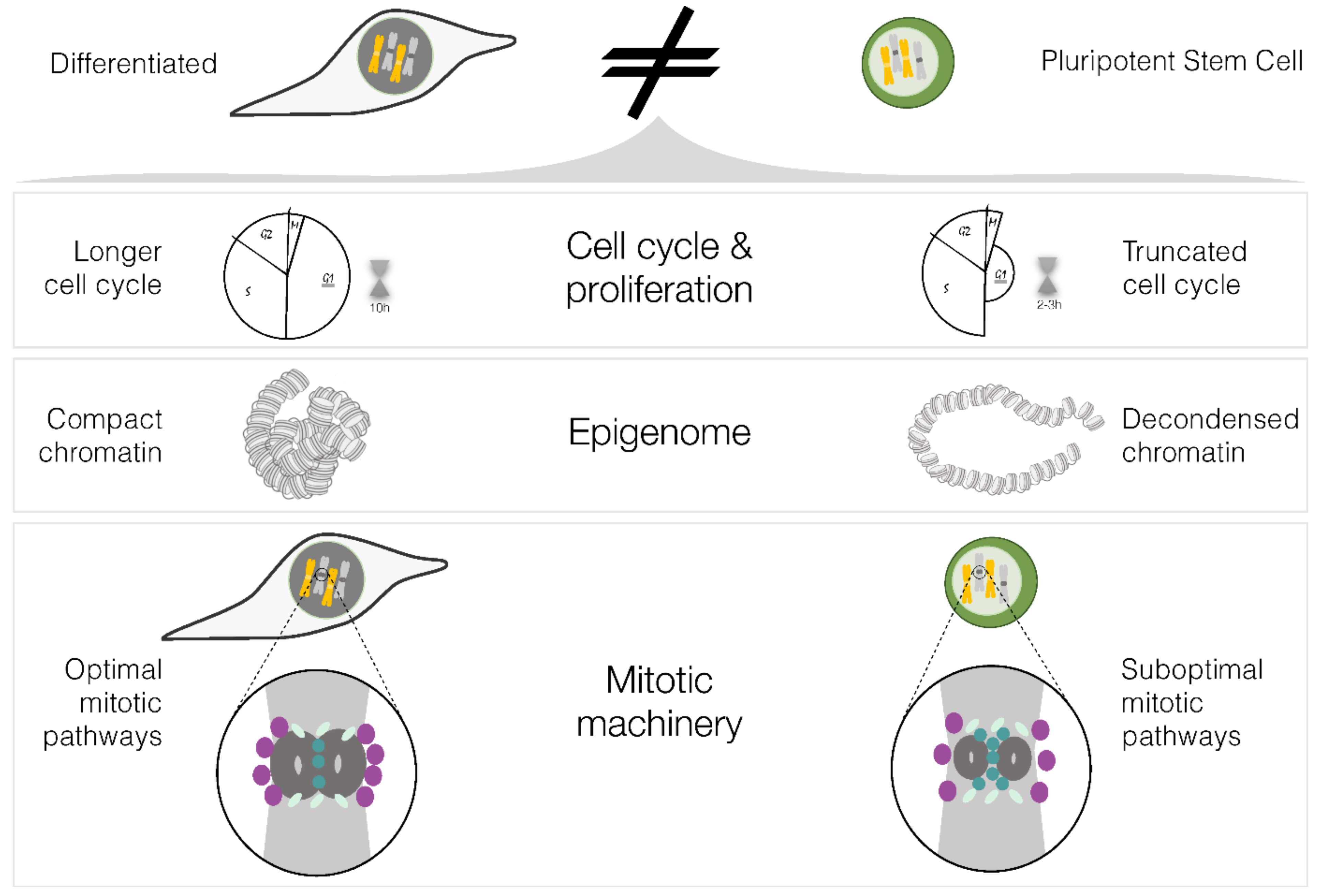
Figure 2.
Distinct physiological characteristics of differentiated and pluripotent stem cells. Schematic of the physiological properties of pluripotent stem cells which can affect mitotic fidelity and cause the observed increased aneuploidy in PSCs (please refer to the main text for a detailed explanation). Figure designed in Inkscape v1.2.2.
Figure 2.
Distinct physiological characteristics of differentiated and pluripotent stem cells. Schematic of the physiological properties of pluripotent stem cells which can affect mitotic fidelity and cause the observed increased aneuploidy in PSCs (please refer to the main text for a detailed explanation). Figure designed in Inkscape v1.2.2.

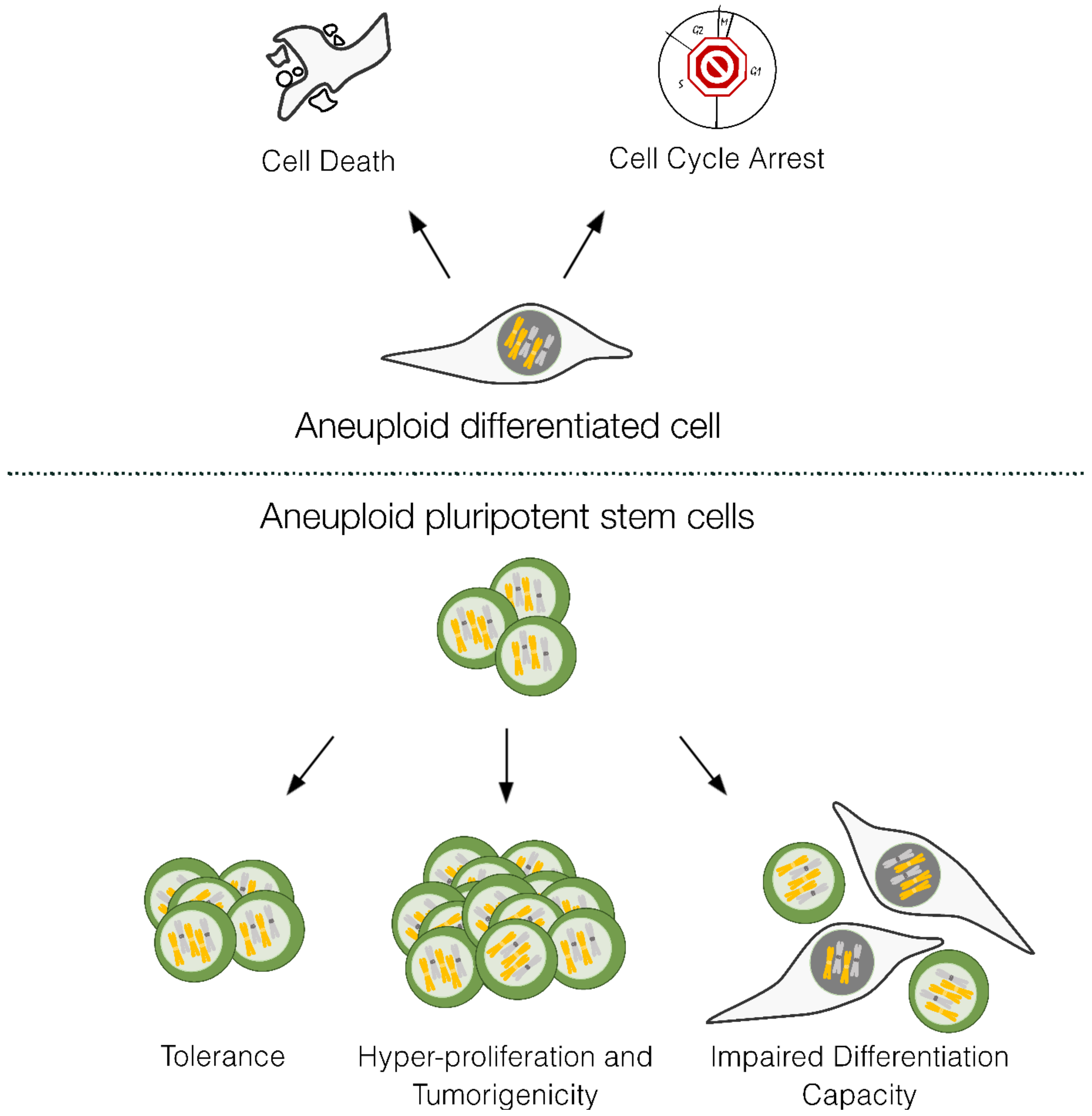
Figure 3.
Response to aneuploidy in differentiated versus pluripotent stem cells. Differentiated aneuploid cells tend to activate cell cycle checkpoints which leads to arrest and can result in apoptosis. PSCs have higher tolerance to aneuploidy, can become hyperproliferative and have impaired differentiation capacity. Figure designed in Inkscape v1.2.2.
Figure 3.
Response to aneuploidy in differentiated versus pluripotent stem cells. Differentiated aneuploid cells tend to activate cell cycle checkpoints which leads to arrest and can result in apoptosis. PSCs have higher tolerance to aneuploidy, can become hyperproliferative and have impaired differentiation capacity. Figure designed in Inkscape v1.2.2.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the key references proving PSC have compromised mitosis and the cell types used in these studies.
Table 1.
Summary of the key references proving PSC have compromised mitosis and the cell types used in these studies.
| Main Findings | Human ESCs | Human iPSCs |
|---|---|---|
| PSCs have recurrent chromosomal aberrations, including whole chromosome aneuploidies | [10,11,12,13,15,16] | [10,11,12,14,16] |
| PSCs have erroneous mitosis | [23,24,25,26] | [26,27] |
| PSCs undergo high replication stress during S phase | [23,27,28,29] | [23,27,28,29,30] |
| Aneuploid PSCs have problems in chromosome condensation | [23] | [23] |
| PSCs have weaker centromeres | [24] | [24] |
| PSCs have high number of erroneous kinetochore–microtubule attachments | [26,31] | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Milagre, I.; Pereira, C.; Oliveira, R.A. Compromised Mitotic Fidelity in Human Pluripotent Stem Cells. Int. J. Mol. Sci. 2023, 24, 11933. https://doi.org/10.3390/ijms241511933
AMA Style
Milagre I, Pereira C, Oliveira RA. Compromised Mitotic Fidelity in Human Pluripotent Stem Cells. International Journal of Molecular Sciences. 2023; 24(15):11933. https://doi.org/10.3390/ijms241511933
Chicago/Turabian StyleMilagre, Inês, Carolina Pereira, and Raquel A. Oliveira. 2023. "Compromised Mitotic Fidelity in Human Pluripotent Stem Cells" International Journal of Molecular Sciences 24, no. 15: 11933. https://doi.org/10.3390/ijms241511933
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.