
Assessment of the Contribution of a Thermodynamic and Mechanical Destabilization of Myosin-Binding Protein C Domain C2 to the Pathomechanism of Hypertrophic Cardiomyopathy-Causing Double Mutation MYBPC3Δ25bp/D389V



Abstract
:1. Introduction
2. Results
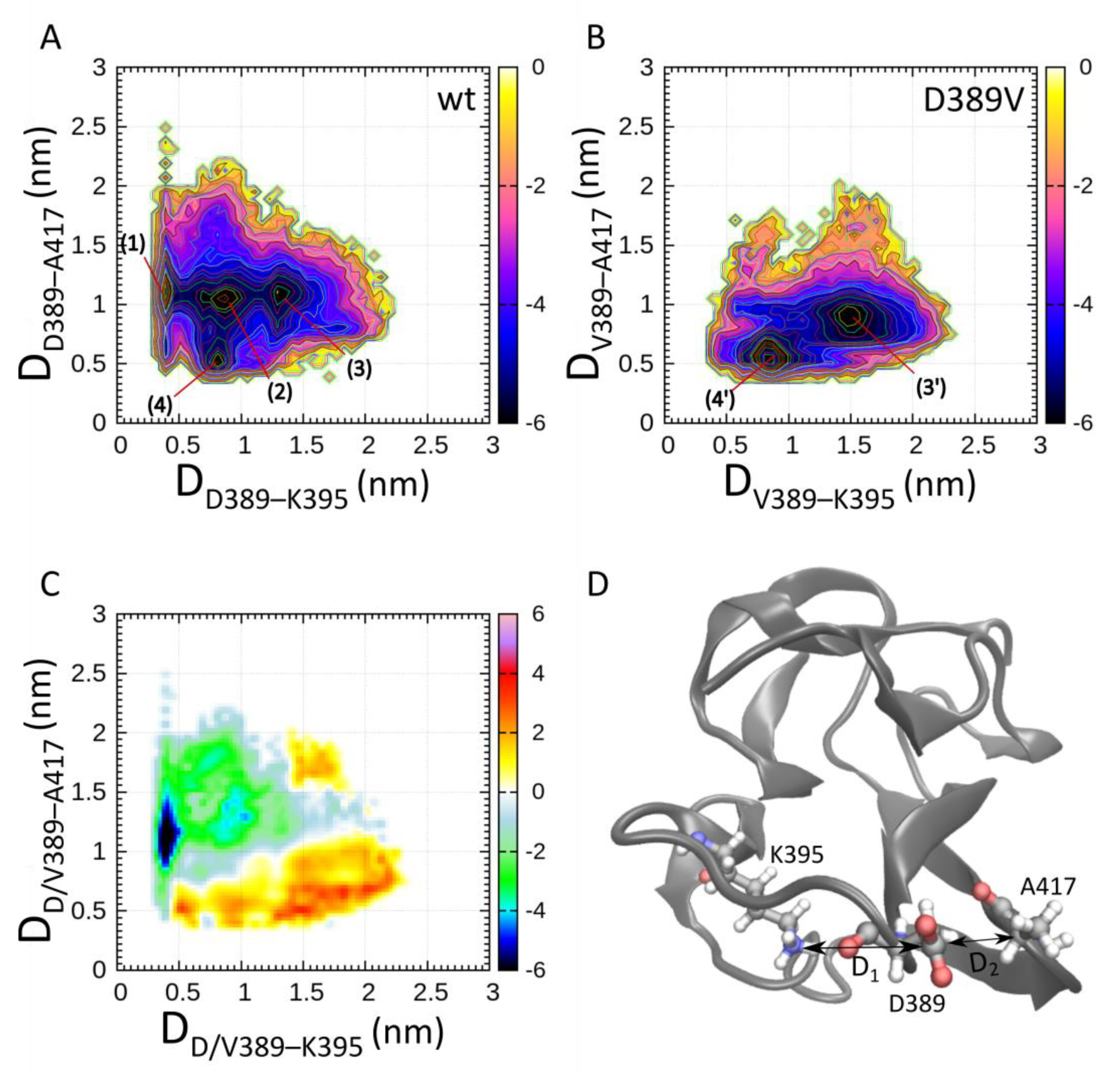
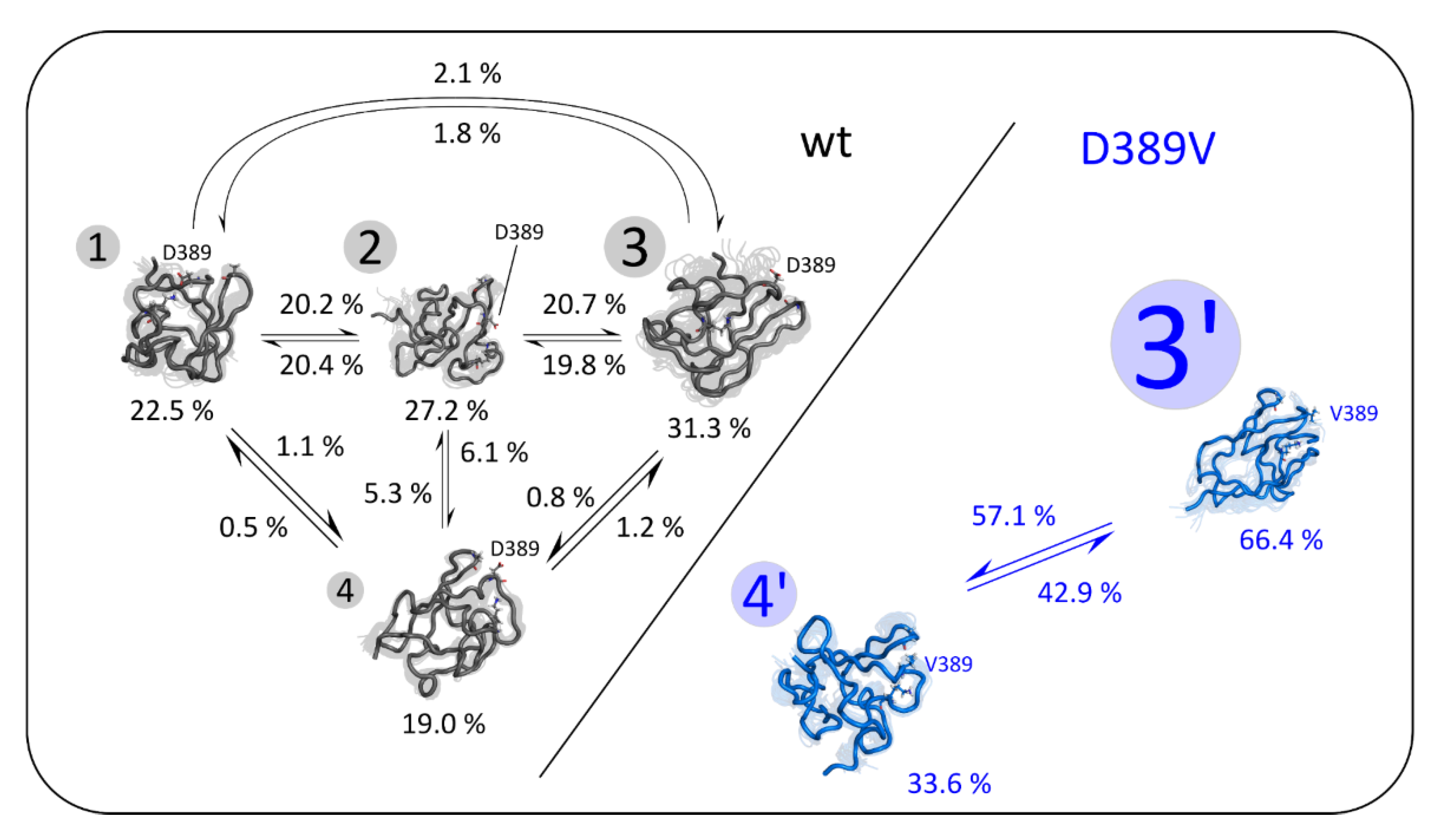
2.1. Enhanced CORE-MD II Simulations of the Wild Type and the D389V C2 Domain
2.2. Structural Integrity and Thermodynamic Stability of N-Terminal MyBPC Domains
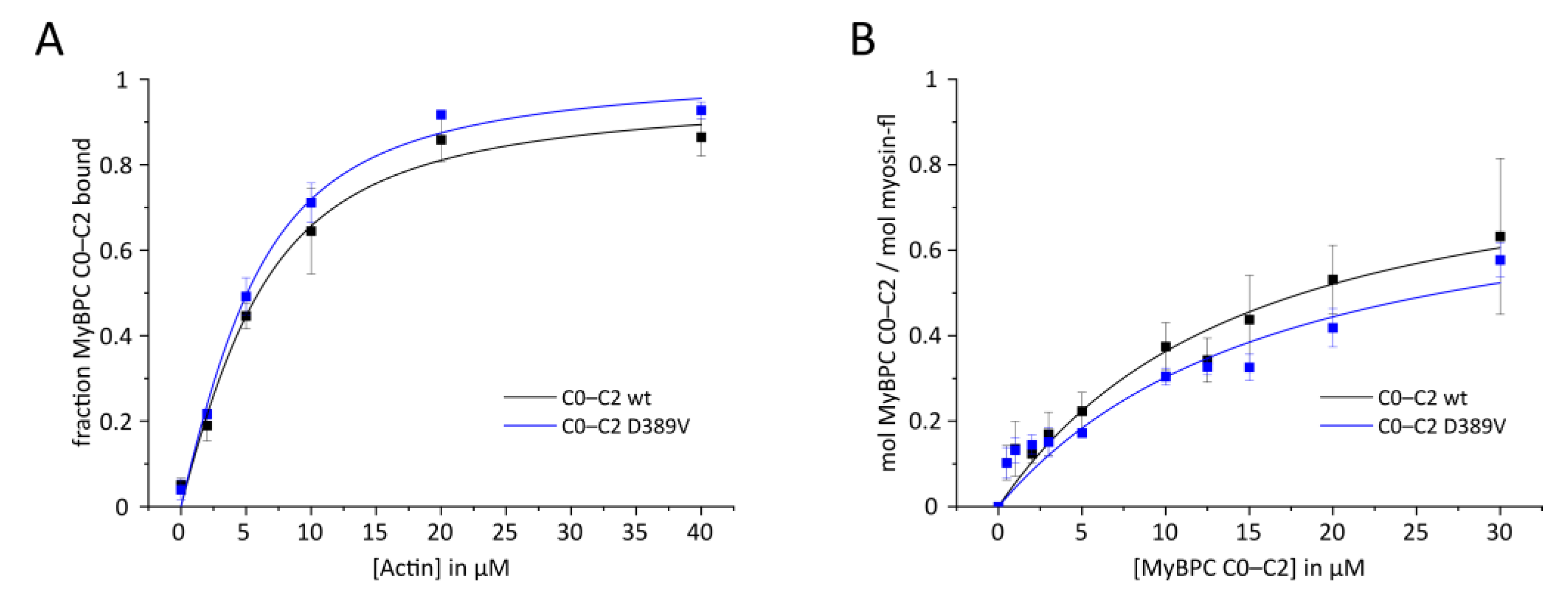
2.3. Interaction of N-Terminal Domains of MyBPC with Key Sarcomeric Proteins
2.4. Simulation of C2 Domain Unfolding Dynamics
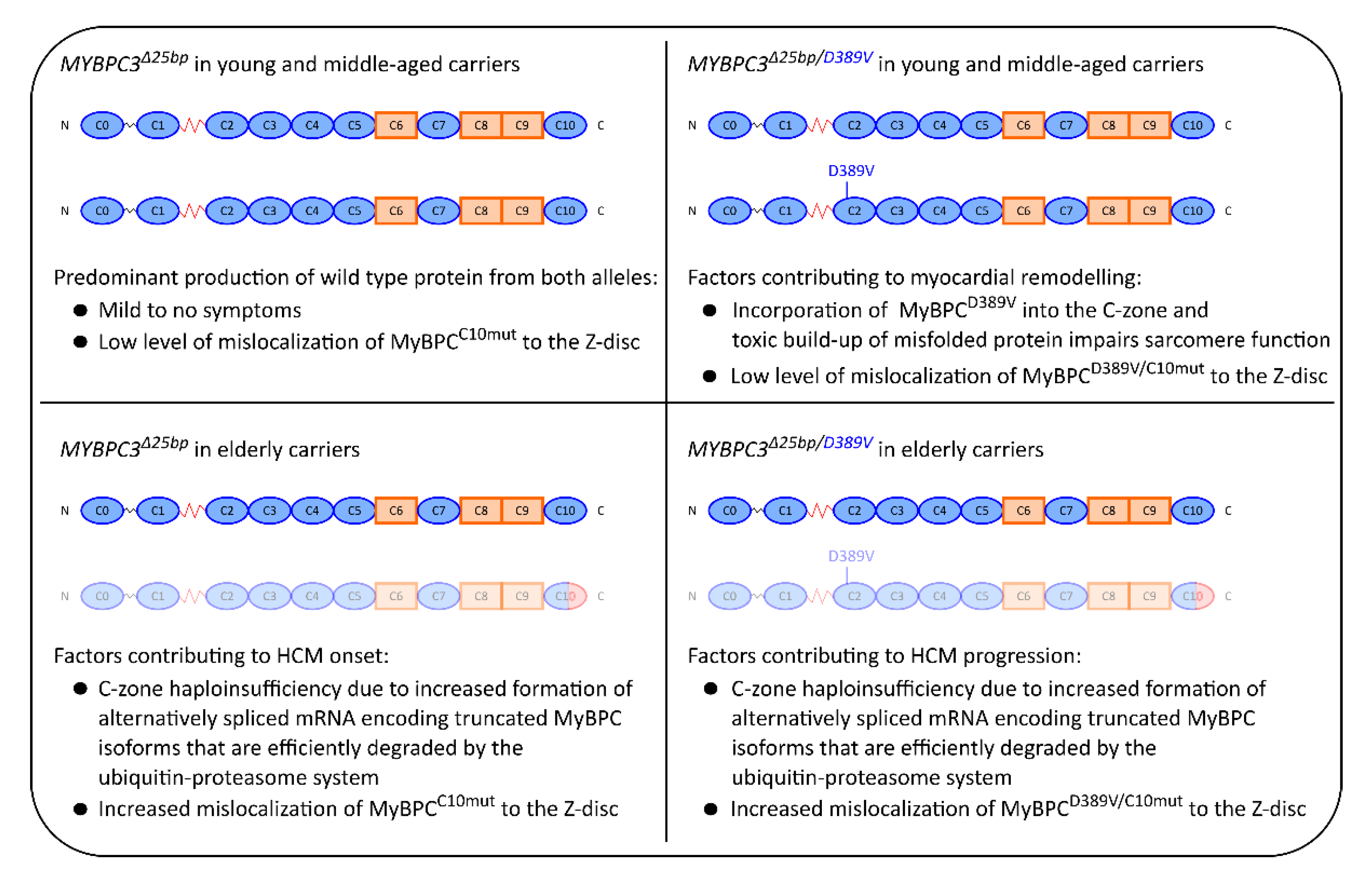
3. Discussion
4. Materials and Methods
4.1. Homology Modeling of the C2 Domain
4.2. CORE-MD II Simulation
4.3. MD Trajectory Analysis
4.4. Plasmid Construction
4.5. Protein Purification
4.6. Thermal Stability of MyBPC Subdomains
4.7. Circular Dichroism Measurements
4.8. High-Speed Cosedimentation Assay
4.9. Steered Molecular Dynamics Simulation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| β-CM | β-Cardiac myosin |
| CD | Circular dichroism |
| cvSMD | Constant velocity steered molecular dynamics |
| DSC | Differential scanning calorimetry |
| DTT | Dithiothreitol |
| GBSA | Generalized Born and surface area continuum solvation |
| IPTG | Isopropyl β-d-1-thiogalactopyranoside |
| MyBPC | Cardiac myosin-binding protein C |
| PBS | Phosphate buffered saline |
| SDS-PAGE | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| SMD | Steered molecular dynamics |
| TAME | 1,1,1-Tris(aminomethyl)ethane |
| Tm | Protein melting temperature |
| TPCK | Tosyl phenylalanyl chloromethyl ketone |
| TSA | Thermal shift assay |
| UPS | Ubiquitin–proteasome system |
| vdW | Van der Waals |
| Wt | Wild type |
References
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef]
- Barsheshet, A.; Brenyo, A.; Moss, A.J.; Goldenberg, I. Genetics of sudden cardiac death. Curr. Cardiol. Rep. 2011, 13, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Seidman, J.G.; Seidman, C.E. Narrative review: Harnessing molecular genetics for the diagnosis and management of hypertrophic cardiomyopathy. Ann. Intern. Med. 2010, 152, 513–520. [Google Scholar] [CrossRef]
- Maron, B.J.; Sato, N.; Roberts, W.C.; Edwards, J.E.; Chandra, R.S. Quantitative analysis of cardiac muscle cell disorganization in the ventricular septum. Comparison of fetuses and infants with and without congenital heart disease and patients with hypertrophic cardiomyopathy. Circulation 1979, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrier, L.; Mearini, G.; Stathopoulou, K.; Cuello, F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 2015, 573, 188–197. [Google Scholar] [CrossRef]
- Heling, L.W.H.J.; Geeves, M.A.; Kad, N.M. MyBP-C: One protein to govern them all. J. Muscle Res. Cell Motil. 2020, 41, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barefield, D.; Sadayappan, S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 2010, 48, 866–875. [Google Scholar] [CrossRef]
- Kuster, D.W.D.; Bawazeer, A.C.; Zaremba, R.; Goebel, M.; Boontje, N.M.; van der Velden, J. Cardiac myosin binding protein C phosphorylation in cardiac disease. J. Muscle Res. Cell Motil. 2012, 33, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Freiburg, A.; Gautel, M. A molecular map of the interactions between titin and myosin-binding protein C. Eur. J. Biochem. 1996, 235, 317–323. [Google Scholar] [CrossRef]
- Furst, D.O.; Vinkemeier, U.; Weber, K. Mammalian skeletal muscle c-protein: Purification from bovine muscle, binding to titin and the characterization of a full-length human CDNA. J. Cell Sci. 1992, 102, 769–778. [Google Scholar] [CrossRef]
- Offer, G.; Moos, C.; Starr, R. A new protein of the thick filaments of vertebrate skeletal myofibrils: Extraction, purification and characterization. J. Mol. Biol. 1973, 74, 653–676. [Google Scholar] [CrossRef]
- Flashman, E.; Redwood, C.; Moolman-Smook, J.; Watkins, H. Cardiac myosin binding protein C. Circ. Res. 2004, 94, 1279–1289. [Google Scholar] [CrossRef]
- Moos, C.; Mason, C.M.; Besterman, J.M.; Feng, I.-N.M.; Dubin, J.H. The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1. J. Mol. Biol. 1978, 124, 571–586. [Google Scholar] [CrossRef]
- Razumova, M.V.; Shaffer, J.F.; Tu, A.-Y.; Flint, G.V.; Regnier, M.; Harris, S.P. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J. Biol. Chem. 2006, 281, 35846–35854. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, J.F.; Kensler, R.W.; Harris, S.P. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 2009, 284, 12318–12327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitten, A.E.; Jeffries, C.M.; Harris, S.P.; Trewhella, J. Cardiac myosin-binding protein C decorates F-actin: Implications for cardiac function. Proc. Natl. Acad. Sci. USA 2008, 105, 18360–18365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruen, M.; Gautel, M. Mutations in β-Myosin S2 That cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J. Mol. Biol. 1999, 286, 933–949. [Google Scholar] [CrossRef]
- Gruen, M.; Prinz, H.; Gautel, M. CAPK-Phosphorylation Controls the Interaction of the Regulatory Domain of Cardiac Myosin Binding Protein C with Myosin-S2 in an on-off Fashion. FEBS Lett. 1999, 453, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Starr, R.; Offer, G. The interaction of C-protein with heavy meromyosin and subfragment-2. Biochem. J. 1978, 171, 813–816. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.W.; Li, A.; Smith, N.J.; Lal, S.; Graham, R.M.; Kooiker, K.B.; van Dijk, S.J.; Remedios, C.G.D.; Harris, S.P.; Cooke, R. Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J. Mol. Cell. Cardiol. 2016, 94, 65–71. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.W.; Singh, R.R.; Sadayappan, S. Cardiac myosin binding protein-C phosphorylation regulates the super-relaxed state of myosin. Proc. Natl. Acad. Sci. USA 2019, 116, 11731–11736. [Google Scholar] [CrossRef] [Green Version]
- Toepfer, C.N.; Wakimoto, H.; Garfinkel, A.C.; McDonough, B.; Liao, D.; Jiang, J.; Tai, A.C.; Gorham, J.M.; Lunde, I.G.; Lun, M.; et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Kampourakis, T.; Ponnam, S.; Sun, Y.-B.; Sevrieva, I.; Irving, M.M. Structural and functional effects of myosin-binding protein-C phosphorylation in heart muscle are not mimicked by serine-to-aspartate substitutions. J. Biol. Chem. 2018, 293, 14270–14275. [Google Scholar] [CrossRef] [Green Version]
- Inchingolo, A.V.; Previs, S.B.; Previs, M.J.; Warshaw, D.M.; Kad, N.M. Revealing the mechanism of how cardiac myosin-binding protein C N-terminal fragments sensitize thin filaments for myosin binding. Proc. Natl. Acad. Sci. USA 2019, 116, 6828–6835. [Google Scholar] [CrossRef] [Green Version]
- Karsai, Á.; Kellermayer, M.S.Z.; Harris, S.P. Mechanical unfolding of cardiac myosin binding protein-C by atomic force microscopy. Biophys. J. 2011, 101, 1968–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsai, Á.; Kellermayer, M.S.Z.; Harris, S.P. Cross-species mechanical fingerprinting of cardiac myosin binding protein-C. Biophys. J. 2013, 104, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Weith, A.; Sadayappan, S.; Gulick, J.; Previs, M.J.; VanBuren, P.; Robbins, J.; Warshaw, D.M. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. J. Mol. Cell. Cardiol. 2012, 52, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ababou, A.; Gautel, M.; Pfuhl, M. Dissecting the N-terminal myosin binding site of human cardiac myosin-binding protein C. structure and myosin binding of domain C2. J. Biol. Chem. 2007, 282, 9204–9215. [Google Scholar] [CrossRef] [Green Version]
- Helms, A.S.; Thompson, A.D.; Glazier, A.A.; Hafeez, N.; Kabani, S.; Rodriguez, J.; Yob, J.M.; Woolcock, H.; Mazzarotto, F.; Lakdawala, N.K.; et al. Spatial and functional distribution of MYBPC3 pathogenic variants and clinical outcomes in patients with hypertrophic cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 396–405. [Google Scholar] [CrossRef]
- Marston, S.; Copeland, O.; Jacques, A.; Livesey, K.; Tsang, V.; McKenna, W.J.; Jalilzadeh, S.; Carballo, S.; Redwood, C.; Watkins, H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 2009, 105, 219–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, T.S.; Snyder, J.; Sadayappan, S.; Day, S.M.; Previs, M.J. MYBPC3 Truncation mutations enhance actomyosin contractile mechanics in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 127, 165–173. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.J.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.M.; et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy. Circulation 2009, 119, 1473–1483. [Google Scholar] [CrossRef] [Green Version]
- Barefield, D.; Kumar, M.; Gorham, J.; Seidman, J.G.; Seidman, C.E.; de Tombe, P.P.; Sadayappan, S. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J. Mol. Cell. Cardiol. 2015, 79, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Glazier, A.A.; Thompson, A.; Day, S.M. Allelic imbalance and haploinsufficiency in MYBPC3-Linked hypertrophic cardiomyopathy. Pflugers Arch.-Eur. J. Physiol. 2019, 471, 781–793. [Google Scholar] [CrossRef]
- Helms, A.S.; Davis, F.M.; Coleman, D.; Bartolone, S.N.; Glazier, A.A.; Pagani, F.; Yob, J.M.; Sadayappan, S.; Pedersen, E.; Lyons, R.; et al. Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2014, 7, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Nadvi, N.A.; Michie, K.A.; Kwan, A.H.; Guss, J.M.; Trewhella, J. Clinically linked mutations in the central domains of cardiac myosin-binding protein C with distinct phenotypes show differential structural effects. Structure 2016, 24, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezold, K.L.; Shaffer, J.F.; Khosa, J.K.; Hoye, E.R.; Harris, S.P. A gain-of-function mutation in the m-domain of cardiac myosin-binding protein-C increases binding to actin. J. Biol. Chem. 2013, 288, 21496–21505. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, S.J.; Bezold Kooiker, K.; Mazzalupo, S.; Yang, Y.; Kostyukova, A.S.; Mustacich, D.J.; Hoye, E.R.; Stern, J.A.; Kittleson, M.D.; Harris, S.P. The A31P missense mutation in cardiac myosin binding protein C alters protein structure but does not cause haploinsufficiency. Arch. Biochem. Biophys. 2016, 601, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Smelter, D.F.; Lange, W.J.D.; Cai, W.; Ge, Y.; Ralphe, J.C. The HCM-Linked W792R mutation in cardiac myosin binding protein-C reduces C6 FnIII domain stability. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H1179–H1191. [Google Scholar] [CrossRef] [PubMed]
- Sakthievel, S.; Waldmüller, S.; Saadi, A.; Joseph, P.K.; Rakesh, P.G.; Padmakumar, R.; Jagan, M.T.; Richard, P.; Schwartz, K.; Rajamanickam, C.; et al. Novel mutations in MYH7 and MYBPC3 of an Indian family causing hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2001, 33, A105. [Google Scholar] [CrossRef]
- Waldmüller, S.; Sakthivel, S.; Saadi, A.V.; Selignow, C.; Rakesh, P.G.; Golubenko, M.; Joseph, P.K.; Padmakumar, R.; Richard, P.; Schwartz, K.; et al. Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2003, 35, 623–636. [Google Scholar] [CrossRef]
- Dhandapany, P.S.; Sadayappan, S.; Xue, Y.; Powell, G.T.; Rani, D.S.; Nallari, P.; Rai, T.S.; Khullar, M.; Soares, P.; Bahl, A.; et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat. Genet. 2009, 41, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Kuster, D.W.D.; Govindan, S.; Springer, T.I.; Martin, J.L.; Finley, N.L.; Sadayappan, S. A hypertrophic cardiomyopathy-associated MYBPC3 mutation common in populations of South Asian descent causes contractile dysfunction. J. Biol. Chem. 2015, 290, 5855–5867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuster, D.W.D.; Lynch, T.L.; Barefield, D.Y.; Sivaguru, M.; Kuffel, G.; Zilliox, M.J.; Lee, K.H.; Craig, R.; Namakkal-Soorappan, R.; Sadayappan, S. Altered C10 domain in cardiac myosin Binding protein-C results in hypertrophic cardiomyopathy. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Viswanathan, S.K.; Puckelwartz, M.J.; Mehta, A.; Ramachandra, C.J.A.; Jagadeesan, A.; Fritsche-Danielson, R.; Bhat, R.V.; Wong, P.; Kandoi, S.; Schwanekamp, J.A.; et al. Association of cardiomyopathy with MYBPC3 D389V and MYBPC3 Δ25bp intronic deletion in South Asian descendants. JAMA Cardiol. 2018, 3, 481–488. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Peter, E.K.; Manstein, D.J.; Shea, J.-E.; Schug, A. CORE-MD II: A fast, adaptive, and accurate enhanced sampling method. J. Chem. Phys. 2021, 155, 104114. [Google Scholar] [CrossRef] [PubMed]
- Guarnera, E.; Berezovsky, I.N. Toward comprehensive allosteric control over protein activity. Structure 2019, 27, 866–878.e1. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Shen, Q.; Zhang, J. Allosteric methods and their applications: Facilitating the discovery of allosteric drugs and the investigation of allosteric mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef]
- Pricolo, M.R.; Herrero-Galán, E.; Mazzaccara, C.; Losi, M.A.; Alegre-Cebollada, J.; Frisso, G. Protein thermodynamic destabilization in the assessment of pathogenicity of a variant of uncertain significance in cardiac myosin binding protein C. J. Cardiovasc. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Suay-Corredera, C.; Pricolo, M.R.; Velázquez-Carreras, D.; Pathak, D.; Nandwani, N.; Pimenta-Lopes, C.; Sánchez-Ortiz, D.; Urrutia-Irazabal, I.; Vilches, S.; Dominguez, F.; et al. Nanomechanical phenotypes in cardiac myosin-binding protein C mutants that cause hypertrophic cardiomyopathy. ACS Nano 2021, 15, 10203–10216. [Google Scholar] [CrossRef]
- Ratti, J.; Rostkova, E.; Gautel, M.; Pfuhl, M. Structure and interactions of myosin-binding protein C domain C0: Cardiac-specific regulation of myosin at its neck? J. Biol. Chem. 2011, 286, 12650–12658. [Google Scholar] [CrossRef] [Green Version]
- Kunst, G.; Kress, K.R.; Gruen, M.; Uttenweiler, D.; Gautel, M.; Fink, R.H.A. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ. Res. 2000, 86, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.L.; Shaffer, J.F.; Harris, S.P.; Dawson, J.F. Altered interactions between cardiac myosin binding protein-c and α-cardiac actin variants associated with cardiomyopathies. Arch. Biochem. Biophys. 2014, 550–551, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.R.; McNamara, J.W.; Sadayappan, S. Mutations in myosin S2 alter cardiac myosin-binding protein-C interaction in hypertrophic cardiomyopathy in a phosphorylation-dependent manner. J. Biol. Chem. 2021, 297, 100836. [Google Scholar] [CrossRef] [PubMed]
- Calaghan, S.C.; Trinick, J.; Knight, P.J.; White, E. A Role for C-protein in the regulation of contraction and intracellular Ca2+ in intact rat ventricular myocytes. J. Physiol. 2000, 528, 151–156. [Google Scholar] [CrossRef]
- Colson, B.A.; Bekyarova, T.; Locher, M.R.; Fitzsimons, D.P.; Irving, T.C.; Moss, R.L. Protein kinase A–Mediated phosphorylation of CMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ. Res. 2008, 103, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, R.W.; Craig, R.; Moss, R.L. Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc. Natl. Acad. Sci. USA 2017, 114, E1355–E1364. [Google Scholar] [CrossRef] [Green Version]
- Luther, P.K.; Winkler, H.; Taylor, K.; Zoghbi, M.E.; Craig, R.; Padrón, R.; Squire, J.M.; Liu, J. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc. Natl. Acad. Sci. USA 2011, 108, 11423–11428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woody, M.S.; Winkelmann, D.A.; Capitanio, M.; Ostap, E.M.; Goldman, Y.E. Single molecule mechanics resolves the earliest events in force generation by cardiac myosin. Elife 2019, 8, e49266. [Google Scholar] [CrossRef]
- Bhuiyan, M.S.; Gulick, J.; Osinska, H.; Gupta, M.; Robbins, J. Determination of the critical residues responsible for cardiac myosin binding protein C’s interactions. J. Mol. Cell. Cardiol. 2012, 53, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11, 224–230. [Google Scholar] [CrossRef]
- Izrailev, S.; Stepaniants, S.; Isralewitz, B.; Kosztin, D.; Lu, H.; Molnar, F.; Wriggers, W.; Schulten, K. Steered molecular dynamics. In Proceedings of the Computational Molecular Dynamics: Challenges, Methods, Ideas, Berlin, Germany, 21–24 May 1997; Deuflhard, P., Hermans, J., Leimkuhler, B., Mark, A.E., Reich, S., Skeel, R.D., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; pp. 39–65. [Google Scholar]
- Lu, H.; Isralewitz, B.; Krammer, A.; Vogel, V.; Schulten, K. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys. J. 1998, 75, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Previs, M.J.; Previs, S.B.; Gulick, J.; Robbins, J.; Warshaw, D.M. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science 2012, 337, 1215–1218. [Google Scholar] [CrossRef] [Green Version]
- Brenner, B.; Seebohm, B.; Tripathi, S.; Montag, J.; Kraft, T. Familial hypertrophic cardiomyopathy: Functional variance among individual cardiomyocytes as a trigger of FHC-phenotype development. Front. Physiol. 2014, 5, 392. [Google Scholar] [CrossRef] [Green Version]
- Kraft, T.; Montag, J.; Radocaj, A.; Brenner, B. Hypertrophic cardiomyopathy. Circ. Res. 2016, 119, 992–995. [Google Scholar] [CrossRef] [Green Version]
- Kraft, T.; Montag, J. Altered force generation and cell-to-cell contractile imbalance in hypertrophic cardiomyopathy. Pflugers Arch.-Eur. J. Physiol. 2019, 471, 719–733. [Google Scholar] [CrossRef]
- Sadayappan, S.; Puckelwartz, M.J.; McNally, E.M. South Asian–Specific MYBPC3Δ25bp intronic deletion and its role in cardiomyopathies and heart failure. Circ. Genom. Precis. Med. 2020, 13, e002986. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamud, E.; Moult, J. Stochastic noise in splicing machinery. Nucleic Acids Res. 2009, 37, 4873–4886. [Google Scholar] [CrossRef]
- Ito, K.; Patel, P.N.; Gorham, J.M.; McDonough, B.; DePalma, S.R.; Adler, E.E.; Lam, L.; MacRae, C.A.; Mohiuddin, S.M.; Fatkin, D.; et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, 7689–7694. [Google Scholar] [CrossRef] [Green Version]
- Dainis, A.; Tseng, E.; Clark, T.A.; Hon, T.; Wheeler, M.; Ashley, E. Targeted long-read rna sequencing demonstrates transcriptional diversity driven by splice-site variation in MYBPC3. Circ. Genom. Precis. Med. 2019, 12, e002464. [Google Scholar] [CrossRef]
- Da’as, S.I.; Fakhro, K.; Thanassoulas, A.; Krishnamoorthy, N.; Saleh, A.; Calver, B.L.; Safieh-Garabedian, B.; Toft, E.; Nounesis, G.; Lai, F.A.; et al. Hypertrophic cardiomyopathy-linked variants of cardiac myosin-binding protein C3 display altered molecular properties and actin interaction. Biochem. J. 2018, 475, 3933–3948. [Google Scholar] [CrossRef] [Green Version]
- Doh, C.Y.; Li, J.; Mamidi, R.; Stelzer, J. The HCM-Causing Y235S CMyBPC Mutation accelerates contractile function by altering C1 domain structure. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Li, R.H.L.; Stern, J.A.; Ho, V.; Tablin, F.; Harris, S.P. Platelet activation and clopidogrel effects on ADP-induced platelet activation in cats with or without the A31P mutation in MYBPC3. J. Vet. Intern. Med. 2016, 30, 1619–1629. [Google Scholar] [CrossRef] [Green Version]
- Holly, A.C.; Melzer, D.; Pilling, L.C.; Fellows, A.C.; Tanaka, T.; Ferrucci, L.; Harries, L.W. Changes in splicing factor expression are associated with advancing age in man. Mech. Ageing Dev. 2013, 134, 356–366. [Google Scholar] [CrossRef] [Green Version]
- Latorre, E.; Harries, L.W. Splicing regulatory factors, ageing and age-related disease. Ageing Res. Rev. 2017, 36, 165–170. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Ma, T.; Wei, G.; Ni, T. Alternative splicing in aging and age-related diseases. Transl. Med. Aging 2017, 1, 32–40. [Google Scholar] [CrossRef]
- Roux, J.; Robinson-Rechavi, M. Age-dependent gain of alternative splice forms and biased duplication explain the relation between splicing and duplication. Genome Res. 2011, 21, 357–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Wu, D.; Zhang, H.; Das, A.; Basu, M.; Malin, J.; Cao, K.; Hannenhalli, S. Comprehensive map of age-associated splicing changes across human tissues and their contributions to age-associated diseases. Sci. Rep. 2018, 8, 10929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michie, K.A.; Kwan, A.H.; Tung, C.-S.; Guss, J.M.; Trewhella, J. A highly conserved yet flexible linker is part of a polymorphic protein-binding domain in myosin-binding protein C. Structure 2016, 24, 2000–2007. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Kollman, P.A. Advances and continuing challenges in achieving realistic and predictive simulations of the properties of organic and biological molecules. Acc. Chem. Res. 1996, 29, 461–469. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. the method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy Server. In The Proteomics Protocols Handbook; Human Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Margossian, S.S.; Lowey, S. [7] Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982, 85, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.S.; Kerwar, G. Intrinsic fluorescence of actin. Biochemistry 1972, 11, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.-C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of Fluorescence-Based Thermal Shift Assays for Hit Identification in Drug Discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Lobley, A.; Whitmore, L.; Wallace, B.A. DICHROWEB: An interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 2002, 18, 211–212. [Google Scholar] [CrossRef] [Green Version]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Taft, M.H.; Behrmann, E.; Munske-Weidemann, L.-C.; Thiel, C.; Raunser, S.; Manstein, D.J. Functional characterization of human myosin-18A and its interaction with F-actin and GOLPH3. J. Biol. Chem. 2013, 288, 30029–30041. [Google Scholar] [CrossRef] [Green Version]
- Latham, S.L.; Weiß, N.; Schwanke, K.; Thiel, C.; Croucher, D.R.; Zweigerdt, R.; Manstein, D.J.; Taft, M.H. Myosin-18B regulates higher-order organization of the cardiac sarcomere through thin filament cross-linking and thick filament dynamics. Cell Rep. 2020, 32, 108090. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 All-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct | α (%) | β (%) | T (%) | U (%) | Σ (%) | Tm (TSA)/°C | Tm (DSC)/°C |
|---|---|---|---|---|---|---|---|
| C2 wt | 7 | 37 | 22 | 34 | 100 | 59.3 ± 0.1 | 56.5 ± 0.3 |
| C2 D389V | 7 | 36 | 22 | 35 | 100 | 52.1 ± 0.3 | 52.3 ± 0.2 |
| C0–C2 wt | 7 | 36 | 24 | 32 | 99 | 51.8 ± 0.5 | 50.2 ± 0.1 |
| C0–C2 D389V | 8 | 40 | 22 | 30 | 100 | 48.9 ± 0.6 | 47.6 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwäbe, F.V.; Peter, E.K.; Taft, M.H.; Manstein, D.J. Assessment of the Contribution of a Thermodynamic and Mechanical Destabilization of Myosin-Binding Protein C Domain C2 to the Pathomechanism of Hypertrophic Cardiomyopathy-Causing Double Mutation MYBPC3Δ25bp/D389V. Int. J. Mol. Sci. 2021, 22, 11949. https://doi.org/10.3390/ijms222111949
Schwäbe FV, Peter EK, Taft MH, Manstein DJ. Assessment of the Contribution of a Thermodynamic and Mechanical Destabilization of Myosin-Binding Protein C Domain C2 to the Pathomechanism of Hypertrophic Cardiomyopathy-Causing Double Mutation MYBPC3Δ25bp/D389V. International Journal of Molecular Sciences. 2021; 22(21):11949. https://doi.org/10.3390/ijms222111949
Chicago/Turabian StyleSchwäbe, Frederic V., Emanuel K. Peter, Manuel H. Taft, and Dietmar J. Manstein. 2021. "Assessment of the Contribution of a Thermodynamic and Mechanical Destabilization of Myosin-Binding Protein C Domain C2 to the Pathomechanism of Hypertrophic Cardiomyopathy-Causing Double Mutation MYBPC3Δ25bp/D389V" International Journal of Molecular Sciences 22, no. 21: 11949. https://doi.org/10.3390/ijms222111949