
Cyanobacteria, Cyanotoxins, and Neurodegenerative Diseases: Dangerous Liaisons

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Cyanobacterial and Dinoflagellates Neurotoxins
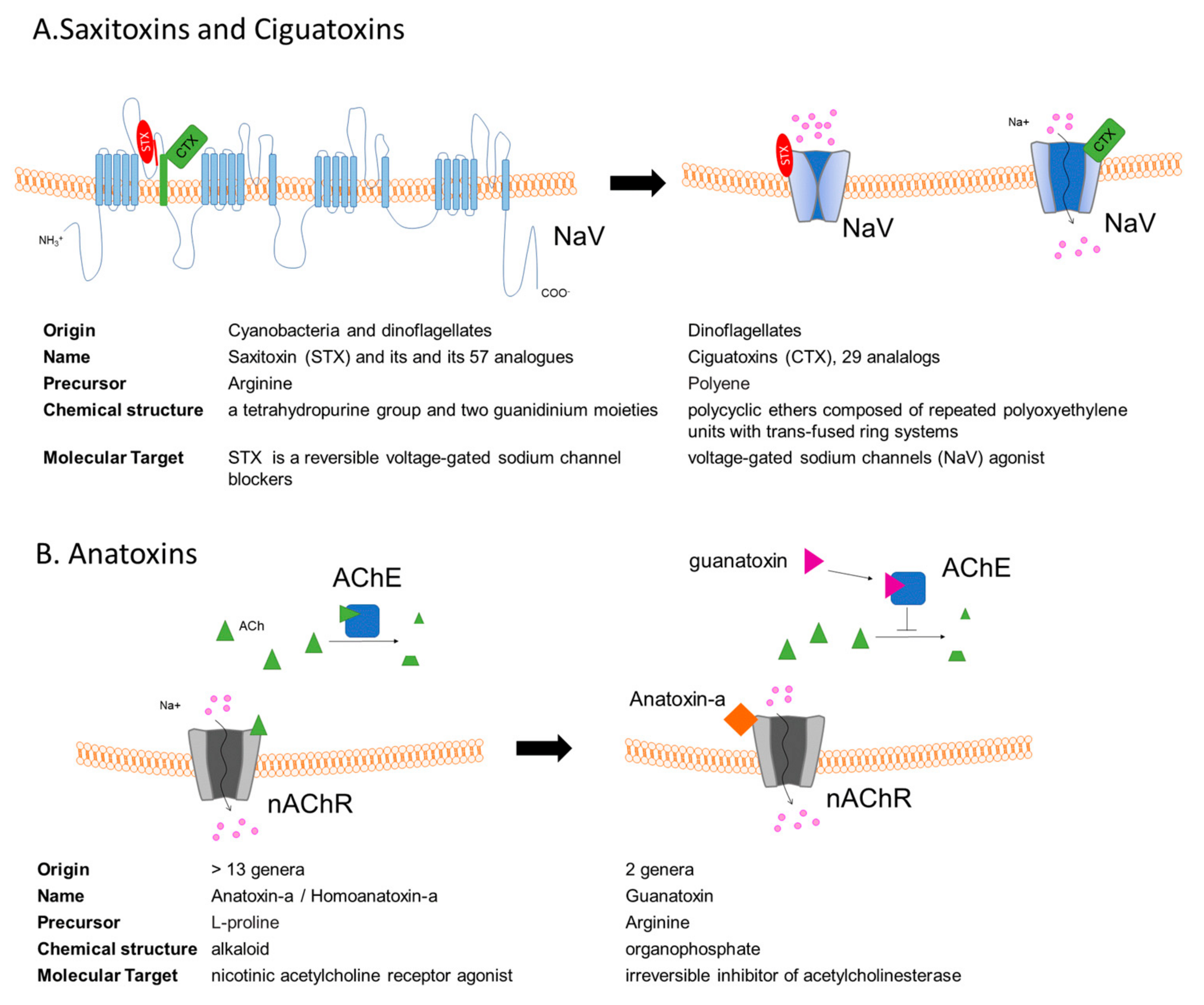
3. Saxitoxins and the Paralytic Shellfish Poisoning
4. Ciguatoxins
5. Anatoxins
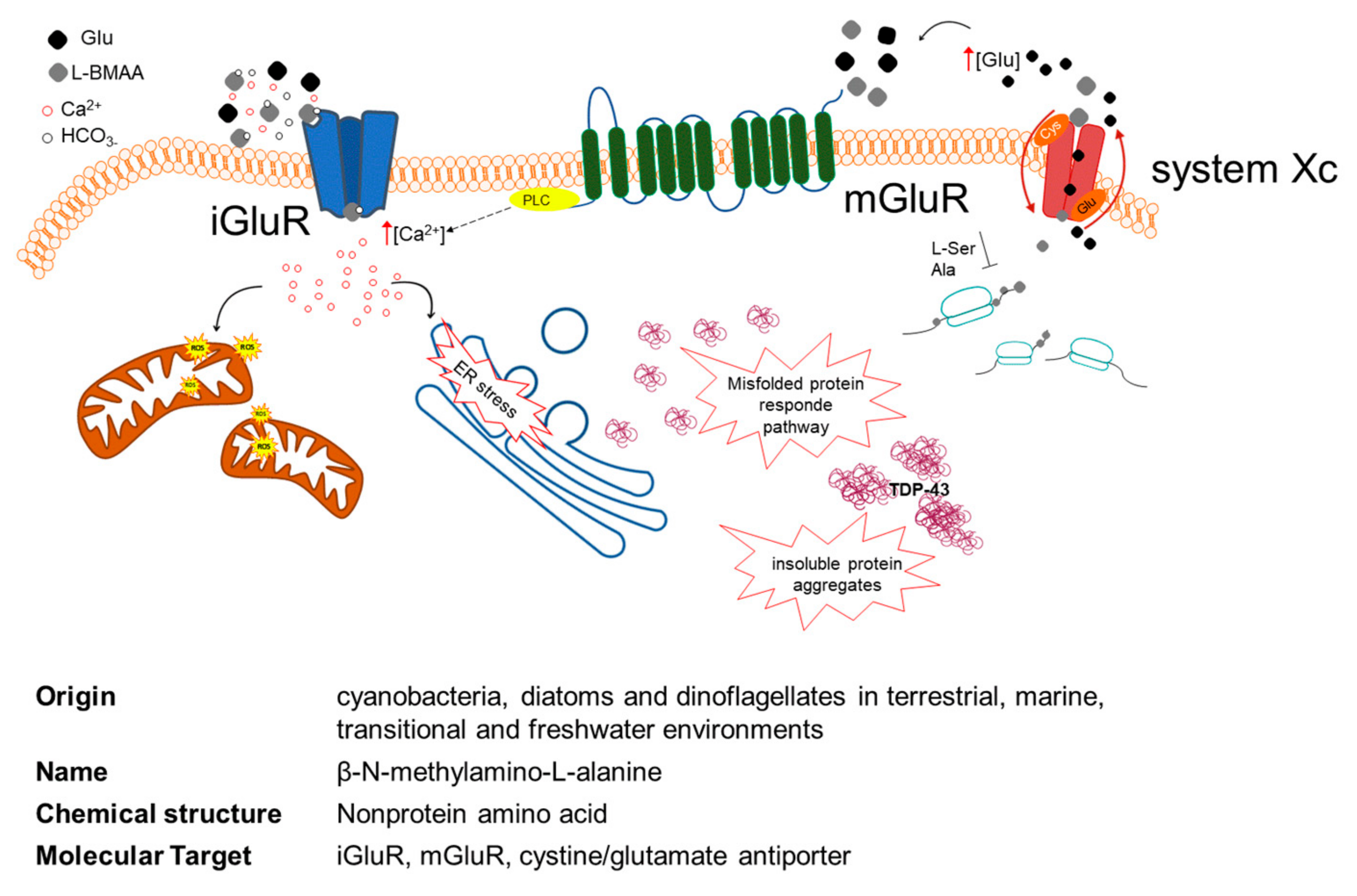
6. Role of L-BMAA in Neurodegenerative Diseases
7. Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Ruffini, N.; Klingenberg, S.; Schweiger, S.; Gerber, S. Common Factors in Neurodegeneration: A Meta-Study Revealing Shared Patterns on a Multi-Omics Scale. Cells 2020, 9, 2642. [Google Scholar] [CrossRef]
- Gunnarsson, L.-G.; Bodin, L. Amyotrophic Lateral Sclerosis and Occupational Exposures: A Systematic Literature Review and Meta-Analyses. Int. J. Environ. Res. Public Health 2018, 15, 2371. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yi, J.; Li, X.; Xiao, Y.; Dhakal, K.; Zhou, J. ALS-Associated Mutation SOD1(G93A) Leads to Abnormal Mitochondrial Dynamics in Osteocytes. Bone 2018, 106, 126–138. [Google Scholar] [CrossRef]
- Killin, L.O.J.; Starr, J.M.; Shiue, I.J.; Russ, T.C. Environmental Risk Factors for Dementia: A Systematic Review. BMC Geriatr. 2016, 16, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Signorelli, S.S.; Ferrante, M.; Zappia, M.; Nicoletti, A. Metals and Neurodegenerative Diseases. A Systematic Review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef]
- Racette, B.A.; Nielsen, S.S.; Criswell, S.R.; Sheppard, L.; Seixas, N.; Warden, M.N.; Checkoway, H. Dose-Dependent Progression of Parkinsonism in Manganese-Exposed Welders. Neurology 2017, 88, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, F.-C.; Goutman, S.A.; Chernay, S.; Mukherjee, B.; Callaghan, B.C.; Batterman, S.; Feldman, E.L. Association of Environmental Toxins With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016, 73, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, Paraquat, and Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Ascherio, A.; Schwarzschild, M.A. The Epidemiology of Parkinson’s Disease: Risk Factors and Prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Sánchez-Santed, F.; Colomina, M.T.; Herrero Hernández, E. Organophosphate Pesticide Exposure and Neurodegeneration. Cortex 2016, 74, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, L.-G.; Bodin, L. Occupational Exposures and Neurodegenerative Diseases-A Systematic Literature Review and Meta-Analyses. Int. J. Environ. Res. Public Health 2019, 16, 337. [Google Scholar] [CrossRef] [Green Version]
- Gunnarsson, L.-G.; Bodin, L. Alzheimer’s Disease and Occupational Exposures: A Systematic Literature Review and Meta-Analyses; 2015; Volume 17. In Alzheimer’s Disease & Treatment. Available online: http://openaccessebooks.com/alzheimers-disease-treatment/alzheimers-disease-and-occupational-exposures-a-systematic-literature-review-and-meta-analyses.pdf (accessed on 2 August 2021).
- Riancho, J.; Sanchez de la Torre, J.R.; Paz-Fajardo, L.; Limia, C.; Santurtun, A.; Cifra, M.; Kourtidis, K.; Fdez-Arroyabe, P. The Role of Magnetic Fields in Neurodegenerative Diseases. Int. J. Biometeorol. 2021, 65, 107–117. [Google Scholar] [CrossRef]
- Jacobs, B.M.; Belete, D.; Bestwick, J.; Blauwendraat, C.; Bandres-Ciga, S.; Heilbron, K.; Dobson, R.; Nalls, M.A.; Singleton, A.; Hardy, J.; et al. Parkinson’s Disease Determinants, Prediction and Gene–Environment Interactions in the UK Biobank. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1046–1054. [Google Scholar] [CrossRef]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-Analysis of Early Nonmotor Features and Risk Factors for Parkinson Disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Wallin, C.; Sholts, S.B.; Österlund, N.; Luo, J.; Jarvet, J.; Roos, P.M.; Ilag, L.; Gräslund, A.; Wärmländer, S.K.T.S. Alzheimer’s Disease and Cigarette Smoke Components: Effects of Nicotine, PAHs, and Cd(II), Cr(III), Pb(II), Pb(IV) Ions on Amyloid-β Peptide Aggregation. Sci. Rep. 2017, 7, 14423. [Google Scholar] [CrossRef] [Green Version]
- Menounos, S.; Hansbro, P.M.; Diwan, A.D.; Das, A. Pathophysiological Correlation between Cigarette Smoking and Amyotrophic Lateral Sclerosis. NeuroSci 2021, 2, 120–134. [Google Scholar] [CrossRef]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P.A. Environmental Risk Factors and Parkinson’s Disease: An Umbrella Review of Meta-Analyses. Parkinsonism Relat. Disord. 2016, 23, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaar, B.J.H.; de Leeuw, F.A.; Doorduijn, A.S.; Fieldhouse, J.L.P.; van de Rest, O.; Teunissen, C.E.; van Berckel, B.N.M.; Barkhof, F.; Visser, M.; de van der Schueren, M.A.E.; et al. Nutritional Status and Structural Brain Changes in Alzheimer’s Disease: The NUDAD Project. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12063. [Google Scholar] [CrossRef]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia Muciniphila in Overweight and Obese Human Volunteers: A Proof-of-Concept Exploratory Study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential Roles of Gut Microbiome and Metabolites in Modulating ALS in Mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The Gut Microbiome in Neurological Disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson Disease--the Gut-Brain Axis and Environmental Factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of Brain Amyloidosis with Pro-Inflammatory Gut Bacterial Taxa and Peripheral Inflammation Markers in Cognitively Impaired Elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Cox, P.A.; Kostrzewa, R.M.; Guillemin, G.J. BMAA and Neurodegenerative Illness. Neurotox. Res. 2018, 3, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M.F.; de Lima, S.T.; Carmichael, W.W.; McKinnie, S.M.K.; Chekan, J.R.; Moore, B.S. Guanitoxin, Re-Naming a Cyanobacterial Organophosphate Toxin. Harmful Algae 2020, 92, 101737. [Google Scholar] [CrossRef]
- Buratti, F.M.; Manganelli, M.; Vichi, S.; Stefanelli, M.; Scardala, S.; Testai, E.; Funari, E. Cyanotoxins: Producing Organisms, Occurrence, Toxicity, Mechanism of Action and Human Health Toxicological Risk Evaluation. Arch Toxicol. 2017, 91, 1049–1130. [Google Scholar] [CrossRef] [PubMed]
- Plaas, H.E.; Paerl, H.W. Toxic Cyanobacteria: A Growing Threat to Water and Air Quality. Environ. Sci. Technol. 2021, 55, 44–64. [Google Scholar] [CrossRef]
- Berntzon, L.; Ronnevi, L.O.; Bergman, B.; Eriksson, J. Detection of BMAA in the Human Central Nervous System. Neuroscience 2015, 292, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, R.A.; Banack, S.A.; Bishop, E.S.; Metcalf, J.S.; Murch, S.J.; Davis, D.A.; Stommel, E.W.; Karlsson, A.; Bribetto, A.; Chatziefthimiou, A.; et al. Is Exposure to BMAA a Risk Factor for Neurodegenerative Diseases? A Response to a Critical Review of the BMAA Hypothesis. Neurotox. Res. 2021, 39, 81–106. [Google Scholar] [CrossRef]
- Brooks, B.W.; Lazorchak, J.M.; Howard, M.D.; Johnson, M.V.; Morton, S.L.; Perkins, D.A.; Reavie, E.D.; Scott, G.I.; Smith, S.A.; Steevens, J.A. Are Harmful Algal Blooms Becoming the Greatest Inland Water Quality Threat to Public Health and Aquatic Ecosystems? Environ. Toxicol. Chem. 2016, 35, 6–13. [Google Scholar] [CrossRef]
- Anderson, D.M.; Fensin, E.; Gobler, C.J.; Hoeglund, A.E.; Hubbard, K.A.; Kulis, D.M.; Landsberg, J.H.; Lefebvre, K.A.; Provoost, P.; Richlen, M.L.; et al. Marine Harmful Algal Blooms (HABs) in the United States: History, Current Status and Future Trends. Harmful Algae 2021, 102, 101975. [Google Scholar] [CrossRef]
- Carpenter, S.R.; Caraco, N.F.; Correll, D.L.; Howarth, R.W.; Sharpley, A.N.; Smith, V.H. Nonpoint Pollution of Surface Waters with Phosphorus and Nitrogen. Ecol. Appl. 1998, 8, 559–568. [Google Scholar] [CrossRef]
- Smith, V.H.; Tilman, G.D.; Nekola, J.C. Eutrophication: Impacts of Excess Nutrient Inputs on Freshwater, Marine, and Terrestrial Ecosystems. Environ. Pollut. 1999, 100, 179–196. [Google Scholar] [CrossRef]
- Dodds, W.K.; Bouska, W.W.; Eitzmann, J.L.; Pilger, T.J.; Pitts, K.L.; Riley, A.J.; Schloesser, J.T.; Thornbrugh, D.J. Eutrophication of U.S. Freshwaters: Analysis of Potential Economic Damages. Environ. Sci. Technol. 2009, 43, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Assmy, P.; Smetacek, V. Algal Blooms. In Encyclopedia of Microbiology; Elsevier: New York, NY, USA, 2009; pp. 27–41. ISBN 978-0-12-373944-5. [Google Scholar]
- Paerl, H.W.; Otten, T.G. Harmful Cyanobacterial Blooms: Causes, Consequences, and Controls. Microb. Ecol. 2013, 65, 995–1010. [Google Scholar] [CrossRef]
- Merel, S.; Walker, D.; Chicana, R.; Snyder, S.; Baurès, E.; Thomas, O. State of Knowledge and Concerns on Cyanobacterial Blooms and Cyanotoxins. Environ. Int. 2013, 59, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, S.E.; International Lake Environment Committee and United Nations Environmental Programme. Lakes and Reservoirs. Volume 3, Water Quality: The Impact of Eutrophication; UNEP-IETC/ILEC. 2001. Available online: https://www.ilec.or.jp/wp-content/uploads/pub/Vol.3.pdf (accessed on 2 August 2021).
- Padedda, B.M.; Sechi, N.; Lai, G.G.; Mariani, M.A.; Pulina, S.; Sarria, M.; Satta, C.T.; Virdis, T.; Buscarinu, P.; Lugliè, A. Consequences of Eutrophication in the Management of Water Resources in Mediterranean Reservoirs: A Case Study of Lake Cedrino (Sardinia, Italy). Glob. Ecol. Conserv. 2017, 12, 21–35. [Google Scholar] [CrossRef]
- Mariani, M.A.; Padedda, B.M.; Kastovsky, J.; Buscarinu, P.; Sechi, N.; Virdis, T.; Luglie, A. Effects of Trophic Status on Microcystin Production and the Dominance of Cyanobacteria in the Phytoplankton Assemblage of Mediterranean Reservoirs. Sci. Rep. 2015, 5, 17964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, M.A.; Lai, G.G.; Padedda, B.M.; Pulina, S.; Sechi, N.; Virdis, T.; Lugliè, A. Long-Term Ecological Studies on Phytoplankton in Mediterranean Reservoirs: A Case Study from Sardinia (Italy). Inland Waters 2015, 5, 339–354. [Google Scholar] [CrossRef]
- Oliver, R.; Ganf, G. Freshwater blooms. In The Ecology of Cyanobacteria; Kluwer Academic Publishers: Dordrecht, The Netherland, 2000; pp. 149–194. [Google Scholar]
- Schindler, D.; Hecky, R.; Findlay, D.; Stainton, M.; Parker, B.; Paterson, M.; Beaty, K.; Lyng, M.; Kasian, S. Eutrophication of Lakes Cannot Be Controlled by Reducing Nitrogen Input: Results of a 37-Year Whole-Ecosystem Experiment|PNAS. Available online: https://www.pnas.org/content/105/32/11254 (accessed on 29 July 2021).
- Padedda, B.M.; Lugliè, A.; Ceccherelli, G.; Trebini, F.; Sechi, N. Nutrient-Flux Evaluation by the LOICZ Biogeochemical Model in Mediterranean Lagoons: The Case of Cabras Lagoon (Central-Western Sardinia). Chem. Ecol. 2010, 26, 147–162. [Google Scholar] [CrossRef]
- Seitzinger, S.P.; Kroeze, C. Global Distribution of Nitrous Oxide Production and N Inputs in Freshwater and Coastal Marine Ecosystems. Glob. Biogeochem. Cycles 1998, 12, 93–113. [Google Scholar] [CrossRef]
- Van Drecht, G.; Bouwman, A.F.; Knoop, J.M.; Beusen, A.H.W.; Meinardi, C.R. Global Modeling of the Fate of Nitrogen from Point and Nonpoint Sources in Soils, Groundwater, and Surface Water. Glob. Biogeochem. Cycles 2003, 17. [Google Scholar] [CrossRef]
- Tilman, D. Resource Competition between Plankton Algae: An Experimental and Theoretical Approach. Ecology 1977, 58, 338–348. [Google Scholar] [CrossRef]
- Smayda, T.J. Harmful Algal Blooms: Their Ecophysiology and General Relevance to Phytoplankton Blooms in the Sea. Limnol. Oceanogr. 1997, 42, 1137–1153. [Google Scholar] [CrossRef]
- Smayda, T.; Graneli, E.; Sundstrom, B.; Edler, L.; Anderson, D.; Smayda, T. Novel and Nuisance Phytoplankton Blooms in the Sea: Evidence for a Global Epidemic. Toxic Mar. Phytoplankton 1990, 1, 29–40. [Google Scholar]
- Anderson, D.M.; Glibert, P.M.; Burkholder, J.M. Harmful Algal Blooms and Eutrophication: Nutrient Sources, Composition, and Consequences. Estuaries 2002, 25, 704–726. [Google Scholar] [CrossRef]
- Glibert, P.M.; Harrison, J.; Heil, C.; Seitzinger, S. Escalating Worldwide Use of Urea—A Global Change Contributing to Coastal Eutrophication. Biogeochemistry 2006, 77, 441–463. [Google Scholar] [CrossRef]
- Burkholder, J.M.; Glibert, P.M.; Skelton, H.M. Mixotrophy, a Major Mode of Nutrition for Harmful Algal Species in Eutrophic Waters. Harmful Algae 2008, 8, 77–93. [Google Scholar] [CrossRef]
- Fadda, A.; Marková, S.; Kotlík, P.; Lugliè, A.; Padedda, B.; Buscarinu, P.; Sechi, N.; Manca, M. First Record of Planktonic Crustaceans in Sardinian Reservoirs. Biologia 2011, 66, 856–865. [Google Scholar] [CrossRef]
- Padedda, B.M.; Sechi, N.; Lai, G.G.; Mariani, M.A.; Pulina, S.; Satta, C.T.; Bazzoni, A.M.; Virdis, T.; Buscarinu, P.; Lugliè, A. A Fast-Response Methodological Approach to Assessing and Managing Nutrient Loads in Eutrophic Mediterranean Reservoirs. Ecol. Eng. 2015, 85, 47–55. [Google Scholar] [CrossRef]
- Zingone, A.; Escalera, L.; Aligizaki, K.; Fernández-Tejedor, M.; Ismael, A.; Montresor, M.; Mozetič, P.; Taş, S.; Totti, C. Toxic Marine Microalgae and Noxious Blooms in the Mediterranean Sea: A Contribution to the Global HAB Status Report. Harmful Algae 2021, 102, 101843. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Costa, D.; Magalhães, J.D.; G-Fernandes, M.; Cardoso, S.M.; Empadinhas, N. Microbial BMAA and the Pathway for Parkinson’s Disease Neurodegeneration. Front. Aging Neurosci. 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, P.S.; Palmer, V.S.; Kisby, G.E. Cycad β-N-Methylamino-L-Alanine (BMAA), Methylazoxymethanol, Genotoxicity, and Neurodegeneration. Toxicon 2018, 155, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Luglie, A.; Pulina, S.; Bruno, M.; Padedda, B.M.; Satta, C.T.; Sechi, N. Marine Toxins and Climate Change: The Case of PSP from Cyanobacteria in Coastal Lagoons. In Phycotoxins: Chemistry and Biochemistry, 2nd ed.; Botana, L.M., Alfonso, A., Eds.; Wiley-Blackwell: Malden, MA, USA, 2015; ISBN 978-1-118-50033-0. [Google Scholar]
- Rastogi, R.P.; Madamwar, D.; Incharoensakdi, A. Bloom Dynamics of Cyanobacteria and Their Toxins: Environmental Health Impacts and Mitigation Strategies. Front. Microbiol. 2015, 6, 1254. [Google Scholar] [CrossRef] [Green Version]
- Sha, J.; Xiong, H.; Li, C.; Lu, Z.; Zhang, J.; Zhong, H.; Zhang, W.; Yan, B. Harmful Algal Blooms and Their Eco-Environmental Indication. Chemosphere 2021, 274, 129912. [Google Scholar] [CrossRef]
- Moreira, C.; Vasconcelos, V.; Antunes, A. Phylogeny and Biogeography of Cyanobacteria and Their Produced Toxins. Mar. Drugs 2013, 11, 4350–4369. [Google Scholar] [CrossRef] [Green Version]
- Paerl, H.W.; Huisman, J. Climate. Blooms like It Hot. Science 2008, 320, 57–58. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, K.; Musgrave, I.F.; Humpage, A. Extended Low-Dose Exposure to Saxitoxin Inhibits Neurite Outgrowth in Model Neuronal Cells. Basic Clin. Pharmacol. Toxicol. 2017, 120, 390–397. [Google Scholar] [CrossRef] [Green Version]
- Drobac, D.; Tokodi, N.; Simeunovic, J.; Baltic, V.; Stanic, D.; Svircev, Z. Human Exposure to Cyanotoxins and Their Effects on Health. Arh. Hig. Rada Toksikol. 2013, 64, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, B.Y.; Zhu, X.; Sotto, P.; Paulino, Y. Oral Exposure to Environmental Cyanobacteria Toxins: Implications for Cancer Risk. Environ. Int. 2021, 148, 106381. [Google Scholar] [CrossRef]
- Rutkowska, M.; Płotka-Wasylka, J.; Majchrzak, T.; Wojnowski, W.; Mazur-Marzec, H.; Namieśnik, J. Recent Trends in Determination of Neurotoxins in Aquatic Environmental Samples. TrAC Trends Anal. Chem. 2019, 112, 112–122. [Google Scholar] [CrossRef]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic Alkaloids: Saxitoxin and Its Analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Kanamori, M.; Yoshida, H.; Okumura, Y.; Uchida, H.; Matsushima, R.; Oikawa, H.; Suzuki, T. Development of Ultra-Performance Liquid Chromatography with Post-Column Fluorescent Derivatization for the Rapid Detection of Saxitoxin Analogues and Analysis of Bivalve Monitoring Samples. Toxins 2019, 11, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batoréu, M.C.C.; Dias, E.; Pereira, P.; Franca, S. Risk of Human Exposure to Paralytic Toxins of Algal Origin. Environ. Toxicol. Pharmacol. 2005, 19, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Li, Z.; Jiang, Y.; Pan, X.; Wu, J.; Cristofori-Armstrong, B.; Smith, J.J.; Chin, Y.K.Y.; Lei, J.; Zhou, Q.; et al. Structural Basis for the Modulation of Voltage-Gated Sodium Channels by Animal Toxins. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Cervantes Cianca, R.C.; Pallares, M.A.; Durán Barbosa, R.; Vidal Adan, L.; Leão Martins, J.M.; Gago-Martínez, A. Application of Precolumn Oxidation HPLC Method with Fluorescence Detection to Evaluate Saxitoxin Levels in Discrete Brain Regions of Rats. Toxic. Off. J. Int. Soc. Toxinol. 2007, 49, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Wetzel, A.; Dombert, B.; Yadav, P.; Havlicek, S.; Jablonka, S.; Nassar, M.A.; Blum, R.; Sendtner, M. Role of Na(v)1.9 in Activity-Dependent Axon Growth in Motoneurons. Hum. Mol. Genet. 2012, 21, 3655–3667. [Google Scholar] [CrossRef]
- Da Silva, C.A.; de Morais, E.C.P.; Costa, M.D.M.; Ribas, J.L.C.; Guiloski, I.C.; Ramsdorf, W.A.; Zanata, S.M.; Cestari, M.M.; Ribeiro, C.A.O.; Magalhães, V.F.; et al. Saxitoxins Induce Cytotoxicity, Genotoxicity and Oxidative Stress in Teleost Neurons in Vitro. Toxic. Off. J. Int. Soc. Toxinol. 2014, 86, 8–15. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, X.; Chen, H.; Wang, Y. Comparative Studies of Saxitoxin (STX) -Induced Cytotoxicity in Neuro-2a and RTG-2 Cell Lines: An Explanation with Respect to Changes in ROS. Chemosphere 2018, 192, 66–74. [Google Scholar] [CrossRef]
- D’Mello, F.; Braidy, N.; Marcal, H.; Guillemin, G.; Rossi, F.; Chinian, M.; Laurent, D.; Teo, C.; Neilan, B.A. Cytotoxic Effects of Environmental Toxins on Human Glial Cells. Neurotox. Res. 2017, 31, 245–258. [Google Scholar] [CrossRef]
- Chen, G.; Jia, Z.; Wang, L.; Hu, T. Effect of Acute Exposure of Saxitoxin on Development of Zebrafish Embryos (Danio Rerio). Environ. Res. 2020, 185, 109432. [Google Scholar] [CrossRef] [PubMed]
- Lima-Filho, C.M.; Nogaroli, L.; Hedin-Pereira, C.; Azevedo, S.M.F.O.; Soares, R.M. Effects of Saxitoxins Exposure on Oligodendrocyte Development in Mouse Neonates. Toxicon 2020, 188, 89–94. [Google Scholar] [CrossRef] [PubMed]
- L’Herondelle, K.; Talagas, M.; Mignen, O.; Misery, L.; Le Garrec, R. Neurological Disturbances of Ciguatera Poisoning: Clinical Features and Pathophysiological Basis. Cells 2020, 9, 2291. [Google Scholar] [CrossRef] [PubMed]
- Vilarino, N.; Louzao, M.C.; Abal, P.; Cagide, E.; Carrera, C.; Vieytes, M.R.; Botana, L.M. Human Poisoning from Marine Toxins: Unknowns for Optimal Consumer Protection. Toxins 2018, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Au, N.P.; Kumar, G.; Asthana, P.; Tin, C.; Mak, Y.L.; Chan, L.L.; Lam, P.K.; Ma, C.H. Ciguatoxin Reduces Regenerative Capacity of Axotomized Peripheral Neurons and Delays Functional Recovery in Pre-Exposed Mice after Peripheral Nerve Injury. Sci. Rep. 2016, 6, 26809. [Google Scholar] [CrossRef] [Green Version]
- Inserra, M.C.; Israel, M.R.; Caldwell, A.; Castro, J.; Deuis, J.R.; Harrington, A.M.; Keramidas, A.; Garcia-Caraballo, S.; Maddern, J.; Erickson, A.; et al. Multiple Sodium Channel Isoforms Mediate the Pathological Effects of Pacific Ciguatoxin-1. Sci. Rep. 2017, 7, 42810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touska, F.; Sattler, S.; Malsch, P.; Lewis, R.J.; Reeh, P.W.; Zimmermann, K. Ciguatoxins Evoke Potent CGRP Release by Activation of Voltage-Gated Sodium Channel Subtypes NaV1.9, NaV1.7 and NaV1.1. Mar. Drugs 2017, 15, 269. [Google Scholar] [CrossRef] [Green Version]
- Russell, F.A.; King, R.; Smillie, S.-J.; Kodji, X.; Brain, S.D. Calcitonin Gene-Related Peptide: Physiology and Pathophysiology. Physiol. Rev. 2014, 94, 1099–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringer, C.; Tune, S.; Bertoune, M.A.; Schwarzbach, H.; Tsujikawa, K.; Weihe, E.; Schütz, B. Disruption of Calcitonin Gene-Related Peptide Signaling Accelerates Muscle Denervation and Dampens Cytotoxic Neuroinflammation in SOD1 Mutant Mice. Cell. Mol. Life Sci. CMLS 2017, 74, 339–358. [Google Scholar] [CrossRef]
- Ratliff, W.A.; Saykally, J.N.; Kane, M.J.; Citron, B.A. Neuromuscular Junction Morphology and Gene Dysregulation in the Wobbler Model of Spinal Neurodegeneration. J. Mol. Neurosci. MN 2018, 66, 114–120. [Google Scholar] [CrossRef]
- Morabito, S.; Silvestro, S.; Faggio, C. How the Marine Biotoxins Affect Human Health. Nat. Prod. Res. 2018, 32, 621–631. [Google Scholar] [CrossRef]
- Rubiolo, J.A.; Vale, C.; Boente-Juncal, A.; Hirama, M.; Yamashita, S.; Camiña, M.; Vieytes, M.R.; Botana, L.M. Transcriptomic Analysis of Ciguatoxin-Induced Changes in Gene Expression in Primary Cultures of Mice Cortical Neurons. Toxins 2018, 10, 192. [Google Scholar] [CrossRef] [Green Version]
- Coccini, T.; Caloni, F.; De Simone, U. Human Neuronal Cell Based Assay: A New in Vitro Model for Toxicity Evaluation of Ciguatoxin. Environ. Toxicol. Pharmacol. 2017, 52, 200–213. [Google Scholar] [CrossRef]
- Alonso, E.; Vieira, A.C.; Rodriguez, I.; Alvariño, R.; Gegunde, S.; Fuwa, H.; Suga, Y.; Sasaki, M.; Alfonso, A.; Cifuentes, J.M.; et al. Tetracyclic Truncated Analogue of the Marine Toxin Gambierol Modifies NMDA, Tau, and Amyloid β Expression in Mice Brains: Implications in AD Pathology. ACS Chem. Neurosci. 2017, 8, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Touska, F.; Hess, A.; Hinsbey, R.; Sattler, S.; Lampert, A.; Sergejeva, M.; Sharov, A.; Collins, L.S.; Eberhardt, M.; et al. Ciguatoxins Activate Specific Cold Pain Pathways to Elicit Burning Pain from Cooling. EMBO J. 2012, 31, 3795–3808. [Google Scholar] [CrossRef] [Green Version]
- Asthana, P.; Zhang, N.; Kumar, G.; Chine, V.B.; Singh, K.K.; Mak, Y.L.; Chan, L.L.; Lam, P.K.S.; Ma, C.H.E. Pacific Ciguatoxin Induces Excitotoxicity and Neurodegeneration in the Motor Cortex Via Caspase 3 Activation: Implication for Irreversible Motor Deficit. Mol. Neurobiol. 2018, 55, 6769–6787. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Liu, H.; Yuan, L.; Wang, Y.; Ma, Y.; Wang, R.; Chen, X.; Losiewicz, M.D.; Guo, H.; Zhang, H. The Diversity of Cyanobacterial Toxins on Structural Characterization, Distribution and Identification: A Systematic Review. Toxins 2019, 11, 530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, C.E.; Witkop, B.; Albuquerque, E.X. Anatoxin-a: A Novel, Potent Agonist at the Nicotinic Receptor. Mol. Pharmacol. 1980, 18, 384–394. [Google Scholar]
- Posadas, I.; López-Hernández, B.; Ceña, V. Nicotinic Receptors in Neurodegeneration. Curr. Neuropharmacol. 2013, 11, 298–314. [Google Scholar] [CrossRef] [Green Version]
- Koukouli, F.; Changeux, J.-P. Do Nicotinic Receptors Modulate High-Order Cognitive Processing? Trends Neurosci. 2020, 43, 550–564. [Google Scholar] [CrossRef]
- Moss, D.E. Improving Anti-Neurodegenerative Benefits of Acetylcholinesterase Inhibitors in Alzheimer’s Disease: Are Irreversible Inhibitors the Future? Int. J. Mol. Sci. 2020, 21, 3438. [Google Scholar] [CrossRef] [PubMed]
- Takser, L.; Benachour, N.; Husk, B.; Cabana, H.; Gris, D. Cyanotoxins at Low Doses Induce Apoptosis and Inflammatory Effects in Murine Brain Cells: Potential Implications for Neurodegenerative Diseases. Toxicol. Rep. 2016, 3, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunn, P.B. 50 Years of Research on α-Amino-β-Methylaminopropionic Acid (β-Methylaminoalanine). Phytochemistry 2017, 144, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, N.; Hill, D.J.; Diggs, D.L.; Faison, B.D.; Francis, B.M.; Lang, J.R.; Larue, M.M.; Le, T.-T.; Loftin, K.A.; Lugo, J.N.; et al. A Critical Review of the Postulated Role of the Non-Essential Amino Acid, β-N-Methylamino-L-Alanine, in Neurodegenerative Disease in Humans. J. Toxicol. Environ. Health Part B 2017, 20, 183–229. [Google Scholar] [CrossRef] [PubMed]
- Pablo, J.; Banack, S.A.; Cox, P.A.; Johnson, T.E.; Papapetropoulos, S.; Bradley, W.G.; Buck, A.; Mash, D.C. Cyanobacterial Neurotoxin BMAA in ALS and Alzheimer’s Disease. Acta Neurol. Scand. 2009, 120, 216–225. [Google Scholar] [CrossRef]
- Spencer, P.S.; Nunn, P.B.; Hugon, J.; Ludolph, A.C.; Robertson, R.C. Guam Amyotrophic Lateral Sclerosis-Parkinsonism- Dementia Linked to a Plant Excitant Neurotoxin. Science 1987, 237, 517–522. [Google Scholar] [CrossRef]
- Cox, P.A.; Banack, S.A.; Murch, S.J.; Rasmussen, U.; Tien, G.; Bidigare, R.R.; Metcalf, J.S.; Morrison, L.F.; Codd, G.A.; Bergman, B. Diverse Taxa of Cyanobacteria Produce Beta-N-Methylamino-L-Alanine, a Neurotoxic Amino Acid. Proc. Natl. Acad. Sci. USA 2005, 102, 5074–5078. [Google Scholar] [CrossRef] [Green Version]
- Cox, P.A.; Banack, S.A.; Murch, S.J. Biomagnification of Cyanobacterial Neurotoxins and Neurodegenerative Disease among the Chamorro People of Guam. Proc. Natl. Acad. Sci. USA 2003, 100, 13380–13383. [Google Scholar] [CrossRef] [Green Version]
- Lage, S.; Annadotter, H.; Rasmussen, U.; Rydberg, S. Biotransfer of β-N-Methylamino-L-Alanine (BMAA) in a Eutrophicated Freshwater Lake. Mar. Drugs 2015, 13, 1185–1201. [Google Scholar] [CrossRef] [Green Version]
- Samardzic, K.; Steele, J.R.; Violi, J.P.; Colville, A.; Mitrovic, S.M.; Rodgers, K.J. Toxicity and Bioaccumulation of Two Non-Protein Amino Acids Synthesised by Cyanobacteria, β-N-Methylamino-L-Alanine (BMAA) and 2,4-Diaminobutyric Acid (DAB), on a Crop Plant. Ecotoxicol. Environ. Saf. 2021, 208, 111515. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Basile, M.; Mash, D.C. Cerebral Uptake and Protein Incorporation of Cyanobacterial Toxin β-N-Methylamino-L-Alanine. Neuroreport 2013, 24, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.J.; Cox, P.A.; Banack, S.A.; Steele, J.C.; Sacks, O.W. Occurrence of Beta-Methylamino-l-Alanine (BMAA) in ALS/PDC Patients from Guam. Acta Neurol. Scand. 2004, 110, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Bradley, W.G.; Mash, D.C. Beyond Guam: The Cyanobacteria/BMAA Hypothesis of the Cause of ALS and Other Neurodegenerative Diseases. Amyotroph. Lateral Scler. 2009, 10 (Suppl. 2), 7–20. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.A.; Guillemin, G.J. The Cyanotoxin and Non-Protein Amino Acid β-Methylamino-L-Alanine (L-BMAA) in the Food Chain: Incorporation into Proteins and Its Impact on Human Health. Neurotox. Res. 2019, 36, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Lobner, D. Mechanisms of Beta-N-Methylamino-L-Alanine Induced Neurotoxicity. Amyotroph. Lateral Scler. 2009, 10 (Suppl. 2), 56–60. [Google Scholar] [CrossRef]
- Chiu, A.S.; Gehringer, M.M.; Braidy, N.; Guillemin, G.J.; Welch, J.H.; Neilan, B.A. Excitotoxic Potential of the Cyanotoxin β-Methyl-Amino-l-Alanine (BMAA) in Primary Human Neurons. Toxicon 2012, 60, 1159–1165. [Google Scholar] [CrossRef]
- Weiss, J.H.; Choi, D.W. Beta-N-Methylamino-L-Alanine Neurotoxicity: Requirement for Bicarbonate as a Cofactor. Sci. New Ser. 1988, 241, 973–975. [Google Scholar] [CrossRef]
- Delcourt, N.; Claudepierre, T.; Maignien, T.; Arnich, N.; Mattei, C. Cellular and Molecular Aspects of the β-N-Methylamino-l-Alanine (BMAA) Mode of Action within the Neurodegenerative Pathway: Facts and Controversy. Toxins 2017, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Primer 2017, 3, 17071. [Google Scholar] [CrossRef]
- Liu, X.; Rush, T.; Zapata, J.; Lobner, D. β-N-Methylamino-l-Alanine Induces Oxidative Stress and Glutamate Release through Action on System Xc−. Exp. Neurol. 2009, 217, 429–433. [Google Scholar] [CrossRef]
- Dunlop, R.A.; Cox, P.A.; Banack, S.A.; Rodgers, K.J. The Non-Protein Amino Acid BMAA Is Misincorporated into Human Proteins in Place of l-Serine Causing Protein Misfolding and Aggregation. PLoS ONE 2013, 8, e75376. [Google Scholar] [CrossRef] [Green Version]
- Glover, W.B.; Mash, D.C.; Murch, S.J. The Natural Non-Protein Amino Acid N-β-Methylamino-l-Alanine (BMAA) Is Incorporated into Protein during Synthesis. Amino Acids 2014, 46, 2553–2559. [Google Scholar] [CrossRef] [PubMed]
- Sanna, S.; Esposito, S.; Masala, A.; Sini, P.; Nieddu, G.; Galioto, M.; Fais, M.; Iaccarino, C.; Cestra, G.; Crosio, C. HDAC1 Inhibition Ameliorates TDP-43-Induced Cell Death in Vitro and in Vivo. Cell Death Dis. 2020, 11, 369. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The Role of TDP-43 Propagation in Neurodegenerative Diseases: Integrating Insights from Clinical and Experimental Studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- De Munck, E.; Munoz-Saez, E.; Miguel, B.G.; Solas, M.T.; Ojeda, I.; Martinez, A.; Gil, C.; Arahuetes, R.M. Beta-N-Methylamino-l-Alanine Causes Neurological and Pathological Phenotypes Mimicking Amyotrophic Lateral Sclerosis (ALS): The First Step towards an Experimental Model for Sporadic ALS. Environ. Toxicol. Pharmacol. 2013, 36, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.; Downing, T. Dose-Dependent Adult Neurodegeneration in a Rat Model After Neonatal Exposure to β-N-Methylamino-l-Alanine. Neurotox. Res. 2019, 35, 711–723. [Google Scholar] [CrossRef]
- Yin, H.Z.; Yu, S.; Hsu, C.-I.; Liu, J.; Acab, A.; Wu, R.; Tao, A.; Chiang, B.J.; Weiss, J.H. Intrathecal Infusion of BMAA Induces Selective Motor Neuron Damage and Astrogliosis in the Ventral Horn of the Spinal Cord. Exp. Neurol. 2014, 261, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ritson, G.P.; Custer, S.K.; Freibaum, B.D.; Guinto, J.B.; Geffel, D.; Moore, J.; Tang, W.; Winton, M.J.; Neumann, M.; Trojanowski, J.Q.; et al. TDP-43 Mediates Degeneration in a Novel Drosophila Model of Disease Caused by Mutations in VCP/P97. J. Neurosci. 2010, 30, 7729–7739. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.M.; Bereman, M.S.; Marsden, K.C. BMAA and MCLR Interact to Modulate Behavior and Exacerbate Molecular Changes Related to Neurodegeneration in Larval Zebrafish. Toxicol. Sci. Off. J. Soc. Toxicol. 2020, 179, 251–261. [Google Scholar] [CrossRef]
- Okle, O.; Stemmer, K.; Deschl, U.; Dietrich, D.R. L-BMAA Induced ER Stress and Enhanced Caspase 12 Cleavage in Human Neuroblastoma SH-SY5Y Cells at Low Nonexcitotoxic Concentrations. Toxicol. Sci. 2013, 131, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Beri, J.; Nash, T.; Martin, R.M.; Bereman, M.S. Exposure to BMAA Mirrors Molecular Processes Linked to Neurodegenerative Disease. PROTEOMICS 2017, 17, 1700161. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunys, J.; Duplan, E.; Checler, F. The Transcription Factor X-Box Binding Protein-1 in Neurodegenerative Diseases. Mol. Neurodegener. 2014, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierozan, P.; Cattani, D.; Karlsson, O. Hippocampal Neural Stem Cells Are More Susceptible to the Neurotoxin BMAA than Primary Neurons: Effects on Apoptosis, Cellular Differentiation, Neurite Outgrowth, and DNA Methylation. Cell Death Dis. 2020, 11, 910. [Google Scholar] [CrossRef]
- Greene, L.A.; Tischler, A.S. Establishment of a Noradrenergic Clonal Line of Rat Adrenal Pheochromocytoma Cells Which Respond to Nerve Growth Factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; McGeer, P.L. Weak BMAA Toxicity Compares with That of the Dietary Supplement Beta-Alanine. Neurobiol. Aging 2012, 33, 1440–1447. [Google Scholar] [CrossRef]
- Munoz-Saez, E.; de Munck Garcia, E.; Arahuetes Portero, R.M.; Vicente, F.; Ortiz-Lopez, F.J.; Cantizani, J.; Gomez Miguel, B. Neuroprotective Role of Sphingosine-1-Phosphate in L-BMAA Treated Neuroblastoma Cells (SH-SY5Y). Neurosci. Lett. 2015, 593, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Main, B.J.; Dunlop, R.A.; Rodgers, K.J. The Use of L-Serine to Prevent Beta-Methylamino-L-Alanine (BMAA)-Induced Proteotoxic Stress in Vitro. Toxicon 2016, 109, 7–12. [Google Scholar] [CrossRef]
- Engskog, M.K.R.; Ersson, L.; Haglöf, J.; Arvidsson, T.; Pettersson, C.; Brittebo, E. β-N-Methylamino-l-Alanine (BMAA) Perturbs Alanine, Aspartate and Glutamate Metabolism Pathways in Human Neuroblastoma Cells as Determined by Metabolic Profiling. Amino Acids 2017, 49, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.A.; Carney, J.M. Mechanisms of L-Serine-Mediated Neuroprotection Include Selective Activation of Lysosomal Cathepsins B and L. Neurotox. Res. 2020, 39, 17–26. [Google Scholar] [CrossRef]
- Silva, D.F.; Candeias, E.; Esteves, A.R.; Magalhães, J.D.; Ferreira, I.L.; Nunes-Costa, D.; Rego, A.C.; Empadinhas, N.; Cardoso, S.M. Microbial BMAA Elicits Mitochondrial Dysfunction, Innate Immunity Activation, and Alzheimer’s Disease Features in Cortical Neurons. J. Neuroinflamm. 2020, 17, 332. [Google Scholar] [CrossRef]
- Van Onselen, R.; Venables, L.; van de Venter, M.; Downing, T.G. β-N-Methylamino-L-Alanine Toxicity in PC12: Excitotoxicity vs. Misincorporation. Neurotox. Res. 2018, 33, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Soto, T.; Buzzi, E.D.; Rotstein, N.P.; German, O.L.; Politi, L.E. Damaging Effects of BMAA on Retina Neurons and Müller Glial Cells. Exp. Eye Res. 2021, 202, 108342. [Google Scholar] [CrossRef]
- Okamoto, S.; Esumi, S.; Hamaguchi-Hamada, K.; Hamada, S. β-N-Methylamino-L-Alanine (BMAA) Suppresses Cell Cycle Progression of Non-Neuronal Cells. Sci. Rep. 2018, 8, 17995. [Google Scholar] [CrossRef] [Green Version]
- Richter, K.E.; Mena, E.E. L-ß-Methylaminoalanine Inhibits [3H]Glutamate Binding in the Presence of Bicarbonate Ions. Brain Res. 1989, 492, 385–388. [Google Scholar] [CrossRef]
- Staton, P.C.; Bristow, D.R. The Dietary Excitotoxins β-N-Methylamino-l-Alanine and β-N-Oxalylamino-l-Alanine Induce Necrotic- and Apoptotic-Like Death of Rat Cerebellar Granule Cells. J. Neurochem. 1997, 69, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Lobner, D.; Piana, P.M.T.; Salous, A.K.; Peoples, R.W. β-N-Methylamino-l-Alanine Enhances Neurotoxicity through Multiple Mechanisms. Neurobiol. Dis. 2007, 25, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Tan, V.X.; Mazzocco, C.; Varney, B.; Bodet, D.; Guillemin, T.A.; Bessede, A.; Guillemin, G.J. Detection of the Cyanotoxins L-BMAA Uptake and Accumulation in Primary Neurons and Astrocytes. Neurotox. Res. 2018, 33, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Tan, V.X.; Lassus, B.; Lim, C.K.; Tixador, P.; Courte, J.; Bessede, A.; Guillemin, G.J.; Peyrin, J.-M. Neurotoxicity of the Cyanotoxin BMAA Through Axonal Degeneration and Intercellular Spreading. Neurotox. Res. 2018, 33, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.E.; King, S.R.; Banack, S.A.; Webster, C.; Callanaupa, W.J.; Cox, P.A. Cyanobacteria (Nostoc Commune) Used as a Dietary Item in the Peruvian Highlands Produce the Neurotoxic Amino Acid BMAA. J. Ethnopharmacol. 2008, 118, 159–165. [Google Scholar] [CrossRef]
- Backer, L.C.; McNeel, S.V.; Barber, T.; Kirkpatrick, B.; Williams, C.; Irvin, M.; Zhou, Y.; Johnson, T.B.; Nierenberg, K.; Aubel, M.; et al. Recreational Exposure to Microcystins during Algal Blooms in Two California Lakes. Toxic. Off. J. Int. Soc. Toxinol. 2010, 55, 909–921. [Google Scholar] [CrossRef]
- Cox, P.A. BMAA, Neurodegeneration, and Neuroprotection. Neurotox. Res. 2021, 39, 1–5. [Google Scholar] [CrossRef]
- Scott, L.L.; Downing, S.; Downing, T.G. The Evaluation of BMAA Inhalation as a Potential Exposure Route Using a Rat Model. Neurotox. Res. 2018, 33, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.D.; Miller, R.G.; Bradley, W.G.; Moore, D.H.; Saperstein, D.S.; Flynn, L.E.; Katz, J.S.; Forshew, D.A.; Metcalf, J.S.; Banack, S.A.; et al. Phase I Clinical Trial of Safety of L-Serine for ALS Patients. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Fridman, V.; Suriyanarayanan, S.; Novak, P.; David, W.; Macklin, E.A.; McKenna-Yasek, D.; Walsh, K.; Aziz-Bose, R.; Oaklander, A.L.; Brown, R.; et al. Randomized Trial of L-Serine in Patients with Hereditary Sensory and Autonomic Neuropathy Type 1. Neurology 2019, 92, e359–e370. [Google Scholar] [CrossRef]
- Brenner, S.R. Blue-Green Algae or Cyanobacteria in the Intestinal Micro-Flora May Produce Neurotoxins Such as Beta-N-Methylamino-L-Alanine (BMAA) Which May Be Related to Development of Amyotrophic Lateral Sclerosis, Alzheimer’s Disease and Parkinson-Dementia-Complex in Humans and Equine Motor Neuron Disease in Horses. Med. Hypotheses 2013, 80, 103. [Google Scholar] [CrossRef]
- Brenner, D.; Hiergeist, A.; Adis, C.; Mayer, B.; Gessner, A.; Ludolph, A.C.; Weishaupt, J.H. The Fecal Microbiome of ALS Patients. Neurobiol. Aging 2018, 61, 132–137. [Google Scholar] [CrossRef]
- Ceppa, F.A.; Izzo, L.; Sardelli, L.; Raimondi, I.; Tunesi, M.; Albani, D.; Giordano, C. Human Gut-Microbiota Interaction in Neurodegenerative Disorders and Current Engineered Tools for Its Modeling. Front. Cell. Infect. Microbiol. 2020, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Palmas, V.; Melis, M.; Pisanu, S.; Cusano, R.; Uva, P.; Perra, D.; Madau, V.; Sarchioto, M.; Oppo, V.; et al. Gut Microbiota and Metabolome Alterations Associated with Parkinson’s Disease. Msystems 2020, 5, e00561-20. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environmental Factors | Effects | Diseases | Reference |
|---|---|---|---|
| Heavy metals | Lead (crosses the blood–brain barrier and accumulates in neuronal and glial cells) | ALS 1 | [3,4] |
| Aluminium | AD 2 | [5,6] | |
| Manganese | PD 3 | [6,7] | |
| Pesticide | Pentachlorobenzene | ALS | [8] |
| Rotenone and paraquat | ALS, PD | [9,10] | |
| Organophosphate pesticides | ALS, PD, AD | [11] | |
| Electromagnetic fields | Contradictory results | ALS, AD | [12,13,14] |
| Smoking | Protective | PD | [15,16] |
| Risk factor | AD, ALS | [17,18] | |
| Physical activity | Protective | PD | [19] |
| Body mass index and nutritional state | Lower nutritional parameters | AD | [20] |
| Microbiota structure and dysfunction of the gut–brain axis | Akkermansia muciniphila reduces symptoms; Ruminococcus torques and Parabacteroides distasonis | ALS | [21,22] |
| Suppression of Prevotellaceae and anti-inflammatory genera; blooming of pro-inflammatory Proteobacteria, Enterococcaceae, and Enterobacteriaceae | PD | [23,24,25] | |
| Suppression of anti-inflammatory taxa such as Eubacterium rectale and a profusion of pro-inflammatory taxa such as Escherichia and Shigella | AD | [24,26] | |
| Cyanobacteria and cyanotoxins | Risk factors | ALS, PD, AD | [27,28] |
| Experimental Model | Saxitoxins Exposure Protocol | Effects | Reference |
|---|---|---|---|
| primary neuron culture from tropical freshwater fish | 0.3–3.0 mg L−1 24h | oxidative stress, neurotoxicity, genotoxicity and apoptosis | [76] |
| murine neuroblastoma N2A | 0–256 nM 24–48 h | high levels of ROS generation mild cytotoxic or apoptotic effects | [77] |
| rainbow trout fish cell line RTG-2 | 0–256 nM 24–48 h | mild cytotoxic or apoptotic effects | [77] |
| human primary astrocytes | high levels of ROS generation reduced cell survival | [78] | |
| zebrafish embryos | 0.05–0.1 µM | adverse effect on development of zebrafish embryos, oxidative stress-induced apoptosis | [79] |
| mouse neonate brain | single intraperitoneal 7.5 μg kg−1 body weight in pregnant mice | increased proliferation of OPCs, but not maturation process of these cells | [80] |
| Experimental Model | Ciguatoxins Exposure Protocol | Molecular Target | effects | Reference |
|---|---|---|---|---|
| SH-SY5Y | 25 pM–100 nM P-CTX-3C short-(4–24 h) and long-term exposure (10 days) | cytotoxic effect, alterations of the mitochondrial metabolism, cell morphology, and [Ca2+]i | [91] | |
| primary cortical neurons | 5 nM CTX3C 6-24- 72 h | gene expression alteration mediated by voltage-gated sodium channel | [90] | |
| C57BL/6 mice | shallow intraplantar (i.pl.) injection of P-CTX-1 (1–10 nM) | Nav 1.8 and TTXs Nav subtypes are effectors of ciguatoxin-induced cold allodynia | spontaneous pain | [93] |
| transgenic mice and rat | 0.01–31 nM P-CTX-1 (>95% purity) isolated from moray eel (Gymnothorax javanicus) liver | NaV1.9 | release of calcitonin-gene related peptide (CGRP) from nerve terminals | [85] |
| C57BL/6 mice | (0.26 ng/g body weight) intraperitoneally on day 0 followed by second exposure on day 3 P-CTX-1 (isolated and purified from moray eels) | irreversible motor deficit in 4-month pre-exposed mice following peripheral nerve injury astrogliosis and excitotoxic neuronal cell death via the activation of caspase 3 in motor cortex | [94] |
| Experimental Model | L-BMAA Exposure Protocol | Molecular Target | Reference |
|---|---|---|---|
| SH-SY5Y | 3 mM plus antagonist for kainate/AMPA receptors 5 days | low neurotoxicity of BMAA and weak action at glutamatergic receptors | [135] |
| 0.1 mM 48h | Low non-excitotoxic BMAA concentrations induce effects on the ubiquitin/proteasome system not ROS-related | [129] | |
| 3–10 mM 48h | decrease cell viability in a dose-response manner and evoke alterations in GSK3β and TDP-43 | [136] | |
| 0.5 mM 24h–48h–72h | Increased caspase-3 activity and cathepsins, ER stress | [137] | |
| 0.05–0.25–1 mM 24 h | alterations in alanine, aspartate, and glutamate metabolism | [138] | |
| 0.1–1 mM 24–48 h | autophagy | [139] | |
| 3 mM 48h | disrupts mitochondrial metabolism | [140] | |
| PC12 | 2 mM 6–12 h | apoptosis and mGluR1 increase | [141] |
| 0.4–1 mM 48h | promoted cell death and axon-like outgrowth | [142] | |
| NSC-34 | 0.1–1 mM 72 h | exposure to BMAA causes protein misfolding, ER stress, induction of the UPR, disruption of the mitochondrial function | [130,141] |
| NIH/3T3 | 1–3 mM 48–96 h | L-BMAA causes arrest of cell cycle progression at the G1/S. No evidence of cell membrane damage, apoptosis, or ROS overproduction | [143] |
| primary cortical neurons | 3 mM 1 h 20 mM HCO3- | L-BMAA activity is dependent on HCO3-, resulting in a destruction of cortical neuronal population. | [115] [144] |
| primary cerebellar granule cells colture, rat | up to 3 mM 24–48h | L-BMAA induced both necrotic- and apoptotic-like cell death | [145] |
| primary neurons and astrocytes cortical cell cultures, fetal mouse | 3–10 mM 3–24h 0.1 mM 48h | enhancement death of cortical neurons damaged by other insults; oxidative stress, Wallerian-Like Degeneration | [146,147,148] |
| neural stem cells | 50 µM–3 mM 24 h | apoptosis, cellular differentiation, neurite outgrowth, and DNA methylation | [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sini, P.; Dang, T.B.C.; Fais, M.; Galioto, M.; Padedda, B.M.; Lugliè, A.; Iaccarino, C.; Crosio, C. Cyanobacteria, Cyanotoxins, and Neurodegenerative Diseases: Dangerous Liaisons. Int. J. Mol. Sci. 2021, 22, 8726. https://doi.org/10.3390/ijms22168726
Sini P, Dang TBC, Fais M, Galioto M, Padedda BM, Lugliè A, Iaccarino C, Crosio C. Cyanobacteria, Cyanotoxins, and Neurodegenerative Diseases: Dangerous Liaisons. International Journal of Molecular Sciences. 2021; 22(16):8726. https://doi.org/10.3390/ijms22168726
Chicago/Turabian StyleSini, Paola, Thi Bang Chau Dang, Milena Fais, Manuela Galioto, Bachisio Mario Padedda, Antonella Lugliè, Ciro Iaccarino, and Claudia Crosio. 2021. "Cyanobacteria, Cyanotoxins, and Neurodegenerative Diseases: Dangerous Liaisons" International Journal of Molecular Sciences 22, no. 16: 8726. https://doi.org/10.3390/ijms22168726