
Structural and Functional Analysis of Disease-Linked p97 ATPase Mutant Complexes

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
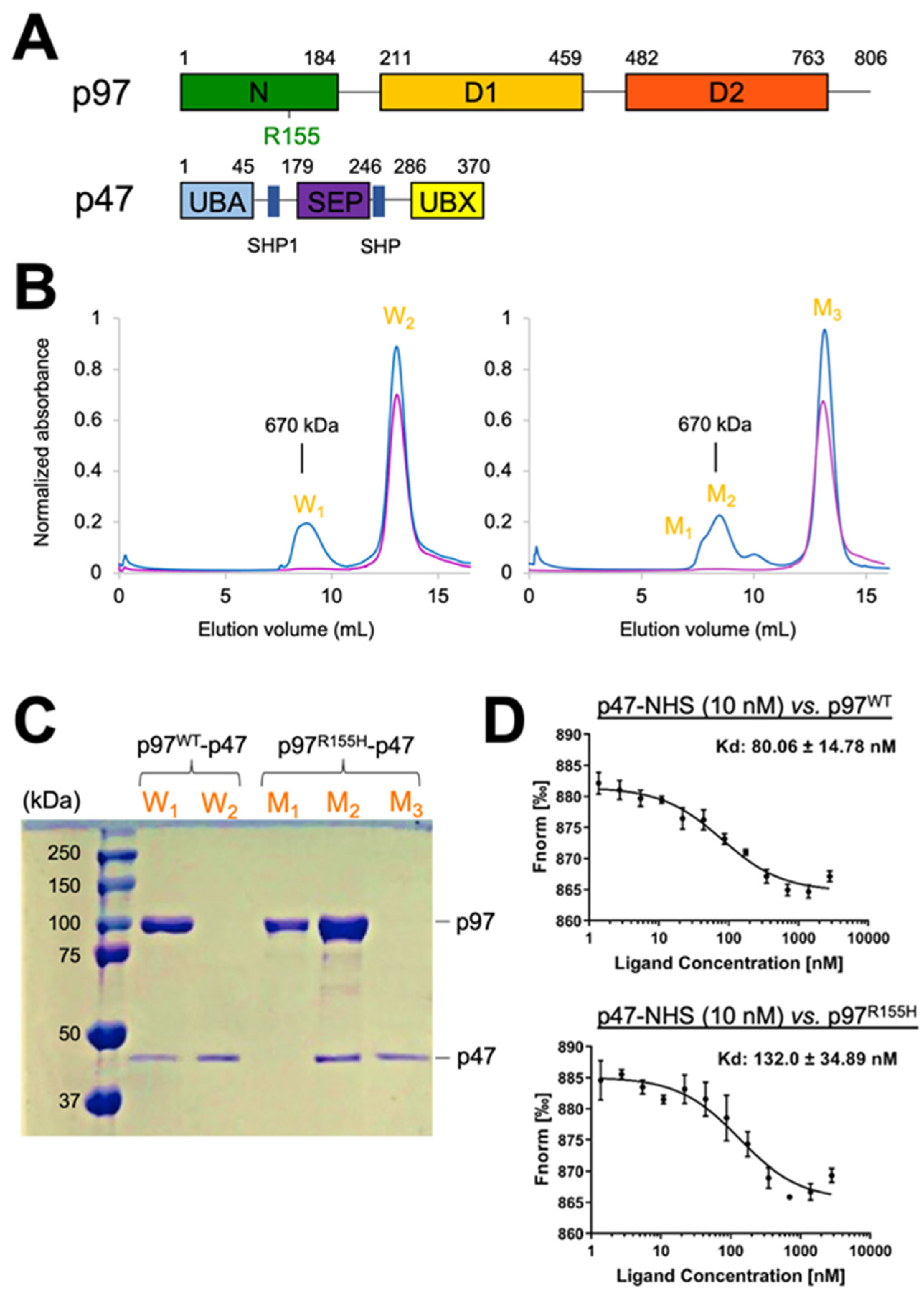
2.1. Two Molecular Populations Are in the p97R155H-p47 Assembly
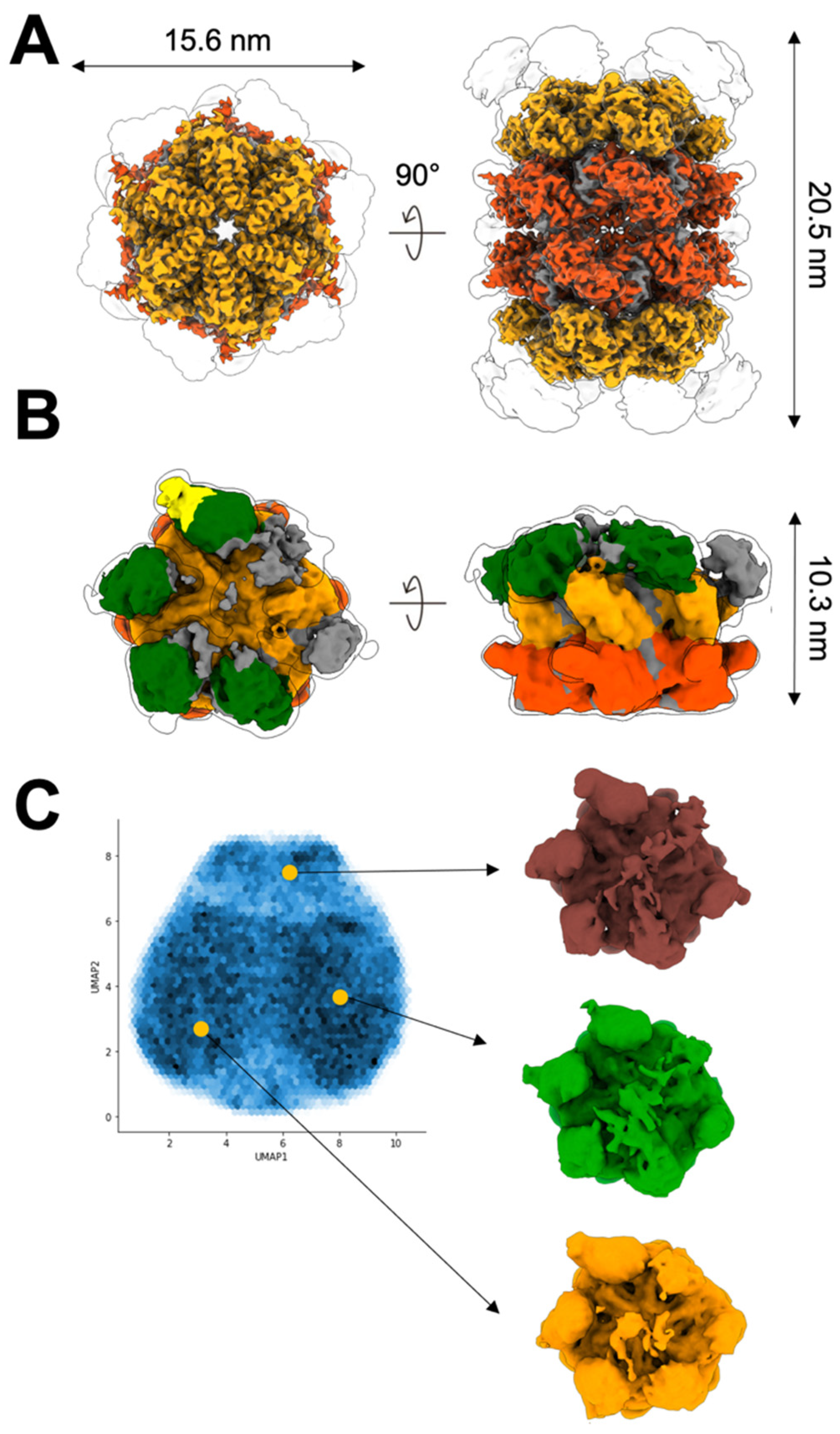
2.2. Up NTDs of the p97R155H Mutant and p47 Are Structurally Disordered
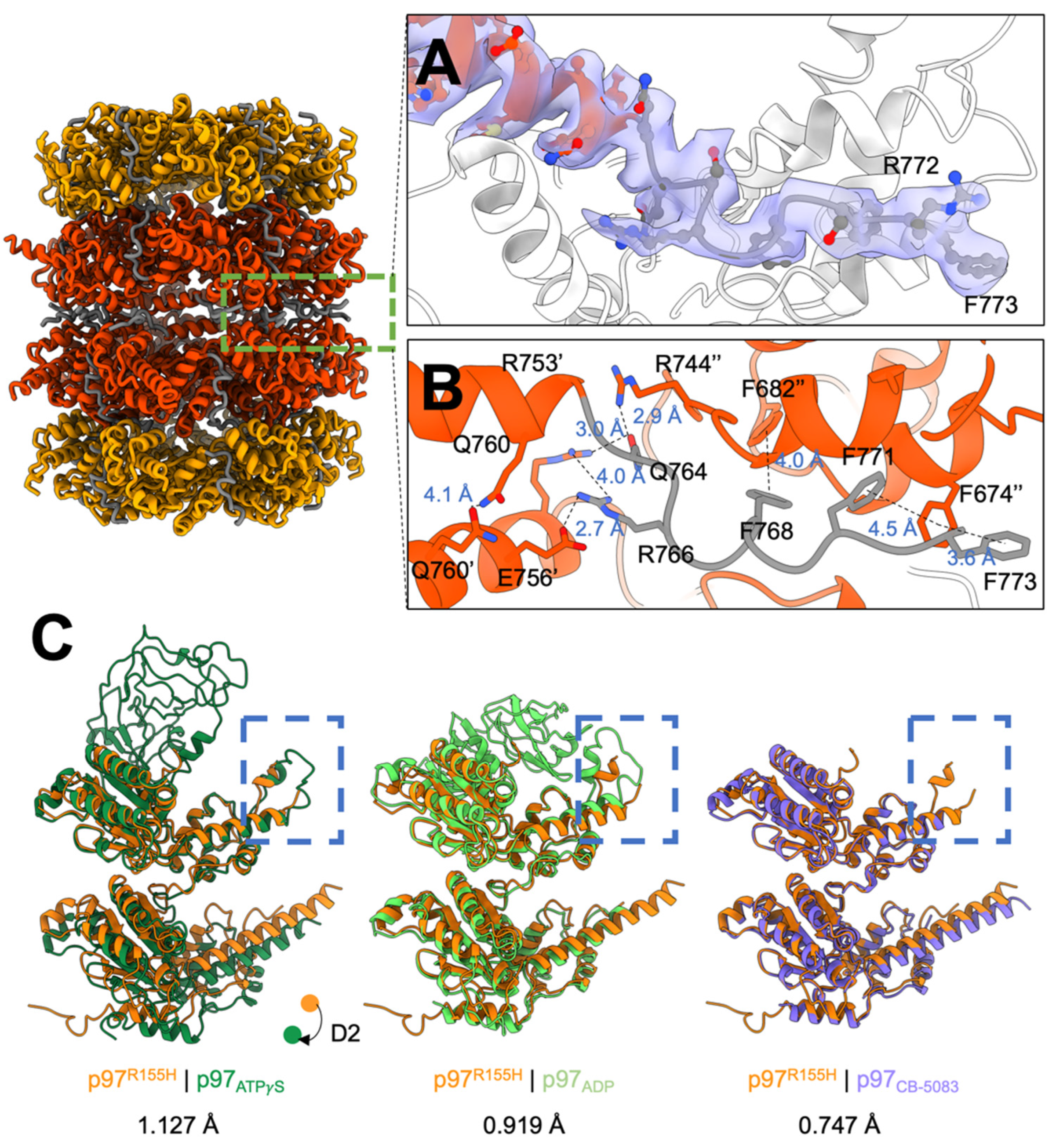
2.3. P97R155H Dodecamer Is Stabilized by the Two Oppositely Stacked D2 Rings
2.4. Nucleotide Binding Destabilizes the p97R155H Dodecameric Formation
2.5. P97R155H Dodecamer Is Likely to Be an Inactive Form
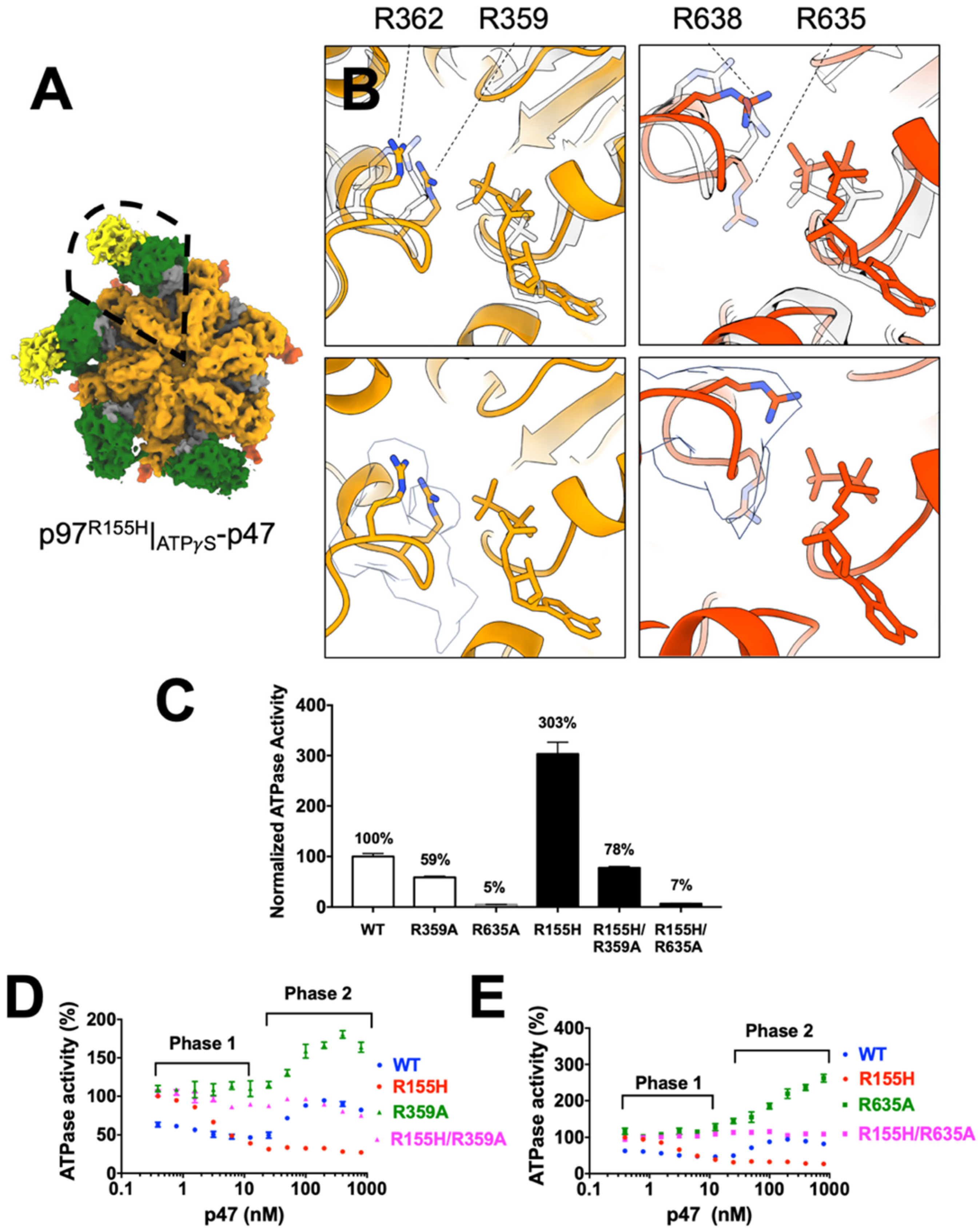
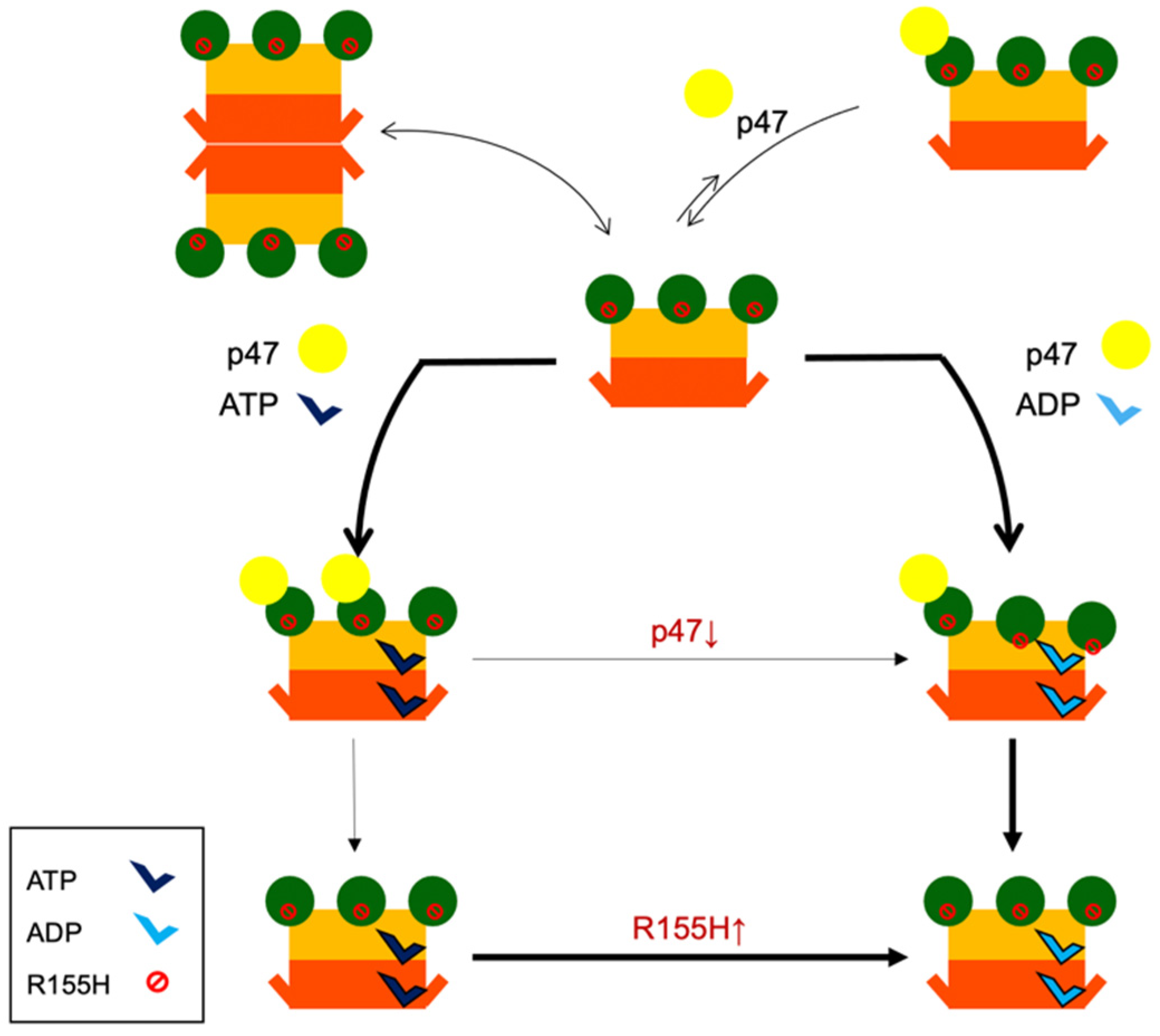
2.6. Nucleotide Binding Influences the p47 Binding onto p97R155H
2.7. P47 Binding Impacts p97R155H Function via an Allosteric Effect on ATPases
3. Discussion
4. Materials and Methods
4.1. Overexpression of the Wild Type p97, p97R155H Disease Mutant, and p47 Proteins
4.2. ATPase Activity Measurements
4.3. P47 Binding Affinity Measurements
4.4. Assembling p97-p47 Complexes
4.5. Negative-Stain Electron Microscopy for Single-Particle Analysis
4.6. Cryo-EM Data Collection
4.7. Image Processing
4.8. Modeling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef]
- Xia, D.; Tang, W.K.; Ye, Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016, 583, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Wendler, P.; Ciniawsky, S.; Kock, M.; Kube, S. Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 2012, 1823, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.H.; Shorter, J.G.; Seemann, J.; Pappin, D.; Warren, G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000, 19, 2181–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetzer, M.; Meyer, H.H.; Walther, T.C.; Bilbao-Cortes, D.; Warren, G.; Mattaj, I.W. Distinct AAA-ATPase p97 complexes function in discrete steps of nuclear assembly. Nat. Cell Biol. 2001, 3, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Snider, J.; Thibault, G.; Houry, W.A. The AAA+ superfamily of functionally diverse proteins. Genome Biol. 2008, 9, 216. [Google Scholar] [CrossRef]
- Kondo, H.; Rabouille, C.; Newman, R.; Levine, T.P.; Pappin, D.; Freemont, P.; Warren, G. p47 is a cofactor for p97-mediated membrane fusion. Nature 1997, 388, 75–78. [Google Scholar] [CrossRef]
- Koller, K.J.; Brownstein, M.J. Use of a cDNA clone to identify a supposed precursor protein containing valosin. Nature 1987, 325, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.M.; Walsh, M.J.; Franke, W.W. An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. EMBO J. 1990, 9, 1757–1767. [Google Scholar] [CrossRef]
- Buchberger, A.; Schindelin, H.; Hänzelmann, P. Control of p97 function by cofactor binding. FEBS Lett. 2015, 589, 2578–2589. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Hilt, W.; Buchberger, A.; Wolf, D.H. Cdc48: A power machine in protein degradation. Trends Biochem. Sci. 2011, 36, 515–523. [Google Scholar] [CrossRef]
- Yeung, H.O.; Kloppsteck, P.; Niwa, H.; Isaacson, R.L.; Matthews, S.; Zhang, X.; Freemont, P.S. Insights into adaptor binding to the AAA protein p97. Biochem. Soc. Trans. 2008, 36, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Abramzon, Y.; Johnson, J.O.; Scholz, S.W.; Taylor, J.P.; Brunetti, M.; Calvo, A.; Mandrioli, J.; Benatar, M.; Mora, G.; Restagno, G.; et al. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2231.e1–2231.e6. [Google Scholar] [CrossRef] [Green Version]
- Chapman, E.; Fry, A.N.; Kang, M. The complexities of p97 function in health and disease. Mol. Biosyst. 2011, 7, 700–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, G.D.J.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Ogura, T.; Wilkinson, A.J. AAA+ superfamily ATPases: Common structure–Diverse function. Genes Cells 2001, 6, 575–597. [Google Scholar] [CrossRef]
- Niwa, H.; Ewens, C.A.; Tsang, C.; Yeung, H.O.; Zhang, X.; Freemont, P.S. The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J. Biol. Chem. 2012, 287, 8561–8570. [Google Scholar] [CrossRef] [Green Version]
- Nishikori, S.; Esaki, M.; Yamanaka, K.; Sugimoto, S.; Ogura, T. Positive cooperativity of the p97 AAA ATPase is critical for essential functions. J. Biol. Chem. 2011, 286, 15815–15820. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.K.; Xia, D. Altered intersubunit communication is the molecular basis for functional defects of pathogenic p97 mutants. J. Biol. Chem. 2013, 288, 36624–36635. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003, 162, 71–84. [Google Scholar] [CrossRef]
- Davies, J.M.; Tsuruta, H.; May, A.P.; Weis, W.I. Conformational changes of p97 during nucleotide hydrolysis determined by small-angle X-Ray scattering. Structure 2005, 13, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreveny, I.; Kondo, H.; Uchiyama, K.; Shaw, A.; Zhang, X.; Freemont, P.S. Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J. 2004, 23, 1030–1039. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, P.; Buchberger, A.; Schindelin, H. Hierarchical binding of cofactors to the AAA ATPase p97. Structure 2011, 19, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.K.; Li, D.; Li, C.-C.; Esser, L.; Dai, R.; Guo, L.; Xia, D. A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 2010, 29, 2217–2229. [Google Scholar] [CrossRef]
- Zhang, X.; Shaw, A.; Bates, P.A.; Newman, R.H.; Gowen, B.; Orlova, E.; Gorman, M.A.; Kondo, H.; Dokurno, P.; Lally, J.; et al. Structure of the AAA ATPase p97. Mol. Cell 2000, 6, 1473–1484. [Google Scholar] [CrossRef]
- Torrecilla, I.; Oehler, J.; Ramadan, K. The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160282. [Google Scholar] [CrossRef] [Green Version]
- van den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Stach, L.; Freemont, P.S. The AAA+ ATPase p97, a cellular multitool. Biochem. J. 2017, 474, 2953–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, I.; Han, H.; Stewart, M.G.; Carson, R.H.; Hansen, D.T.; Iwasa, J.H.; Price, J.C.; Hill, C.P.; Shen, P.S. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science 2019, 365, 502–505. [Google Scholar] [CrossRef]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019, 365. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, N.O.; Rapoport, T.A. Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 2017, 169, 722–735.e9. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Ripstein, Z.A.; Rubinstein, J.L.; Kay, L.E. Cooperative subunit dynamics modulate p97 function. Proc. Natl. Acad. Sci. USA 2019, 116, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Gates, S.N.; Martin, A. Stairway to translocation: AAA+ motor structures reveal the mechanisms of ATP-dependent substrate translocation. Protein Sci. 2020, 29, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, P.; Schindelin, H. The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97. Front. Mol. Biosci. 2017, 4, 21. [Google Scholar] [CrossRef]
- Banerjee, S.; Bartesaghi, A.; Merk, A.; Rao, P.; Bulfer, S.L.; Yan, Y.; Green, N.; Mroczkowski, B.; Neitz, R.J.; Wipf, P.; et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 2016, 351, 871–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blythe, E.E.; Gates, S.N.; Deshaies, R.J.; Martin, A. Multisystem Proteinopathy Mutations in VCP/p97 Increase NPLOC4·UFD1L Binding and Substrate Processing. Structure 2019, 27, 1820–1829.e4. [Google Scholar] [CrossRef] [Green Version]
- Prattes, M.; Loibl, M.; Zisser, G.; Luschnig, D.; Kappel, L.; Rössler, I.; Grassegger, M.; Hromic, A.; Krieger, E.; Gruber, K.; et al. A conserved inter-domain communication mechanism regulates the ATPase activity of the AAA-protein Drg1. Sci. Rep. 2017, 7, 44751. [Google Scholar] [CrossRef] [Green Version]
- Beuron, F.; Dreveny, I.; Yuan, X.; Pye, V.E.; McKeown, C.; Briggs, L.C.; Cliff, M.J.; Kaneko, Y.; Wallis, R.; Isaacson, R.L.; et al. Conformational changes in the AAA ATPase p97-p47 adaptor complex. EMBO J. 2006, 25, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, G.; Lennarz, W.J. Dynamic flexibility of the ATPase p97 is important for its interprotomer motion transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 9792–9797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halawani, D.; LeBlanc, A.C.; Rouiller, I.; Michnick, S.W.; Servant, M.J.; Latterich, M. Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol. Cell. Biol. 2009, 29, 4484–4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saracino, D.; Clot, F.; Camuzat, A.; Anquetil, V.; Hannequin, D.; Guyant-Maréchal, L.; Didic, M.; Guillot-Noël, L.; Rinaldi, D.; Latouche, M.; et al. Novel VCP mutations expand the mutational spectrum of frontotemporal dementia. Neurobiol. Aging 2018, 72, e11–e187. [Google Scholar] [CrossRef]
- Buchberger, A.; Howard, M.J.; Proctor, M.; Bycroft, M. The UBX domain: A widespread ubiquitin-like module. J. Mol. Biol. 2001, 307, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gui, L.; Zhang, X.; Bulfer, S.L.; Sanghez, V.; Wong, D.E.; Lee, Y.; Lehmann, L.; Lee, J.S.; Shih, P.-Y.; et al. Altered cofactor regulation with disease-associated p97/VCP mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E1705–E1714. [Google Scholar] [CrossRef] [Green Version]
- Schuberth, C.; Buchberger, A. UBX domain proteins: Major regulators of the AAA ATPase Cdc48/p97. Cell. Mol. Life Sci. 2008, 65, 2360–2371. [Google Scholar] [CrossRef] [Green Version]
- Ewens, C.A.; Panico, S.; Kloppsteck, P.; McKeown, C.; Ebong, I.-O.; Robinson, C.; Zhang, X.; Freemont, P.S. The p97-FAF1 protein complex reveals a common mode of p97 adaptor binding. J. Biol. Chem. 2014, 289, 12077–12084. [Google Scholar] [CrossRef] [Green Version]
- Arumughan, A.; Roske, Y.; Barth, C.; Forero, L.L.; Bravo-Rodriguez, K.; Redel, A.; Kostova, S.; McShane, E.; Opitz, R.; Faelber, K.; et al. Quantitative interaction mapping reveals an extended UBX domain in ASPL that disrupts functional p97 hexamers. Nat. Commun. 2016, 7, 13047. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kang, W.; Suh, S.W.; Yang, J.K. Crystal structure of FAF1 UBX domain in complex with p97/VCP N domain reveals a conformational change in the conserved FcisP touch-turn motif of UBX domain. Proteins 2011, 79, 2583–2587. [Google Scholar] [CrossRef]
- Kim, S.J.; Cho, J.; Song, E.J.; Kim, S.J.; Kim, H.M.; Lee, K.E.; Suh, S.W.; Kim, E.E. Structural basis for ovarian tumor domain-containing protein 1 (OTU1) binding to p97/valosin-containing protein (VCP). J. Biol. Chem. 2014, 289, 12264–12274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conicella, A.E.; Huang, R.; Ripstein, Z.A.; Nguyen, A.; Wang, E.; Löhr, T.; Schuck, P.; Vendruscolo, M.; Rubinstein, J.L.; Kay, L.E. An intrinsically disordered motif regulates the interaction between the p47 adaptor and the p97 AAA+ ATPase. Proc. Natl. Acad. Sci. USA 2020, 117, 26226–26236. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, A.K.; Kay, L.E. A Dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife 2016, 5, e20143. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.K.; Xia, D. Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front. Mol. Biosci. 2016, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.K.; Odzorig, T.; Jin, W.; Xia, D. Structural Basis of p97 Inhibition by the Site-Selective Anticancer Compound CB-5083. Mol. Pharmacol. 2019, 95, 286–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoq, M.R.; Vago, F.S.; Li, K.; Kovaliov, M.; Nicholas, R.J.; Huryn, D.M.; Wipf, P.; Jiang, W.; Thompson, D.H. Affinity capture of p97 with small-molecule ligand bait reveals a 3.6 Å double-hexamer cryoelectron microscopy structure. ACS Nano 2021, 15, 8376–8385. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, P.; Schindelin, H. Structural Basis of ATP Hydrolysis and Intersubunit Signaling in the AAA+ ATPase p97. Structure 2016, 24, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Simpson, P.; McKeown, C.; Kondo, H.; Uchiyama, K.; Wallis, R.; Dreveny, I.; Keetch, C.; Zhang, X.; Robinson, C.; et al. Structure, dynamics and interactions of p47, a major adaptor of the AAA ATPase, p97. EMBO J. 2004, 23, 1463–1473. [Google Scholar] [CrossRef] [Green Version]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Magnaghi, P.; D’Alessio, R.; Valsasina, B.; Avanzi, N.; Rizzi, S.; Asa, D.; Gasparri, F.; Cozzi, L.; Cucchi, U.; Orrenius, C.; et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 2013, 9, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.C.; Baldwin, G.S.; Miyata, N.; Kondo, H.; Zhang, X.; Freemont, P.S. Analysis of nucleotide binding to P97 reveals the properties of a tandem AAA hexameric ATPase. J. Biol. Chem. 2008, 283, 13745–13752. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-F.; Bulfer, S.L.; Weihl, C.C.; Li, K.; Lis, L.G.; Walters, M.A.; Schoenen, F.J.; Lin, H.J.; Deshaies, R.J.; Arkin, M.R. Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. J. Mol. Biol. 2014, 426, 2886–2899. [Google Scholar] [CrossRef] [Green Version]
- Bulfer, S.L.; Chou, T.-F.; Arkin, M.R. p97 Disease Mutations Modulate Nucleotide-Induced Conformation to Alter Protein-Protein Interactions. ACS Chem. Biol. 2016, 11, 2112–2116. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, G.; Schindelin, H.; Lennarz, W.J. Tyrosine phosphorylation of ATPase p97 regulates its activity during ERAD. Biochem. Biophys. Res. Commun. 2008, 375, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Ewens, C.A.; Kloppsteck, P.; Förster, A.; Zhang, X.; Freemont, P.S. Structural and functional implications of phosphorylation and acetylation in the regulation of the AAA+ protein p97. Biochem. Cell Biol. 2010, 88, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Manno, A.; Noguchi, M.; Fukushi, J.; Motohashi, Y.; Kakizuka, A. Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Genes Cells 2010, 15, 911–922. [Google Scholar] [CrossRef]
- Trusch, F.; Matena, A.; Vuk, M.; Koerver, L.; Knævelsrud, H.; Freemont, P.S.; Meyer, H.; Bayer, P. The N-terminal Region of the Ubiquitin Regulatory X (UBX) Domain-containing Protein 1 (UBXD1) Modulates Interdomain Communication within the Valosin-containing Protein p97. J. Biol. Chem. 2015, 290, 29414–29427. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Driver, D.; Zhao, Z.; Zheng, Z.; Chan, C.; Cheng, X. Controlling the Revolving and Rotating Motion Direction of Asymmetric Hexameric Nanomotor by Arginine Finger and Channel Chirality. ACS Nano 2019, 13, 6207–6223. [Google Scholar] [CrossRef] [Green Version]
- Ogura, T.; Whiteheart, S.W.; Wilkinson, A.J. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J. Struct. Biol. 2004, 146, 106–112. [Google Scholar] [CrossRef]
- Wang, Q.; Song, C.; Irizarry, L.; Dai, R.; Zhang, X.; Li, C.-C.H. Multifunctional roles of the conserved Arg residues in the second region of homology of p97/valosin-containing protein. J. Biol. Chem. 2005, 280, 40515–40523. [Google Scholar] [CrossRef] [Green Version]
- Bersano, A.; Del Bo, R.; Lamperti, C.; Ghezzi, S.; Fagiolari, G.; Fortunato, F.; Ballabio, E.; Moggio, M.; Candelise, L.; Galimberti, D.; et al. Inclusion body myopathy and frontotemporal dementia caused by a novel VCP mutation. Neurobiol. Aging 2009, 30, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Stach, L.; Morgan, R.M.; Makhlouf, L.; Douangamath, A.; von Delft, F.; Zhang, X.; Freemont, P.S. Crystal structure of the catalytic D2 domain of the AAA+ ATPase p97 reveals a putative helical split-washer-type mechanism for substrate unfolding. FEBS Lett. 2020, 594, 933–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Shaw, A.; Zhang, X.; Kondo, H.; Lally, J.; Freemont, P.S.; Matthews, S. Solution structure and interaction surface of the C-terminal domain from p47: A major p97-cofactor involved in SNARE disassembly. J. Mol. Biol. 2001, 311, 255–263. [Google Scholar] [CrossRef]
- Ripstein, Z.A.; Huang, R.; Augustyniak, R.; Kay, L.E.; Rubinstein, J.L. Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 2017, 6. [Google Scholar] [CrossRef]
- Blythe, E.E.; Olson, K.C.; Chau, V.; Deshaies, R.J. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. USA 2017, 114, E4380–E4388. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Tsuchiya, H.; Yamagata, A.; Okatsu, K.; Tanaka, K.; Saeki, Y.; Fukai, S. Structural insights into ubiquitin recognition and Ufd1 interaction of Npl4. Nat. Commun. 2019, 10, 5708. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, K.; Jokitalo, E.; Kano, F.; Murata, M.; Zhang, X.; Canas, B.; Newman, R.; Rabouille, C.; Pappin, D.; Freemont, P.; et al. VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J. Cell Biol. 2002, 159, 855–866. [Google Scholar] [CrossRef]
- Shukla, A.K.; Westfield, G.H.; Xiao, K.; Reis, R.I.; Huang, L.-Y.; Tripathi-Shukla, P.; Qian, J.; Li, S.; Blanc, A.; Oleskie, A.N.; et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 2014, 512, 218–222. [Google Scholar] [CrossRef]
- Ohi, M.; Li, Y.; Cheng, Y.; Walz, T. Negative Staining and Image Classification—Powerful Tools in Modern Electron Microscopy. Biol. Proced. Online 2004, 6, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H.W. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Eng, E.T.; Alink, L.; Rice, W.J.; Jordan, K.D.; Kim, L.Y.; Potter, C.S.; Carragher, B. High resolution single particle cryo-electron microscopy using beam-image shift. J. Struct. Biol. 2018, 204, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Scheres, S.H.W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 2020, 7, 253–267. [Google Scholar] [CrossRef] [Green Version]
- Zivanov, J.; Nakane, T.; Scheres, S.H.W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 2019, 6, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Scheres, S.H.W.; Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 2012, 9, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Bepler, T.; Morin, A.; Rapp, M.; Brasch, J.; Shapiro, L.; Noble, A.J.; Berger, B. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 2019, 16, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nandi, P.; Li, S.; Columbres, R.C.A.; Wang, F.; Williams, D.R.; Poh, Y.-P.; Chou, T.-F.; Chiu, P.-L. Structural and Functional Analysis of Disease-Linked p97 ATPase Mutant Complexes. Int. J. Mol. Sci. 2021, 22, 8079. https://doi.org/10.3390/ijms22158079
Nandi P, Li S, Columbres RCA, Wang F, Williams DR, Poh Y-P, Chou T-F, Chiu P-L. Structural and Functional Analysis of Disease-Linked p97 ATPase Mutant Complexes. International Journal of Molecular Sciences. 2021; 22(15):8079. https://doi.org/10.3390/ijms22158079
Chicago/Turabian StyleNandi, Purbasha, Shan Li, Rod Carlo A. Columbres, Feng Wang, Dewight R. Williams, Yu-Ping Poh, Tsui-Fen Chou, and Po-Lin Chiu. 2021. "Structural and Functional Analysis of Disease-Linked p97 ATPase Mutant Complexes" International Journal of Molecular Sciences 22, no. 15: 8079. https://doi.org/10.3390/ijms22158079