
Co-Targeting PIM Kinase and PI3K/mTOR in NSCLC

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Drugs
2.2. Patient Samples for Immunohistochemistry
2.3. Immunohistochemical Detection Methods
2.4. Western Blotting
2.5. Cell Proliferation and Cell Viability Assays
2.6. Ex Vivo Cultured Lung Tumor Explants
2.7. Phospho-Protein Profiling
2.8. IL6/STAT3 Signaling Gene Array
2.9. Real Time PCR Analysis
2.10. miRNA Expression Profiling
2.11. MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead 10-Plex Panel
2.12. In-Silico Data Analysis
2.13. Statistical Analysis
3. Results
3.1. PIM1, PIM2 and PIM3 Expression in HCC827, H460 and H1975 Cell Lines and Investigating PIM1 Expression as a Prognostic Marker in NSCLC
3.2. IBL-301
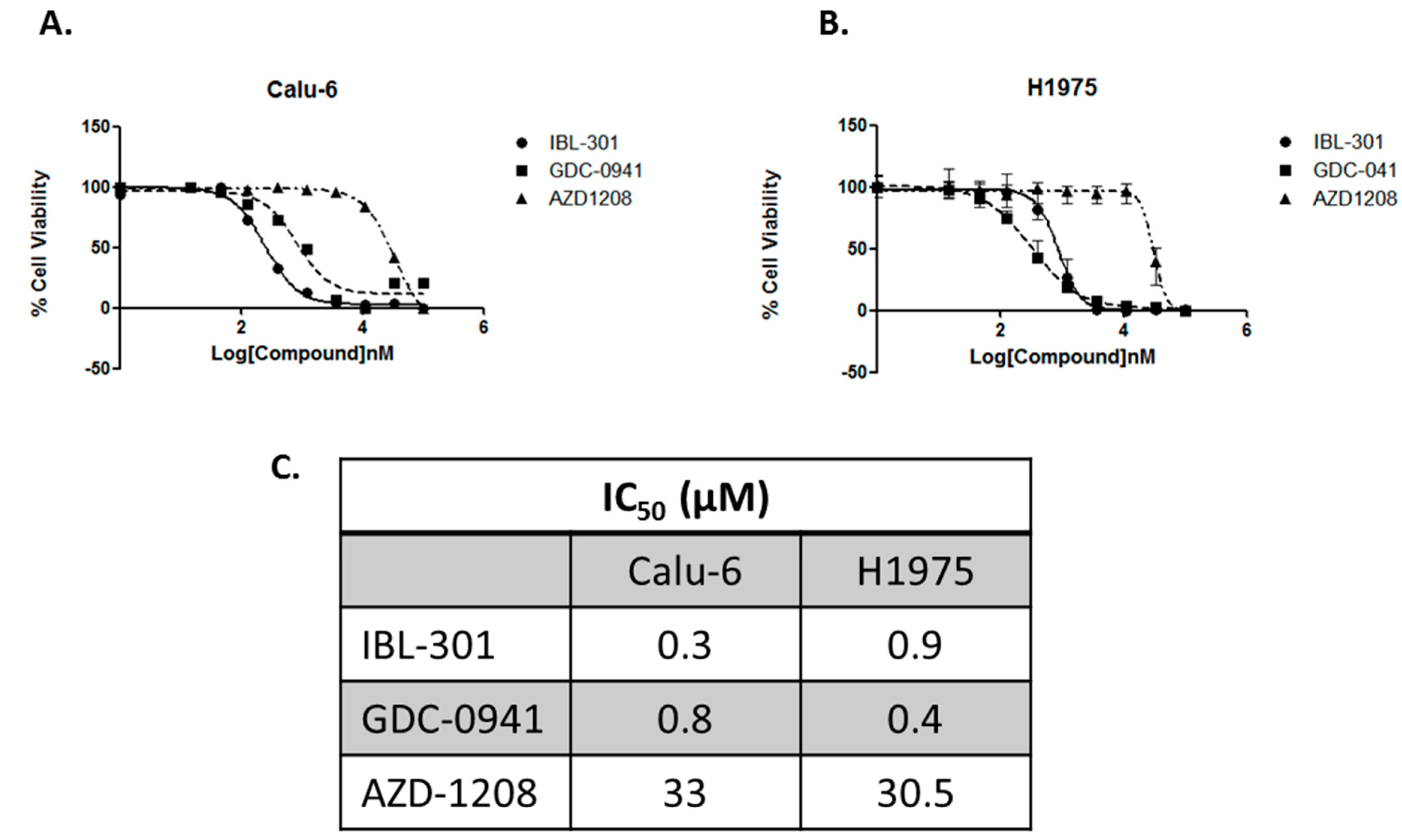
3.2.1. Comparing the Efficacy of pan-PI3K Inhibitor (GDC-0941), pan-PIM Kinase Inhibitor (AZD1208), to Triple Targeted PI3K/mTOR/PIM Inhibitor (IBL-301)
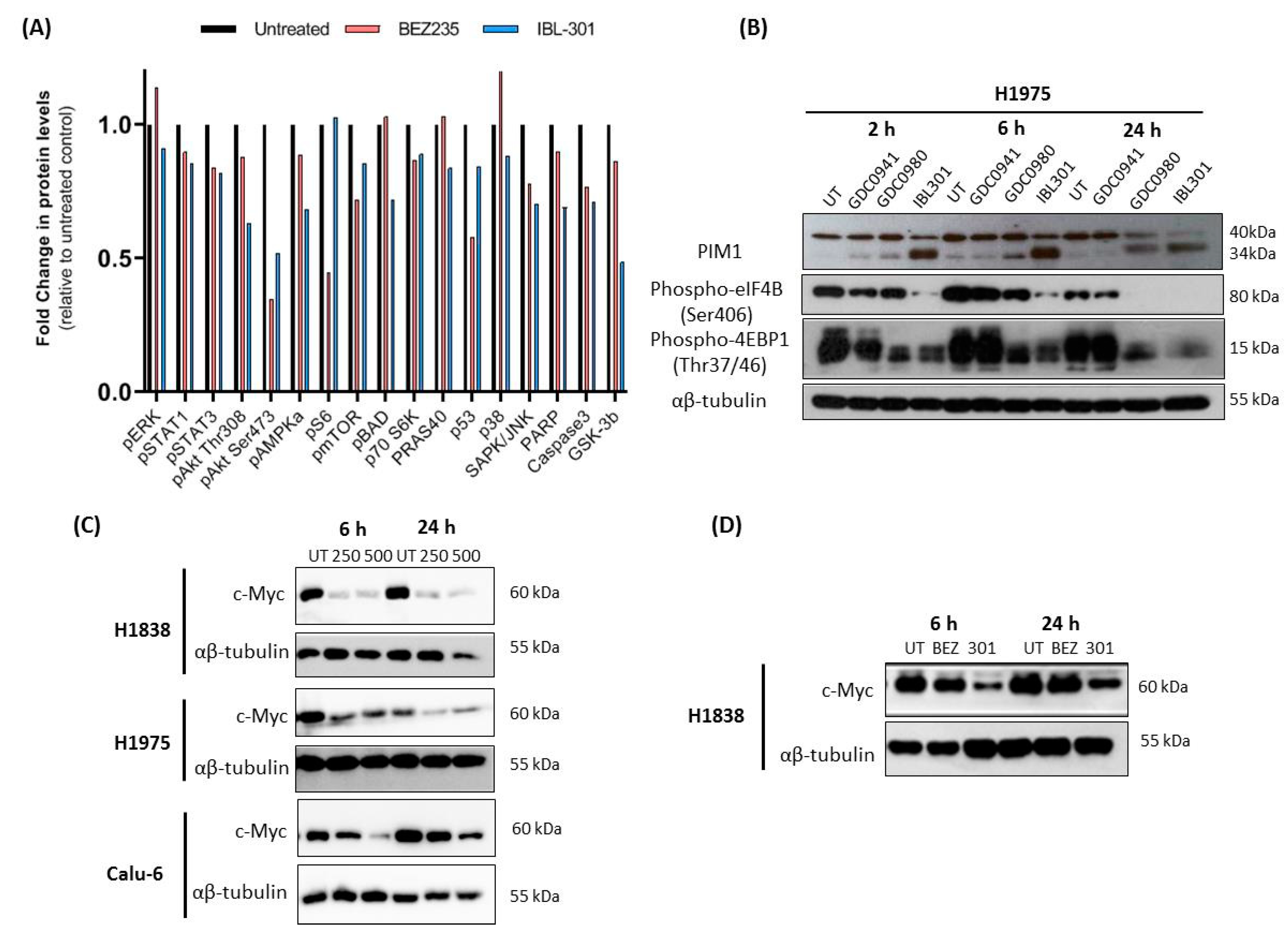
3.2.2. The Effect of Triple Targeted Inhibitor IBL-301 on Intracellular Signaling Pathways In-Vitro
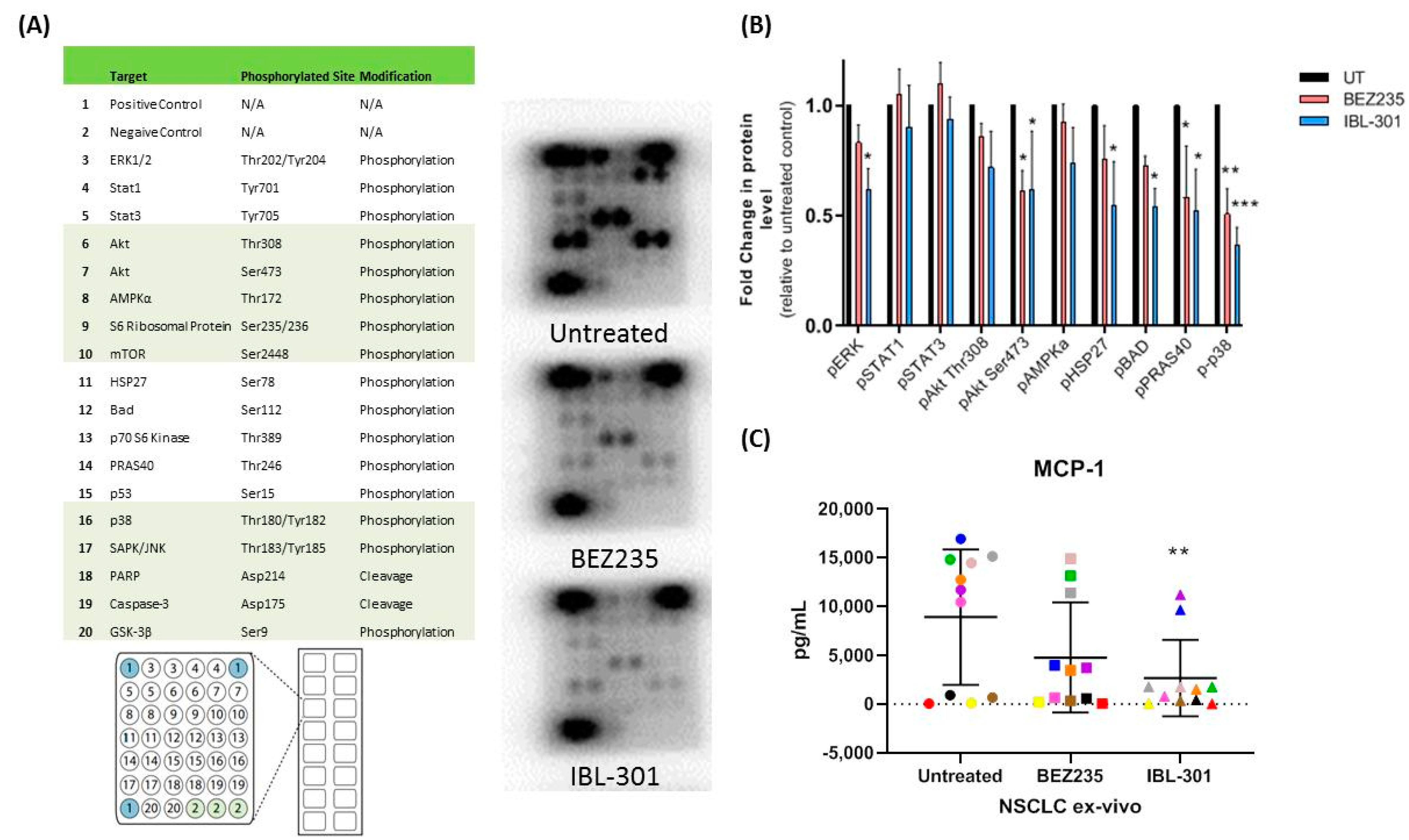
3.2.3. The Effect of Triple Targeted Inhibitor IBL-301 on Intracellular Signaling Pathways in Cultured NSCLC Tumor Explants
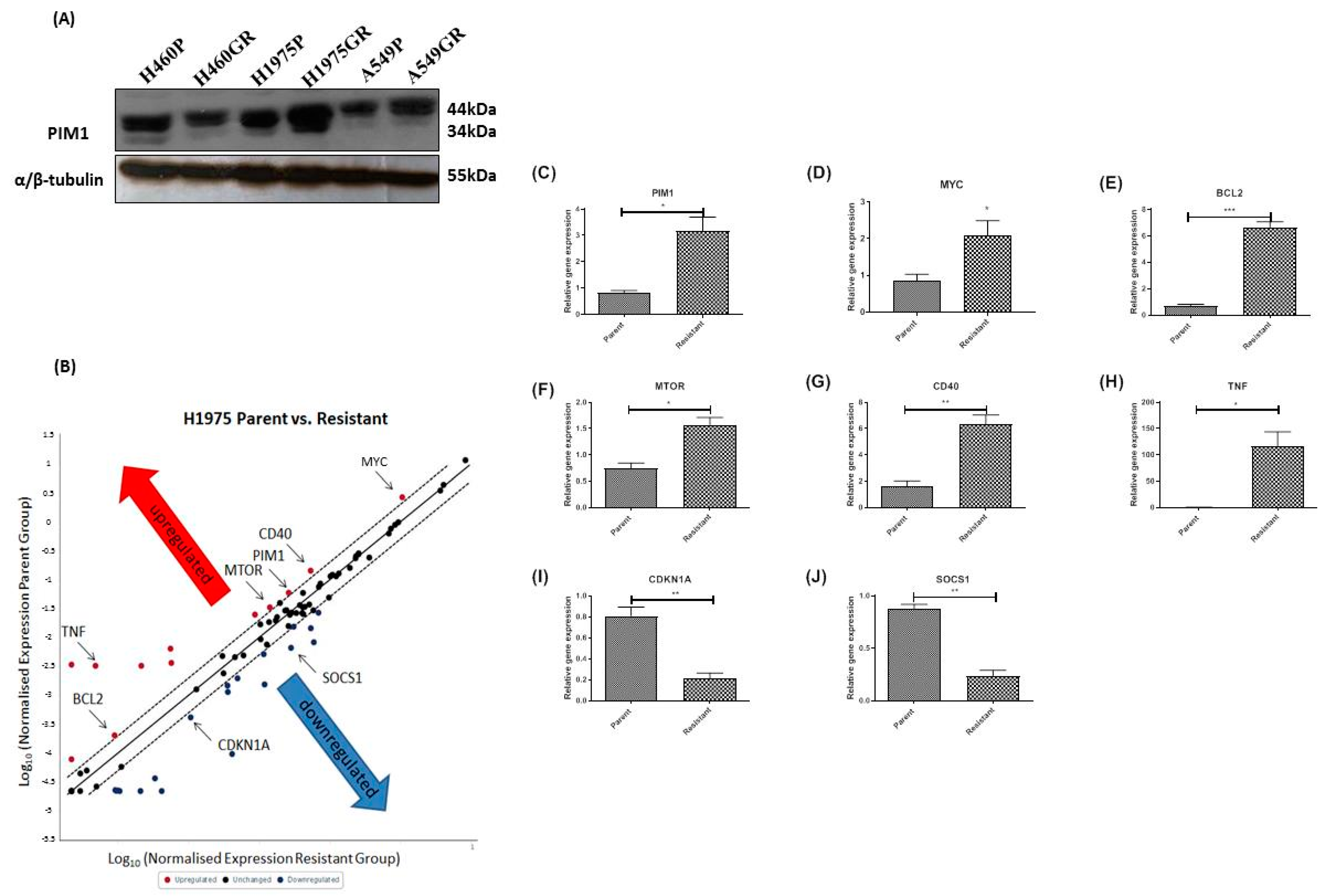
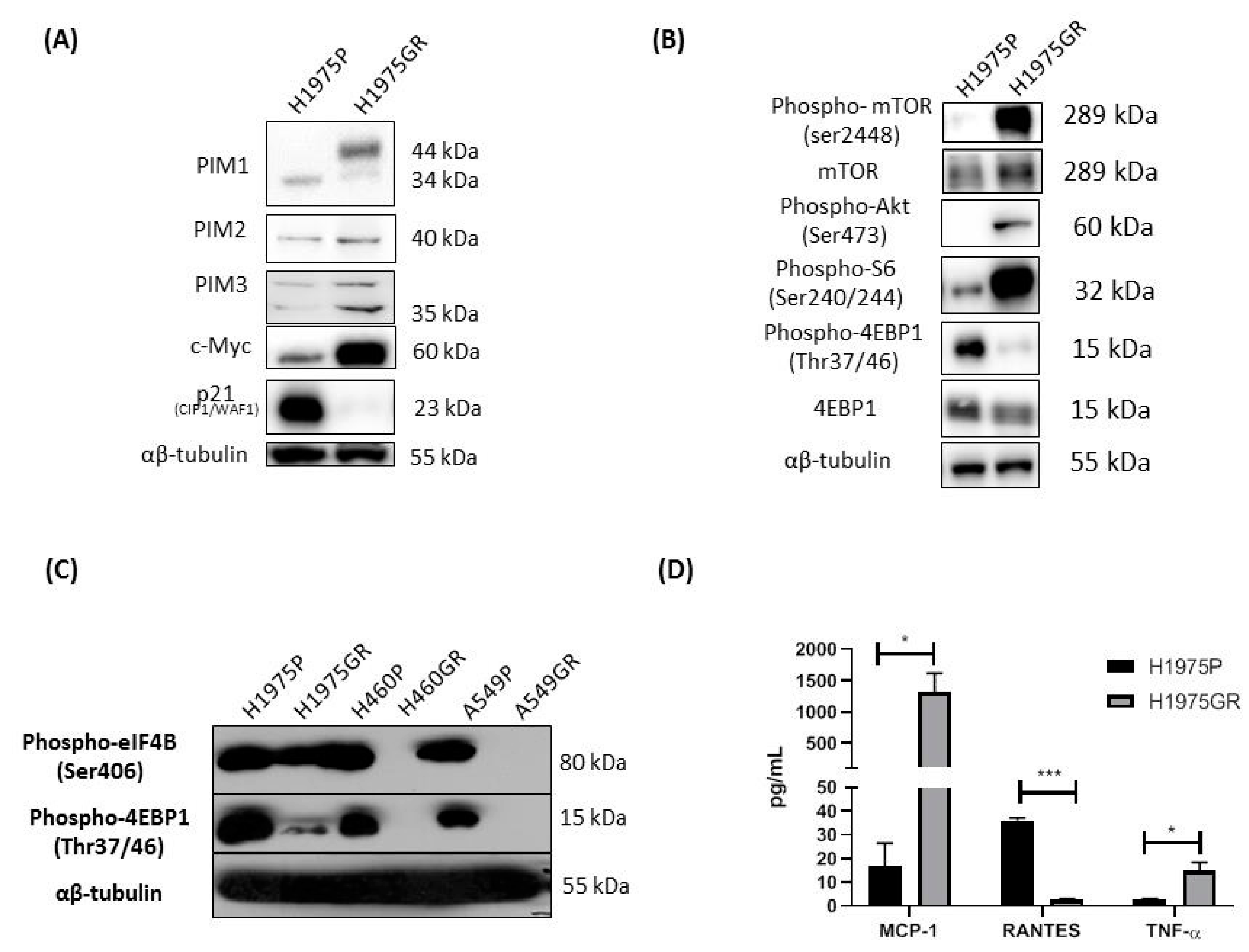
3.3. Acquired Resistance to PI3K/mTOR Inhibitors Alters the Protein Expression Profile of PIM1, and Gene Expression of IL-6/STAT3 Signaling Pathway Members Including PIM1 and MYC in the H1975GR Cells
3.4. Protein Evaluation of Identified Genes Associated with PI3K/mTOR Inhibitor Resistance in H1975GR Cells
3.5. Testing the Efficacy of IBL-301 in H1975GR and H1975P Cell Lines
3.6. Investigation of Expression of a PIM1 and c-Myc Associated microRNA Signature in PI3K/mTOR Inhibitor Resistant H1975GR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, R.; Wang, X.; Jin, Z.; Li, K. Association of Nuclear PIM1 Expression with Lymph Node Metastasis and Poor Prognosis in Patients with Lung Adenocarcinoma and Squamous Cell Carcinoma. J. Cancer 2016, 7, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, W.; Tian, X.; Bai, F.; Han, R.; Wang, J.; Shen, H.; Zhang, X.; Liu, Y.; Yan, X.; Jiang, F.; et al. Pim-1 kinase is a target of miR-486-5p and eukaryotic translation initiation factor 4E, and plays a critical role in lung cancer. Mol. Cancer 2014, 13, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Xu, K.; Dai, B.; Guo, Z.; Jiang, T.; Chen, H.; Qiu, Y. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene 2006, 25, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musiani, D.; Hammond, D.E.; Cirillo, L.; Erriquez, J.; Olivero, M.; Clague, M.J.; Di Renzo, M.F. PIM2 kinase is induced by cisplatin in ovarian cancer cells and limits drug efficacy. J. Proteome Res. 2014, 13, 4970–4982. [Google Scholar] [CrossRef]
- Cen, B.; Mahajan, S.; Wang, W.; Kraft, A.S. Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1. Cancer Res. 2013, 73, 3402–3411. [Google Scholar] [CrossRef] [Green Version]
- An, N.; Xiong, Y.; LaRue, A.C.; Kraft, A.S.; Cen, B. Activation of Pim Kinases Is Sufficient to Promote Resistance to MET Small-Molecule Inhibitors. Cancer Res. 2015, 75, 5318–5328. [Google Scholar] [CrossRef] [Green Version]
- Bracht, J.W.P.; Karachaliou, N.; Berenguer, J.; Fernandez-Bruno, M.; Filipska, M.; Pedraz-Valdunciel, C.; Codony-Servat, C.; Codony-Servat, J.; Rosell, R. PIM-1 inhibition with AZD1208 to prevent osimertinib-induced resistance in EGFR-mutation positive non-small cell lung cancer. J. Cancer Metastasis Treat. 2019, 5, 22. [Google Scholar] [CrossRef]
- Wang, J.; Kim, J.; Roh, M.; Franco, O.E.; Hayward, S.W.; Wills, M.L.; Abdulkadir, S.A. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene 2010, 29, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Liu, X.; Lu, Y.; Wang, Y.; Cao, W.; Liu, X.; Hu, H.; Wang, H. PIM1 is responsible for IL-6-induced breast cancer cell EMT and stemness via c-myc activation. Breast Cancer 2019, 26, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Luszczak, S.; Simpson, B.S.; Stopka-Farooqui, U.; Sathyadevan, V.K.; Echeverria, L.M.C.; Kumar, C.; Costa, H.; Haider, A.; Freeman, A.; Jameson, C.; et al. Co-targeting PIM and PI3K/mTOR using multikinase inhibitor AUM302 and a combination of AZD-1208 and BEZ235 in prostate cancer. Sci. Rep. 2020, 10, 14380. [Google Scholar] [CrossRef]
- Balsara, B.R.; Pei, J.; Mitsuuchi, Y.; Page, R.; Klein-Szanto, A.; Wang, H.; Unger, M.; Testa, J.R. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis 2004, 25, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Heavey, S.; Dowling, P.; Moore, G.; Barr, M.P.; Kelly, N.; Maher, S.G.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Gately, K. Development and characterisation of a panel of phosphatidylinositide 3-kinase—Mammalian target of rapamycin inhibitor resistant lung cancer cell lines. Sci. Rep. 2018, 8, 1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Gao, Q.; Han, S.; Pan, F.; Fan, W. The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-Twist signaling. Tumour Biol. 2014, 36, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Shien, K.; Papadimitrakopoulou, V.A.; Ruder, D.; Behrens, C.; Shen, L.; Kalhor, N.; Song, J.; Lee, J.J.; Wang, J.; Tang, X.; et al. JAK1/STAT3 Activation through a Proinflammatory Cytokine Pathway Leads to Resistance to Molecularly Targeted Therapy in Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2017, 16, 2234–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Delineation of prognostic biomarkers in prostate cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef]
- Reiser-Erkan, C.; Erkan, M.; Pan, Z.; Bekasi, S.; Giese, N.A.; Streit, S.; Michalski, C.W.; Friess, H.; Kleeff, J. Hypoxia-inducible proto-oncogene Pim-1 is a prognostic marker in pancreatic ductal adenocarcinoma. Cancer Biol. Ther. 2008, 7, 1352–1359. [Google Scholar] [CrossRef] [Green Version]
- Warnecke-Eberz, U.; Bollschweiler, E.; Drebber, U.; Pohl, A.; Baldus, S.E.; Hoelscher, A.H.; Metzger, R. Frequent down-regulation of pim-1 mRNA expression in non-small cell lung cancer is associated with lymph node metastases. Oncol. Rep. 2008, 20, 619–624. [Google Scholar] [PubMed] [Green Version]
- Brunen, D.; de Vries, R.C.; Lieftink, C.; Beijersbergen, R.L.; Bernards, R. PIM Kinases Are a Potential Prognostic Biomarker and Therapeutic Target in Neuroblastoma. Mol. Cancer Ther. 2018, 17, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Holder, S.L.; Abdulkadir, S.A. PIM1 kinase as a target in prostate cancer: Roles in tumorigenesis, castration resistance, and docetaxel resistance. Curr. Cancer Drug Targets 2014, 14, 105–114. [Google Scholar] [CrossRef]
- Liao, Y.; Feng, Y.; Shen, J.; Gao, Y.; Cote, G.; Choy, E.; Harmon, D.; Mankin, H.; Hornicek, F.; Duan, Z. Clinical and biological significance of PIM1 kinase in osteosarcoma. J. Orthop. Res. 2015, 34, 1185–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braso-Maristany, F.; Filosto, S.; Catchpole, S.; Marlow, R.; Quist, J.; Francesch-Domenech, E.; Plumb, D.A.; Zakka, L.; Gazinska, P.; Liccardi, G.; et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat. Med. 2016, 22, 1303–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Xiong, G.; Cao, Z.; Huang, H.; Wang, T.; You, L.; Zhou, L.; Zheng, L.; Hu, Y.; Zhang, T.; et al. PIM-1 contributes to the malignancy of pancreatic cancer and displays diagnostic and prognostic value. J. Exp. Clin. Cancer Res. 2016, 35, 133. [Google Scholar] [CrossRef] [Green Version]
- Malone, T.; Schfer, L.; Simon, N.; Heavey, S.; Cuffe, S.; Finn, S.; Moore, G.; Gately, K. Current perspectives on targeting PIM kinases to overcome mechanisms of drug resistance and immune evasion in cancer. Pharmacol. Ther. 2020, 207, 107454. [Google Scholar] [CrossRef]
- Mohlin, S.; Hansson, K.; Radke, K.; Martinez, S.; Blanco-Apiricio, C.; Garcia-Ruiz, C.; Welinder, C.; Esfandyari, J.; O’Neill, M.; Pastor, J.; et al. Anti-tumor effects of PIM/PI3K/mTOR triple kinase inhibitor IBL-302 in neuroblastoma. EMBO Mol. Med. 2019, 11, e10058. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.P.; O’Neill, M.; Cunningham, D.; Morris, P.G.; Toomey, S.; Blanco-Aparicio, C.; Martinez, S.; Pastor, J.; Eustace, A.J.; Hennessy, B.T. Preclinical evaluation of a novel triple-acting PIM/PI3K/mTOR inhibitor, IBL-302, in breast cancer. Oncogene 2020, 39, 3028–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebello, R.J.; Huglo, A.V.; Furic, L. PIM activity in tumours: A key node of therapy resistance. Adv. Biol. Regul. 2018, 67, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Padi, S.K.; Luevano, L.A.; Minden, M.D.; DeAngelo, D.J.; Hardiman, G.; Ball, L.E.; Warfel, N.A.; Kraft, A.S. Insulin receptor substrate 1 is a substrate of the Pim protein kinases. Oncotarget 2016, 7, 20152–20165. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Yang, J.; Li, J.; Wang, X.; Chen, Y.; Huang, S.; Chen, J.L. eIF4B is a convergent target and critical effector of oncogenic Pim and PI3K/Akt/mTOR signaling pathways in Abl transformants. Oncotarget 2016, 7, 10073–10089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, A.U.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer. Biomolecules 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.K.; Obiang-Obounou, B.W.; Lee, K.B.; Choi, J.S.; Jang, B.C. AZD1208, a pan-Pim kinase inhibitor, inhibits adipogenesis and induces lipolysis in 3T3-L1 adipocytes. J. Cell Mol. Med. 2018, 22, 2488–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuz, S.; Holmes, K.B.; Tooze, R.M.; Lefevre, P.F. Loss of PIM2 enhances the antiproliferative effect of the pan-PIM kinase inhibitor AZD1208 in non-Hodgkin lymphomas. Mol. Cancer 2015, 14, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Z.; Li, X.; Magnuson, N.S. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008, 27, 4809–4819. [Google Scholar] [CrossRef] [Green Version]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Roh, M.; Abdulkadir, S.A. Pim1 promotes human prostate cancer cell tumorigenicity and c-MYC transcriptional activity. BMC Cancer 2010, 10, 248. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.D.; Zhan, Y.T.; Zhu, Y.H.; Li, Y.; Xie, D.; Guan, X.Y. High levels of CCL2 or CCL4 in the tumor microenvironment predict unfavorable survival in lung adenocarcinoma. Thorac. Cancer 2018, 9, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Tursynbay, Y.; Zhang, J.; Li, Z.; Tokay, T.; Zhumadilov, Z.; Wu, D.; Xie, Y. Pim-1 kinase as cancer drug target: An update. Biomed. Rep. 2016, 4, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Xu, K.; Linn, D.E.; Yang, X.; Guo, Z.; Shimelis, H.; Nakanishi, T.; Ross, D.D.; Chen, H.; Fazli, L.; et al. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J. Biol. Chem. 2008, 283, 3349–3356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Beharry, Z.M.; Harris, T.E.; Lilly, M.B.; Smith, C.D.; Mahajan, S.; Kraft, A.S. PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol. Ther. 2009, 8, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.J.; Hammerman, P.S.; Cinalli, R.M.; Master, S.R.; Chodosh, L.A.; Thompson, C.B. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003, 17, 1841–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muranen, T.; Selfors, L.M.; Worster, D.T.; Iwanicki, M.P.; Song, L.; Morales, F.C.; Gao, S.; Mills, G.B.; Brugge, J.S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012, 21, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Johannes, G.; Carter, M.S.; Eisen, M.B.; Brown, P.O.; Sarnow, P. Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc. Natl. Acad. Sci. USA 1999, 96, 13118–13123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiluiza, D.; Bargo, S.; Callahan, R.; Rhoads, R.E. Expression of truncated eukaryotic initiation factor 3e (eIF3e) resulting from integration of mouse mammary tumor virus (MMTV) causes a shift from cap-dependent to cap-independent translation. J. Biol. Chem. 2011, 286, 31288–31296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eischen, C.M.; Packham, G.; Nip, J.; Fee, B.E.; Hiebert, S.W.; Zambetti, G.P.; Cleveland, J.L. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene 2001, 20, 6983–6993. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Conrad, S.E. c-Myc suppresses p21WAF1/CIP1 expression during estrogen signaling and antiestrogen resistance in human breast cancer cells. J. Biol. Chem. 2005, 280, 17617–17625. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Cetinkaya, C.; Munoz-Alonso, M.J.; von der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; Leon, J.; Larsson, L.G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Liu, H.; Yuan, X.; Wang, Y.; Li, L.; Wang, G.; Song, J.; Shao, Z.; Fu, R. Downregulation of Pim-2 induces cell cycle arrest in the G0/G1 phase via the p53-non-dependent p21 signaling pathway. Oncol. Lett. 2018, 15, 4079–4086. [Google Scholar] [CrossRef] [Green Version]
- Ghafouri-Fard, S.; Oskooei, V.K.; Azari, I.; Taheri, M. Suppressor of cytokine signaling (SOCS) genes are downregulated in breast cancer. World J. Surg. Oncol. 2018, 16, 226. [Google Scholar] [CrossRef]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [Green Version]
- Moran, C.J.; Arenberg, D.A.; Huang, C.C.; Giordano, T.J.; Thomas, D.G.; Misek, D.E.; Chen, G.; Iannettoni, M.D.; Orringer, M.B.; Hanash, S.; et al. RANTES expression is a predictor of survival in stage I lung adenocarcinoma. Clin. Can. Res. 2002, 8, 3803–3812. [Google Scholar]
- Yao, M.; Brummer, G.; Acevedo, D.; Cheng, N. Cytokine Regulation of Metastasis and Tumorigenicity. Adv. Cancer Res. 2016, 132, 265–367. [Google Scholar] [PubMed]
- Fridlender, Z.G.; Kapoor, V.; Buchlis, G.; Cheng, G.; Sun, J.; Wang, L.C.; Singhal, S.; Snyder, L.A.; Albelda, S.M. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küper, C.; Beck, F.-X.; Neuhofer, W. Autocrine MCP-1/CCR2 signaling stimulates proliferation and migration of renal carcinoma cells. Oncol. Lett. 2016, 12, 2201–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Zhan, Q.; Peng, X.; Qiu, Z.; Zhao, T. CCL2 influences the sensitivity of lung cancer A549 cells to docetaxel. Oncol. Lett. 2018, 16, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.H.; Singh, N.; Luevano, L.A.; Padi, S.K.R.; Okumura, K.; Olive, V.; Black, S.M.; Warfel, N.A.; Goodrich, D.W.; Kraft, A.S. Mechanisms Behind Resistance to PI3K Inhibitor Treatment Induced by the PIM Kinase. Mol. Cancer Ther. 2018, 17, 2710–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Pan, C.; Guo, L.; Wu, M.; Guo, J.; Peng, S.; Wu, Q.; Zuo, Q. A new mechanism of trastuzumab resistance in gastric cancer: MACC1 promotes the Warburg effect via activation of the PI3K/AKT signaling pathway. J. Hematol. Oncol. 2016, 9, 76. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, B.; Sun, L.; Yan, Q.; Zhang, Y.; Zhang, Z.; Su, Y.; Wang, C. Cisplatin resistance in lung cancer is mediated by MACC1 expression through PI3K/AKT signaling pathway activation. Acta Biochim. Biophys Sin. 2018, 50, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, H.; Zhao, M.Y.; Hei, K.W.; Yang, B.C.; Sun, L.; Du, X.; Li, Y.M. Aberrant expression of pim-3 promotes proliferation and migration of ovarian cancer cells. Asian Pac. J. Cancer Prev. 2015, 16, 3325–3331. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Krysan, K.; Kusko, R.; Grogan, T.; O’Hearn, J.; Reckamp, K.L.; Walser, T.C.; Garon, E.B.; Lenburg, M.E.; Sharma, S.; Spira, A.E.; et al. PGE2-driven expression of c-Myc and oncomiR-17-92 contributes to apoptosis resistance in NSCLC. Mol. Can. Res. 2014, 12, 765–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Bo, M.; Bomben, R.; Hernandez, L.; Gattei, V. The MYC/miR-17-92 axis in lymphoproliferative disorders: A common pathway with therapeutic potential. Oncotarget 2015, 6, 19381–19392. [Google Scholar] [CrossRef] [PubMed]
- Benhamou, D.; Labi, V.; Getahun, A.; Benchetrit, E.; Dowery, R.; Rajewsky, K.; Cambier, J.C.; Melamed, D. The c-Myc/miR17-92/PTEN Axis Tunes PI3K Activity to Control Expression of Recombination Activating Genes in Early B Cell Development. Front. Immunol. 2018, 9, 2715. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Lange-Grunweller, K.; Hartmann, D.; Golde, L.; Schlereth, J.; Streng, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. Analysis of transcriptional regulation of the human miR-17-92 cluster; evidence for involvement of Pim-1. Int. J. Mol. Sci. 2013, 14, 12273–12296. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ji, F. Downregulation of microRNA-17-5p inhibits drug resistance of gastric cancer cells partially through targeting p21. Oncol. Lett. 2018, 15, 4585–4591. [Google Scholar] [CrossRef]
- Sokolova, V.; Fiorino, A.; Zoni, E.; Crippa, E.; Reid, J.F.; Gariboldi, M.; Pierotti, M.A. The Effects of miR-20a on p21: Two Mechanisms Blocking Growth Arrest in TGF-beta-Responsive Colon Carcinoma. J. Cell Physiol. 2015, 230, 3105–3114. [Google Scholar] [CrossRef]
- Kim, K.T.; Carroll, A.P.; Mashkani, B.; Cairns, M.J.; Small, D.; Scott, R.J. MicroRNA-16 is down-regulated in mutated FLT3 expressing murine myeloid FDC-P1 cells and interacts with Pim-1. PLoS ONE 2012, 7, e44546. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Banerjee, J.; Gnyawali, S.C.; Khanna, S.; He, G.; Pfeiffer, D.; Zweier, J.L.; Sen, C.K. Suppression of Induced microRNA-15b Prevents Rapid Loss of Cardiac Function in a Dicer Depleted Model of Cardiac Dysfunction. PLoS ONE 2013, 8, e66789. [Google Scholar] [CrossRef]
- Weirauch, U.; Beckmann, N.; Thomas, M.; Grunweller, A.; Huber, K.; Bracher, F.; Hartmann, R.K.; Aigner, A. Functional role and therapeutic potential of the pim-1 kinase in colon carcinoma. Neoplasia 2013, 15, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Xue, G.; Yan, H.L.; Zhang, Y.; Hao, L.Q.; Zhu, X.T.; Mei, Q.; Sun, S.H. c-Myc-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2alpha and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene 2015, 34, 1393–1406. [Google Scholar] [CrossRef]
- Lohcharoenkal, W.; Harada, M.; Loven, J.; Meisgen, F.; Landen, N.X.; Zhang, L.; Lapins, J.; Mahapatra, K.D.; Shi, H.; Nissinen, L.; et al. MicroRNA-203 Inversely Correlates with Differentiation Grade, Targets c-MYC, and Functions as a Tumor Suppressor in cSCC. J. Investig. Dermatol. 2016, 136, 2485–2494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lei, W.; Yan, W.; Li, X.; Wang, X.; Zhao, Z.; Hui, J.; Shen, Z.; Yang, J. microRNA-206 is involved in survival of hypoxia preconditioned mesenchymal stem cells through targeting Pim-1 kinase. Stem. Cell Res. Ther. 2016, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Lange-Grunweller, K.; Weirauch, U.; Gutsch, D.; Aigner, A.; Grunweller, A.; Hartmann, R.K. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 2012, 31, 918–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Yu, X.J.; Guo, X.Z.; Sun, M.H.; Wang, Z.; Song, Y.; Ni, Q.X.; Li, H.Y.; Mukaida, N.; Li, Y.Y. MicroRNA-33a-mediated downregulation of Pim-3 kinase expression renders human pancreatic cancer cells sensitivity to gemcitabine. Oncotarget 2015, 6, 14440–14455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Zhang, L.; Chen, J.; Li, Y.; Kang, K.; Qu, J.; Wang, Z.; Zhai, Y.; Li, L.; Gou, D. MiR-328 targeting PIM-1 inhibits proliferation and migration of pulmonary arterial smooth muscle cells in PDGFBB signaling pathway. Oncotarget 2016, 7, 54998–55011. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, G.; Lightner, C.; Elbai, S.; Brady, L.; Nicholson, S.; Ryan, R.; O’Sullivan, K.E.; O’Byrne, K.J.; Blanco-Aparicio, C.; Cuffe, S.; et al. Co-Targeting PIM Kinase and PI3K/mTOR in NSCLC. Cancers 2021, 13, 2139. https://doi.org/10.3390/cancers13092139
Moore G, Lightner C, Elbai S, Brady L, Nicholson S, Ryan R, O’Sullivan KE, O’Byrne KJ, Blanco-Aparicio C, Cuffe S, et al. Co-Targeting PIM Kinase and PI3K/mTOR in NSCLC. Cancers. 2021; 13(9):2139. https://doi.org/10.3390/cancers13092139
Chicago/Turabian StyleMoore, Gillian, Clara Lightner, Samira Elbai, Lauren Brady, Siobhan Nicholson, Ronan Ryan, Katie E. O’Sullivan, Kenneth J. O’Byrne, Carmen Blanco-Aparicio, Sinead Cuffe, and et al. 2021. "Co-Targeting PIM Kinase and PI3K/mTOR in NSCLC" Cancers 13, no. 9: 2139. https://doi.org/10.3390/cancers13092139