

Metabolic Syndrome Programming and Reprogramming: Mechanistic Aspects of Oxidative Stress


1
Department of Pediatrics, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung 833, Taiwan
2
College of Medicine, Chang Gung University, Taoyuan 333, Taiwan
3
Department of Pharmacy, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung 833, Taiwan
4
School of Pharmacy, Kaohsiung Medical University, Kaohsiung 807, Taiwan
*
Author to whom correspondence should be addressed.
Antioxidants 2022, 11(11), 2108; https://doi.org/10.3390/antiox11112108
Submission received: 15 September 2022
/
Revised: 6 October 2022
/
Accepted: 21 October 2022
/
Published: 26 October 2022
(This article belongs to the Special Issue Oxidative Stress and Inflammation in Metabolic Syndrome)
Abstract
:Metabolic syndrome (MetS) is a worldwide public health issue characterized by a set of risk factors for cardiovascular disease. MetS can originate in early life by developmental programming. Increasing evidence suggests that oxidative stress, which is characterized as an imbalance between reactive oxygen species (ROS), nitric oxide (NO), and antioxidant systems, plays a decisive role in MetS programming. Results from human and animal studies indicate that maternal-derived insults induce MetS later in life, accompanied by oxidative stress programming of various organ systems. On the contrary, perinatal use of antioxidants can offset oxidative stress and thereby prevent MetS traits in adult offspring. This review provides an overview of current knowledge about the core mechanisms behind MetS programming, with particular focus on the occurrence of oxidative-stress-related pathogenesis as well as the use of potential oxidative-stress-targeted interventions as a reprogramming strategy to avert MetS of developmental origins. Future clinical studies should provide important proof of concept for the effectiveness of these reprogramming interventions to prevent a MetS epidemic.
1. Introduction
Emerging evidence suggests that early life environment may negatively affect long-term health and result in increased risk for developing chronic diseases later in life. In a series of studies, David Barker and his colleagues showed that low birth weight (LBW) is associated with increased rates of heart disease, diabetes, and many other features of metabolic syndrome (MetS) in adult life [1,2,3,4]. Based on these findings, David Barker and colleagues proposed the concept of fetal origins of adult disease [5]. It soon became clear that adverse environmental insults also occur during a critical developmental window that produces long-term alterations in tissue structure or function by what is now called developmental programming [6], as well as predisposition to future illness. These developments led to the emergence of the field known as ‘The Developmental Origins of Health and Disease’ (DOHaD) [7]. Notably, the DOHaD concept also provides a novel way to avert adult disease by reprogramming therapy [8,9], that is, by switching therapy prior to illness onset from adulthood to fetal or fetal life. For that reason, reprogramming can potentially serve as an innovative preventive strategy to reduce the global burden of disease.
Non-communicable diseases (NCDs) are of increasing global concern due to their high mortality rate [10]. Importantly, MetS and associated disorders account for two-thirds of NCD deaths [11]. Also important is the prevalence of MetS, which continues to rise globally because of a lack of specific therapeutic regimens [11]. Based on this, the pursuit of a DOHaD approach that can better understand metabolic programming and develop efficient reprogramming strategies has the potential to reduce global burden of MetS.
MetS is a collection of medical conditions that occur together and that increase risk of cardiovascular disease (CVD) [12]. The main components of MetS comprise insulin resistance, obesity, hypertension, non-alcoholic fatty-liver disease (NAFLD), dyslipidemia, and accumulation of adipose tissue. Although the pathogenesis of MetS is highly complex and not yet clear, increasing evidence suggests that oxidative stress has a decisive role in its manifestations [13].
Oxidative stress is a phenomenon caused by an imbalance in overproduction of deleterious reactive oxygen and nitrogen species (ROS and RNS) that overwhelm the capacity of cellular antioxidant defense [14]. Novel research findings increasingly support the importance of oxidative stress in various components of MetS, including hypertension [15], obesity [16], insulin resistance [17], NAFLD [18], etc. Conversely, treatment with antioxidants has been suggested to aid in the prevention of MetS-related disorders [19,20,21].
Despite the evidence showing the impact of oxidative stress and antioxidant therapy in MetS, little attention has been paid to their implications for the developmental programming of MetS. The aim of the current review is to map the best available evidence onto the interplay between oxidative stress and developmental programming of MetS. Our review also tends to highlight the common mechanisms behind MetS programming, their interactions with oxidative stress, and the potential of oxidative-stress-targeted therapy as a reprogramming strategy for MetS of developmental origins.
We used the PubMed, Medline, and Embase databases to search studies written in English using the following keywords: “metabolic syndrome”, “hypertension”, “dyslipidemia”, “hyperlipidemia”, “obesity”, “diabetes”, “insulin resistance”, “hyperglycemia”, “developmental programming”, “DOHaD”, “free radicals”, “offspring”, “progeny”, “mother”, “prenatal”, “nitric oxide”, “oxidative stress”, “pregnancy”, “reprogramming”, “reactive oxygen species”, “reactive nitrogen species”, and “antioxidant”. Additional studies were selected based on references from eligible articles. The search was ended by 23 August 2022.
2. Current Evidence Supporting the Developmental Origins of MetS
2.1. Human Research
Currently, several lines of epidemiological evidence suggest that adverse intrauterine conditions coincide with the risk of developing MetS throughout the lifetime. Existing human studies mainly come from natural history famine birth cohorts. The studies on the Dutch famine showed that pregnant women under famine had children who developed several features of MetS later in life, such as hypertension, dyslipidemia, obesity, and insulin resistance [22,23]. Studies in other famines also support the notion that early-life famine exposure appears to be a risk factor for obesity, hypertension, and coronary heart disease [22,23,24,25]. Also, data from twin studies suggest that LBW is related to an increased risk of adult cardiometabolic disorders [26,27].
In 1989, Barker and colleagues reported that LBW was associated with an increased risk of death from CVD [1]. Likewise, there have been many studies showing an association between LBW and hypertension [28], impaired glucose tolerance [29], and obesity [30] in later life. Much of the observational research on risk factors for MetS traits represent another line of evidence to support developmental origins of MetS. Risk factors now known to have such effects include maternal malnutrition [22,23], maternal obesity [31,32], gestational diabetes [33], maternal smoking [34], environmental toxins [35], maternal stress [36], etc. Finally, postnatal overnutrition is detrimental for infants with LBW who attain “catch-up growth”, being related to obesity and cardiometabolic risks [37,38]. A systematic review summarizing 39 studies revealed that rapid weight gain in infants with LBW was linked to an 80% greater risk for CVDs [39].
A number of hypotheses, such as thrifty phenotype [40], catch-up growth hypothesis [41], and predictive adaptive responses [42] have been developed to explain the epidemiological observations of an association between early life insults and later adult diseases. Despite these human studies supporting a connection between early-life environmental exposure and developmental origins of MetS traits in later life, these clinical studies seem unable to provide molecular mechanisms underlying developmental origins of MetS for the creation of reprogramming interventions. As a result, the consideration of biological plausibility when assessing causality and the creation of potential reprogramming strategies rely heavily upon evidence derived from animal models.
2.2. Animal Models
A number of previous studies address the importance of animal models being used to understand MetS programming, and this has been reviewed elsewhere [43,44,45,46]. Bearing in mind the complexity of MetS, developmental origin studies of MetS are mostly conducted using models that display some, but not all, features of MetS in most investigations [43,44,45,46]. Many animal models are derived from a variety of early-life risk factors to elicit certain characteristics of MetS in adult offspring. Similar to human studies, these early-life insults contribute to the developmental origins of MetS, including maternal nutrition imbalance, maternal illness, environmental toxins, maternal stress, medication use, etc. Although rats are the most frequently used animals [43,44,45,46], other species like mice [47], sheep [48], rabbits [49], pigs [50], and non-human primate [51] have also been used for comparisons of major components of MetS development during the lifetime. As we primarily focus on oxidative stress in this review, and for the sake of brevity, we have limited the animal models of oxidative-stress-related MetS with developmental origins; these are discussed in detail in the following section.
3. Oxidative-Stress-Related Developmental Origins of MetS
3.1. ROS/NO Disequilibrium
Oxidative stress results from a state of disequilibrium in the ROS/NO balance and a limited biological antioxidant capability. Both ROS and RNS are damaging biological molecules [14]. ROS are highly reactive chemicals formed from oxygen, including free radicals such as superoxide anion (O2−) and hydroxyl anion (OH−) as well as non-radical molecules such as hydrogen peroxide (H2O2). Among them, the superoxide anion radical initiates a cascade of reactions, resulting in the generation of other ROS species.
RNS that bear nitrogen atoms include the nitric oxide radical (NO−), the nitrogen dioxide radical (NO2−), and peroxynitrite (ONOO−). Much of RNS-dependent cytotoxicity resides in peroxynitrite, which is produced by the reaction between NO and superoxide [52]. In contrast, NO physiologically functions as a gasotransmitter, participating in cardiometabolic health at an optimal level [53]. Asymmetric dimethylarginine (ADMA) is an endogenous competitive inhibitor of NOS [54]. High ADMA can uncouple NOS isoenzymes to generate peroxynitrite, further contributing to reduced NO bioavailability and increased oxidative stress [55].
On the other hand, several antioxidants can counteract the harmful effects of ROS/RNS. Superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), glutathione reductase, etc. are enzymatic antioxidants. There are also quite a few non-enzymatic antioxidants, which include glutathione (GSH) and vitamins [56]. The discrepancy between excessive ROS/RNS and weak endogenous antioxidant defense leads to damaged DNA, lipids, proteins, and cellular structures.
3.2. Oxidative Stress and NO Signaling during Pregnancy
During pregnancy, the balance between ROS and antioxidants should be maintained to provide an appropriate environment for the fetus [57]. The physiological generation of ROS positively impacts a variety of developmental processes, ranging from oocyte maturation [58], embryo implantation [59], and placental differentiation [60] to fetal development. The fetus needs oxygen early in pregnancy, but the oxygen consumption differs at different trimesters of pregnancy [61]. Fetal oxygen levels are low during the first trimester. During the second and third trimesters, increasing oxygen needs are in response to rapid fetal weight gain and establishment of fetal–placental circulation [62]. Increased production of ROS occurs because of high consumption of oxygen, enhanced metabolism, and utilization of fatty acids, while abnormal overproduction of ROS disrupts these processes, resulting in compromised pregnancy [57]. Oxidative damage arises due to the failure of defensive antioxidant mechanisms in responding to excessive ROS and RNS [56]. Adverse conditions in pregnancy that are now known to induce oxidative stress include preeclampsia, diabetes, maternal smoking, obesity, and intrauterine growth retardation (IUGR) [63].
NO has a crucial role in governing feto-placental blood flow. Along with the main vasodilator in the placenta, NO is involved in vascular reactivity regulation, placental bed vascular resistance, and angiogenesis [64]. Circulating ADMA levels, an endogenous inhibitor of NOS, are reduced in the first trimester but increase as the gestational age increases [65,66]. In early pregnancy, low ADMA and concomitant high NO may result in hemodynamic adaptation, a greater need of organ perfusion, and uterine relaxation to allow for fetal growth. In contrast, increased ADMA levels in later pregnancy aid in the higher uterine muscle contractile activity that is required for successful delivery [67]. In compromised pregnancies, such as in pre-eclampsia [67], gestational diabetes [68], and maternal undernutrition [69], ADMA levels rise to levels higher than those seen in normal pregnancy. Summarily, imbalances between ROS and ADMA/NO pathway result in oxidative stress, which is a condition that contributes to fetal programming in compromised pregnancies.
3.3. Animal Models of Oxidative-Stress-Related Developmental Origins of MetS
Although mounting evidence indicates the pathogenic interrelationship between oxidative stress and MetS [13], there is a relative paucity of information regarding the impact of oxidative stress in early life on offspring MetS traits. Hence, this section mainly covers evidence regarding animal models used to study oxidative-stress-related developmental origins of MetS. These animal models are summarized in Table 1 [57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118]. Since there is a large amount of available information for single components of MetS, for the sake of brevity, we limited our study to those animal models that display at least two of the components of MetS in offspring. Additionally, this review was restricted to rat models to facilitate appropriate comparisons of major features of MetS as they appear throughout a lifetime. In rats, one month of life is equivalent to 3 human years in adulthood [119]. Table 1 lists the timing of offspring outcomes, ranging from one week to one year of age in rats, which corresponds to humans from infancy to middle adulthood.
3.3.1. Maternal-Derived Insults
Various environmental insults have been examined in animal models, including maternal nutritional imbalance [69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85], pregnancy complications [86,87,88,89,90,91,92,93,94], maternal illness [95,96,97,98,99,100,101,102,103,104,105,106], and toxin/chemical exposure [107,108,109,110,111,112,113,114,115,116,117,118]. Maternal nutritional imbalance can induce nutritional programming. Following the observational studies evaluating exposure to severe famine [22,23,24], maternal caloric or protein restriction models have been conducted to mimic malnutrition in pregnant women exposed to severe famine at that time. Adult rat progeny born to dams exposed to 50% caloric restriction develop insulin resistance and hypertension [69,70,71].
Similarly, protein restriction (8–9%) during pregnancy and/or lactation leads to offspring hypertension and insulin resistance [72,73]. Offspring MetS traits can also be programmed by maternal overnutrition. A maternal high-fat diet has been commonly used as an animal model for studying MetS of developmental origins [120]. Mother rats receiving a high-fat diet saw an elevation in BP, body weight, blood lipids, and insulin level in their offspring [74,75,76,77]. Likewise, hypertension, abnormal regulation of lipid metabolism, and insulin signaling can be programmed by a maternal high-fructose diet [80,81,82].
Additionally, complications during pregnancy and maternal illness are able to cause MetS programming. Bilateral uterine artery ligation induced maternal uteroplacental insufficiency that led to hypertension, dyslipidemia, and insulin resistance in adult male rat offspring [86,87]. In addition, adult male offspring born to dams exposed to hypoxia developed hypertension, obesity, and insulin resistance [89,90]. Likewise, offspring hypertension and insulin resistance can be induced by maternal inflammation in a lipopolysaccharide (LPS) exposure model or a surgically induced periodontitis model [92,93].
Several components of MetS such as hypertension, obesity, insulin resistance, and dyslipidemia in adult offspring induced by maternal diabetes are also demonstrable in animal models [95,96,97]. Though many models have been used for diabetes research, only streptozotocin (STZ)-induced diabetes has been modeled for MetS of developmental origins [95,96,97]. Both type 1 and type 2 diabetes can be induced by STZ when given to adult [95] or neonate rats [95,96,97]. Previous reports also demonstrated that adult male offspring in a rat model with maternal continuous light exposure had hypertension and insulin resistance [100,101]. Another common pregnancy complication is maternal stress. A developing fetus is prone to being exposed to excessive glucocorticoid due to a stressed pregnancy. Dexamethasone exposure during pregnancy was shown to induce hypertension, obesity, insulin resistance, and liver steatosis in adult male rat offspring [103,104,105].
Moreover, maternal exposures to toxin/chemical have also been associated with the developmental programming of MetS. Several of the studies listed in Table 1 indicated that maternal exposure to di-n-butyl phthalate (DEHP) [108,109] or bisphenol A (BPA) [111,112] can lead to hypertension and insulin resistance in adult rat offspring. Additionally, maternal nicotine administration during lactation was shown to cause hypertension, hyperlipidemia, and steatosis in adult offspring [113,114,115]. Furthermore, administration of 1 g ethanol/kg on gestational days 13 and 14 in mother rats induced MetS programming, resulting in hypertension and insulin resistance in offspring of both sexes by 6 months of age [116,117].
3.3.2. Mechanisms behind Oxidative Stress
Oxidative-stress-mediated mechanisms involved in the pathogenesis of developmental MetS include increased ROS generation enzymes [84,106,113,118], increased ROS [85,88,89,102,110], decreased expression and/or activity of antioxidant enzymes [72,78,99,106,115,118], increased peroxynitrite [70,98,113], increased oxidative damage [69,72,78,79,83,84,88,94,98,111,113,115], and dysregulated ADMA-NO pathway [69,70,83,95,99,103,107,111]. Notably, most studies have focused on the renal and cardiovascular systems: investigators generally paid less attention to oxidative stress programming on other organ systems, such as the brain [84,102,118], spleen [85], liver [104,105,115], and adrenal gland [106].
Over the years, many oxidative stress biomarkers have been proposed, mainly reflecting the assessment of oxidative damage in biological molecules: lipids, proteins, and DNA. Among these, lipid peroxidation biomarkers are the most commonly used. Table 1 shows how several biomarkers of lipid peroxidation have been utilized to determine oxidative damage in different models of programmed MetS, including F2-isoprostanes [72,88], malondialdehyde (MDA) [78,84,94,113], thiobarbituric acid reactive substances (TBARS) [99], and 4-hydroxynonenal (4-NHE) [115]. Notably, MetS of developmental origins programmed by different maternal insults accompanies organ-specific lipid peroxidation in the kidney [72,78,88,94], vessels [99,113], brain [84], and liver [115].
Additionally, 8-hydroxydeoxyguanosine (8-OHdG) is a biomarker used to detect oxidized nucleoside of DNA [121]. Several studies support the idea that oxidative stress with increased renal 8-OHdG expression is involved in the pathogenesis of MetS programming in models of caloric restriction [69], high-fat diet [78], high-fructose diet [83], prenatal dexamethasone exposure [107], and prenatal bisphenol A exposure [111]. Another biomarker of oxidative stress is 3-nitrotyrosine (3-NT), which represents the nitration of protein-bound and free tyrosine residues by reactive peroxynitrite molecules [122]. Prior work revealed that increased 3-NT in the vessels [70,113] and kidneys [98] is related to MetS of developmental origins.
Decreased antioxidant capacities can also be involved in oxidative-stress-related MetS programming. Impaired enzymatic and non-enzymatic antioxidant defenses, including SOD [78,99], glutathione peroxidase 1 [106,115,118], catalase [118], and glutathione [72], have been shown in several models of MetS programming.
Prior reviews support the notion that ADMA-related NO-ROS imbalance in early life induces offspring hypertension, a hallmark of MetS. Table 1 illustrates how ADMA is a key risk factor for oxidative stress programming in several animal models, such as caloric restriction [69], diabetes [95], prenatal dexamethasone exposure [107], and prenatal bisphenol A exposure [111]. Moreover, NO deficiency in the vessels [70,84] and kidneys [69,99,103,111] is also relevant to MetS of developmental origins. A summary of the interaction between maternal-derived insults implicated in oxidative stress and the major organ systems involved in MetS of developmental origins is depicted in Figure 1.
3.3.3. Other Mechanisms Related to MetS Programming
In addition to oxidative stress, several core mechanisms may participate in MetS programming [45], including the glucocorticoid effect [123], dysregulated nutrient-sensing signals [124], aberrant activation of the renin–angiotensin aldosterone system (RAAS) [125], gut microbiota dysbiosis [126], etc. Oxidative stress acts a molecular hub facilitating a wide range of functional interactions among the above-mentioned core mechanisms behind MetS programming (Figure 2). Several of the studies presented in Table 1 have linked maternal glucocorticoid exposure to MetS programming [103,104,105,106,107]. As a product of the activation of the hypothalamic–pituitary–adrenal (HPA) axis, glucocorticoids have potent programming effects on fetal development [127]. Also, the interplay of oxidative stress and nutrient-sensing signals has been implicated in maternal high-fructose diet-induced offspring hypertension [84,128]. Further, it is known that RAAS intrinsic to tissues modulates BP, metabolic homeostasis, adiposity, and insulin sensitivity [125,129]. The aberrant activation of the RAAS and oxidative stress concurrently exist in several models of MetS programming [84,103,130]. Moreover, disruption in gut microbiota is tightly connected to MetS and associated disorders [131], such as obesity [132], insulin resistance [133], dyslipidemia [134], cardiovascular disease [135], etc. An imbalanced redox state induces gut microbiota dysbiosis, while gut microbial communities regulate redox signaling to preserve host–microbiota homeostasis [136,137].
4. Reprogramming Strategies: Oxidative-Stress-Targeting Therapies
Although the role of oxidative stress in the pathogenesis of many diseases is undoubted, the beneficial effects of antioxidant therapy, based on available clinical evidence, remain inconclusive. So far, the majority of epidemiological studies have not confirmed any evidence of proven benefits from antioxidant supplementation, especially in the cardiovascular field [138,139]. These controversial findings may be due to the type of antioxidant, the single versus multiple approach, supplement timing and dosage, the population suitable to be treated, etc. Accordingly, it is vital to target specific critical redox pathways and increase the selectivity of these oxidative-stress-targeted approaches in animal models before clinical translation.
As for our contemporary knowledge of the DOHaD concept, it turns out that prevention and management of MetS can be started earlier, even before disease occurs, by reprogramming [8,9]. In the above sections, we illustrated the critical roles that oxidative stress plays in the pathogenesis of MetS programming. On that basis, antioxidants and other oxidative-stress-targeted interventions hold promise for the early-life prevention of MetS in adult progeny.
Non-enzymatic antioxidants could be natural and synthetic antioxidants [140]. Examples of natural non-enzymatic antioxidants are glutathione, polyphenols, carotenoids, flavonoids, vitamins A, C, and E, etc. [141]. Apart from natural antioxidants, several synthetic antioxidants have also been implemented in MetS. This section discusses the reprogramming role of oxidative-stress-targeted therapies that are involved in the main redox reactions and avert MetS of developmental origins. There are several different types of oxidative-stress-targeted intervention. These are grouped together, depending on which mechanism of oxidative stress they mediate. Overall, these interventions can be classified as targeting ROS with enzymatic antioxidants, targeting ROS with non-enzymatic antioxidants, and targeting NO. These potential oxidative-stress-targeted interventions used as reprogramming therapies for MetS of developmental origins are illustrated in Figure 3.
4.1. Targeting ROS with Enzymatic Antioxidants
The NOX family, as a key enzymatic source of ROS, can employ NADPH as an electron donor and then drive molecular oxygen to convert into superoxide [142]. Therefore, agents that would efficaciously target NOXs to scavenge ROS might hold significant promise for reducing oxidative stress [143]. There are two types of NOXs inhibitors: small-molecule inhibitors and peptidic inhibitors [143]. However, neither of them have been examined in MetS of developmental origins.
On the other hand, SOD can eliminate superoxides with a dismutation mechanism. SOD mimetics have also been explored as a potential treatment for many oxidative-stress-related disorders [144]. It has long been known that SOD modulates metabolism. Prior work indicates that several types of SOD mimics show therapeutic potential against dyslipidemia [145], obesity [146], insulin resistance [146], and hypertension [147]. Although administration of SOD mimetic tempol in pregnancy has been reported to reduce BP in spontaneously hypertensive rat offspring [148], none of the SOD mimetics have been approved in models of MetS programming to date.
4.2. Targeting ROS with Non-Enzymatic Antioxidants
Several non-enzymatic antioxidants applied during gestation and lactation have been utilized as reprogramming strategies to prevent the development of MetS in animal models, including vitamins, amino acids, melatonin, polyphenol, N-acetylcysteine (NAC), and synthetic antioxidants.
4.2.1. Vitamins
The most widely explored nutraceuticals are vitamins C and E. Vitamin C is a potent water-soluble antioxidant with the ability to quench ROS [149]. Vitamin E is a lipid-soluble antioxidant that inhibits NADPH oxidase, cyclooxygenase, and lipoxygenase [150]. Our prior review summarizes current evidence supporting perinatal use of vitamins C and E, alone or combined with other antioxidants, for protecting rat offspring hypertension [151]. Disruption of epigenetic regulation can result in oxidative stress in relation to MetS programming [152]. Despite a recognized role of vitamins B6, B12, and folate as methyl donors for DNA methylation [153], whether their supplementations in pregnancy can avert offspring MetS via regulation of epigenetics remain largely unknown. Although several vitamins exert advantageous effects on oxidative-stress-related disorders, less attention has been paid to determine their reprogramming effects on MetS of developmental origins.
4.2.2. Amino Acids
Several amino acids have antioxidant properties [154]. It is well-known that amino acids participate in body fat composition [155], insulin signaling [155], and BP regulation [156]. Previous research indicates that amino acid supplementation during gestation and lactation can avert offspring hypertension in several animal models. Examples of amino acids are taurine, arginine, citrulline, cysteine, and branched-chain amino acids (BCAAs). BCAA supplementation in pregnancy does not only prevent maternal caloric-restriction-induced offspring hypertension [157]: gestational supplementation of BCAAs also benefits obesity-associated insulin resistance programmed by maternal high-fat diet [158].
Even though there are other amino acids showing reprogramming potential for hypertension of developmental origins [156], their reprogramming effects in other MetS traits remain largely unclear. Importantly, amino acid metabolism between the mother and the fetus in pregnancy is crucial for fetal development. We must elucidate the pathophysiologic roles of specific amino acids and their connections in the developmental programming of MetS to avoid unintentional adverse consequences.
4.2.3. Melatonin
Melatonin (N-acetyl-5-methoxytryptamine) is a pleiotropic hormone essential for pregnancy and fetal development [159]. Melatonin and its metabolites, acting as naturally occurring antioxidants, can scavenge ROS/RNS, enhance expression of antioxidant enzymes, and increase NO bioavailability [160,161]. Perinatal use of melatonin has been proposed as a reprogramming strategy for many DOHaD-related adult diseases [162].
As shown in Table 1, the beneficial effects of maternal melatonin therapy are expressed in different models against offspring hypertension [100], insulin resistance [101], and liver steatosis [106]. Perinatal use of melatonin can have beneficial effects against rat offspring hypertension via restoration of the ROS/NO balance in a maternal caloric restriction model [163] and a high-fructose model [164]. Additionally, prior studies have demonstrated interplay between melatonin and several core mechanisms underlying MetS programming, such as aberrant RAAS, dysregulated nutrient-sensing signaling, and glucocorticoid programming [161]. These observations support the notion that perinatal use of melatonin may act in diverse ways to avert MetS programming-induced disorders in later life [161]. Melatonin is also involved in epigenetic regulation [165]. Melatonin can regulate antioxidant and pro-inflammatory genes via epigenetic on/off mechanisms [166]. While maternal melatonin therapy can epigenetically alter more than 450 transcripts in the 1-week-old offspring kidney [165], whether epigenetic regulation of melatonin has a role in its protective effect in MetS programming remains to be elucidated.
Of note is that melatonin is a quite safe supplement in humans [167]. Although the clinical use of melatonin during pregnancy remains inconclusive, it has nonetheless been clinically used for several neonatal diseases [168]. Therefore, there is a desperate need for further translational research into the long-term MetS-associated outcomes of perinatal melatonin use.
4.2.4. Polyphenols
Polyphenols are the widespread phytochemical antioxidants in food [169]. Prior work has revealed the valuable effect of polyphenols in the counterbalance of oxidative stress by working as free-radical scavengers, NOS activators, metal chelators, and stimulator of antioxidant enzymes [170,171]. Accordingly, polyphenols have shown beneficial effects in MetS [172,173]. Though several systematic reviews have shown that dietary polyphenol intake reduces CVD risk [174,175,176,177], only a few polyphenols have been tested in animal models of MetS programming.
Polyphenols are commonly categorized as flavonoids and nonflavonoids [169]. Several flavonoids are potent antioxidants [169]. As an antioxidant, quercetin has been used in pregnancy to protect adult rat progeny against hypertension programmed by maternal high-fat diet [106]. In another antenatal dexamethasone exposure rat model, maternal treatment with epigallocatechin gallate moderated the developmental programming of hypertension [106].
Resveratrol is a nonflavonoid polyphenol that is commonly used as a nutritional supplement [170,178]. Resveratrol can act as an antioxidant against oxidative stress. Currently, there is accumulating evidence that suggests a reprogramming effect of resveratrol for the prevention of offspring MetS [179]. The use of resveratrol in early life has been reported to protect rat offspring against hypertension [107,111], hyperlipidemia [75], obesity [76,180], and insulin resistance [181] in various developmental programming models.
Also, genistein, curcumin, and resveratrol have been demonstrated to trigger the antioxidant and anti-inflammatory machinery and ameliorate MetS traits via epigenetic mechanisms [182]. However, further research is needed to understand whether the beneficial effects of polyphenols in MetS programming are directly related to epigenetic changes [183].
One major issue that limits the clinical translation of polyphenols is their low bioavailability in vivo [184]. Considering the complexity and inter-individual variability of polyphenol pharmacokinetics, further research is required to better elucidate the differential impact of various polyphenols on the MetS of developmental origins.
4.2.5. N-acetylcysteine
N-acetylcysteine, an antioxidant naturally found in Allium plant, is a precursor to glutathione [185]. Also, NAC is a stable L-cysteine analogue and can be used for H2S synthesis [186]. Perinatal NAC therapy averts rat offspring hypertension as induced by a number of early-life insults, such as maternal nicotine exposure [114], maternal hypertension [187], maternal L-NAME exposure [188], suramin-induced pre-eclampsia [189], and prenatal dexamethasone and postnatal high-fat diet [190].
Using a maternal L-NAME exposure model, perinatal NAC therapy was shown to protect rat offspring hypertension, accompanied by enhancement of H2S-generating enzyme expression and activity in offspring kidneys [188]. In another study [190], the advantageous effects of NAC against offspring hypertension were associated with an increase in plasma glutathione level, reduction of oxidative stress, and upregulation of H2S-generating enzymes. Furthermore, maternal NAC therapy was able to avert rat offspring hypertension programmed by maternal suramin administration, which coincided with increased glutathione levels, restoration of NO bioavailability, and augmentation of H2S pathways [189].
4.2.6. Synthetic Antioxidants
In addition to natural antioxidants, some synthetic antioxidants have been applied to reduce oxidative stress in animal models to study MetS programming. The transcription factor NRF2 is a master regulator of various homeostatic genes that defend against oxidative stress [191]. In response to oxidative stress, NRF2 is released from its principal negative regulator Kelch-like ECH-associated protein 1 (KEAP1) and translocated to the nucleus, where NRF2 promotes the expression of several antioxidant genes via binding to antioxidant response element (ARE) [192]. Accordingly, NRF2 activators are considered as potential agents to protect oxidative-stress-related damage [193].
Dimethyl fumarate (DMF), an NRF2 activator, has been used to prevent rat offspring hypertension in a combined maternal dexamethasone exposure and postnatal high-fat diet model [194,195]. In addition, maternal lazaroid therapy, an inhibitor of lipid peroxidation [196], prevented the elevation of BP in adult rat progeny born to dams that received a protein-restricted diet [72]. Although certain synthetic antioxidants have been explored in several animal models of oxidative stress, little is known regarding their ability to protect adult offspring against MetS programming.
4.3. Targeting NO
A number of NO-targeted approaches have been utilized to increase NO bioavailability, such as NO donors, supplementation of NO substrate, enhancement of the expression and/or activity of NOS, ADMA-lowering agents, etc. So far, some of them have been examined for therapeutic prevention of MetS programming.
While NO donors, molsidomine, and pentaerythritol tetranitrate have shown beneficial effects against the development of hypertension [197,198], their reprogramming effects on MetS traits deserve further clarification.
As the substrate for NOS isoenzymes, L-arginine supplementation has been applied to augment NO bioavailability in several diseases [199], while the beneficial effects of L-arginine from human trials remain inconclusive [200]. As the main precursor of L-arginine, oral l-citrulline supplementation has been utilized to increase l-arginine production and bypass hepatic metabolism to raise NO levels [201]. To date, gestational L-citrulline supplementation has shown benefits against offspring hypertension in rat models of maternal caloric restriction [69], streptozotocin-induced diabetes [95], and prenatal dexamethasone exposure [103]. Along with averting hypertension, L-citrulline supplementation has also attenuated liver fat accumulation and prevented hypertriglyceridemia in adult rat offspring born to dams that received a high-fructose diet [202].
The use of ADMA-lowering agents is another way to increase NO. Though a specific ADMA-lowering agent remains inaccessible at the time of this paper, a number of clinically used drugs have been shown to restore ROS/NO balance throughout lowering ADMA levels [203]. Telmisartan, glucagon-like peptide-1 receptor agonist, rosuvastatin, and epigallocatechin-3-gallate can lower ADMA levels via reduced expression of ADMA-generating enzyme. On the other hand, metformin, NAC, melatonin, atorvastatin, salvianolic acid A, telmisartan, oxymatrine, and rosuvastatin can augment the activity and/or expression of ADMA-metabolizing enzymes and thus decrease ADMA levels [203]. So far, only a few ADMA-lowering agents have been studied in developmental programming models to prevent offspring hypertension, including NAC [191], melatonin [204], and metformin [205]. Metformin also showed benefits against liver steatosis in a maternal high-fat diet rat model [206]. Moreover, supplementing melinjo (Gnetum gnemon) seed extract during lactation protected adult female rat offspring hypertension by enhancing eNOS expression in a maternal high-fructose diet model [207].
4.4. Pros and Cons
Although animal studies implicate oxidative stress as an attractive target for MetS prevention and therapy, their efficacy still awaits validation in human trials. Considering the difficulties of recruiting pregnant or lactating women in medication research, the use of breastmilk as a reprogramming strategy would be a good start. Breastmilk has a powerful antioxidant composition [208]. There are reports that suggest that there is a relationship between premature infants fed with breastmilk and lower rates of MetS in young adult life [209]. As breastfeeding is recommended for infants during the first 6 months after birth [210], the antioxidant protection offered by breastfeeding against MetS programming is a key issue that deserve further study.
On the other hand, oxidative-stress-targeted therapy can also be disadvantageous. Most oxidative-stress-targeted therapies such as antioxidants are administered orally or intravenously, which eventually enter the circulation and reach the targeted organ. However, healthy tissues other than the targeted organs, which have not experienced oxidative stress damage, may be non-specifically targeted by the antioxidant [211]. As a result, healthy tissues/organs may be affected negatively, as their levels of ROS may fall below their physiologically normal limit. As homeostasis of ROS is one of the mandatory requirements for normal pregnancy and fetal development [57], antioxidant supplementation during pregnancy and breastfeeding would only apply in the case of deficits, but not as a usual dietary supplement.
Moreover, excessive antioxidant supplement may shift oxidative stress to an opposite state, namely antioxidant stress [212]. However, it currently remains unclear which pathogenetic mechanism should be targeted, which timing for reprogramming should be appropriate, and which kind of antioxidants should be used. Further studies are required to establish the particular developmental window (e.g., prenatal or pre-weaning stage), to elucidate organ-specific redox-sensitive signaling responsible for different maternal-derived insults underlying MetS programming, and to determine the ‘right’ oxidative-stress-targeted therapy with the ‘right’ dose at the ‘right’ time for reprogramming.
5. Concluding Remarks and Perspectives
There is substantial evidence that suggests that oxidative stress is involved in MetS programming, and oxidative-stress-targeted therapy is a potential preventive strategy. Our review highlights how targeting ROS with enzymatic antioxidants, targeting ROS with non-enzymatic antioxidants, and targeting NO might represent promising tools for the prevention of MetS and associated disorders. However, with all the obvious benefits of oxidative-stress-targeted therapy in MetS programming, we have to be mindful of timing, dosage, and target organ for various pathologies, since the heterogeneity of MetS has to be a central consideration. Although several oxidative-stress-targeted strategies were explored in animal studies and some of them revealed promising data, their efficacy still awaits future translation into human investigations.
While there has been significant progress in establishing animal models for studying MetS of developmental origins, only a few models present all components of MetS. While some oxidative-stress-targeted therapies offer substantial progress in certain characteristics of MetS, it remains unclear whether their effects are beneficial for other MetS traits or if they should be translated from one model into other models. Importantly, MetS of developmental origins, along with oxidative stress, is associated with other core mechanisms. Therefore, it remains to be determined whether the protective effects of antioxidant therapy in pregnancy are related to the common mechanisms behind MetS programming.
A deeper understanding of the molecular and biochemical mechanisms of abnormalities associated with oxidative stress in MetS programming will facilitate the development of preventive therapeutics. Such efforts might prove effective in the prevention of a global epidemic of MetS.
Author Contributions
Y.-L.T. contributed to data interpretation, concept generation, methodology, critical revision of the manuscript, drafting of the manuscript, and approval of the article; C.-N.H. contributed to methodology, concept generation, critical revision of the manuscript, drafting of the manuscript, and approval of the article. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Chang Gung Memorial Hospital, Kaohsiung, Taiwan, grants CMRPG8M0721, CORPG8M0201, CORPG8M0151, and CORPG8M0081.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 1, 1077–1081. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Hales, C.N.; Fall, C.H.D.; Osmond, C.; Phipps, K.; Clark, P.M.S. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidemia (syndrome X): Relation to reduced fetal growth. Diabetologia 1993, 36, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J.P.; Osmond, C.; Winter, P.D.; Margetts, B.M.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 2, 577–580. [Google Scholar] [CrossRef]
- Barker, D.J.; Eriksson, J.G.; Forsen, T.; Osmond, C. Fetal origins of adult disease: Strength of effects and biological basis. Int. J. Epidemiol. 2002, 31, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, V.; Cardoso, R.C.; Puttabyatappa, M. Developmental Programming, a Pathway to Disease. Endocrinology 2016, 157, 1328–1340. [Google Scholar] [CrossRef] [Green Version]
- Haugen, A.C.; Schug, T.T.; Collman, G.; Heindel, J.J. Evolution of DOHaD: The impact of environmental health sciences. J. Dev. Orig. Health Dis. 2015, 6, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Joles, J.A. Reprogramming: A Preventive Strategy in Hypertension Focusing on the Kidney. Int. J. Mol. Sci. 2015, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Paauw, N.D.; Van Rijn, B.B.; Lely, A.T.; Joles, J.A. Pregnancy as a critical window for blood pressure regulation in mother and child: Programming and reprogramming. Acta Physiol. 2016, 219, 241–259. [Google Scholar] [CrossRef]
- Zarocostas, J. Need to increase focus on non-communicable diseases in global health, says WHO. Br. Med. J. 2010, 341, c7065. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [PubMed] [Green Version]
- Spahis, S.; Borys, J.M.; Levy, E. Metabolic Syndrome as a Multifaceted Risk Factor for Oxidative Stress. Antioxid. Redox Signal. 2017, 26, 445–461. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Sinha, N.; Dabla, P.K. Oxidative stress and antioxidants in hypertension-a current review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Bondia-Pons, I.; Ryan, L.; Martinez, J.A. Oxidative stress and inflammation interactions in human obesity. J. Physiol. Biochem. 2012, 68, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Ahmad, K.A.; Yuan Yuan, D.; Nawaz, W.; Ze, H.; Zhuo, C.X.; Talal, B.; Taleb, A.; Mais, E.; Qilong, D. Antioxidant therapy for management of oxidative stress induced hypertension. Free Radic. Res. 2017, 51, 428–438. [Google Scholar] [CrossRef]
- Abdali, D.; Samson, S.E.; Grover, A.K. How effective are antioxidant supplements in obesity and diabetes? Med. Princ. Pract. 2015, 24, 201–215. [Google Scholar] [CrossRef]
- Gregório, B.M.; De Souza, D.B.; de Morais Nascimento, F.A.; Pereira, L.M.; Fernandes-Santos, C. The potential role of antioxidants in metabolic syndrome. Curr. Pharm. Des. 2016, 22, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Schulz, L.C. The Dutch Hunger Winter and the Developmental Origins of Health and Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16757–16758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumey, L.H. Reproductive outcomes in women prenatally exposed to undernutrition: A review of findings from the Dutch famine birth cohort. Proc. Nutr. Soc. 1998, 57, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanner, S.A.; Yudkin, J.S. Fetal programming and the Leningrad Siege study. Twin Res. 2001, 4, 287–292. [Google Scholar] [CrossRef]
- Hult, M.; Tornhammar, P.; Ueda, P.; Chima, C.; Bonamy, A.K.; Ozumba, B.; Norman, M. Hypertension, diabetes and overweight: Looming legacies of the Biafran famine. PLoS ONE 2010, 5, e13582. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, R.; Yang, W.; Xu, H.; Song, R.; Qi, X.; Xu, W. Association of low birth weight with cardiometabolic diseases in Swedish twins: A population-based cohort study. BMJ Open 2021, 11, e048030. [Google Scholar] [CrossRef]
- Bo, S.; Cavallo-Perin, P.; Ciccone, G.; Scaglione, L.; Pagano, G. The metabolic syndrome in twins: A consequence of low birth weight or of being a twin? Exp. Clin. Endocrinol. Diabetes 2001, 109, 135–140. [Google Scholar] [CrossRef]
- Huxley, R.; Neil, A.; Collins, R. Unravelling the fetal origins hypothesis: Is there really an inverse association between birthweight and subsequent blood pressure? Lancet 2002, 360, 659–665. [Google Scholar] [CrossRef]
- Phipps, K.; Barker, D.J.P.; Hales, C.N.; Fall, C.H.D.; Osmond, C.; Clark, P.M.S. Fetal growth and impaired glucose tolerance in men and women. Diabetologia 1993, 36, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Parsons, T.J.; Power, C.; Manor, O. Fetal and early life growth and body mass index from birth to early adulthood in 1958 British cohort: Longitudinal study. Bone Miner. J. 2001, 323, 1331–1335. [Google Scholar] [CrossRef]
- Hrudey, E.J.; Reynolds, R.M.; Oostvogels, A.J.; Brouwer, I.A.; Vrijkotte, T.G. The association between maternal 25-hydroxyvitamin D concentration during gestation and early childhood cardio-metabolic outcomes: Is there interaction with pre-pregnancy BMI? PLoS ONE 2015, 10, e0133313. [Google Scholar] [CrossRef]
- Boney, C.M.; Verma, A.; Tucker, R.; Vohr, B.R. Metabolic syndrome in childhood: Association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005, 115, e290–e296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, W.H.; Ma, R.C.W.; Ozaki, R.; Li, A.M.; Chan, M.H.M.; Yuen, L.Y.; Lao, T.T.H.; Yang, X.; Ho, C.S.; Tutino, G.E.; et al. In utero exposure to maternal hyperglycemia increases childhood cardiometabolic risk in offspring. Diabetes Care 2017, 40, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.M. Smoking and pregnancy: Epigenetics and developmental origins of the metabolic syndrome. Birth Defects Res. 2019, 111, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, Z.; Bartell, T.; Wang, X. Early Life Origins of Metabolic Syndrome: The Role of Environmental Toxicants. Curr. Environ. Health Rep. 2014, 1, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Eberle, C.; Fasig, T.; Brüseke, F.; Stichling, S. Impact of maternal prenatal stress by glucocorticoids on metabolic and cardiovascular outcomes in their offspring: A systematic scoping review. PLoS ONE 2021, 16, e0245386. [Google Scholar] [CrossRef] [PubMed]
- Remacle, C.; Bieswal, F.; Reusens, B. Programming of obesity and cardiovascular disease. Int. J. Obes. Relat. Metab. Disord. 2004, 28 (Suppl. 3), S46–S53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojha, S.; Saroha, V.; Symonds, M.E.; Budge, H. Excess nutrient supply in early life and its later metabolic consequences. Clin. Exp. Pharmacol. Physiol. 2013, 40, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Kelishadi, R.; Haghdoost, A.A.; Jamshidi, F.; Aliramezany, M.; Moosazadeh, M. Low birthweight or rapid catch-up growth: Which is more associated with cardiovascular disease and its risk factors in later life? A systematic review and cryptanalysis. Paediatr. Int. Child Health 2015, 35, 110–123. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef]
- Cianfarani, S.; Germani, D.; Branca, F. Low birthweight and adult insulin resistance: The “catch-up growth” hypothesis. Arch. Dis. Child. Fetal Neonatal. 1999, 81, F71–F73. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A. Living with the past: Evolution, development, and patterns of disease. Science 2004, 305, 1733–1736. [Google Scholar] [CrossRef] [Green Version]
- McMillen, I.C.; Robinson, J.S. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol. Rev. 2005, 85, 571–633. [Google Scholar] [CrossRef]
- Armitage, J.A.; Khan, I.Y.; Taylor, P.D.; Nathanielsz, P.W.; Poston, L. Developmental programming of the metabolic syndrome by maternal nutritional imbalance: How strong is the evidence from experimental models in mammals? J. Physiol. 2004, 561, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Hou, C.Y.; Hsu, W.H.; Tain, Y.L. Early-Life Origins of Metabolic Syndrome: Mechanisms and Preventive Aspects. Int. J. Mol. Sci. 2021, 22, 11872. [Google Scholar] [CrossRef] [PubMed]
- de Gusmão Correia, M.L.; Volpato, A.M.; Águila, M.B.; Mandarim-de-Lacerda, C.A. Developmental origins of health and disease: Experimental and human evidence of fetal programming for metabolic syndrome. J. Hum. Hypertens. 2012, 26, 405–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, J.; Nakagawa, K.; Kato, S.; Miyazawa, T.; Kimura, F.; Miyazawa, T. The combination of maternal and offspring high-fat diets causes marked oxidative stress and development of metabolic syndrome in mouse offspring. Life Sci. 2016, 151, 70–75. [Google Scholar] [CrossRef]
- Pankey, C.L.; Walton, M.W.; Odhiambo, J.F.; Smith, A.M.; Ghnenis, A.B.; Nathanielsz, P.W.; Ford, S.P. Intergenerational impact of maternal overnutrition and obesity throughout pregnancy in sheep on metabolic syndrome in grandsons and granddaughters. Domest. Anim. Endocrinol. 2017, 60, 67–74. [Google Scholar] [CrossRef]
- Rousseau-Ralliard, D.; Richard, C.; Hoarau, P.; Lallemand, M.S.; Morillon, L.; Aubrière, M.C.; Valentino, S.A.; Dahirel, M.; Guinot, M.; Fournier, N.; et al. Prenatal air pollution exposure to diesel exhaust induces cardiometabolic disorders in adulthood in a sex-specific manner. Environ. Res. 2021, 200, 111690. [Google Scholar] [CrossRef]
- Arentson-Lantz, E.J.; Buhman, K.K.; Ajuwon, K.; Donkin, S.S. Excess pregnancy weight gain leads to early indications of metabolic syndrome in a swine model of fetal programming. Nutr. Res. 2014, 34, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Puppala, S.; Li, C.; Glenn, J.P.; Saxena, R.; Gawrieh, S.; Quinn, A.; Palarczyk, J.; Dick, E.J., Jr.; Nathanielsz, P.W.; Cox, L.A. Primate fetal hepatic responses to maternal obesity: Epigenetic signalling pathways and lipid accumulation. J. Physiol. 2018, 596, 5823–5837. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlström, M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat. Rev. Nephrol. 2021, 17, 575–590. [Google Scholar] [CrossRef]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar]
- Cardounel, A.J.; Cui, H.; Samouilov, A.; Johnson, W.; Kearns, P.; Tsai, A.L.; Berka, V.; Zweier, J.L. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J. Biol. Chem. 2007, 282, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, C.; Wilson, R.; Roberts, J.; Miller, H.; McKillop, J.H.; Walker, J.J. Antioxidants: Their role in pregnancy and miscarriage. Antioxid. Redox Signal. 2000, 2, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Murtaza, G.; Metwally, E.; Kalhoro, D.H.; Kalhoro, M.S.; Rahu, B.A.; Sahito, R.G.A.; Yin, Y.; Yang, H.; Chughtai, M.I.; et al. The Role of Oxidative Stress and Antioxidant Balance in Pregnancy. Mediators Inflamm. 2021, 2021, 9962860. [Google Scholar] [CrossRef]
- Shkolnik, K.; Tadmor, A.; Ben-Dor, S.; Nevo, N.; Galiani, D.; Dekel, N. Reactive oxygen species are indispensable in ovulation. Proc. Natl. Acad. Sci. USA 2011, 108, 1462–1467. [Google Scholar] [CrossRef] [Green Version]
- Guérin, P.; El Mouatassim, S.; Ménézo, Y. Oxidative stress and protection against reactive oxygen species in the pre-implantation embryo and its surroundings. Hum. Reprod. Update 2001, 7, 175–189. [Google Scholar] [CrossRef]
- Myatt, L. Review: Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta 2010, 31, S66–S69. [Google Scholar] [CrossRef] [Green Version]
- Dennery, P.A. Oxidative stress in development: Nature or nurture? Free Radic. Biol. Med. 2010, 49, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Placental oxygen consumption. Part, I. In vivo studies—A review. Placenta 2000, 21, S31–S37. [Google Scholar] [CrossRef]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef]
- Myatt, L. Placental adaptive responses and fetal programming. J. Physiol. 2006, 572, 25–30. [Google Scholar] [CrossRef]
- Fickling, S.A.; Williams, D.; Vallance, P.; Nussey, S.S.; Whitley, G.S. Plasma of endogenous inhibitor of nitric oxide synthesis in normal pregnancy and pre-eclampsia. Lancet 1993, 342, 242–243. [Google Scholar] [CrossRef]
- Holden, D.P.; Fickling, S.A.; Whitley, G.S.; Nussey, S.S. Plasma concentrations of asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy and preeclampsia. Am. J. Obstet. Gynecol. 1998, 178, 551–556. [Google Scholar] [CrossRef]
- Vida, G.; Sulyok, E.; Ertl, T.; Martens-Lobenhoffer, J.; Bode-Böger, S.M. Birth by cesarean section is associated with elevated neonatal plasma levels of dimethylarginines. Pediatr. Int. 2012, 54, 476–479. [Google Scholar] [CrossRef]
- Akturk, M.; Altinova, A.; Mert, I.; Dincel, A.; Sargin, A.; Buyukkagnici, U.; Arslan, M.; Danisman, N. Asymmetric dimethylarginine concentrations are elevated in women with gestational diabetes. Endocrine 2010, 38, 134–141. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal L-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric Oxide 2010, 23, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Franco Mdo, C.; Ponzio, B.F.; Gomes, G.N.; Gil, F.Z.; Tostes, R.; Carvalho, M.H.; Fortes, Z.B. Micronutrient prenatal supplementation prevents the development of hypertension and vascular endothelial damage induced by intrauterine malnutrition. Life Sci. 2009, 85, 327–333. [Google Scholar] [CrossRef]
- Holemans, K.; Verhaeghe, J.; Dequeker, J.; Van Assche, F.A. Insulin sensitivity in adult female rats subjected to malnutrition during the perinatal period. J. Soc. Gynecol. Investig. 1996, 3, 71–77. [Google Scholar] [CrossRef]
- Cambonie, G.; Comte, B.; Yzydorczyk, C.; Ntimbane, T.; Germain, N.; Lê, N.L.; Pladys, P.; Gauthier, C.; Lahaie, I.; Abran, D.; et al. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1236–R1245. [Google Scholar] [CrossRef]
- Ozanne, S.E.; Smith, G.D.; Tikerpae, J.; Hales, C.N. Altered regulation of hepatic glucose output in the male offspring of protein-malnourished rat dams. Am. J. Physiol. 1996, 270, E559–E564. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal Garlic Oil Supplementation Prevents High-Fat Diet-Induced Hypertension in Adult Rat Offspring: Implications of H2S-Generating Pathway in the Gut and Kidneys. Mol. Nutr. Food Res. 2021, 65, e2001116. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.A.; Tsai, C.K.; Huang, L.T.; Sheen, J.M.; Tiao, M.M.; Tain, Y.L.; Chen, C.C.; Lin, I.C.; Lai, Y.J.; Tsai, C.C.; et al. Maternal Resveratrol Treatment Re-Programs and Maternal High-Fat Diet-Induced Retroperitoneal Adiposity in Male Offspring. Int. J. Environ. Res. Public Health 2020, 17, 2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheen, J.M.; Yu, H.R.; Tain, Y.L.; Tsai, W.L.; Tiao, M.M.; Lin, I.C.; Tsai, C.C.; Lin, Y.J.; Huang, L.T. Combined maternal and postnatal high-fat diet leads to metabolic syndrome and is effectively reversed by resveratrol: A multiple-organ study. Sci. Rep. 2018, 8, 5607. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Zhao, J.; Xu, H.; Lyv, Y.; Feng, X.; Fang, Y.; Xu, Y. Maternal quercetin administration during gestation and lactation decrease endoplasmic reticulum stress and related inflammation in the adult offspring of obese female rats. Eur. J. Nutr. 2014, 53, 1669–1683. [Google Scholar] [CrossRef]
- Tsai, W.L.; Hsu, C.N.; Tain, Y.L. Whether AICAR in Pregnancy or Lactation Prevents Hypertension Programmed by High Saturated Fat Diet: A Pilot Study. Nutrients 2020, 12, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do Nascimento, L.C.P.; Neto, J.P.R.C.; de Andrade Braga, V.; Lagranha, C.J.; de Brito Alves, J.L. Maternal exposure to high-fat and high-cholesterol diet induces arterial hypertension and oxidative stress along the gut-kidney axis in rat offspring. Life Sci. 2020, 261, 118367. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, Y.J.; Hou, C.Y.; Tain, Y.L. Maternal Administration of Probiotic or Prebiotic Prevents Male Adult Rat Offspring against Developmental Programming of Hypertension Induced by High Fructose Consumption in Pregnancy and Lactation. Nutrients 2018, 10, 1229. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.M.; Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Developmental programming of the metabolic syndrome: Next generation sequencing analysis of transcriptome expression in a rat model of maternal high fructose intake. Sheng Li Xue Bao 2016, 68, 557–567. [Google Scholar] [PubMed]
- Saad, A.F.; Dickerson, J.; Kechichian, T.B.; Yin, H.; Gamble, P.; Salazar, A.; Patrikeev, I.; Motamedi, M.; Saade, G.R.; Costantine, M.M. High-fructose diet in pregnancy leads to fetal programming of hypertension, insulin resistance, and obesity in adult offspring. Am. J. Obstet. Gynecol. 2016, 215, 378.e1–378.e6. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Wu, K.L.H.; Leu, S.; Chan, J.Y.H. Targeting arachidonic acid pathway to prevent programmed hypertension in maternal fructose-fed male adult rat offspring. J. Nutr. Biochem. 2016, 38, 86–92. [Google Scholar] [CrossRef]
- Chao, Y.M.; Wu, K.L.H.; Tsai, P.C.; Tain, Y.L.; Leu, S.; Lee, W.C.; Chan, J.Y.H. Anomalous AMPK-regulated angiotensin AT1R expression and SIRT1-mediated mitochondrial biogenesis at RVLM in hypertension programming of offspring to maternal high fructose exposure. J. Biomed. Sci. 2020, 27, 68. [Google Scholar] [CrossRef]
- Tsai, P.C.; Chao, Y.M.; Chan, J.Y.H. Sympathetic activation of splenic T-lymphocytes in hypertension of adult offspring programmed by maternal high fructose exposure. Chin. J. Physiol. 2020, 63, 263–275. [Google Scholar] [PubMed]
- Wlodek, M.E.; Westcott, K.; Siebel, A.L.; Owens, J.A.; Moritz, K.M. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008, 74, 187–195. [Google Scholar] [CrossRef]
- Nüsken, K.D.; Dötsch, J.; Rauh, M.; Rascher, W.; Schneider, H. Uteroplacental insufficiency after bilateral uterine artery ligation in the rat: Impact on postnatal glucose and lipid metabolism and evidence for metabolic programming of the offspring by sham operation. Endocrinology 2008, 149, 1056–1063. [Google Scholar] [CrossRef]
- Ojeda, N.B.; Hennington, B.S.; Williamson, D.T.; Hill, M.L.; Betson, N.E.; Sartori-Valinotti, J.C.; Reckelhoff, J.F.; Royals, T.P.; Alexander, B.T. Oxidative stress contributes to sex differences in blood pressure in adult growth-restricted offspring. Hypertension 2012, 60, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Giussani, D.A.; Camm, E.J.; Niu, Y.; Richter, H.G.; Blanco, C.E.; Gottschalk, R.; Blake, E.Z.; Horder, K.A.; Thakor, A.S.; Hansell, J.A.; et al. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS ONE 2012, 7, e31017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, W.; Ciriello, J. Effect of maternal chronic intermittent hypoxia during gestation on offspring growth in the rat. Am. J. Obstet. Gynecol. 2013, 209, 564.e1–564.e9. [Google Scholar] [CrossRef]
- Chen, L.; Zadi, Z.H.; Zhang, J.; Scharf, S.M.; Pae, E.K. Intermittent hypoxia in utero damages postnatal growth and cardiovascular function in rats. J. Appl. Physiol. 2018, 124, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, N.; Deng, Y.; Wei, Y.; Huang, Y.; Pu, X.; Li, L.; Zheng, Y.; Guo, J.; Yu, J.; et al. Ascorbic Acid Protects against Hypertension through Downregulation of ACE1 Gene Expression Mediated by Histone Deacetylation in Prenatal Inflammation Induced Offspring. Sci. Rep. 2016, 6, 39469. [Google Scholar] [CrossRef]
- Tsosura, T.V.S.; Chiba, F.Y.; Mattera, M.S.L.C.; Pereira, R.F.; Cintra, L.T.A.; Conti, L.C.; Santos, R.M.D.; Mateus, J.H.P.; Garbin, C.A.S.; Sumida, D.H. Maternal apical periodontitis is associated with insulin resistance in adult offspring. Int. Endod. J. 2019, 52, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.D.; Farias, J.S.; de Queiroz, D.B.; Cabral, E.V.; Lima-Filho, M.M.; Sant’Helena, B.R.M.; Aires, R.S.; Ribeiro, V.S.; SantosRocha, J.; Xavier, F.E.; et al. Oxidative stress induced by prenatal LPS leads to endothelial dysfunction and renal haemodynamic changes through angiotensin II/NADPH oxidase pathway: Prevention by early treatment with α-tocopherol. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3577–3587. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Andreotti, S.; Chimin, P.; Sertié, R.A.; Farias Tda, S.; Torres-Leal, F.L.; de Proença, A.R.; Campaña, A.B.; D’Avila, L.S.; Oliveira, K.A.; et al. Neonatal streptozotocin-induced diabetes in mothers promotes metabolic programming of adipose tissue in male rat offspring. Life Sci. 2015, 136, 151–156. [Google Scholar] [CrossRef]
- Thaeomor, A.; Teangphuck, P.; Chaisakul, J.; Seanthaweesuk, S.; Somparn, N.; Roysommuti, S. Perinatal Taurine Supplementation Prevents Metabolic and Cardiovascular Effects of Maternal Diabetes in Adult Rat Offspring. Adv. Exp. Med. Biol. 2017, 975, 295–305. [Google Scholar]
- Martínez Gascón, L.E.; Ortiz, M.C.; Galindo, M.; Sanchez, J.M.; Sancho-Rodriguez, N.; Albaladejo Otón, M.D.; Rodriguez Mulero, M.D.; Rodriguez, F. Role of heme oxygenase in the regulation of the renal hemodynamics in a model of sex dependent programmed hypertension by maternal diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2022, 322, R181–R191. [Google Scholar] [CrossRef]
- Yu, C.; Chen, S.; Wang, X.; Wu, G.; Zhang, Y.; Fu, C.; Hu, C.; Liu, Z.; Luo, X.; Wang, J.; et al. Exposure to maternal diabetes induces endothelial dysfunction and hypertension in adult male rat offspring. Microvasc. Res. 2021, 133, 104076. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lin, Y.J.; Chan, J.Y.H.; Lee, C.T.; Hsu, C.N. Maternal melatonin or agomelatine therapy prevents programmed hypertension in male offspring of mother exposed to continuous light. Biol. Reprod. 2017, 97, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.S.; Amaral, F.G.; Mesquita, C.C.; Barbosa, A.P.; Lellis-Santos, C.; Turati, A.O.; Santos, L.R.; Sollon, C.S.; Gomes, P.R.; Faria, J.A.; et al. Maternal melatonin programs the daily pattern of energy metabolism in adult offspring. PLoS ONE 2012, 7, e38795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voiculescu, S.E.; Le Duc, D.; Roșca, A.E.; Zeca, V.; Chiţimuș, D.M.; Arsene, A.L.; Drăgoi, C.M.; Nicolae, A.C.; Zăgrean, L.; Schöneberg, T.; et al. Behavioral and molecular effects of prenatal continuous light exposure in the adult rat. Brain Res. 2016, 1650, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Sheen, J.M.; Chen, C.C.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic. Res. 2014, 48, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Tiao, M.M.; Sheen, J.M.; Huang, L.T.; Tain, Y.L.; Lin, I.C.; Lin, Y.J.; Lai, Y.J.; Chen, C.C.; Chang, K.A.; et al. Obesity programmed by prenatal dexamethasone and postnatal high-fat diet leads to distinct alterations in nutrition sensory signals and circadian-clock genes in visceral adipose tissue. Lipids Health Dis. 2019, 18, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiao, M.M.; Huang, L.T.; Chen, C.J.; Sheen, J.M.; Tain, Y.L.; Chen, C.C.; Kuo, H.C.; Huang, Y.H.; Tang, K.S.; Chu, E.W.; et al. Melatonin in the regulation of liver steatosis following prenatal glucocorticoid exposure. BioMed Res. Int. 2014, 2014, 942172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamothe, J.; Khurana, S.; Tharmalingam, S.; Williamson, C.; Byrne, C.J.; Lees, S.J.; Khaper, N.; Kumar, A.; Tai, T.C. Oxidative Stress Mediates the Fetal Programming of Hypertension by Glucocorticoids. Antioxidants 2021, 10, 531. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Maternal resveratrol therapy protects male rat offspring against programmed hypertension induced by TCDD and dexamethasone exposures: Is it relevant to aryl hydrocarbon receptor? Int. J. Mol. Sci. 2018, 19, 2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Song, L.; Wei, J.; Chen, T.; Chen, J.; Lin, Y.; Xia, W.; Xu, B.; Li, X.; Chen, X.; et al. Maternal exposure to di-(2-ethylhexyl) phthalate alters kidney development through the renin-angiotensin system in offspring. Toxicol. Lett. 2012, 212, 212–221. [Google Scholar] [CrossRef]
- Rajagopal, G.; Bhaskaran, R.S.; Karundevi, B. Maternal di-(2-ethylhexyl) phthalate exposure alters hepatic insulin signal transduction and glucoregulatory events in rat F1 male offspring. J. Appl. Toxicol. 2019, 39, 751–763. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Chen, L.; Wang, X.J.; Jiang, Q.H.; Bei, X.Y.; Sun, W.L.; Xia, S.J.; Jiang, J.T. Maternal exposure to di-n-butyl phthalate (DBP) induces renal fibrosis in adult rat offspring. Oncotarget 2017, 8, 31101–31111. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Tain, Y.L. Maternal exposure to bisphenol A combined with high-fat diet-induced programmed hypertension in adult male rat offspring: Effects of resveratrol. Int. J. Mol. Sci. 2019, 20, 4382. [Google Scholar] [CrossRef] [Green Version]
- Galyon, K.D.; Farshidi, F.; Han, G.; Ross, M.G.; Desai, M.; Jellyman, J.K. Maternal bisphenol A exposure alters rat offspring hepatic and skeletal muscle insulin signaling protein abundance. Am. J. Obstet. Gynecol. 2017, 216, 290.e1–290.e9. [Google Scholar] [CrossRef]
- Xiao, D.; Huang, X.; Yang, S.; Zhang, L. Antenatal nicotine induces heightened oxidative stress and vascular dysfunction in rat offspring. Br. J. Pharmacol. 2011, 164, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Huang, X.; Li, Y.; Dasgupta, C.; Wang, L.; Zhang, L. Antenatal Antioxidant Prevents Nicotine-Mediated Hypertensive Response in Rat Adult Offspring. Biol. Reprod. 2015, 93, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conceição, E.P.; Peixoto-Silva, N.; Pinheiro, C.R.; Oliveira, E.; Moura, E.G.; Lisboa, P.C. Maternal nicotine exposure leads to higher liver oxidative stress and steatosis in adult rat offspring. Food Chem. Toxicol. 2015, 78, 52–59. [Google Scholar] [CrossRef]
- Gray, S.P.; Denton, K.M.; Cullen-McEwen, L.; Bertram, J.F.; Moritz, K.M. Prenatal exposure to alcohol reduces nephron number and raises blood pressure in progeny. J. Am. Soc. Nephrol. 2010, 21, 1891–1902. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.M.T.; Steane, S.E.; Moritz, K.M.; Akison, L.K. Prenatal alcohol exposure programmes offspring disease: Insulin resistance in adult males in a rat model of acute exposure. J. Physiol. 2019, 597, 5619–5637. [Google Scholar] [CrossRef]
- Contreras, M.L.; de la Fuente-Ortega, E.; Vargas-Roberts, S.; Muñoz, D.C.; Goic, C.A.; Haeger, P.A. NADPH Oxidase Isoform 2 (NOX2) Is Involved in Drug Addiction Vulnerability in Progeny Developmentally Exposed to Ethanol. Front. Neurosci. 2017, 11, 338. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar] [PubMed]
- Williams, L.; Seki, Y.; Vuguin, P.M.; Charron, M.J. Animal models of in utero exposure to a high fat diet: A review. Biochim. Biophys. Acta 2014, 1842, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, E.C.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, T.; Powell, T.L. Role of placental nutrient sensing in developmental programming. Clin. Obstet. Gynecol. 2013, 56, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Targeting the Renin-Angiotensin-Aldosterone System to Prevent Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef]
- Jašarevic´, E.; Bale, T.L. Prenatal and postnatal contributions of the maternal microbiome on offspring programming. Front. Neuroendocrinol. 2019, 55, 100797. [Google Scholar] [CrossRef]
- Fowden, A.L.; Valenzuela, O.A.; Vaughan, O.R.; Jellyman, J.K.; Forhead, A.J. Glucocorticoid programming of intrauterine development. Domest. Anim. Endocrinol. 2016, 56, S121–S132. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, W.C.; Wu, K.L.H.; Leu, S.; Chan, J.Y.H. Resveratrol Prevents the Development of Hypertension Programmed by Maternal Plus Post-Weaning High-Fructose Consumption through Modulation of Oxidative Stress, Nutrient-Sensing Signals, and Gut Microbiota. Mol. Nutr. Food Res. 2018, 30, e1800066. [Google Scholar] [CrossRef]
- Favre, G.A.; Esnault, V.L.; Van Obberghen, E. Modulation of glucose metabolism by the renin-angiotensin-aldosterone system. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E435–E449. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Lin, Y.J.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Tsai, C.C.; Huang, L.T.; Hsu, C.N. High fat diets sex-specifically affect the renal transcriptome and program obesity, kidney injury, and hypertension in the offspring. Nutrients 2017, 9, 357. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The microbiome in early life: Implications for health outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef]
- Guimarães, K.S.L.; Braga, V.A.; Noronha, S.I.S.R.; Costa, W.K.A.D.; Makki, K.; Cruz, J.C.; Brandão, L.R.; Chianca Junior, D.A.; Meugnier, E.; Leulier, F.; et al. Lactiplanti bacillus plantarum WJL administration during pregnancy and lactation improves lipid profile, insulin sensitivity and gut microbiota diversity in dyslipidemic dams and protects male offspring against cardiovascular dysfunction in later life. Food Funct. 2020, 11, 8939–8950. [Google Scholar] [CrossRef]
- De Oliveira, Y.; Cavalcante, R.G.S.; Cavalcanti Neto, M.P.; Magnani, M.; Braga, V.A.; de Souza, E.L.; de Brito Alves, J.L. Oral administration of Lactobacillus fermentum post-weaning improves the lipid profile and autonomic dysfunction in rat offspring exposed to maternal dyslipidemia. Food Funct. 2020, 11, 5581–5594. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.L.; Colgan, S.P. Control and dysregulation of redox signalling in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 106–120. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Maternal High-Fat Diet and Offspring Hypertension. Int. J. Mol. Sci. 2022, 23, 8179. [Google Scholar] [CrossRef]
- Goszcz, K.; Deakin, S.J.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Antioxidants in Cardiovascular Therapy: Panacea or False Hope? Front. Cardiovasc. Med. 2015, 2, 29. [Google Scholar] [CrossRef]
- Katsiki, N.; Manes, C. Is there a role for supplemented antioxidants in the prevention of atherosclerosis? Clin. Nutr. 2009, 28, 3–9. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Nimse, S.B.; Palb, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Konate, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the Activity of NADPH Oxidase in Cancer. Antioxid. Redox Signal. 2020, 33, 435–454. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, M.E.; Meijles, D.N.; Pagano, P.J. Nox Inhibitors & Therapies: Rational Design of Peptidic and Small Molecule Inhibitors. Curr. Pharm. Des. 2015, 21, 6023–6035. [Google Scholar]
- Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules 2021, 26, 1844. [Google Scholar] [CrossRef]
- Natarajan, G.; Perriotte-Olson, C.; Bhinderwala, F.; Powers, R.; Desouza, C.V.; Talmon, G.A.; Yuhang, J.; Zimmerman, M.C.; Kabanov, A.V.; Saraswathi, V. Nanoformulated copper/zinc superoxide dismutase exerts differential effects on glucose vs lipid homeostasis depending on the diet composition possibly via altered AMPK signaling. Transl. Res. 2017, 188, 10–26. [Google Scholar] [CrossRef]
- Carillon, J.; Knabe, L.; Montalban, A.; Stévant, M.; Keophiphath, M.; Lacan, D.; Cristol, J.P.; Rouanet, J.M. Curative diet supplementation with a melon superoxide dismutase reduces adipose tissue in obese hamsters by improving insulin sensitivity. Mol. Nutr. Food Res. 2014, 58, 842–850. [Google Scholar] [CrossRef]
- Cao, P.; Ito, O.; Ito, D.; Rong, R.; Zheng, Y.; Kohzuki, M. Combination of Exercise Training and SOD Mimetic Tempol Enhances Upregulation of Nitric Oxide Synthase in the Kidney of Spontaneously Hypertensive Rats. Int. J. Hypertens. 2020, 2020, 2142740. [Google Scholar] [CrossRef]
- Koeners, M.P.; Braam, B.; Joles, J.A. Perinatal inhibition of NF-kappaB has long-term antihypertensive effects in spontaneously hypertensive rats. J. Hypertens. 2011, 29, 1160–1166. [Google Scholar] [CrossRef]
- Said, H.M.; Nexo, E. Gastrointestinal Handling of Water-Soluble Vitamins. Compr. Physiol. 2018, 8, 1291–1311. [Google Scholar]
- Niki, E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Early Origins of Hypertension: Should Prevention Start before Birth Using Natural Antioxidants? Antioxidants 2020, 9, 1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, Z.; Li, D.; Li, N.; Dindot, S.V.; Satterfield, M.C.; Bazer, F.W.; Wu, G. Nutrition, epigenetics, and metabolic syndrome. Antioxid. Redox Signal. 2012, 17, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wahlqvist, M.L.; Li, D. Nutrition, One-Carbon Metabolism and Neural Tube Defects: A Review. Nutrients 2016, 8, 741. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.S.; Ahsan, H.; Zia, M.K.; Siddiqui, T.; Khan, F.H. Understanding oxidants and antioxidants: Classical team with new players. J. Food Biochem. 2020, 44, e13145. [Google Scholar] [CrossRef] [PubMed]
- Simonson, M.; Boirie, Y.; Guillet, C. Protein, amino acids and obesity treatment. Rev. Endocr. Metab. Disord. 2020, 21, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Poggiogalle, E.; Fontana, M.; Giusti, A.M.; Pinto, A.; Iannucci, G.; Lenzi, A.; Donini, L.M. Amino Acids and Hypertension in Adults. Nutrients 2019, 11, 1459. [Google Scholar] [CrossRef] [Green Version]
- Fujii, T.; Yura, S.; Tatsumi, K.; Kondoh, E.; Mogami, H.; Fujita, K.; Kakui, K.; Aoe, S.; Itoh, H.; Sagawa, N.; et al. Branched-chain amino acid supplemented diet during maternal food restriction prevents developmental hypertension in adult rat offspring. J. Dev. Orig. Health Dis. 2011, 2, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H.; Nakamura, Y.; Terron, M.P.; Flores, L.J.; Manchester, L.C.; Tan, D.X.; Sugino, N.; Reiter, R.J. Melatonin and pregnancy in the human. Reprod. Toxicol. 2008, 25, 291–303. [Google Scholar] [CrossRef]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Terron, M.P.; Flores, L.J.; Czarnocki, Z. Melatonin and its metabolites: New findings regarding their production and their radical scavenging actions. Acta Biochim. Pol. 2007, 54, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N. Developmental Programming of Adult Disease: Reprogramming by Melatonin? Int. J. Mol. Sci. 2017, 18, 426. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N.; Lee, C.T. Melatonin therapy prevents programmed hypertension and nitric oxide deficiency in offspring exposed to maternal caloric restriction. Oxid. Med. Cell Longev. 2014, 2014, 283180. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: Roles of nitric oxide and arachidonic acid metabolites. J. Pineal Res. 2014, 57, 80–89. [Google Scholar] [CrossRef]
- Tain, Y.L.; Huang, L.T.; Chan, J.Y. Transcriptional regulation of programmed hypertension by melatonin: An epigenetic perspective. Int. J. Mol. Sci. 2014, 15, 18484–18495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkmaz, A.; Rosales-Corral, S.; Reiter, R.J. Gene regulation by melatonin linked to epigenetic phenomena. Gene 2012, 503, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.P.; Gögenur, I.; Rosenberg, J.; Reiter, R.J. The Safety of Melatonin in Humans. Clin. Drug Investig. 2016, 36, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Tain, Y.L.; Sheen, J.M.; Huang, L.T. Melatonin utility in neonates and children. J. Formos. Med. Assoc. 2012, 111, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durazzo, A.; Lucarini, M.; Souto, E.B.; Cicala, C.; Caiazzo, E.; Izzo, A.A.; Novellino, E.; Santini, A. Polyphenols: A concise overview on the chemistry, occurrence, and human health. Phytother. Res. 2019, 33, 2221–2243. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef]
- Urquiaga, I.; Leighton, F. Plant polyphenol antioxidants and oxidative stress. Biol. Res. 2000, 33, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, M.; Zhang, W.; Liu, C.; Chen, S. Natural Polyphenols in Metabolic Syndrome: Protective Mechanisms and Clinical Applications. Int. J. Mol. Sci. 2021, 22, 6110. [Google Scholar] [CrossRef]
- Chiva-Blanch, G.; Badimon, L. Effects of Polyphenol Intake on Metabolic Syndrome: Current Evidences from Human Trials. Oxid. Med. Cell Longev. 2017, 2017, 5812401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.M.; Zhou, B.; Wang, Y.S.; Gong, Q.Y.; Wang, Q.M.; Yan, J.J.; Gao, W.; Wang, L.S. Black and green tea consumption and the risk of coronary artery disease: A meta-analysis. Am. J. Clin. Nutr. 2011, 93, 506–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, L.; Flowers, N.; Holmes, J.; Clarke, A.; Stranges, S.; Hooper, L.; Rees, K. Green and black tea for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, 2013, CD009934. [Google Scholar] [CrossRef]
- Ellwood, L.; Torun, G.; Bahar, Z.; Fernandez, R. Effects of flavonoid-rich fruits on hypertension in adults: A systematic review. JBI Database Syst. Rev. Implement. Rep. 2019, 17, 2075–2105. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Oest, M.E.; Prater, M.R. Intrauterine exposure to high saturated fat diet elevates risk of adult-onset chronic diseases in C57BL/6 mice. Birth Defects Res. B Dev. Reprod. Toxicol. 2009, 86, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gerevini, G.T.; Repossi, G.; Dain, A.; Tarres, M.C.; Das, U.N.; Eynard, A.R. Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Developmental Programming of the Metabolic Syndrome: Can We Reprogram with Resveratrol? Int. J. Mol. Sci. 2018, 19, 2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, T.; Chen, D.; Yang, Q.; Wang, B.; Zhu, M.J.; Nathanielsz, P.W.; Du, M. Resveratrol supplementation of high-fat diet-fed pregnant mice promotes brown and beige adipocyte development and prevents obesity in male offspring. J. Physiol. 2017, 595, 1547–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, C.C.; Reyes-Castro, L.A.; Rodríguez-González, G.L.; Bautista, C.J.; Vázquez-Martínez, M.; Larrea, F.; Chamorro-Cevallos, G.A.; Nathanielsz, P.W.; Zambrano, E. Resveratrol partially prevents oxidative stress and metabolic dysfunction in pregnant rats fed a low protein diet and their offspring. J. Physiol. 2016, 594, 1483–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milagro, F.I.; Mansego, M.L.; De Miguel, C.; Martínez, J.A. Dietary factors, epigenetic modifications and obesity outcomes: Progresses and perspectives. Mol. Asp. Med. 2013, 34, 782–812. [Google Scholar] [CrossRef] [PubMed]
- Remely, M.; Lovrecic, L.; de la Garza, A.L.; Migliore, L.; Peterlin, B.; Milagro, F.I.; Martinez, A.J.; Haslberger, A.G. Therapeutic perspectives of epigenetically active nutrients. Br. J. Pharmacol. 2015, 172, 2756–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Šalamon, Š.; Kramar, B.; Marolt, T.P.; Poljšak, B.; Milisav, I. Medical and Dietary Uses of N-Acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goor, H.; van den Born, J.C.; Hillebrands, J.L.; Joles, J.A. Hydrogen sulfide in hypertension. Curr. Opin. Nephrol. Hypertens. 2016, 25, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal N-Acetylcysteine Therapy Prevents Hypertension in Spontaneously Hypertensive Rat Offspring: Implications of Hydrogen Sulfide-Generating Pathway and Gut Microbiota. Antioxidants 2020, 9, 856. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal NG-Nitro-L-arginine-methyl ester (L-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636.e1–636.e72. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Lee, C.T.; Lin, Y.J.; Tsai, C.C. N-Acetylcysteine prevents programmed hypertension in male rat offspring born to suramin-treated mothers. Biol. Reprod. 2016, 95, 8. [Google Scholar] [CrossRef]
- Tai, I.H.; Sheen, J.M.; Lin, Y.J.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Tain, Y.L. Maternal N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and prevents programmed hypertension in male offspring exposed to prenatal dexamethasone and postnatal high-fat diet. Nitric Oxide 2016, 53, 6–12. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Yu, H.R.; Lin, I.C.; Sheen, J.M.; Huang, L.T.; Tain, Y.L. Protection of Male Rat Offspring against Hypertension Programmed by Prenatal Dexamethasone Administration and Postnatal High-Fat Diet with the Nrf2 Activator Dimethyl Fumarate during Pregnancy. Int. J. Mol. Sci. 2019, 20, 3957. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Lin, I.C.; Yu, H.R.; Sheen, J.M.; Huang, L.T.; Tain, Y.L. Early Postweaning Treatment with Dimethyl Fumarate Prevents Prenatal Dexamethasone- and Postnatal High-Fat Diet-Induced Programmed Hypertension in Male Rat Offspring. Oxid. Med. Cell Longev. 2018, 2018, 5343462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.H.; Leonard, S.; Shi, X.; Goins, M.R.; Vallyathan, V. Antioxidant activity of lazaroid (U-75412E) and its protective effects against crystalline silica-induced cytotoxicity. Free Radic. Biol. Med. 1998, 24, 529–536. [Google Scholar] [CrossRef]
- Wesseling, S.; Essers, P.B.; Koeners, M.P.; Pereboom, T.C.; Braam, B.; van Faassen, E.E.; Macinnes, A.W.; Joles, J.A. Perinatal exogenous nitric oxide in fawn-hooded hypertensive rats reduces renal ribosomal biogenesis in early life. Front. Genet. 2011, 2, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Münzel, T.; Förstermann, U.; Li, H. Maternal treatment of spontaneously hypertensive rats with pentaerythritoltetranitrate reduces blood pressure in female offspring. Hypertension 2015, 65, 232–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luiking, Y.C.; Ten Have, G.A.M.; Wolfe, R.R.; Deutz, N.E.P. Arginine de novo and nitric oxide production in disease states. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1177–E1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues-Krause, J.; Krause, M.; Rocha, I.M.G.D.; Umpierre, D.; Fayh, A.P.T. Association of l-arginine supplementation with markers of endothelial function in patients with cardiovascular or metabolic disorders: A systematic review and meta-analysis. Nutrients 2018, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Cynober, L.; Moinard, C.; De Bandt, J.P. The 2009 ESPEN Sir David Cuthbertson. Citrulline: A new major signaling molecule or just another player in the pharmaconutrition game? Clin. Nutr. 2010, 29, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Ventura, G.; Sarfati, G.; Nubret, E.; Kapel, N.; Waligora-Dupriet, A.J.; Bergheim, I.; Cynober, L.; De-Bandt, J.P. Effect of specific amino acids on hepatic lipid metabolism in fructose-induced non-alcoholic fatty liver disease. Clin. Nutr. 2016, 35, 175–182. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Targeting on asymmetric dimethylarginine related nitric oxide-reactive oxygen species imbalance to reprogram the development of hypertension. Int. J. Mol. Sci. 2016, 17, 2020. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Leu, S.; Lee, W.C.; Wu, K.L.H.; Chan, J.Y.H. Maternal melatonin therapy attenuated maternal high-fructose combined with post-weaning high-salt diets-induced hypertension in adult male rat offspring. Molecules 2018, 23, 886. [Google Scholar] [CrossRef]
- Tain, Y.L.; Wu, K.L.H.; Lee, W.C.; Leu, S.; Chan, J.Y.H. Prenatal Metformin Therapy Attenuates Hypertension of Developmental Origin in Male Adult Offspring Exposed to Maternal High-Fructose and Post-Weaning High-Fat Diets. Int. J. Mol. Sci. 2018, 19, 1066. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.W.; Ou, Y.C.; Tang, K.S.; Yu, H.R.; Huang, L.T.; Tain, Y.L.; Lin, I.C.; Sheen, J.M.; Hou, C.Y.; Tsai, C.C.; et al. Metformin ameliorates maternal high-fat diet-induced maternal dysbiosis and fetal liver apoptosis. Lipids Health Dis. 2021, 20, 100. [Google Scholar] [CrossRef]
- Uson-Lopez, R.A.; Kataoka, S.; Mukai, Y.; Sato, S.; Kurasaki, M. Melinjo (Gnetum gnemon) Seed Extract Consumption during Lactation Improved Vasodilation and Attenuated the Development of Hypertension in Female Offspring of Fructose-Fed Pregnant Rats. Birth Defects Res. 2018, 110, 27–34. [Google Scholar] [CrossRef]
- Yuksel, S.; Yigit, A.A.; Cinar, M.; Atmaca, N.; Onaran, Y. Oxidant and Antioxidant Status of Human Breast Milk during Lactation Period. Dairy Sci. Technol. 2015, 95, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Singhal, A.; Cole, T.J.; Lucas, A. Early Nutrition in Preterm Infants and Later Blood Pressure: Two Cohorts after Randomised Trials. Lancet 2001, 357, 413–419. [Google Scholar] [CrossRef]
- Section on Breastfeeding. Breastfeeding and the use of human milk. Pediatrics 2012, 129, e827–e841. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. The antioxidant paradox. Lancet 2000, 355, 1179–1180. [Google Scholar] [CrossRef]
- Villanueva, C.; Kross, R.D. Antioxidant-induced stress. Int. J. Mol. Sci. 2012, 13, 2091–2109. [Google Scholar] [CrossRef]
Figure 1.
Schema outlining how maternal-derived insults induce MetS in later life via oxidative stress programming of various organ systems. Maternal-dervied insults that induce oxidative stress are related to increases in reactive oxygen/nitrogen species (ROS/RNS), decreases in nitric oxide (NO), and reductions in antioxidants.
Figure 1.
Schema outlining how maternal-derived insults induce MetS in later life via oxidative stress programming of various organ systems. Maternal-dervied insults that induce oxidative stress are related to increases in reactive oxygen/nitrogen species (ROS/RNS), decreases in nitric oxide (NO), and reductions in antioxidants.

Figure 2.
The interconnection between oxidative stress and other common mechanisms underlying metabolic syndromes of developmental origins.
Figure 2.
The interconnection between oxidative stress and other common mechanisms underlying metabolic syndromes of developmental origins.

Figure 3.
Schema outlining the potential oxidative-stress-targeted interventions as a reprogramming strategy to prevent metabolic syndrome of developmental origins.
Figure 3.
Schema outlining the potential oxidative-stress-targeted interventions as a reprogramming strategy to prevent metabolic syndrome of developmental origins.


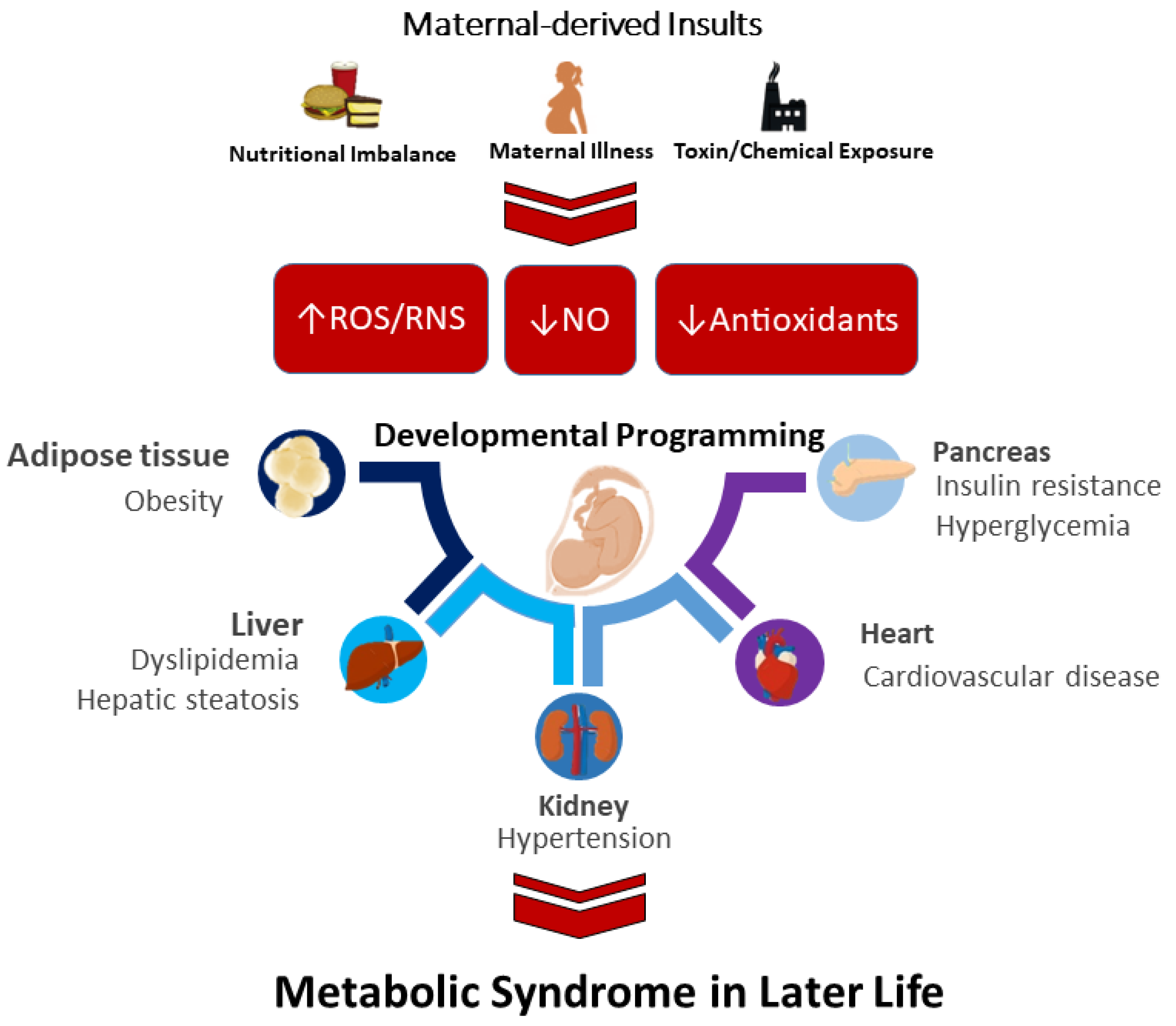{kind=link}
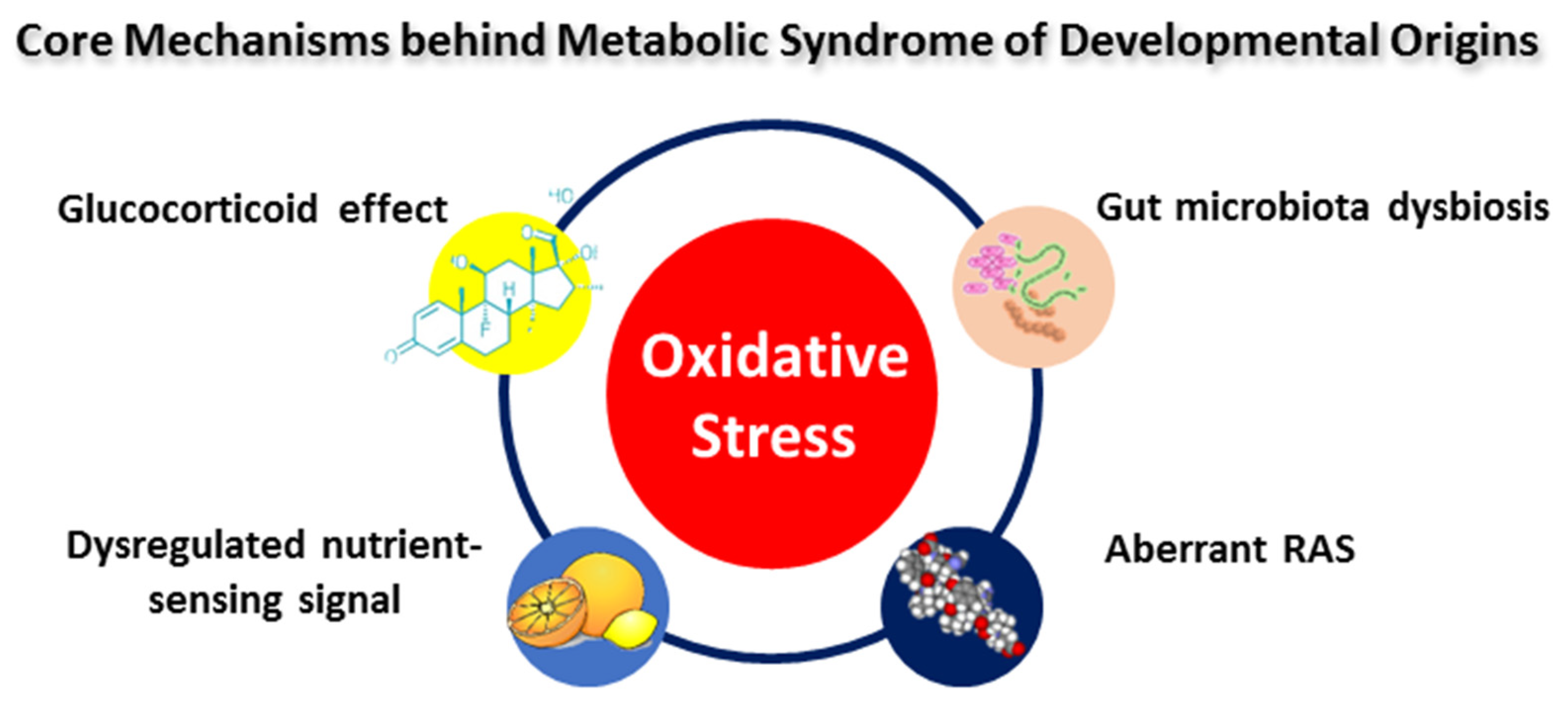{kind=link}
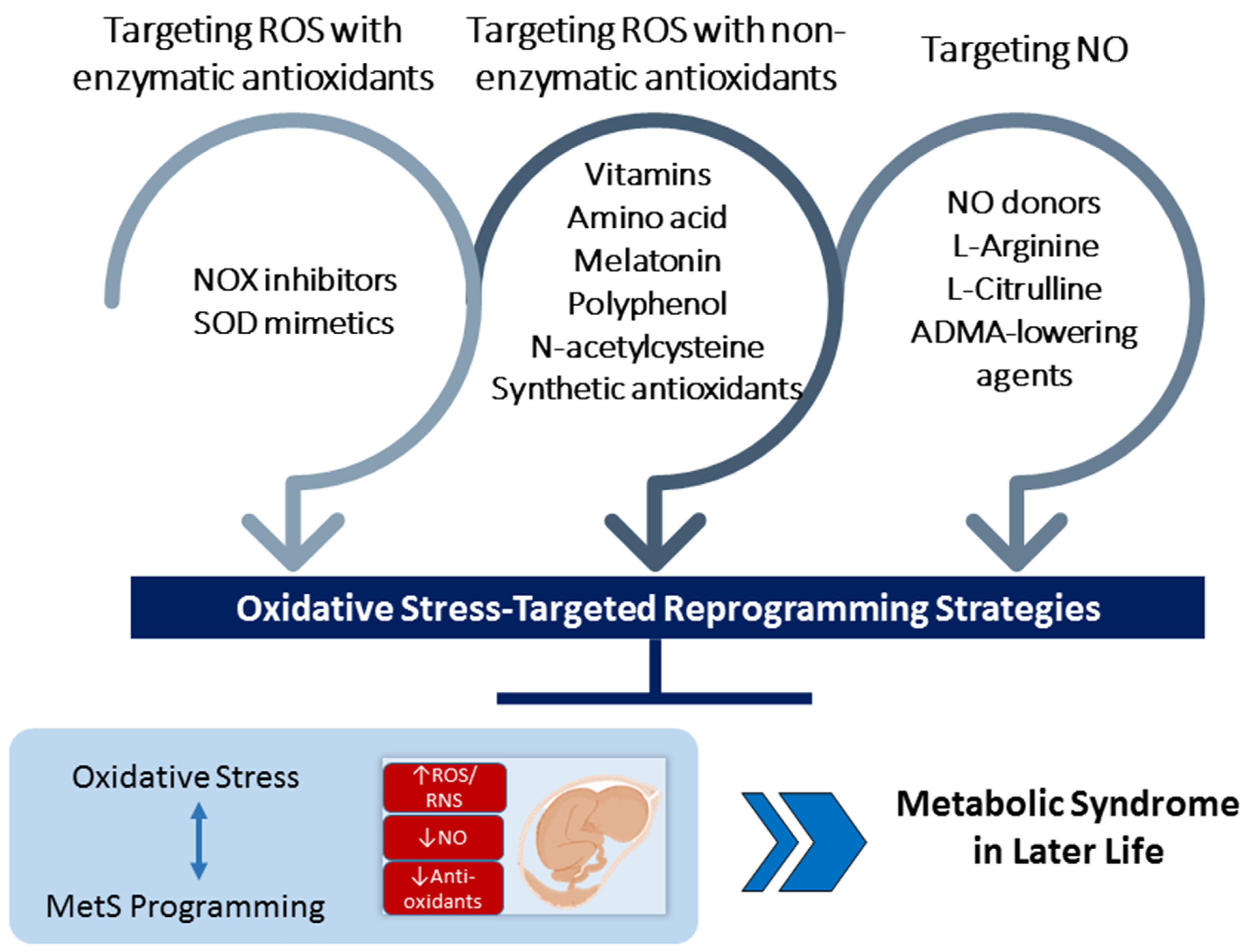{kind=link}
Table 1.
Summary of oxidative-stress-related developmental origins of MetS in rodent animal models.
| Animal Models | Timing and Dose | Offspring Species/Gender | MetS-Related Outcomes in Offspring | Mechanisms of Oxidative Stress | Programmed Organ System |
|---|---|---|---|---|---|
| Caloric restriction | 50% caloric restriction during pregnancy and lactation | SD rats/M [69,70]; Wistar rats/M [71] | Hypertension: 12–16 weeks [69,70]; insulin resistance: 14 weeks [71] | ↑ ADMA, ↓ NO, ↑ renal 8-OHdG expression [69]; ↑3-NT, ↓ NO [70] | Kidney [69], vessel [70] |
| Protein restriction | 9% low-protein diet during pregnancy [72]; 8% low-protein diet during pregnancy and lactation [73] | Wistar rats/M [72,73] | Hypertension: 12 weeks [72]; insulin resistance: 12 weeks [73] | ↑ F2-isoprostane, ↓ glutathione [72] | Kidney [72] |
| Maternal high-fat diet | 58% high-fat diet during pregnancy and lactation [74,75,76,77,78]; 31% high-fat high-cholesterol diet during pregnancy [79] | SD rats/M [74,75,76,77,78]; Wistar rats/M & F [79] | Hypertension: 16 weeks [74]; ↑adiposity: 16 weeks [75]; dyslipidemia: 16 weeks [76]; obesity, dyslipidemia, and hyperinsulinemia: 100 days [77] | ↓ SOD activity in M; ↑ renal MDA level in F [78]; ↑ renal 8-OHdG expression [79] | Kidney [78,79] |
| Maternal high-fructose consumption | 60% high-fructose diet during pregnancy and lactation [80,81]; 10% wt/vol fructose solution during pregnancy [82] | SD rats/M [80,81]; C57BL/6J/M & F [82] | Hypertension, insulin resistance, and dyslipidemia: 12 weeks [80,81]; hypertension, insulin resistance, and obesity: 1 year [82] | ↑ Renal 8-OHdG expression, ↓ NO [83]; ↑brain NADPH-oxidase expression and MDA [84]; ↑ ROS [85] | Kidney [83], brain [84], spleen [85] |
| Uteroplacental insufficiency | Bilateral uterine artery ligation on day 18 [86] or 19 [87] of pregnancy | Wistar–Kyoto rats/M [86]; Wistar rats/M [87] | Hypertension: 22 weeks [86]; dyslipidemia and insulin resistance: 30 weeks [87] | ↑ Urinary F2-isoprostane level & renal NADPH-oxidase-dependent superoxide [88] | Kidney [86,88] |
| Maternal hypoxia | Hypoxia exposure (13% O2) from day 6 to 20 of gestation [89]; alternating cycles of normoxic (room air; 120 s) and hypoxic (6.5% O2; 80 s) exposure during pregnancy [90] | Wistar rats/M [89]; SD rats/M [90] | Hypertension: 4 months [89]; obesity and insulin resistance: 12 weeks [90] | ↑ Lipid peroxidation [91] | Heart [91] |
| Maternal inflammation | Intraperitoneally administered 0.79 mg/kg LPS on gestational day 8, 10, and 12 [92]; surgically induced periodontitis 13 days before mating [93] | SD rats/M & F [92]; Wistar rats/M [93] | Hypertension: 12 weeks [92]; insulin resistance: 75 days [93] | ↑ Renal MDA [94] | Kidney [94] |
| Maternal diabetes | Intraperitoneally administered 45 mg/kg STZ on gestational day 0 [95]; intraperitoneally administered 50 mg/kg STZ on postnatal day 1 [95]; intraperitoneally administered 120 mg/kg STZ on postnatal day 5 [96,97] | SD rats/M [95]; Wistar rats/M [96,97] | Hypertension: 12 week [95]; obesity: 12 weeks [96]; insulin resistance and dyslipidemia: 16 weeks [97] | ↑ ADMA,↓ NO [95]; ↑ renal TBARS and 3-NT [98]; ↑ ROS,↓ NO,↓ SOD activity [99] | Kidney [95,98], vessel [99] |
| Maternal chronodisruption | Continuous light exposure during pregnancy and lactation [100]; continuous light exposure from day 12 to 21 of gestation [101] | SD rats/M [100]; Wistar rats/M [101] | Hypertension: 12 weeks [100], insulin resistance: 18 weeks [101] | ↑ Brain ROS [102] | Brain [102] |
| Maternal stress | Intraperitoneally administrated 0.2 mg/kg dexamethasone daily on gestational days 15 and 16 [103]; intraperitoneally administered 0.1 mg/kg dexamethasone from 14 to 20 of gestation [104,105] | SD rats/M [103,104,105] | Hypertension: 16 weeks [103]; obesity, insulin resistance, and hypertension: 6 months [104]; liver steatosis: 1 week [105] | ↓ Renal NO [103]; ↑ NADPH-oxidase, ↓ Gpx1 expression [106]; ↑ renal 8-OHdG expression, ↑ ADMA [107] | Kidney [103,106], liver [104,105], adrenal gland [106] |
| Maternal di-n-butyl phthalate (DEHP) exposure | Oral gavage with 6.25 mg/kg DEHP during pregnancy and lactation [108]; oral gavage with 100 mg/kg DEHP from gestational day 9 to postnatal day 21 [109] | Wistar rats/M [108]; SD rats/M [109] | Hypertension: 21 weeks [108]; insulin resistance: 80 days [109] | ↑ Renal ROS [110] | Kidney [110] |
| Prenatal bisphenol A (BPA) exposure | Oral gavage with 50 μg/kg BPA during pregnancy and lactation [111]; oral 240 μg/kg BPA from 2 weeks prior to mating and through pregnancy and lactation [112] | SD rats/M [111]; SD rats/M & F [112] | Hypeertension: 16 weeks [111]; insulin resistance: 6 months [112] | ↑ Renal 8-OHdG expression, ↑ ADMA, ↓ NO [111] | Kidney [111] |
| Maternal nicotine exposure | Nicotine administration through an osmotic minipump at 4 µg/kg/min from day 4 of pregnancy to postnatal day 10 [113,114]; nicotine administration through an osmotic minipump at 6 mg/kg/day from postnatal days 2 to 16 [115] | SD rats/M [113,114]; Wistar rats/M & F [115] | Hypertension: 5–8 months [113,114]; hyperlipidemia and steatosis: 6 months [115] | ↑ 3-NT, MDA, and NADPH oxidase [113]; ↑ MDA and 4-NHE levels, ↓ GPx1 activity [115] | Vessel [113], liver [115] |
| Maternal ethanol exposure | Oral gavage with 1 g of ethanol/kg on gestational day 13 and 14 [116,117] | SD rats/M & F [116,117] | Hypertension: 6 months [116], insulin resistance: 6 months [117] | ↓ SOD1, CAT, and Gpx1; ↑ NOX2 [118] | Brain [118] |
SD = Sprague–Dawley rat; M = Male; F = Female; LPS = lipopolysaccharide; STZ = streptozotocin; ADMA = asymmetric dimethylarginine; NO = nitric oxide; 8-OHdG = 8-hydroxy-2′–deoxyguanosine; ROS = reactive oxygen species; TBARS = thiobarbituric acid; 3-NT = 3-nitrotyrosine; 4-NHE = 4-hydroxynonenal; Gpx1 = glutathione peroxidase 1; MDA = malondialdehyde; SOD = superoxidase dismutase; CAT = catalase; NOX2 = NADPH oxidase 2.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tain, Y.-L.; Hsu, C.-N. Metabolic Syndrome Programming and Reprogramming: Mechanistic Aspects of Oxidative Stress. Antioxidants 2022, 11, 2108. https://doi.org/10.3390/antiox11112108
AMA Style
Tain Y-L, Hsu C-N. Metabolic Syndrome Programming and Reprogramming: Mechanistic Aspects of Oxidative Stress. Antioxidants. 2022; 11(11):2108. https://doi.org/10.3390/antiox11112108
Chicago/Turabian StyleTain, You-Lin, and Chien-Ning Hsu. 2022. "Metabolic Syndrome Programming and Reprogramming: Mechanistic Aspects of Oxidative Stress" Antioxidants 11, no. 11: 2108. https://doi.org/10.3390/antiox11112108
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.