Rett Syndrome and Fragile X Syndrome: Different Etiology With Common Molecular Dysfunctions
 Snow Bach
Snow Bach Stephen Shovlin
Stephen Shovlin Michael Moriarty
Michael Moriarty Barbara Bardoni
Barbara Bardoni Daniela Tropea
Daniela Tropea- 1School of Mathematical Sciences, Dublin City University, Dublin, Ireland
- 2Neuropsychiatric Genetics, Department of Psychiatry, School of Medicine, Trinity College Dublin, Trinity Translational Medicine Institute, St James’s Hospital, Dublin, Ireland
- 3School of Medicine, Trinity College Dublin, Dublin, Ireland
- 4Inserm, CNRS UMR 7275, Institute of Molecular and Cellular Pharmacology, Université Côte d’Azur, Valbonne, France
- 5Trinity College Institute of Neuroscience, Trinity College Dublin, Dublin, Ireland
- 6FutureNeuro, The SFI Research Centre for Chronic and Rare Neurological Diseases, Dublin, Ireland
Rett syndrome (RTT) and Fragile X syndrome (FXS) are two monogenetic neurodevelopmental disorders with complex clinical presentations. RTT is caused by mutations in the Methyl-CpG binding protein 2 gene (MECP2) altering the function of its protein product MeCP2. MeCP2 modulates gene expression by binding methylated CpG dinucleotides, and by interacting with transcription factors. FXS is caused by the silencing of the FMR1 gene encoding the Fragile X Mental Retardation Protein (FMRP), a RNA binding protein involved in multiple steps of RNA metabolism, and modulating the translation of thousands of proteins including a large set of synaptic proteins. Despite differences in genetic etiology, there are overlapping features in RTT and FXS, possibly due to interactions between MeCP2 and FMRP, and to the regulation of pathways resulting in dysregulation of common molecular signaling. Furthermore, basic physiological mechanisms are regulated by these proteins and might concur to the pathophysiology of both syndromes. Considering that RTT and FXS are disorders affecting brain development, and that most of the common targets of MeCP2 and FMRP are involved in brain activity, we discuss the mechanisms of synaptic function and plasticity altered in RTT and FXS, and we consider the similarities and the differences between these two disorders.
Introduction
Rett syndrome (RTT) and Fragile X syndrome (FXS) are neurodevelopmental disorders associated with mutations in genes located on the X chromosome. Hence, in both RTT and FXS, the presentation is more severe in male patients, and the reduced severity in females is to be attributed to the presence of two copies of the genes, although X-inactivation influences the number of copies that remain active.
Patients with RTT show apparent normal development up until 18 months, after which there is a regression in motor and language skills accompanied to behavioral and autonomic deficits. Diagnosis of RTT in males is rare as patients do not often survive past infancy (Reichow et al., 2015), except for rare cases reported beyond early childhood (Pitzianti et al., 2019; Takeguchi et al., 2020). Consequently, the majority of patients with RTT are females with a prevalence of one in every 10,000 births. RTT is caused by mutations in the MECP2 gene (Amir et al., 1999) which codes for Methyl-CpG binding protein 2 (MeCP2), a protein that binds to methylated DNA to regulate transcription. MeCP2 also interacts with other important molecules by regulating microRNA (miRNA) (Wu et al., 2010). Although ubiquitously present, MeCP2 is expressed mostly in the brain, and it is an important regulator in brain development. Its altered functionality leads to neurodevelopmental deficits including impaired modulation of brain cell connectivity.
Fragile X syndrome affects one in every 4000 males, and one in every 7000 females. The symptoms in male patients with FXS are more severe than in females, and include developmental delays, behavioral and social deficits. FXS patients may show some degree of intellectual disabilities, while females may have normal intelligence to mild intellectual disability (Maurin et al., 2014; Dahlhaus, 2018).
Fragile X syndrome is caused by the silencing of the Fragile X Mental Retardation gene 1 (FMR1), which is associated to CGG repeat expansion in its 5′UTR region (Pieretti et al., 1991; Verkerk et al., 1991; Dahlhaus, 2018). As result of the expansion, the 5’UTR and the promoter of the FMR1 gene are hypermethylated, FMR1 expression is silenced and its encoded protein, the Fragile X Mental Retardation Protein (FMRP) is not expressed. FMRP is an RNA-binding protein and a component of ribonucleoprotein complexes involved in shuttling between nucleus and cytoplasm, transport along dendrites and association to polyribosomes (Maurin et al., 2014; Richter and Zhao, 2021). Indeed, FMRP modulates the subcellular localization (Xing and Bassell, 2013) and expression of thousands of its target mRNAs (Maurin and Bardoni, 2018; Richter and Zhao, 2021). The role of FMRP in translational regulation - being both repressor and enhancer of translation – is to date its most studied function (Bechara et al., 2009; Darnell et al., 2011; Maurin and Bardoni, 2018; Richter and Zhao, 2021).
Because of their monogenic origin, it has been possible for researchers to develop animal models to study RTT (Na et al., 2012; Pietri et al., 2013; Ezeonwuka and Rastegar, 2014; Cortelazzo et al., 2020) and FXS (Dahlhaus, 2018). These models provide the necessary framework to study the function of MeCP2 and FMRP and extrapolate mechanisms of action and putative targets. In addition, these animal models allow pre-clinical testing to set up therapeutic approaches. Many animal models are now available to study RTT, and several of them have the same mutation present in patients. Additionally, Mecp2 mutant models can also be generated for studying MeCP2 overexpression or MECP2 duplication syndrome (Collins et al., 2004; Na et al., 2012; Bodda et al., 2013). Mecp2 duplication syndrome is marked by features of autism and it is distinct from typical RTT (Ramocki et al., 2009, 2010).
Multiple models for FXS have been generated in flies (Drozd et al., 2018), zebrafish (Vaz et al., 2019), rat (Kulkarni and Sevilimedu, 2020), and mice (Bakker et al., 1994; Mientjes et al., 2006; Dahlhaus, 2018). The mouse model is the most used, as it recapitulates the main phenotype of the disorder. The similarities are present even if silencing of the gene was obtained by a classical knockout (KO) approach and not by CGG expansion: the KO-1 (Bakker et al., 1994) with the neomycin cassette in the exon 5 of the Fmr1-gene and the KO-2 (Mientjes et al., 2006) that was generated from a conditional Fmr1 KO by flanking the promoter and first exon of Fmr1 with lox P site. In addition, two Fmr1 knockin (KI) model mice have been generated and they reproduced sporadic missense mutations identified in the FMR1 gene in FXS patients (Zang et al., 2009; Prieto et al., 2021).
Despite different molecular etiology and severity of clinical presentation, there are several overlapping symptoms between the disorders: intellectual disabilities, seizures, communication deficits, attention deficits, and defects in the skeletal apparatus. At the cellular level, the impairment of synaptic function and plasticity is recognized in both diseases.
These commonalities can be explained by the interplay between MeCP2 and FMRP, and by the common targets between both molecules. Recently, it was reported that MeCP2 expression is elevated in Fmr1 KO mice cerebral cortex, while FMRP levels are reduced in mice mutants for Mecp2 (Arsenault et al., 2020). This reciprocal relationship was confirmed using MeCP2 knockdown mouse N2A, and human HEK-293 cells lines (Arsenault et al., 2020). MeCP2 association with FMR1 gene has been shown in silico (Bach et al., 2020). Moreover, MeCP2 and FMRP influence the expression of brain derived neurotrophic factor (BDNF), and, on the other hand, alteration in BDNF signaling affects the expression of Fmr1 (Castrén and Castrén, 2014; Vicario et al., 2015).
Several authors, to clarify the neurobiology of RTT and FXS, investigated the molecular targets of MeCP2 and of FMRP (Skene et al., 2010; Darnell et al., 2011; Baubec et al., 2013; Maxwell et al., 2013; Gabel et al., 2015; Rube et al., 2016; Maurin et al., 2018; Sawicka et al., 2019). These studies show that many of the targets are involved in neurodevelopment and modulate brain function. With this premise, here we discuss the similarities and differences between mechanisms of synaptic function and plasticity in RTT and FXS as well as common molecular factors modulated in both disorders.
Altered Synaptic Functioning in Rett Syndrome and Fragile X Syndrome
Alterations in synaptic function have been reported in both RTT and FXS (Sidorov et al., 2013; Bagni and Zukin, 2019; Banerjee et al., 2019) with functional consequences on the balance between excitation and inhibition (E/I ratio) and the mechanisms of synaptic plasticity (Table 1). The E/I ratio has been found to be altered in several neuropsychiatric disorders. While it represents a change in the extent of excitatory and inhibitory transmission, the underlying activity is complicated and multifaceted (Sohal and Rubenstein, 2019). Several sources report a shift in the E-I ratio in both RTT and FXS, however the alterations may differ depending on the brain region or cell-type investigated.
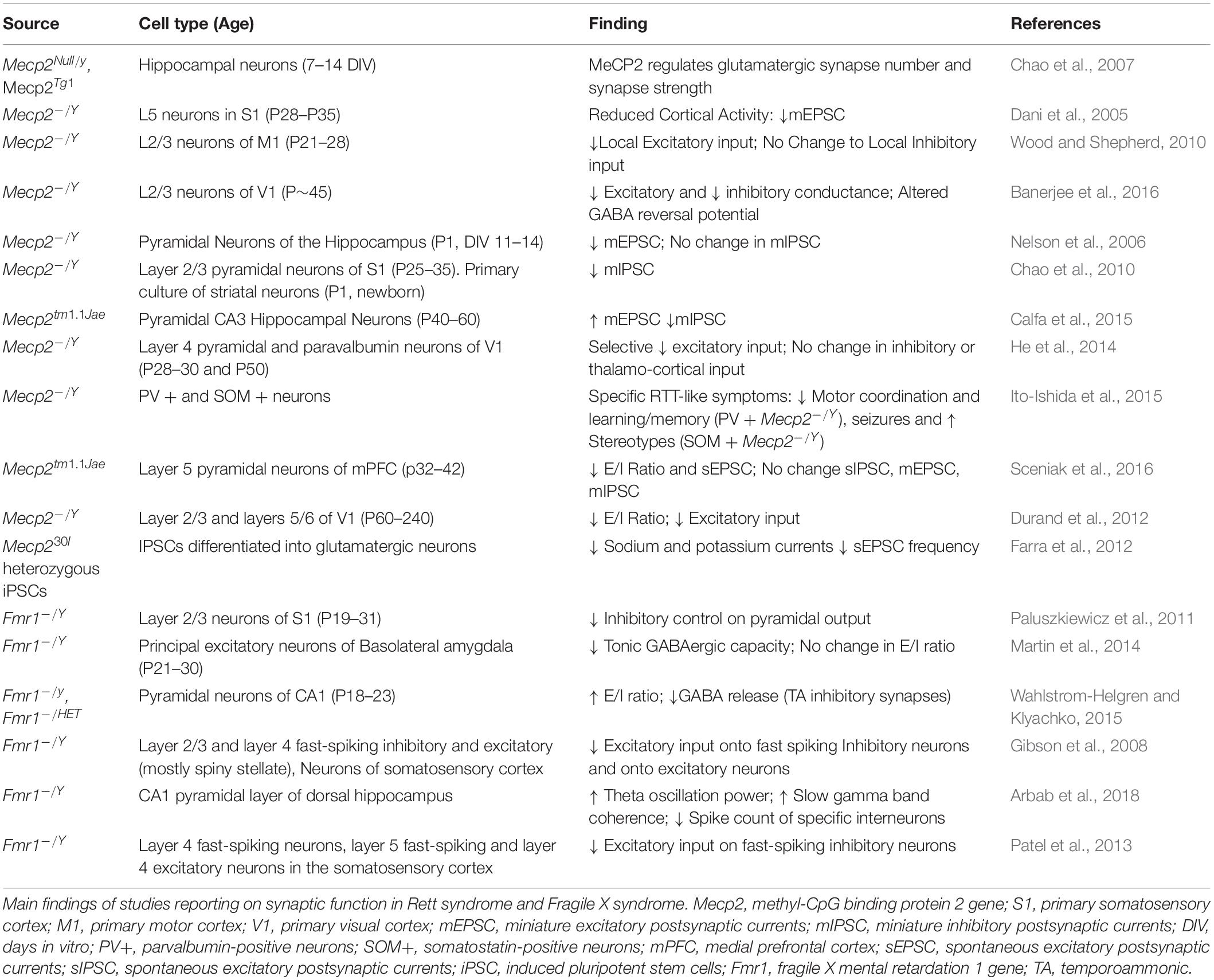
Table 1. Altered synaptic function in Rett syndrome and Fragile X syndrome.
In symptomatic Mecp2 KO mice there is a shift in E/I ratio in favor of inhibition present in a number of brain regions including S1, V1, and mPFC (Dani et al., 2005; Durand et al., 2012; Sceniak et al., 2016). However, at birth Mecp2 mutant mice show an increased glutamatergic transmission, due to the altered shift in GABA function, which is excitatory during the first phases of development, but reverses during the first weeks of postnatal development. In Mecp2 KO mice the GABA shift is postponed, and treatment with bumetanide, an inhibitor of the chloride channel NKCC1 reduces the effects of the delay. When bumetanide is administered prenatally, some of the symptoms of RTT are decreased, but the respiratory dysfunction and the mortality remain (Lozovaya et al., 2019). Symptomatic Mecp2 KO mice display decreased dendritic spine density (Kishi and Macklis, 2004; Tropea et al., 2009; Castro et al., 2014) and MeCP2 deficit has been found to decrease glutamatergic synapse number and strength (Chao et al., 2007).
Deletion of Mecp2 in particular cell neuronal subtypes in mice, reveal the complicated nature of shifts in E/I ratio. Conditional deletion of Mecp2 in parvalbuminergic (PV) neurons removes the experience dependent critical period of plasticity in the visual cortex, and produces only a partial Rett-like phenotype, specifically motor dysfunction (He et al., 2014; Ito-Ishida et al., 2015). A more general effect of GABAergic neurons has been observed in the brainstem, with a reduction in the number of GABAergic synapses. In patients with RTT, abnormality of inhibitory transmission is associated with respiratory dysfunction, and blockade of GABA reuptake decreased the breathing dysfunction in Mecp2 KO mice (Abdala et al., 2016).
Studies in patients-derived cells confirm the decrease excitatory synaptic transmission in iPS cells derived from mice. These results were also observed in mouse preparations in vivo and in vitro (Farra et al., 2012).
In FXS mouse models changes to E/I ratio appear to be more specific compared to the Mecp2 KO mice. Increased intrinsic excitability is observed in FXS at cellular, circuit and behavioral level. The cellular excitability stays with altered ion channels activity, dependent both on the translational activity of FMRP, but also on the direct interaction between FMRP and ion channels (Contractor et al., 2015). In FXS, one aspect of hyperexcitability is linked to the delayed switch in GABA polarity. As for RTT, this timeline is delayed in FXS, where the GABA transmission remains excitatory for longer. The overall increased excitability of the circuits influences also synaptic excitability and spike-timing dependent plasticity, and it is dependent on the chloride transporter NKCC1.
Another aspect of the increased excitability in FXS is due to a reduced expression of GABAA receptors’ subunits, with reduced frequency of IPSCs, but not amplitude of GABA currents. The general imbalance in favor of excitation in FXS reflects on the hyper-reactivity to stimuli, anxiety, and seizures in animal models and patients.
Studies in human-derived cells confirmed the morphological findings in FMRP deprived cultures, but they not always confirmed the altered excitability, especially at early stages of development (Telias et al., 2015). This discrepancy can be due to an un-matched decrease in FMRP expression during the in vitro development (Linda et al., 2018).
The frequency of the brain waves is also affected in FXS. In Fmr1 KO mice, increased synchronization of local field potentials occurs and may underlie deficits to information processing in hippocampal circuitry. This hyper-synchronization was characterized in a freely moving mouse, as increased theta power and coherence of slow gamma oscillations (Sohal et al., 2009; Arbab et al., 2018). More recently, the single-cell analyses of a subpopulation of interneurons in Fmr1-KO brain at Post-natal day (PND) 18 highlighted the increased levels of inhibitory markers in the absence of FMRP (Castagnola et al., 2020).
Morphological Correlates of Synaptic Functions
Alterations in synaptic functions are reflected also in morphological differences between the two disorders. Reduced neuronal cell size and numbers of dendrites have been observed in patients with RTT alongside short dendrites and a decrease in dendritic spines (Armstrong, 2005; Belichenko et al., 2008). In RTT, there is an overall decrease in brain volume differences and altered brain structures that are also present in mice studies (Chen et al., 2001; Ballas et al., 2009; Xu et al., 2014).
In contrast to RTT and MeCP2 deficiency, a lack of FRMP is associated with increases spine density in human patients and mice models (Levenga et al., 2011; Hodges et al., 2017). Both in Fmr1 KO mice, and in brain from patients, it has been reported an increase in the number of elongated spines (filopodia) in absence of functional FMRP (Comery et al., 1997), suggesting a reduction in the pruning of spines (Irwin et al., 2000). However, recent studies on spine functionality (Thomazeau et al., 2020) and spine compartmentalization over time (Wijetunge et al., 2014), find that the altered morphology does not correlate with abnormal function between Fmr1 KO mice and controls. These findings challenge the view of immature status of connections in FXS (He and Portera-Cailliau, 2013), and even the turnover of the spines seems to be invariant to that of matched controls. However, all these measures can be dependent on the brain area and on the stage of development (He and Portera-Cailliau, 2013).
In fact, the presence of the filopodia is dependent on the stage of development and on the preparation. Nimchinsky et al. (2001) analyzed mutant and control mice at several developmental stages and showed that differences in spine morphology and density decreased between 1 and 4 weeks of age. The closing of these phenotypic gaps originally suggested that FMRP played a role in coordinating synaptogenesis in a time-dependant manner. Different cell populations of the hippocampus also show that long and irregular spines are also present in juvenile and adult Fmr1 KO mice, while in vitro and in vivo studies show discrepancies for spine densities as with LTP and LTD function (as reviewed by Bostrom et al., 2016). A summary of the studies investigating morphological correlates of synaptic functions in RTT and FXS is reported in Table 2.
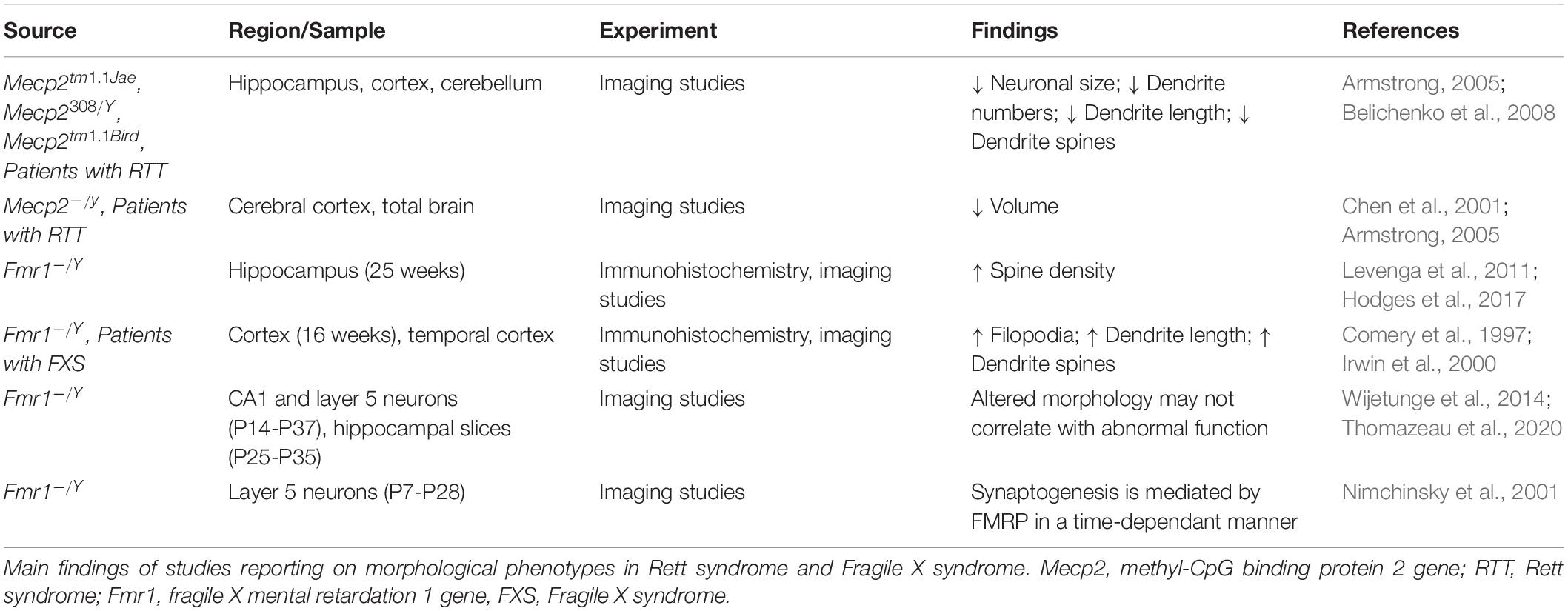
Table 2. Morphological correlates in Rett syndrome and Fragile X syndrome.
Common Forms of Synaptic Plasticity Disrupted in Rett Syndrome and Fragile X Syndrome
The long-term activity-dependent variation in synaptic connectivity and the associated molecular changes are defined as synaptic plasticity (Citri and Malenka, 2008). Several forms of synaptic plasticity have been reported to be disrupted in RTT and FXS (Huber et al., 2002; Blackman et al., 2012; Wang et al., 2012; Na et al., 2013; Wondolowski and Dickman, 2013).
Alterations in Long-Term Potentiation and Long-Term Depression in Rett Syndrome and Fragile X Syndrome
Both RTT and FXS display endophenotypes that signal aberrant Long-term potentiation (LTP) and long-term depression (LTD), which are Hebbian Forms of plasticity.
In RTT, deficits have been reported in excitatory synapses and LTP. Hippocampal slices show reduced LTP in Mecp2 KO (Asaka et al., 2006) and Mecp2308/Y mice (Moretti et al., 2006). Reduced potentiation is present around onset of symptoms while pre-symptomatic mice maintain the same level of activity observed in matching controls, suggesting that synaptic dysfunction and decline in plasticity is an early event in RTT. Pyramidal neuron synapses in the hippocampus are indeed potentiated in Mecp2 KO mice, however there is a failure to regulate AMPA receptors post-activation and an overall deficit in LTP (Li et al., 2016). Mecp2 KO mice display a lack of structural plasticity, and enlarged spines are observed regardless of stimulation or sham (Li et al., 2016). AMPA receptor-related transmission is enhanced at hippocampal synapses and over time, the decrease in internalization fails to counterbalance excessive accumulation. In addition, the lack of receptor trafficking prevents activated synapses from becoming plastic, affecting both LTP and LTD.
Long-term depression is also altered in RTT, however to a lesser extent than LTP. Moretti et al. (2006) induced LTD with two different stimulus paradygms in the hippocampus of Mecp2308/Y mice and show that when LTD was induced by administration of the mGluR agonist 3,5-dihydroxyphenylglycine (DHPG), the response was comparable in mutant and wildtype mice. However, when paired pulse stimulation was applied, LTD was observed in wild type mice, but not in Mecp2308/Y mice. Since both stimulation paradigms affect the post-synaptic sites, but only the paired pulse stimulation protocol is dependent on the presynaptic site, the authors conclude that the impairments in plasticity are due to altered presynaptic terminals, while there is some preservation of LTD. It has also been shown that hippocampal slices of symptomatic mice with an Mecp2 KO show no NMDAR-dependent LTD by low frequency stimulation (Asaka et al., 2006). More recently, in younger P15 Mecp2 KO mice, LTD has been induced by DHPG at two different stages: early and late (Lozovaya et al., 2019). Only early induced LTD displays a significant decrease in the amplitude of the response, implicating even earlier synaptic impairment onset in RTT mice.
In FXS LTP was reported to be reduced in several brain regions (Desai et al., 2006; Lauterborn et al., 2007; Suvrathan and Chattarji, 2011; Seese et al., 2012), however, the main form of plasticity studied in FXS is LTD.
The first to show alterations in synaptic plasticity in the absence of FMRP were Huber and colleagues (Huber et al., 2002), who showed an increase in mGluR-dependent LTD in the hippocampus of Fmr1 KO mice. The dysregulation of mGluR signaling was further confirmed by other authors (Weiler et al., 1997; Gross et al., 2012; Tian et al., 2017), and all the data support the theory that FMRP controls the translation of specific proteins involved in synaptic function, including glutamatergic receptors.
Similarly to what reported in RTT, AMPA aberrant receptors trafficking is driven by a lack of FMRP and consequently associated with cognitive deficits (Nakamoto et al., 2007). PSD-95 synthesis increases in response to DHPG-activation of mGluR, and is co-translated with FMRP (Todd et al., 2003). Further studies in other brain areas showed that LTD in enhanced in multiple cell populations, including cerebellum, where it controls abnormal motor behavior and development of synaptic circuitry in the somatosensory cortex (Greenough et al., 2001; Huber et al., 2002; Koekkoek et al., 2005).
Overall, LTP deficits have been shown in RTT and to a lesser extent in FXS. On the other hand, FXS tends to display enhanced LTD, while RTT shows some LTD preservation.
A summary of the experiments exploring Hebbian forms of plasticity in RTT and FXS is reported in Table 3. Beside LTP and LTD, other forms of plasticity are affected in RTT and FXS.
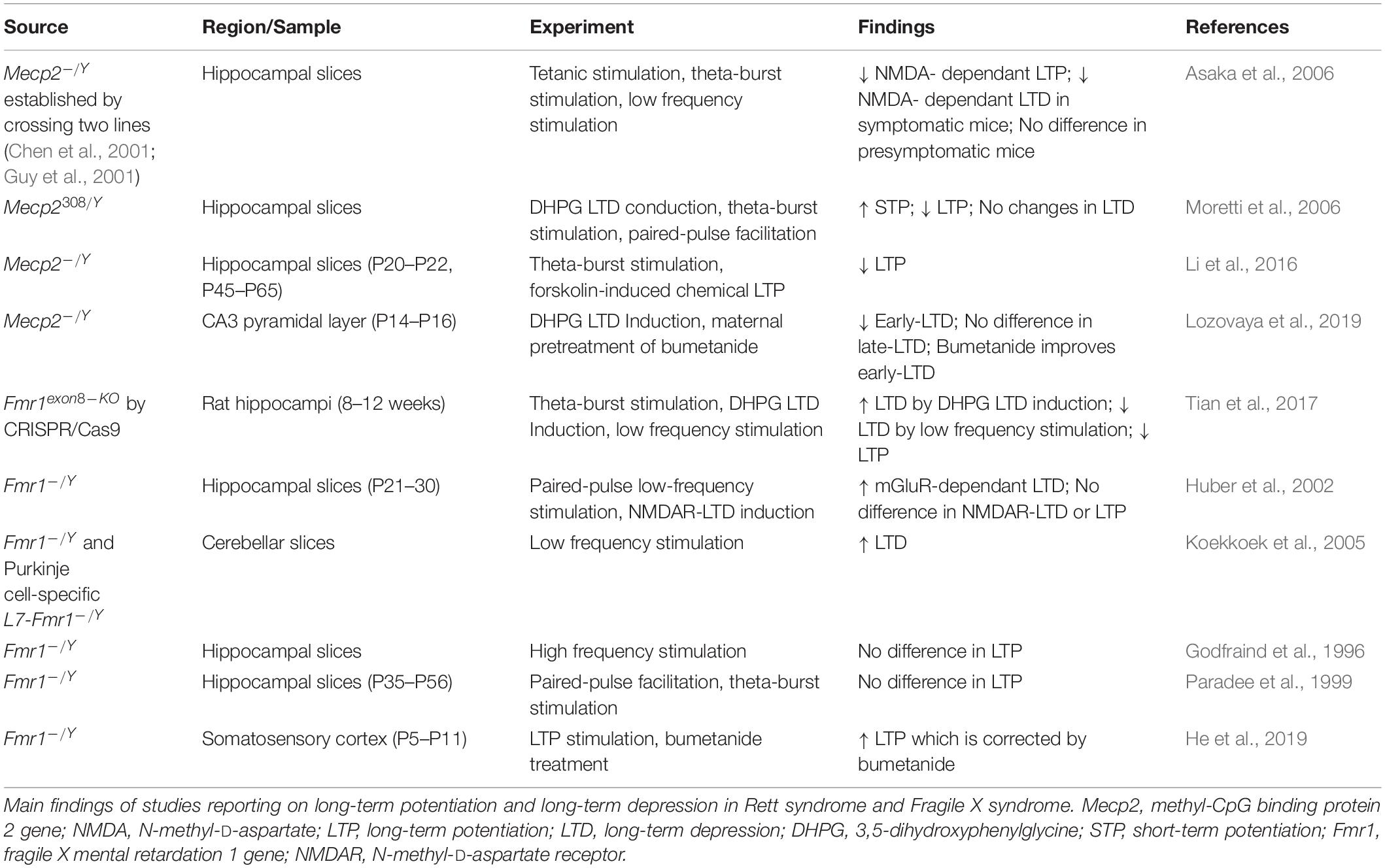
Table 3. Alteration of long-term potentiation and long-term depression in Rett syndrome and Fragile X syndrome.
Alterations in Homeostatic Plasticity in Rett Syndrome and Fragile X Syndrome
Homeostatic plasticity controls the changes in synaptic strength that individual neurons operate in response to prolonged changes of neuronal stimulation, and it also mediates the balance of excitation and inhibition. Both RTT and FXS have shown alterations in mechanisms of homeostatic plasticity, and these alterations may explain the general changes in neuronal activity in patients and animal models with RTT and FXS (Table 4).
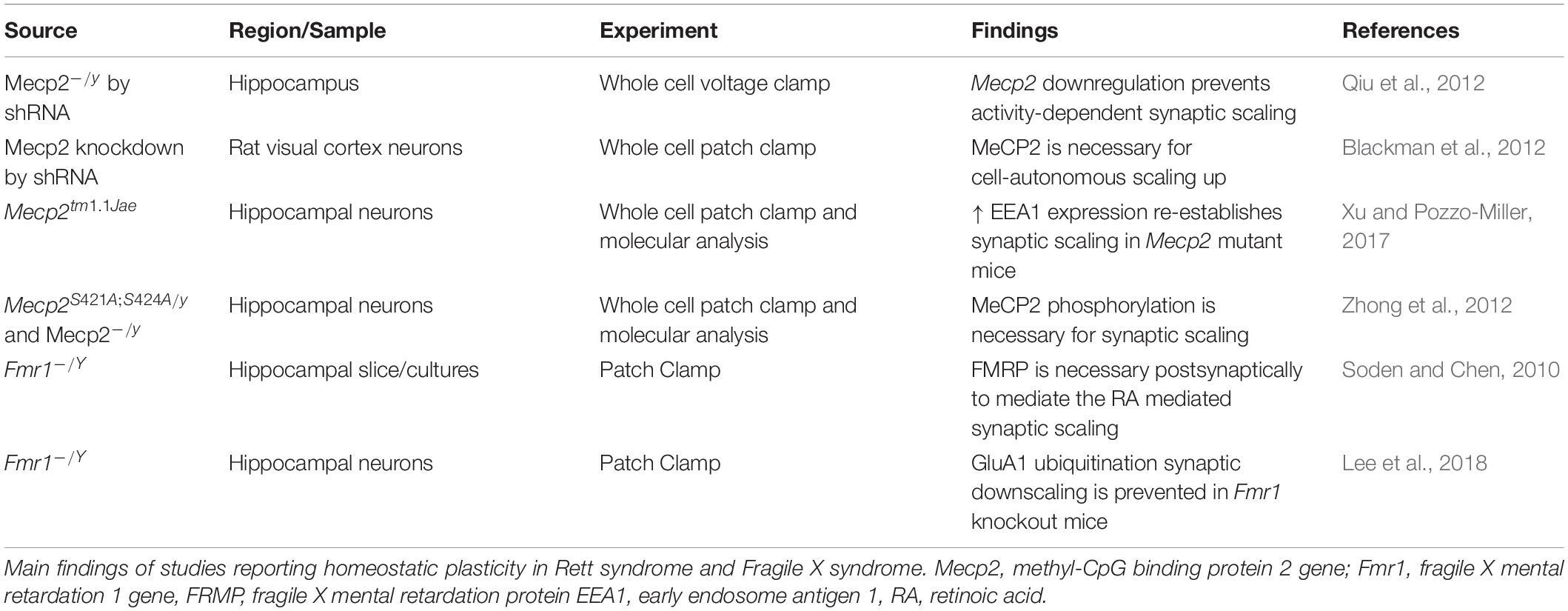
Table 4. Altered homeostatic plasticity in Rett syndrome and Fragile X syndrome.
One of the main mechanisms controlling homeostatic changes is synaptic scaling, and it relies on the synthesis, trafficking, and function of AMPA receptors.
Defects in functional levels of Mecp2 alter homeostatic plasticity. Hence, in RTT, where there is a loss of function of MeCP2, there is impaired ability to respond to homeostatic changes (Na et al., 2013). One of the first studies of homeostatic plasticity in RTT was performed in neuronal cultures. In this study, stimulation with bicuculline produced an increase in neuronal activity, with a sequential decrease in the expression of the AMPA receptors subunit GluR2 and a reduction of mEPSC amplitude. However, the expression of GluR2 is under the control of the transcriptional repressor MeCP2, whose levels also increase during bicuculline stimulation. As a result, the bicuculline treatment in Mecp2 KO neurons, does not lead to the expected decrease of the GluR2 receptor subunit at the synapse. These experiments were among the first to show the importance of Mecp2 in the control of synaptic scaling (Qiu et al., 2012), and they were confirmed by Blackman et al. (2012), who showed that reduced neuronal drive in Mecp2 KO preparations does not produce the expected increase in synaptic scaling as observed in WT controls. One of the factors controlling homeostatic plasticity in RTT is the Early Endosome Antigen 1 (EEA1), which regulates AMPA receptor endocytosis. EEA1 expression is reduced in Mecp2 KO mice, where the synaptic scaling is reduced. However, the increased expression of EEA1 in Mecp2 KO cultures, reinstates the ability to scale the synapses in response to changes in activity (Xu and Pozzo-Miller, 2017). It is important to remember that not only Mecp2 expression, but also its activation, is important for the proper functioning of the protein. Indeed, post translational modifications of MeCP2 have also been found to be implicated in synaptic function and homeostasis (Bellini et al., 2014) and the phosphorylated MeCP2 modulates synaptic scaling -down through mGluR5 (Zhong et al., 2012).
Fragile X mental retardation protein has been involved in synaptic scaling in response to both increased and decreased activity- as observed in Fmr1 KO mice and in neuronal cultures derived from patients with FXS. The increased activity of AMPA receptors consequent to a decrease in activity, is mediated by the Retinoic Acid (RA), which is produced in response to the altered activity, and promotes the synthesis of new AMPA receptors. However, in FXS, while the transcription of the AMPA receptor remains unchanged, the RA- mediated translation of AMPA is reduced, and only restoration of the proper full length functional FMRP re-establishes synaptic scaling in primary cultures derived from Fmr1KO mice (Soden and Chen, 2010). These results suggest that FMRP is essential for the postsynaptic response in RA-mediated synaptic scaling. However, RA is required for the combined action of TTX and NMDA blockade- as there are no changes in scaling with TTX alone. In this regard it is interesting to note that the RA does not affect spine morphology and number. Zhang and colleagues confirmed the impaired synaptic scaling in neuronal cultures derived from patients with FXS (Zhang et al., 2018) and the role of RA as mediator of synaptic scaling. The RA action is effective both on excitatory and inhibitory synapses. Interestingly, RA is one of the mediators of the post- to presynaptic communication and could be involved in linking the post-synaptic events to pre-synaptic adjustments. In FXS, the imbalance in protein synthesis determines a dysregulation in RA which is important at postsynaptic level, but also at the presynaptic level with the regulation of EPSC frequencies (Wang et al., 2011; McCarthy et al., 2012).
Synaptic downscaling – the scaling down in response to increased synaptic activity- is also altered in Fmr1 KO mice. One of the mechanisms of downscaling is the degradation of AMPA receptor through ubiquitination (Lee et al., 2018). The ubiquitination of AMPA receptors is mediated by a complex cascade of molecules, which includes cell-cycle molecules and phosphatases, and FMRP interferes with the mechanism that leads to homeostatic-dependent ubiquitination.
It is worth of note that the Neuroligin-Neurexin complexes, involved in circuitry development and function is also essential for presynaptic homeostatic plasticity (Sons et al., 2006). Several molecular studies in RTT and FXS report that these molecules are targets of FMRP and MeCP2 (Darnell et al., 2011; Gulmez Karaca et al., 2018; Maurin et al., 2018; Raman et al., 2018), suggesting that impaired homeostatic plasticity in these syndromes may be mediated by deficits in Neuroligin or Neurexin.
Molecules and Pathways Controlling Changes in Synaptic Strength and Connectivity in Rett Syndrome and Fragile X Syndrome
The alteration in several forms of plasticity observed in RTT and FXS can be explained considering that many molecular targets of MeCP2 and FMRP are involved in the regulation of synaptic function. The identification of these molecular regulators can shed light on the neurobiology of RTT and FXS and can suggest strategies for treatment. In this section we will examine several of these molecules and pathways (Table 5).
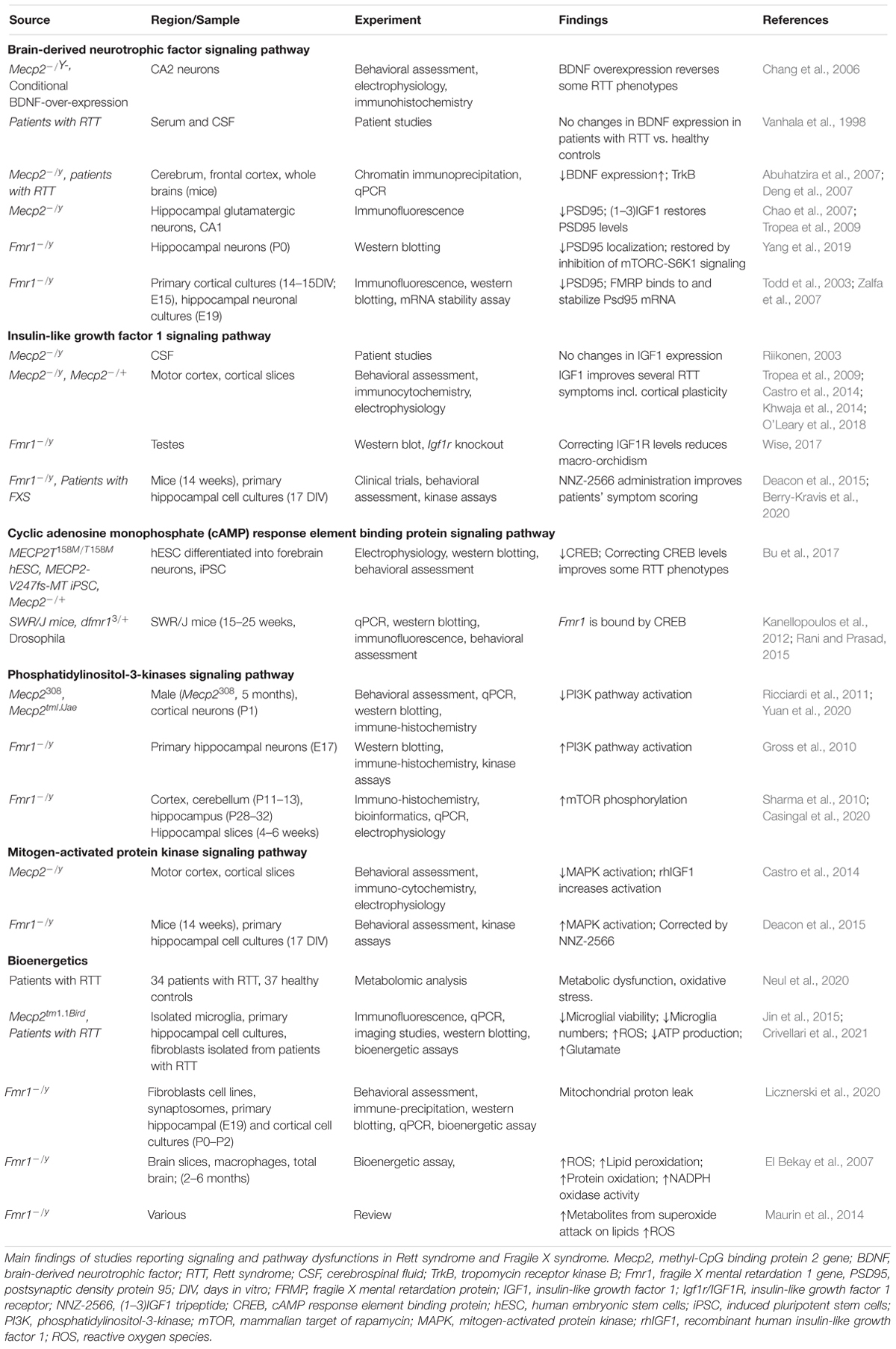
Table 5. Pathways implicated in Rett syndrome and Fragile X syndrome.
Brain derived neurotrophic factor (BDNF) controls brain development and function, and it is involved in activity dependent plasticity. Its expression is highly specific and functionally defined in the mammalian brain (Kowiański et al., 2018). BDNF expression is dependent on the neurodevelopmental stage, and it is present in several different forms that bind different receptors. Pre-Pro-BDNF is concentrated at the endoplasmic reticulum before becoming pro-BDNF at the Golgi apparatus (Foltran and Diaz, 2016). Pro-BDNF is highly expressed during early postnatal development and binds p75 Neurotrophin Receptor (p75NTR) and Sortilin receptor, with a particular polymorphism Val66Met dictating the receptor binding properties (Anastasia et al., 2013). Meanwhile the mature BDNF protein (mBDNF) is present more during adulthood and binds the tyrosine kinase B receptor (TrkB) (Reichardt, 2006).
All these receptors are located on the membranes and in intracellular compartments. The complex resulting from pro-BDNF, Sortilin receptor, and p75NTR signals to RhoA, NFKB and JNK related pathways. These pathways have roles in neurodevelopment, survival and apoptosis respectively (Reichardt, 2006). mBDNF binding to TrkB results in receptor dimerization and phosphorylation on membrane lipid rafts. This complex is also associated with a number of downstream signaling pathways including PI3K, mitogen-activated protein kinase (MAPK), PLC-gamma and GTPases of the Rho family all with a range of neuronal functions (Suzuki et al., 2004). The amount of neuronal cellular processes that BDNF effects means that its expression is highly relevant to synaptic plasticity and functioning. Indeed decrease in BDNF concentration inhibits synaptogenesis and dendritic arborization (Wang et al., 2015).
Brain derived neurotrophic factor administration in control mice increases the synaptic localization of PSD95. This effect is suppressed in Fmr1 KO mice, but it can be retrieved by inhibition of mTORC-S6K1 signaling (Yang et al., 2019). PSD95 is one of the core postsynaptic proteins that functions by regulating activity of excitatory neurotransmitter receptors (Keith and El-Husseini, 2008), and its expression is modulated by FMRP (Muddashetty et al., 2011; Xing and Bassell, 2013; Ifrim et al., 2015).
Brain derived neurotrophic factor level is deficient in Mecp2 KO mouse brain and, consistently, its overexpression has been shown to reverse some RTT phenotypes (Chang et al., 2006). In patients with RTT, BDNF serum and cerebrospinal fluid (CSF) protein levels have been found to be no different from healthy controls (Vanhala et al., 1998), while in the brain the BDNF level is decreased and TrkB level is increased (Abuhatzira et al., 2007; Deng et al., 2007).
Insulin-Like Growth Factor 1
Another factor influencing the intracellular pathways controlling synaptic strength is IGF1, which is a protein involved in growth, maturation, and neuronal development. Although CSF levels of Insulin-Like Growth Factor 1 (IGF1) have been found to be unchanged in RTT (Riikonen, 2003), both the full IGF1 molecule, and its functionally active cleavage product (1-3)IGF1 have been shown to ameliorate symptoms of the RTT pathophysiology (Tropea et al., 2009; Castro et al., 2014; Khwaja et al., 2014; O’Leary et al., 2018). IGF1 and (1-3)IGF1 administration in a RTT mouse model increases PSD95, dendritic arborization and excitatory current. Interestingly, these treatments also appear to re-establish cortical plasticity in Mecp2 KO mice to levels observed in controls (Tropea et al., 2009; Castro et al., 2014). IGF1 signaling occurs primarily through PI3K-AKT and MAPK pathways (Fernandez and Torres-Alemán, 2012). IGF1R activates PI3K-AKT functions to increase neuronal survival (Dudek et al., 1997), while prolonged administration of IGF1 with growth hormone, determines pro-inflammatory responses via activation of MAPK (Wolters et al., 2017). IGF signaling to a lesser extent has been implicated in FXS, where correcting the low level of IGF1R was found to reduce macro-orchidism (enlarged testes) a phenotype characterizing all the adult male patients with FXS (Wise, 2017). More promising evidence comes from the use of the (1-3)IGF1 analog, NNZ-2566 (Trofinetide) as a treatment for FXS; in fact mouse models of FXS treated with NNZ-2566 showed improvements in cognitive function and hyperactivity. Also patients with FXS treated with NNZ-2566 improved in a number of clinical scoring tools (Deacon et al., 2015; Berry-Kravis et al., 2020), but further clinical tests are required to confirm the benefits of the treatment.
Both BDNF and IGF1 signals involve the activation of intracellular pathways involving the PI3K and MAPK cascades, which are related to both RTT and FXS and are involved in activity-dependent plasticity.
Cyclic Adenosine Monophosphate Response Element Binding Protein
Cyclic adenosine monophosphate (cAMP) response element binding protein is involved in transcriptional changes induced by synaptic plasticity, including increasing neuronal excitability and synapse strengthening associated to LTP induction (Caracciolo et al., 2018). Transcriptional genes expressed by CREB activation include c-Fos, whose protein is linked to memory and learning (Gallo et al., 2018), and the co-localization of CREB and c-Fos is associated with long term synaptic plasticity (Miyashita et al., 2018). This molecule has been found to be decreased in Mecp2 KO mice and rectifying CREB levels can correct some of the RTT symptoms (Bu et al., 2017). The Fmr1 gene is thought to be bound and therefore regulated by CREB (Kanellopoulos et al., 2012; Rani and Prasad, 2015), however the relationship of CREB signaling to FXS synaptic plasticity requires further investigation, especially considering the role that FMRP has in the modulation of cAMP and cGMP levels, two molecules upstream the CREB expression (Delhaye and Bardoni, 2021). It was shown that In the hippocampus, Cilostazol (an inhibitor of Phosphodiesterase 3) increases the levels of c-fos and of insulin-like growth factor 1 (IGF-1) (Zhao et al., 2010) and activates CREB in PC12 cells (Zheng and Quirion, 2006). This result suggests a link between the levels of cAMP and cGMP – both targets of PDE3 – and the levels of IGF-1. Remarkably, the inhibition of PDEs both in FXS (PDE2, PDE4, PDE4D) and in RETT (PDE4) has been shown to improve socio-cognitive deficits in animal models and in patients.
Phosphatidylinositol-3-Kinases
Phosphatidylinositol-3-kinases (PI3K) are a family of intracellular signaling molecules functioning downstream of G protein coupled receptors and tyrosine kinases.
Once activated, PI3K subsequently phosphorylates AKT which regulates cell cycle and apoptosis (Chalhoub and Baker, 2009). Further downstream of AKT phosphorylation, is the mammalian Target of Rapamycin (mTOR) whose activation regulates nutrition, energy sensing and growth (Zoncu et al., 2011). In Mecp2 KO mice the activation of the PI3K/AKT/mTOR pathway is reduced, while in FXS it is increased. The alterations in PI3K/AKT/mTOR level in Mecp2 KO mice can be restored using IGF1, and treatment with PI3K antagonists rescue FXS defects (Gross et al., 2010; Ricciardi et al., 2011; Yuan et al., 2020).
Alterations of FMRP levels affect the mGluR-PI3K-AKT-mTOR cascade. In normal conditions mGluR-dependent LTD requires rapid translation of dendritic mRNA, but in Fmr1 KO mice LTD it is enhanced and insensitive to inhibition of protein synthesis (Huber et al., 2002; Nosyreva and Huber, 2006). This insensitivity is due to the de-repression of FMRP, which causes increased basal levels of mGluR-stimulated checkpoint proteins (Thomazeau et al., 2020). Hence in FXS, mGluR-LTD is decoupled from protein synthesis/mTOR activation. Interestingly in Fmr1 KO mice, mTOR phosphorylation is increased in embryonic neocortex and in postnatal hippocampus samples (Sharma et al., 2010; Casingal et al., 2020).
Mitogen-Activated Protein Kinase
The MAPK signaling pathway includes extracellular signal-Regulated Kinase 1 and 2 (ERK1 and ERK2), which are essential for neuronal transcriptional events, including synaptic plasticity, learning and memory (Thomas and Huganir, 2004). When Mecp2 KO mice are treated with recombinant human IGF1, both AKT, ERK1, and ERK2 levels are increased in conjunction with the increase of the post-synaptic marker PSD95 (Castro et al., 2014). The MAPK pathway is upregulated in Fmr1 KO mice, and is modulated by NNZ-2566 (Deacon et al., 2015), suggesting that mechanisms controlling plasticity may be potential targets of therapeutics in FXS and RTT, although the mechanisms of action of NNZ-2566 requires further investigation.
PSD95
Also at the synaptic level, MeCP2 and FMRP control the expression and localization of PSD95, which is strongly related to synaptic strength in excitatory synapses. In the hippocampus, MeCP2 controls the number of glutamatergic synapses. VGLUT1 and PSD95, which are respectively pre- and postsynaptic markers, and they are both downregulated upon loss of MeCP2 in mutant mice. Conversely, a two-fold increase of MeCP2 expression, determines an increase in density and colocalization of these two markers (Chao et al., 2007). PSD95 levels are rescued by (1-3)IGF1 to levels comparable in wild type animals (Tropea et al., 2009) achieving the same phenotype as double mutants for loss and doubling of MeCP2 (Collins et al., 2004; Chao et al., 2007). PSD-95 expression is also deregulated in Fmr1 KO mice. FMRP binds to PSD95 mRNA in vivo (Todd et al., 2003; Zalfa et al., 2007), suggesting that FMRP stabilizes the PSD95 transcript, leading to adequate expression levels of PSD95 (Zalfa et al., 2007).
Bioenergetics
All energy-demanding activities, including synaptic function and plasticity, are dependent on bioenergetics, which appears to be dysfunctional in both RTT and FXS. Recent works in both FXS (Mithal and Chandel, 2020) and RTT (Neul et al., 2020) pointed at dysfunctions in mitochondria: the organelles devoted to produce the energy necessary for neuronal function. Alteration in bioenergetics has been proposed for RTT (Jin et al., 2015; Crivellari et al., 2021) and FXS (D’Antoni et al., 2019), and reflect the evolutionary need to match the cognitive function with the capacity of producing the adequate supply of energy to fulfill the requests. The link between energy metabolism and cognition has been well reviewed, and it appears clear that several brain disorders are now depending on the capacity of the organism to provide the fuel requested by the brain and to control the number of oxidative species, which are the natural side products of aerobic metabolism. Therefore, the ability to produce energy goes hand in hand with the capacity to control the reactive oxygen species and the possible damage created by an excess of these radicals. Considering the oxidative stress in FXS, it is interesting to underline that El Bekay et al. (2007) found that Fmr1 KO mouse brains have higher levels of reactive oxygen species, nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase activation, lipid peroxidation and protein oxidation compared to wild type mice. In the cortex of Fmr1 KO it was also reported an increased level of metabolites that result from the attack of unsaturated lipids by the superoxide anion (Maurin et al., 2014). In normal conditions, the superoxide anion is detoxified by Sod1, the level of which is reduced in the absence of FMRP (Bechara et al., 2009), thus providing a source of oxidative stress (Maurin et al., 2014).
It is also worth mentioning that there is a two-way interaction between the systems controlling the production of energy in the cell, and the ion homeostasis (Castaldo et al., 2009), and that such interaction controls the onset and progression of neurodegeneration. These additional mechanisms should be taken into account for uncovering the underlying mechanisms in brain disorders and for designing to routes of treatment.
The Contributions of Astrocytes
Considering that non-neuronal cells are involved in synaptic function and plasticity, we now discuss the contribution of astrocytes in cellular mechanisms of RTT and FXS (Table 6).
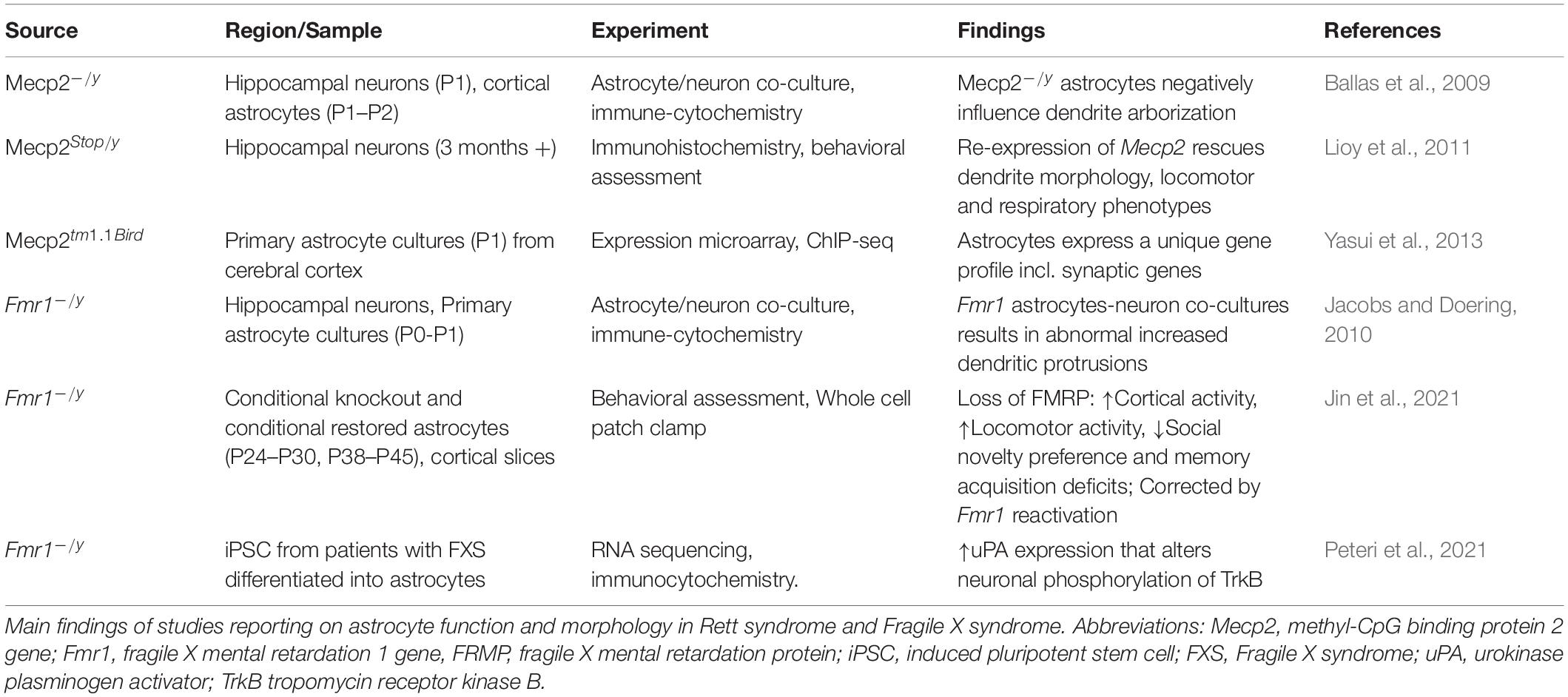
Table 6. Astrocyte function in Rett syndrome and Fragile X syndrome.
Astrocytes have been largely studied in RTT with a smaller body of research carried out in FXS. Astrocytes express both MeCP2 and FMRP. There is clear evidence that glial cells support normal neuronal growth and morphology, and in fact, both in RTT and FXS the co-culturing of astrocytes and neurons influence the morphology of neuronal arborization. In RTT coculturing of Mecp2–/y astrocytes with wildtype hippocampal neurons stunts dendrite arborization and cannot sustain typical cell growth. Conversely, culturing Mecp2 KO neurons with wildtype astrocytes results in typical dendrite morphology (Ballas et al., 2009). Re-expression of Mecp2 in astrocytes of Mecp2 deficient mice improves locomotor and respiratory phenotypes; moreover, re-expression of Mecp2 in astrocytes rescues mutant neuron dendritic morphology (Lioy et al., 2011). Additionally, astrocyte gene expression profiling has identified uniquely dysregulated genes because of Mecp2 deficiency. These genes include Cntn1, Syn2, Gabrg1, and Gria1, which function at the tripartite synapse (Yasui et al., 2013). These studies suggests that MeCP2 deficiency in astrocytes contribute to the RTT phenotype.
Fragile X mental retardation protein is expressed in astrocytes and cocultures of Fmr1 KO astrocytes with wildtype hippocampal neurons result in neurons with abnormal increased dendritic protrusions (Jacobs and Doering, 2010). While there is an increase in dendritic density, there is reduction in overall dendrite length. There is also a significant decrease in pre- and postsynaptic proteins. Interestingly, coculturing Fmr1 KO neurons with wildtype neurons rescues the morphological abnormalities to near wildtype phenotype (Cheng et al., 2012). These results suggest that astrocytes are implicated in neuronal dendritic morphology also in FXS.
Recently, Jin and co-workers (Kang et al., 2021) provided evidence that FMRP mediates synaptic connectivity through astrocytes and therefore controls learning and behavior. By using conditional knockout (cKO) and conditional restored (cON) mice in astrocytes, they find that loss of FMRP results in cortical hyperactivity, increased locomotor activity, reduced social novelty preference and deficit of memory acquisition. Reactivation of the astrocyte Fmr1 rescues these phenotypes (Jin et al., 2021).
Interestingly, in human FXS astrocytes generated from human induced pluripotent stem cells an increased expression of urokinase plasminogen activator (uPA), which modulates degradation of extracellular matrix, was reported. Increased uPA augmented neuronal phosphorylation of TrkB within the docking site for the phospholipase-Cγ1 (PLCγ1), indicating effects of uPA on neuronal plasticity (Peteri et al., 2021) and connecting this molecular alteration to the BDNF pathway.
Conclusion
In recent years it has become clear that neurodevelopmental disorders share common molecular mechanisms, and that their complex clinical presentation results from the interaction of genetic and environmental factors. Genetic studies are growing in power and are showing that genes involved in synaptic function are major risk factors for neurodevelopmental disorders, but other mechanisms, such as neuroimmunity, and mitochondrial functions are also emerging as contributors to the onset and progression of several disorders. In this context, the study of convergent and divergent mechanisms between RTT and FXS can be instructive in understanding the general biological mechanisms that underlie a variety of disorders, including those with multi-genic components. Encouraging results in therapeutic strategies for RTT (Glaze et al., 2019) and FXS (Berry-Kravis et al., 2020) confirm the perspective that some treatments can be effective for multiple conditions, and foster research that uncover overlapping mechanisms across disorders. In line with this perspective, our review suggests that the analysis of common and divergent mechanisms controlling synaptic function and plasticity can instruct new criteria for the classification of neurodevelopmental disorders, with applications to diagnosis, prognosis, and drug discovery.
Author Contributions
DT, SS, and SB contributed to the initial draft of the manuscript. DT, BB, and SB revised the manuscript. All authors discussed and provided the input on the manuscript.
Funding
DT research is partially supported by: IRSF (3507-207417 grant), Meath Foundation (Research award 2019), Fondation Jérôme Lejeune (DT Project#1935), and Science Foundation Ireland (SFI) under Grant Number 16/RC/3948 and co-funded under the European Regional Development Fund and by FutureNeuro industry partners. BB is funded by Agence Nationale de la Recherche ANR-20-CE16-0016 and ANR-15-IDEX-0001, Fondation Jérôme Lejeune (Project #2023). DT and BB acknowledge the Ulysses Project 2019 (Irish Research Council & Ambassade de France en Irlande).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdala, A. P., Toward, M. A., Dutschmann, M., Bissonnette, J. M., and Paton, J. F. R. (2016). Deficiency of GABAergic synaptic inhibition in the Kolliker-Fuse area underlies respiratory dysrhythmia in a mouse model of Rett syndrome. J. Physiol. 594, 223–237. doi: 10.1113/JP270966
Abuhatzira, L., Makedonski, K., Kaufman, Y., Razin, A., and Shemer, R. (2007). MeCP2 deficiency in the brain decreases BDNF levels by REST/CoREST-mediated repression and increases TRKB production. Epigenetics 2, 214–222. doi: 10.4161/epi.2.4.5212
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Anastasia, A., Deinhardt, K., Chao, M. V., Will, N. E., Irmady, K., Lee, F. S., et al. (2013). Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat. Commun. 4:2490. doi: 10.1038/ncomms3490
Arbab, T., Battaglia, F. P., Pennartz, C. M. A., and Bosman, C. A. (2018). Abnormal hippocampal theta and gamma hypersynchrony produces network and spike timing disturbances in the Fmr1-KO mouse model of Fragile X syndrome. Neurobiol. Dis. 114, 65–73. doi: 10.1016/j.nbd.2018.02.011
Armstrong, D. D. (2005). Neuropathology of Rett syndrome. J. Child Neurol. 20, 747–753. doi: 10.1177/08830738050200082401
Arsenault, J., Hooper, A. W. M., Gholizadeh, S., Kong, T., Pacey, L. K., Koxhioni, E., et al. (2020). Interregulation between Fragile X Mental retardation protein and Methyl CpG Binding Protein 2 in the mouse posterior cerebral cortex. Hum. Mol. Genet. 29, 3744–3756.
Asaka, Y., Jugloff, D. G. M., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227. doi: 10.1016/j.nbd.2005.07.005
Bach, S., Ryan, N. M., Guasoni, P., Corvin, A. P., El-Nemr, R. A., Khan, D., et al. (2020). Methyl - CpG - binding protein 2 mediates overlapping mechanisms across brain disorders. Sci. Rep. 10:22255. doi: 10.1038/s41598-020-79268-0
Bagni, C., and Zukin, R. S. (2019). A synaptic perspective of Fragile X Syndrome and autism spectrum disorders. Neuron 101, 1070–1088. doi: 10.1016/j.neuron.2019.02.041
Bakker, C. E., Verheij, C., Willemsen, R., van der Helm, R., Oerlemans, F., Vermey, M., et al. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33. doi: 10.1016/0092-8674(94)90569-X
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
Banerjee, A., Miller, M. T., Li, K., Sur, M., and Kaufmann, W. E. (2019). Towards a better diagnosis and treatment of Rett syndrome: a model synaptic disorder. Brain 142, 239–248. doi: 10.1093/brain/awy323
Banerjee, A., Rikhye, R. V., Breton-Provencher, V., Tangb, X., Li, C., Li, K., et al. (2016). Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, E7287–E7296. doi: 10.1073/pnas.1615330113
Baubec, T., Ivá, R., Lienert, F., and Schü Beler, D. (2013). Methylation-Dependent and -Independent Genomic Targeting Principles of the MBD Protein Family. Cell 153, 480–492. doi: 10.1016/j.cell.2013.03.011
Bechara, E. G., Didiot, M. C., Melko, M., Davidovic, L., Bensaid, M., Martin, P., et al. (2009). A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 7:e1000016. doi: 10.1371/journal.pbio.1000016
Belichenko, N. P., Belichenko, P. V., Hong, H. L., Mobley, W. C., and Francke, U. (2008). Comparative study of brain morphology in Mecp2 Mutant mouse models of Rett syndrome. J. Comp. Neurol. 508, 184–195. doi: 10.1002/cne.21673
Bellini, E., Pavesi, G., Barbiero, I., Bergo, A., Chandola, C., Nawaz, M. S., et al. (2014). MeCP2 post-translational modifications: A mechanism to control its involvement in synaptic plasticity and homeostasis? Front. Cell. Neurosci. 8:236. doi: 10.3389/fncel.2014.00236
Berry-Kravis, E., Horrigan, J. P., Tartaglia, N., Hagerman, R., Kolevzon, A., Erickson, C. A., et al. (2020). A double-blind, randomized, placebo-controlled clinical study of trofinetide in the treatment of Fragile X Syndrome. Pediatr. Neurol. 110, 30–41. doi: 10.1016/j.pediatrneurol.2020.04.019
Blackman, M. P., Djukic, B., Nelson, S. B., and Turrigiano, G. G. (2012). A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J. Neurosci. 32, 13529–13536. doi: 10.1523/JNEUROSCI.3077-12.2012
Bodda, C., Tantra, M., Mollajew, R., Arunachalam, J. P., Laccone, F. A., Can, K., et al. (2013). Mild overexpression of Mecp2 in mice causes a higher susceptibility toward seizures. Am. J. Pathol. 183, 195–210. doi: 10.1016/j.ajpath.2013.03.019
Bostrom, C., Yau, S.-Y., Majaess, N., Vetrici, M., Gil-Mohapel, J., and Christie, B. R. (2016). Hippocampal dysfunction and cognitive impairment in Fragile-X Syndrome. Neurosci. Biobehav. Rev. 68, 563–574. doi: 10.1016/j.neubiorev.2016.06.033
Bu, Q., Wang, A., Hamzah, H., Waldman, A., Jiang, K., Dong, Q., et al. (2017). CREB signaling is involved in rett syndrome pathogenesis. J. Neurosci. 37, 3671–3685. doi: 10.1523/JNEUROSCI.3735-16.2017
Calfa, G., Li, W., Rutherford, J. M., and Pozzo-Miller, L. (2015). Excitation/Inhibition imbalance and impaired synaptic inhibition in hippocampal Area CA3 of Mecp2 Knockout Mice. Hippocampus 25, 159–168. doi: 10.1002/hipo.22360
Caracciolo, L., Marosi, M., Mazzitelli, J., Latifi, S., Sano, Y., Galvan, L., et al. (2018). CREB controls cortical circuit plasticity and functional recovery after stroke. Nat. Commun. 9:2250. doi: 10.1038/s41467-018-04445-9
Casingal, C. R., Kikkawa, T., Inada, H., Sasaki, Y., and Osumi, N. (2020). Identification of FMRP target mRNAs in the developmental brain: FMRP might coordinate Ras/MAPK, Wnt/β-catenin, and mTOR signaling during corticogenesis. Mol. Brain 13:167. doi: 10.1186/s13041-020-00706-1
Castagnola, S., Cazareth, J., Lebrigand, K., Jarjat, M., Magnone, V., Delhaye, S., et al. (2020). Agonist-induced functional analysis and cell sorting associated with single-cell transcriptomics characterizes cell subtypes in normal and pathological brain. Genome Res. 30, 1633–1642. doi: 10.1101/gr.262717.120
Castaldo, P., Cataldi, M., Magi, S., Lariccia, V., Arcangeli, S., and Amoroso, S. (2009). Role of the mitochondrial sodium/calcium exchanger in neuronal physiology and in the pathogenesis of neurological diseases. Prog. Neurobiol. 87, 58–79. doi: 10.1016/j.pneurobio.2008.09.017
Castrén, M. L., and Castrén, E. (2014). BDNF in fragile X syndrome. Neuropharmacology 76, 729–736. doi: 10.1016/j.neuropharm.2013.05.018
Castro, J., Garcia, R. I., Kwok, S., Banerjee, A., Petravicz, J., Woodson, J., et al. (2014). Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, 9941–9946. doi: 10.1073/pnas.1311685111
Chalhoub, N., and Baker, S. J. (2009). PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. Mech. Dis. 4, 127–150. doi: 10.1146/annurev.pathol.4.110807.092311
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348. doi: 10.1016/j.neuron.2005.12.027
Chao, H. T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65. doi: 10.1016/j.neuron.2007.08.018
Chao, H.-T. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Cheng, C., Sourial, M., and Doering, L. C. (2012). Astrocytes and Developmental Plasticity in Fragile X. Neural Plast. 2012:197491. doi: 10.1155/2012/197491
Citri, A., and Malenka, R. C. (2008). Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41. doi: 10.1038/sj.npp.1301559
Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, R., Armstrong, D. L., Noebels, J. L., et al. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689. doi: 10.1093/hmg/ddh282
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J., et al. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U.S.A. 94, 5401–5404. doi: 10.1073/pnas.94.10.5401
Contractor, A., Klyachko, V. A., and Portera-Cailliau, C. (2015). Altered neuronal and circuit excitability in Fragile X syndrome. Neuron 87, 699–715. doi: 10.1016/j.neuron.2015.06.017
Cortelazzo, A., De Felice, C., Guy, J., Timperio, A. M., Zolla, L., Guerranti, R., et al. (2020). Brain protein changes in Mecp2 mouse mutant models: effects on disease progression of Mecp2 brain specific gene reactivation. J. Proteomics 210:103537.
Crivellari, I., Pecorelli, A., Cordone, V., Marchi, S., Pinton, P., Hayek, J., et al. (2021). Impaired mitochondrial quality control in Rett Syndrome. Arch. Biochem. Biophys. 700:108790. doi: 10.1016/j.abb.2021.108790
Dahlhaus, R. (2018). Of men and mice: modeling the fragile X syndrome. Front. Mol. Neurosci. 11:41. doi: 10.3389/fnmol.2018.00041
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565. doi: 10.1073/pnas.0506071102
D’Antoni, S., De Bari, L., Valenti, D., Borro, M., Bonaccorso, C. M., Simmaco, M., et al. (2019). Aberrant mitochondrial bioenergetics in the cerebral cortex of the Fmr1 knockout mouse model of fragile X syndrome. Biol. Chem. 401, 497–503. doi: 10.1515/hsz-2019-0221
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y. S., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Deacon, R. M. J., Glass, L., Snape, M., Hurley, M. J., Altimiras, F. J., Biekofsky, R. R., et al. (2015). NNZ-2566, a Novel Analog of (1–3) IGF-1, as a Potential Therapeutic Agent for Fragile X Syndrome. Neuromol. Med. 17, 71–82. doi: 10.1007/s12017-015-8341-2
Delhaye, S., and Bardoni, B. (2021). Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders. Mol. Psychiatry. doi: 10.1038/s41380-020-00997-9
Deng, V., Matagne, V., Banine, F., Frerking, M., Ohliger, P., Budden, S., et al. (2007). FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum. Mol. Genet. 16, 640–650. doi: 10.1093/hmg/ddm007
Desai, N. S., Casimiro, T. M., Gruber, S. M., and Vanderklish, P. W. (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J. Neurophysiol. 96, 1734–1745. doi: 10.1152/jn.00221.2006
Drozd, M., Bardoni, B., and Capovilla, M. (2018). Modeling fragile X syndrome in drosophila. Front. Mol. Neurosci. 11:124. doi: 10.3389/fnmol.2018.00124
Dudek, H., Datta, S. R., Franke, T. F., Birnbaum, M. J., Yao, R., Cooper, G. M., et al. (1997). Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661–665. doi: 10.1126/science.275.5300.661
Durand, S., Patrizi, A., Quast, K. B., Hachigian, L., Pavlyuk, R., Saxena, A., et al. (2012). NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron 76, 1078–1090. doi: 10.1016/j.neuron.2012.12.004
El Bekay, R., Romero-Zerbo, Y., Decara, J., Sanchez-Salido, L., Del Arco-Herrera, I., Rodríguez-De Fonseca, F., et al. (2007). Enhanced markers of oxidative stress, altered antioxidants and NADPH-oxidase activation in brains from Fragile X mental retardation 1-deficient mice, a pathological model for Fragile X syndrome. Eur. J. Neurosci. 26, 3169–3180. doi: 10.1111/j.1460-9568.2007.05939.x
Ezeonwuka, C., and Rastegar, M. (2014). MeCP2-related diseases and animal models. Diseases 2, 45–70. doi: 10.3390/diseases2010045
Farra, N., Zhang, W. B., Pasceri, P., Eubanks, J. H., Salter, M. W., and Ellis, J. (2012). Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol. Psychiatry 17, 1261–1271. doi: 10.1038/mp.2011.180
Fernandez, A. M. M., and Torres-Alemán, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Foltran, R. B., and Diaz, S. L. (2016). BDNF isoforms: A round trip ticket between neurogenesis and serotonin? J. Neurochem. 138, 204–221. doi: 10.1111/jnc.13658
Gabel, H. W., Kinde, B. Z., Stroud, H., Gilbert, C. S., Harmin, D. A., Kastan, N. R., et al. (2015). Disruption of DNA methylation-dependent long gene repression in Rett syndrome HHS Public Access. Nature 522, 89–93. doi: 10.1038/nature14319
Gallo, F. T., Katche, C., Morici, J. F., Medina, J. H., and Weisstaub, N. V. (2018). Immediate early genes, memory and psychiatric disorders: focus on c-Fos, Egr1 and Arc. Front. Behav. Neurosci. 12:79. doi: 10.3389/fnbeh.2018.00079
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Glaze, D. G., Neul, J. L., Kaufmann, W. E., Berry-Kravis, E., Condon, S., Stoms, G., et al. (2019). Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology 92, E1912–E1925. doi: 10.1212/WNL.0000000000007316
Godfraind, J. M., Reyniers, E., De Boulle, K., D’Hooge, R., De Deyn, P. P., Bakker, C. E., et al. (1996). Long-term potentiation in the hippocampus of fragile X knockout mice. Am. J. Med. Genet. 64, 246–251.
Greenough, W. T., Klintsova, A. Y., Irwin, S. A., Galvez, R., Bates, K. E., and Weiler, I. J. (2001). Synaptic regulation of protein synthesis and the fragile X protein. Proc. Natl. Acad. Sci. U.S.A. 98, 7101–7106. doi: 10.1073/pnas.141145998
Gross, C., Berry-Kravis, E. M., and Bassell, G. J. (2012). Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacol. 37, 178–195. doi: 10.1038/npp.2011.137
Gross, C., Nakamoto, M., Yao, X., Chan, C. B., Yim, S. Y., Ye, K., et al. (2010). Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 30, 10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010
Gulmez Karaca, K., Brito, D. V. C., Zeuch, B., and Oliveira, A. M. M. (2018). Adult hippocampal MeCP2 preserves the genomic responsiveness to learning required for long-term memory formation. Neurobiol. Learn. Mem. 149, 84–97. doi: 10.1016/j.nlm.2018.02.010
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
He, C. X., and Portera-Cailliau, C. (2013). The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251, 120–128. doi: 10.1016/j.neuroscience.2012.03.049
He, L.-J. J., Liu, N., Cheng, T.-L. L., Chen, X.-J. J., Li, Y.-D. D., Shu, Y.-S. S., et al. (2014). Conditional deletion of Mecp2 in parvalbumin-expressing GABAergic cells results in the absence of critical period plasticity. Nat. Commun. 5:5036. doi: 10.1038/ncomms6036
He, Q., Arroyo, E. D., Smukowski, S. N., Xu, J., Piochon, C., Savas, J. N., et al. (2019). Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol. Psychiatry 24, 1732–1747. doi: 10.1038/s41380-018-0048-y
Hodges, J. L., Yu, X., Gilmore, A., Bennett, H., Tjia, M., Perna, J. F., et al. (2017). Astrocytic contributions to synaptic and learning abnormalities in a mouse model of Fragile X Syndrome. Biol. Psychiatry 82, 139–149. doi: 10.1016/j.biopsych.2016.08.036
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750. doi: 10.1073/pnas.122205699
Ifrim, M. F., Williams, K. R., and Bassell, G. J. (2015). Single-molecule imaging of PSD-95 mRNA translation in dendrites and its dysregulation in a mouse model of fragile X syndrome. J. Neurosci. 35, 7116–7130. doi: 10.1523/JNEUROSCI.2802-14.2015
Irwin, S. A., Galvez, R., and Greenough, W. T. (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038–1044. doi: 10.1093/cercor/10.10.1038
Ito-Ishida, A., Ure, K., Chen, H., Swann, J. W., and Zoghbi, H. Y. (2015). Loss of MeCP2 in parvalbumin-and somatostatin-expressing neurons in mice leads to distinct Rett Syndrome-like Phenotypes. Neuron 88, 651–658. doi: 10.1016/j.neuron.2015.10.029
Jacobs, S., and Doering, L. C. (2010). Astrocytes prevent abnormal neuronal development in the fragile X mouse. J. Neurosci. 30, 4508–4514. doi: 10.1523/JNEUROSCI.5027-09.2010
Jin, L. W., Horiuchi, M., Wulff, H., Liu, X. B., Cortopassi, G. A., Erickson, J. D., et al. (2015). Dysregulation of Glutamine Transporter SNAT1 in Rett Syndrome Microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J. Neurosci. 35, 2516–2529. doi: 10.1523/JNEUROSCI.2778-14.2015
Jin, S.-X. X., Higashimori, H., Schin, C., Tamashiro, A., Men, Y., Chiang, M. S. R., et al. (2021). Astroglial FMRP modulates synaptic signaling and behavior phenotypes in FXS mouse model. Glia 69, 594–608. doi: 10.1002/glia.23915
Kanellopoulos, A. K., Semelidou, O., Kotini, A. G., Anezaki, M., and Skoulakis, E. M. C. (2012). Learning and memory deficits consequent to reduction of the fragile X mental retardation protein result from metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J. Neurosci. 32, 13111–13124. doi: 10.1523/JNEUROSCI.1347-12.2012
Kang, Y., Zhou, Y., Li, Y., Han, Y., Xu, J., Niu, W., et al. (2021). A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 24, 1377–1391. doi: 10.1038/s41593-021-00913-6
Keith, D., and El-Husseini, A. (2008). Excitation control: balancing PSD-95 function at the synapse. Front. Mol. Neurosci. 1:4. doi: 10.3389/neuro.02.004.2008
Khwaja, O. S. O. S., Ho, E., Barnes, K. V. K. V., O’Leary, H. M. H. M., Pereira, L. M. L. M., Finkelstein, Y., et al. (2014). Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, 4596–4601. doi: 10.1073/pnas.1311141111
Kishi, N., and Macklis, J. D. (2004). MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 27, 306–321. doi: 10.1016/j.mcn.2004.07.006
Koekkoek, S. K. E., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J. H., Maex, R., et al. (2005). Deletion of FMR1 in purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in fragile X syndrome. Neuron 47, 339–352. doi: 10.1016/j.neuron.2005.07.005
Kowiański, P., Lietzau, G., Czuba, E., Waśkow, M., Steliga, A., and Moryś, J. (2018). BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell. Mol. Neurobiol. 38, 579–593. doi: 10.1007/s10571-017-0510-4
Kulkarni, P., and Sevilimedu, A. (2020). The known unknowns: missing pieces in in vivo models of Fragile X Syndrome. J. Rare Dis. Res. Treat. 5, 1–9. doi: 10.29245/2572-9411/2020/1.1190
Lauterborn, J. C., Rex, C. S., Kramár, E., Chen, L. Y., Pandyarajan, V., Lynch, G., et al. (2007). Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J. Neurosci. 27, 10685–10694. doi: 10.1523/JNEUROSCI.2624-07.2007
Lee, K. Y., Jewett, K. A., Chung, H. J., and Tsai, N. P. (2018). Loss of fragile X protein FMRP impairs homeostatic synaptic downscaling through tumor suppressor p53 and ubiquitin E3 ligase Nedd4-2. Hum. Mol. Genet. 27, 2805–2816. doi: 10.1093/hmg/ddy189
Levenga, J., de Vrij, F. M. S., Buijsen, R. A. M., Li, T., Nieuwenhuizen, I. M., Pop, A., et al. (2011). Subregion-specific dendritic spine abnormalities in the hippocampus of Fmr1 KO mice. Neurobiol. Learn. Mem. 95, 467–472. doi: 10.1016/j.nlm.2011.02.009
Li, W., Xu, X., and Pozzo-Miller, L. (2016). Excitatory synapses are stronger in the hippocampus of Rett syndrome mice due to altered synaptic trafficking of AMPA-type glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 113, E1575–E1584. doi: 10.1073/pnas.1517244113
Licznerski, P., Park, H. A., Rolyan, H., Chen, R., Mnatsakanyan, N., Miranda, P., et al. (2020). ATP Synthase c-subunit leak causes aberrant cellular metabolism in fragile X Syndrome. Cell 182, 1170–1185.e9. doi: 10.1016/j.cell.2020.07.008
Linda, K., Fiuza, C., and Nadif Kasri, N. (2018). The promise of induced pluripotent stem cells for neurodevelopmental disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 84, 382–391. doi: 10.1016/j.pnpbp.2017.11.009
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett-syndrome. Nature 475, 497–500. doi: 10.1038/nature10214
Lozovaya, N., Nardou, R., Tyzio, R., Chiesa, M., Pons-Bennaceur, A., Eftekhari, S., et al. (2019). Early alterations in a mouse model of Rett syndrome: the GABA developmental shift is abolished at birth. Sci. Rep. 9:9276. doi: 10.1038/s41598-019-45635-9
Martin, B. S., Corbin, J. G., and Huntsman, M. M. (2014). Deficient tonic GABAergic conductance and synaptic balance in the fragile X syndrome amygdala. J. Neurophysiol. 112, 890–902. doi: 10.1152/jn.00597.2013
Maurin, T., and Bardoni, B. (2018). Fragile X mental retardation protein: to be or not to be a translational enhancer. Front. Mol. Biosci. 5:113. doi: 10.3389/fmolb.2018.00113
Maurin, T., Lebrigand, K., Castagnola, S., Paquet, A., Jarjat, M., Popa, A., et al. (2018). HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Res. 46, 6344–6355. doi: 10.1093/nar/gky267
Maurin, T., Zongaro, S., and Bardoni, B. (2014). Fragile X Syndrome: from molecular pathology to therapy. Neurosci. Biobehav. Rev. 46, 242–255. doi: 10.1016/j.neubiorev.2014.01.006
Maxwell, S. S., Pelka, G. J., Tam, P. P., and El-Osta, A. (2013). Chromatin context and ncRNA highlight targets of MeCP2 in brain. RNA Biol. 10, 1741–1757. doi: 10.4161/rna.26921
McCarthy, D. J., Chen, Y., and Smyth, G. K. (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. doi: 10.1093/nar/gks042
Mientjes, E. J., Nieuwenhuizen, I., Kirkpatrick, L., Zu, T., Hoogeveen-Westerveld, M., Severijnen, L., et al. (2006). The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol. Dis. 21, 549–555. doi: 10.1016/j.nbd.2005.08.019
Mithal, D. S., and Chandel, N. S. (2020). Mitochondrial dysfunction in Fragile-X Syndrome: plugging the leak may save the ship. Mol. Cell 80, 381–383. doi: 10.1016/j.molcel.2020.10.002
Miyashita, T., Kikuchi, E., Horiuchi, J., and Saitoe, M. (2018). Long-Term Memory Engram Cells Are Established by c-Fos/CREB Transcriptional Cycling. Cell Rep. 25, 2716–2728. doi: 10.1016/j.celrep.2018.11.022
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., et al. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327. doi: 10.1523/JNEUROSCI.2623-05.2006
Muddashetty, R. S., Nalavadi, V. C., Gross, C., Yao, X., Xing, L., Laur, O., et al. (2011). Reversible Inhibition of PSD-95 mRNA Translation by miR-125a, FMRP Phosphorylation, and mGluR Signaling. Mol. Cell 42, 673–688. doi: 10.1016/j.molcel.2011.05.006
Na, E. S., Nelson, E. D., Adachi, M., Autry, A. E., Mahgoub, M. A., Kavalali, E. T., et al. (2012). A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 32, 3109–3117. doi: 10.1523/JNEUROSCI.6000-11.2012
Na, E. S., Nelson, E. D., Kavalali, E. T., and Monteggia, L. M. (2013). The impact of MeCP2 loss-or gain-of-function on synaptic plasticity. Neuropsychopharmacology 38, 212–219. doi: 10.1038/npp.2012.116
Nakamoto, M., Nalavadi, V., Epstein, M. P., Narayanan, U., Bassell, G. J., and Warren, S. T. (2007). Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 15537–15542. doi: 10.1073/pnas.0707484104
Nelson, E. D., Kavalali, E. T., and Monteggia, L. M. (2006). MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol. 16, 710–716. doi: 10.1016/j.cub.2006.02.062
Neul, J. L., Skinner, S. A., Annese, F., Lane, J., Heydemann, P., Jones, M., et al. (2020). Metabolic signatures differentiate Rett syndrome from unaffected siblings. Front. Integr. Neurosci. 14:7. doi: 10.3389/fnint.2020.00007
Nimchinsky, E. A., Oberlander, A. M., and Svoboda, K. (2001). Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. 21, 5139–5146. doi: 10.1523/jneurosci.21-14-05139.2001
Nosyreva, E. D., and Huber, K. M. (2006). Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J. Neurophysiol. 95, 3291–3295. doi: 10.1152/jn.01316.2005
O’Leary, H. M., Kaufmann, W. E., Barnes, K. V., Rakesh, K., Kapur, K., Tarquinio, D. C., et al. (2018). Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann. Clin. Transl. Neurol. 5, 323–332. doi: 10.1002/acn3.533
Paluszkiewicz, S. M., Olmos-Serrano, J. L., Corbin, J. G., and Huntsman, M. M. (2011). Impaired inhibitory control of cortical synchronization in fragile X syndrome. J. Neurophysiol. 106, 2264–2272. doi: 10.1152/jn.00421.2011
Paradee, W., Melikian, H. E., Rasmussen, D. L., Kenneson, A., Conn, P. J., and Warren, S. T. (1999). Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94, 185–192. doi: 10.1016/S0306-4522(99)00285-7
Patel, A. B., Hays, S. A., Bureau, I., Huber, K. M., and Gibson, J. R. (2013). A target cell-specific role for presynaptic Fmr1 in regulating glutamate release onto neocortical fast-spiking inhibitory neurons. J. Neurosci. 33, 2593–2604. doi: 10.1523/JNEUROSCI.2447-12.2013
Peteri, U. K., Pitkonen, J., de Toma, I., Nieminen, O., Utami, K. H., Strandin, T. M., et al. (2021). Urokinase plasminogen activator mediates changes in human astrocytes modeling fragile X syndrome. Glia 69, 2947–2962. doi: 10.1002/glia.24080
Pieretti, M., Zhang, F., Fu, Y. H., Warren, S. T., Oostra, B. A., Caskey, C. T., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. doi: 10.1016/0092-8674(91)90125-I
Pietri, T., Roman, A. C., Guyon, N., Romano, S. A., Washbourne, P., Moens, C. B., et al. (2013). The first mecp2-null zebrafish model shows altered motor behaviors. Front. Neural Circuits 7:118. doi: 10.3389/fncir.2013.00118
Pitzianti, M. B., Palombo, A. S., Esposito, S., and Pasini, A. (2019). Rett syndrome in males: the different clinical course in two brothers with the same microduplication MECP2 Xq28. Int. J. Environ. Res. Public Health 16:3075. doi: 10.3390/ijerph16173075
Prieto, M., Folci, A., Poupon, G., Schiavi, S., Buzzelli, V., Pronot, M., et al. (2021). Missense mutation of Fmr1 results in impaired AMPAR-mediated plasticity and socio-cognitive deficits in mice. Nat. Commun. 12:1557. doi: 10.1038/s41467-021-21820-1
Qiu, Z., Sylwestrak, E. L., Lieberman, D. N., Zhang, Y., Liu, X. Y., and Ghosh, A. (2012). The rett syndrome protein MeCP2 regulates synaptic scaling. J. Neurosci. 32, 989–994. doi: 10.1523/JNEUROSCI.0175-11.2012
Raman, A. T., Pohodich, A. E., Wan, Y. W., Yalamanchili, H. K., Lowry, W. E., Zoghbi, H. Y., et al. (2018). Apparent bias toward long gene misregulation in MeCP2 syndromes disappears after controlling for baseline variations. Nat. Commun. 9:3225. doi: 10.1038/s41467-018-05627-1
Ramocki, M. B., Peters, S. U., Tavyev, Y. J., Zhang, F., Carvalho, C. M. B., Schaaf, C. P., et al. (2009). Autism and other neuropsychiatric symptoms are prevalent in individuals with MECP2 duplication syndrome. Ann. Neurol. 66, 771–782. doi: 10.1002/ana.21715
Ramocki, M. B., Tavyev, Y. J., and Peters, S. U. (2010). The MECP2 Duplication Syndrome. Am. J. Med. Genet. Part A 152A, 1079–1088. doi: 10.1002/ajmg.a.33184
Rani, A., and Prasad, S. (2015). A Special Extract of Bacopa monnieri (CDRI-08)-Restored Memory in CoCl2-Hypoxia Mimetic Mice Is Associated with Upregulation of Fmr-1 Gene Expression in Hippocampus. Evid. Based Complement. Altern. Med. 2015:347978. doi: 10.1155/2015/347978
Reichardt, L. F. (2006). Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B Biol. Sci. 361, 1545–1564. doi: 10.1098/rstb.2006.1894
Reichow, B., George-Puskar, A., Lutz, T., Smith, I. C., and Volkmar, F. R. (2015). Brief report: systematic review of Rett Syndrome in Males. J. Autism Dev. Disord. 45, 3377–3383. doi: 10.1007/s10803-015-2519-1
Ricciardi, S., Boggio, E. M., Grosso, S., Lonetti, G., Forlani, G., Stefanelli, G., et al. (2011). Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 20, 1182–1196. doi: 10.1093/hmg/ddq563
Richter, J. D., and Zhao, X. (2021). The molecular biology of FMRP: new insights into fragile X syndrome. Nat. Rev. Neurosci. 22, 209–222. doi: 10.1038/s41583-021-00432-0
Riikonen, R. (2003). Neurotrophic factors in the pathogenesis of Rett syndrome. J. Child Neurol. 18, 693–697. doi: 10.1177/08830738030180101101
Rube, H. T., Lee, W., Hejna, M., Chen, H., Yasui, D. H., Hess, J. F., et al. (2016). Sequence features accurately predict genome-wide MeCP2 binding in vivo. Nat. Commun. 7:11025. doi: 10.1038/ncomms11025
Sawicka, K., Hale, C. R., Park, C. Y., Fak, J. J., Gresack, J. E., Van Driesche, S. J., et al. (2019). FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. eLife 8:e46919. doi: 10.7554/eLife.46919
Sceniak, M. P., Lang, M., Enomoto, A. C., James Howell, C., Hermes, D. J., Katz, D. M., et al. (2016). Mechanisms of functional hypoconnectivity in the medial prefrontal cortex of Mecp2 null mice. Cereb. Cortex 26, 1938–1956. doi: 10.1093/cercor/bhv002
Seese, R. R., Babayan, A. H., Katz, A. M., Cox, C. D., Lauterborn, J. C., Lynch, G., et al. (2012). LTP induction translocates cortactin at distant synapses in wild-type but not Fmr1 knock-out mice. J. Neurosci. 32, 7403–7413. doi: 10.1523/JNEUROSCI.0968-12.2012
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., McBride, S. M., Klann, E., et al. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702. doi: 10.1523/JNEUROSCI.3696-09.2010
Sidorov, M. S., Auerbach, B. D., and Bear, M. F. (2013). Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 6, 1–11. doi: 10.1186/1756-6606-6-15
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R. W., James, K. D., Turner, D. J., et al. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37, 457–468. doi: 10.1016/j.molcel.2010.01.030
Soden, M. E., and Chen, L. (2010). Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J. Neurosci. 30, 16910–16921. doi: 10.1523/JNEUROSCI.3660-10.2010
Sohal, V. S., and Rubenstein, J. L. R. (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257. doi: 10.1038/s41380-019-0426-0
Sohal, V. S., Zhang, F., Yizhar, O., and Deisseroth, K. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. doi: 10.1038/nature07991
Sons, M. S., Busche, N., Strenzke, N., Moser, T., Ernsberger, U., Mooren, F. C., et al. (2006). A -Neurexins are required for efficient transmitter release and synaptic homeostasis at the mouse neuromuscular junction. Neuroscience 138, 433–446. doi: 10.1016/j.neuroscience.2005.11.040
Suvrathan, A., and Chattarji, S. (2011). Fragile X syndrome and the amygdala. Curr. Opin. Neurobiol. 21, 509–515. doi: 10.1016/J.CONB.2011.04.005
Suzuki, S., Numakawa, T., Shimazu, K., Koshimizu, H., Hara, T., Hatanaka, H., et al. (2004). BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J. Cell Biol. 167, 1205–1215. doi: 10.1083/jcb.200404106
Takeguchi, R., Takahashi, S., Kuroda, M., Tanaka, R., Suzuki, N., Tomonoh, Y., et al. (2020). MeCP2_e2 partially compensates for lack of MeCP2_e1: a male case of Rett syndrome. Mol. Genet. Genomic Med. 8:e1088. doi: 10.1002/mgg3.1088
Telias, M., Mayshar, Y., Amit, A., and Ben-Yosef, D. (2015). Molecular mechanisms regulating impaired neurogenesis of Fragile X syndrome human embryonic stem cells. Stem Cells Dev. 24, 2353–2365. doi: 10.1089/scd.2015.0220
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183. doi: 10.1038/nrn1346
Thomazeau, A., Bosch, M., Essayan-Perez, S., Barnes, S. A., De Jesus-Cortes, H., and Bear, M. F. (2020). Dissociation of functional and structural plasticity of dendritic spines during NMDAR and mGluR-dependent long-term synaptic depression in wild-type and fragile X model mice. Mol. Psychiatry. doi: 10.1038/s41380-020-0821-6 [Epub ahead of print].
Tian, Y., Yang, C., Shang, S., Cai, Y., Deng, X., Zhang, J., et al. (2017). Loss of FMRP impaired hippocampal long-term plasticity and spatial learning in rats. Front. Mol. Neurosci. 10:269. doi: 10.3389/fnmol.2017.00269
Todd, P. K., Mack, K. J., and Malter, J. S. (2003). The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc. Natl. Acad. Sci. U.S.A. 100, 14374–14378. doi: 10.1073/pnas.2336265100
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., et al. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034. doi: 10.1073/pnas.0812394106
Vanhala, R., Korhonen, L., Mikelsaar, M., Lindholm, D., and Riikonen, R. (1998). Neurotrophic factors in cerebrospinal fluid and serum of patients with Rett syndrome. J. Child Neurol. 13, 429–433. doi: 10.1177/088307389801300903
Vaz, R., Hofmeister, W., and Lindstrand, A. (2019). Zebrafish models of neurodevelopmental disorders: limitations and benefits of current tools and techniques. Int. J. Mol. Sci. 20:1296. doi: 10.3390/ijms20061296
Verkerk, A. J. M. H., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P. A., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Vicario, A., Colliva, A., Ratti, A., Davidovic, L., Baj, G., and Gricman, Ł, et al. (2015). Dendritic targeting of short and long 3′ UTR BDNF mRNA is regulated by BDNF or NT-3 and distinct sets of RNA-binding proteins. Front. Mol. Neurosci. 8:62. doi: 10.3389/fnmol.2015.00062
Wahlstrom-Helgren, S., and Klyachko, V. A. (2015). GABAB receptor-mediated feed-forward circuit dysfunction in the mouse model of fragile X syndrome. J. Physiol. 593, 5009–5024. doi: 10.1113/JP271190
Wang, G., Gilbert, J., and Man, H. Y. (2012). AMPA receptor trafficking in homeostatic synaptic plasticity: functional molecules and signaling cascades. Neural Plast. 2012:825364. doi: 10.1155/2012/825364
Wang, H., Xu, H., Wu, L. J., Kim, S. S., Chen, T., Koga, K., et al. (2011). Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci. Transl. Med. 3:65ra3. doi: 10.1126/scitranslmed.3001269
Wang, L., Chang, X., She, L., Xu, D., Huang, W., and Poo, M. M. (2015). Autocrine action of BDNF on dendrite development of adult-born hippocampal neurons. J. Neurosci. 35, 8384–8393. doi: 10.1523/JNEUROSCI.4682-14.2015
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K., et al. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400. doi: 10.1073/pnas.94.10.5395
Wijetunge, L. S., Angibaud, J., Frick, A., Kind, P. C., and Nägerl, U. V. (2014). Stimulated emission depletion (STED) microscopy reveals nanoscale defects in the developmental trajectory of dendritic spine morphogenesis in a mouse model of fragile X syndrome. J. Neurosci. 34, 6405–6412. doi: 10.1523/JNEUROSCI.5302-13.2014
Wise, T. L. (2017). Changes in insulin-like growth factor signaling alter phenotypes in Fragile X Mice. Genes Brain Behav. 16, 241–249. doi: 10.1111/gbb.12340
Wolters, T. L., Netea, M. G., Hermus, A. R. M., Smit, J. W. A., and Netea-Maier, R. T. (2017). IGF1 potentiates the pro-inflammatory response in human peripheral blood mononuclear cells via MAPK. J. Mol. Endocrinol. 59, 129–139. doi: 10.1530/JME-17-0062
Wondolowski, J., and Dickman, D. (2013). Emerging links between homeostatic synaptic plasticity and neurological disease. Front. Cell. Neurosci. 7:223. doi: 10.3389/FNCEL.2013.00223
Wood, L., and Shepherd, G. M. G. (2010). Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in a mutant mouse model of Rett syndrome. Neurobiol. Dis. 38, 281–287. doi: 10.1016/j.nbd.2010.01.018
Wu, H., Tao, J., Chen, P. J., Shahab, A., Ge, W., Hart, R. P., et al. (2010). Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 18161–18166. doi: 10.1073/pnas.1005595107
Xing, L., and Bassell, G. J. (2013). mRNA localization: an orchestration of assembly, traffic and synthesis. Traffic 14, 2–14. doi: 10.1111/tra.12004
Xu, X., Miller, E. C., and Pozzo-Miller, L. (2014). Dendritic spine dysgenesis in Rett syndrome. Front. Neuroanat. 8:97. doi: 10.3389/fnana.2014.00097
Xu, X., and Pozzo-Miller, L. (2017). EEA1 restores homeostatic synaptic plasticity in hippocampal neurons from Rett syndrome mice. J. Physiol. 595, 5699–5712. doi: 10.1113/JP274450
Yang, Y., Geng, Y., Jiang, D., Ning, L., Kim, H. J., Jeon, N. L., et al. (2019). Kinase pathway inhibition restores PSD95 induction in neurons lacking fragile X mental retardation protein. Proc. Natl. Acad. Sci. U.S.A. 116, 12007–12012. doi: 10.1073/pnas.1812056116
Yasui, D. H., Xu, H., Dunaway, K. W., LaSalle, J. M., Jin, L.-W., and Maezawa, I. (2013). MeCP2 modulates gene expression pathways in astrocytes. Mol. Autism 4, 1–11. doi: 10.1186/2040-2392-4-3
Yuan, Z.-F. Z. F., Mao, S. S. S.-S., Shen, J., Jiang, L. H. L.-H., Xu, L., Xu, J.-L. J. L., et al. (2020). Insulin-Like growth factor-1 down-regulates the phosphorylation of FXYD1 and rescues behavioral deficits in a mouse model of Rett syndrome. Front. Neurosci. 14:20. doi: 10.3389/fnins.2020.00020
Zalfa, F., Eleuteri, B., Dickson, K. S., Mercaldo, V., De Rubeis, S., Di Penta, A., et al. (2007). A new function for the Fragile X Mental Retardation Protein in the regulation of PSD-95 mRNA stability Europe PMC Funders Group. Nat. Neurosci. 10, 578–587. doi: 10.1038/nn1893
Zang, J. B., Nosyreva, E. D., Spencer, C. M., Volk, L. J., Musunuru, K., Zhong, R., et al. (2009). A mouse model of the human fragile X syndrome I304N mutation. PLoS Genet. 5:e1000758. doi: 10.1371/journal.pgen.1000758
Zhang, F., Kang, Y., Wang, M., Li, Y., Xu, T., Yang, W., et al. (2018). Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet. 27, 3936–3950. doi: 10.1093/hmg/ddy292
Zhao, J., Harada, N., Kurihara, H., Nakagata, N., and Okajima, K. (2010). Cilostazol improves cognitive function in mice by increasing the production of insulin-like growth factor-I in the hippocampus. Neuropharmacology 58, 774–783. doi: 10.1016/j.neuropharm.2009.12.008
Zheng, W. H., and Quirion, R. (2006). Insulin-like growth factor-1 (IGF-1) induces the activation/phosphorylation of Akt kinase and cAMP response element-binding protein (CREB) by activating different signaling pathways in PC12 cells. BMC Neurosci. 7:51. doi: 10.1186/1471-2202-7-51
Zhong, X., Li, H., and Chang, Q. (2012). MeCP2 phosphorylation is required for modulating synaptic scaling through mGluR5. J. Neurosci. 32, 12841–12847. doi: 10.1523/JNEUROSCI.2784-12.2012
Keywords: Rett syndrome, Fragile X syndrome, synaptic plasticity, FMRP, MeCP2, neurodevelopmental disorders
Citation: Bach S, Shovlin S, Moriarty M, Bardoni B and Tropea D (2021) Rett Syndrome and Fragile X Syndrome: Different Etiology With Common Molecular Dysfunctions. Front. Cell. Neurosci. 15:764761. doi: 10.3389/fncel.2021.764761
Received: 25 August 2021; Accepted: 27 October 2021;
Published: 19 November 2021.
Edited by:
Shuxin Li, Temple University, United StatesReviewed by:
Michael Telias, University of California, Berkeley, United StatesChristina Gross, Cincinnati Children’s Hospital Medical Center, United States
Copyright © 2021 Bach, Shovlin, Moriarty, Bardoni and Tropea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Tropea, tropead@tcd.ie
†These authors have contributed equally to this work