Sphingolipid metabolism and signaling in cardiovascular diseases
 Sonia Borodzicz-Jażdżyk1
Sonia Borodzicz-Jażdżyk1  Piotr Jażdżyk1,2 Wojciech Łysik1 Agnieszka Cudnoch-Jȩdrzejewska1
Piotr Jażdżyk1,2 Wojciech Łysik1 Agnieszka Cudnoch-Jȩdrzejewska1  Katarzyna Czarzasta1*
Katarzyna Czarzasta1*- 1Chair and Department of Experimental and Clinical Physiology, Laboratory of Centre for Preclinical Research, Medical University of Warsaw, Warsaw, Poland
- 2Second Department of Psychiatry, Institute of Psychiatry and Neurology in Warsaw, Warsaw, Poland
Sphingolipids are a structural component of the cell membrane, derived from sphingosine, an amino alcohol. Its sphingoid base undergoes various types of enzymatic transformations that lead to the formation of biologically active compounds, which play a crucial role in the essential pathways of cellular signaling, proliferation, maturation, and death. The constantly growing number of experimental and clinical studies emphasizes the pivotal role of sphingolipids in the pathophysiology of cardiovascular diseases, including, in particular, ischemic heart disease, hypertension, heart failure, and stroke. It has also been proven that altering the sphingolipid metabolism has cardioprotective properties in cardiac pathologies, including myocardial infarction. Recent studies suggest that selected sphingolipids may serve as valuable biomarkers useful in the prognosis of cardiovascular disorders in clinical practice. This review aims to provide an overview of the current knowledge of sphingolipid metabolism and signaling in cardiovascular diseases.
Introduction
Sphingolipids are a structural component of the cell membrane, including cardiomyocytes, neurones, and microglia among others (1, 2). Sphingolipids are derived from sphingosine, which owes its name to its unusual structure, placing it between the amines and alcohols (3). The sphingoid base of sphingosine undergoes various types of enzymatic transformations and this leads to the formation of biologically active compounds, which play a crucial role in the essential pathways of cellular signaling, proliferation, maturation, and death (4). The most important representatives of sphingolipids are ceramide (Cer) and sphingosine (Sph), Sph-1-phosphate (S1P), and Cer-1-phosphate (C1P) (5). It should be noted that Cer can be formed by de novo synthesis, but it can also be formed as a result of the catabolism of complex sphingolipids. Sphingolipids are degraded in lysosomes by the endosomal/lysosomal membrane digestion system (6). Disorders of individual enzymes from the above system lead to the accumulation of the substrate and its distribution to all cell membranes interacting with lysosomes, and these are then called lysosomal storage disorders (LSD) (7). Due to the multidirectional effects of sphingolipids, disruption of their metabolism may be one of the risk factors for the development of diseases, including cardiovascular diseases (1). The aim of this review is to provide an overview of the current knowledge of sphingolipid metabolism and signaling in cardiovascular diseases such as hypertension, coronary artery disease, heart failure, arrhythmias, and stroke.
Sphingolipid metabolism
Ceramide, a representative of biologically active sphingolipids, plays a fundamental role in the sphingolipid metabolic pathway (5). De novo synthesis is one of the three metabolic pathways leading to Cer synthesis (8). This process begins in the endoplasmic reticulum by the action of serine palmitoyltransferase (SPT) in the presence of the substrates: serine and palmitoyl-CoA or also possibly alanine or glycine and stearate or myristate, to produce 3-ketodihydrosphingosine (8). Subsequent reactions of the reduction by 3-ketodihydrosphingosine reductase, and N-acylation by a (dihydro)-ceramide synthase, and desaturation, generate Cer (9).
Apart from de novo biosynthesis, Cer can be generated in the cell through the hydrolytic pathway (5). Sphingomyelin is hydrolyzed by sphingomyelinase into phosphocholine and Cer (5).
And finally, ceramide may be produced from sphingosine via sphinganine N-acyltransferase (ceramide synthase) in the salvage pathway (1). In reverse, Cer may be hydrolyzed by ceramidase enzymes resulting in the product, sphingosine, which can be either phosphorylated by sphingosine kinases (SPHK1 and SPHK2) generating S1P or utilized in the salvage pathway (10). In addition, S1P lyase (SPL) is the enzyme involved in irreversible degradation of S1P into ethanolamine phosphate and hexadecenal (11).
Cer, generated in the endoplasmic reticulum, is then transported to the Golgi apparatus. Upon Cer phosphorylation by Cer kinase, C1P is formed, which in turn can be decomposed by C1P phosphatase or glycosylated by glucosylCer or galactosylCer synthases (10, 12). O-acylation of Cer provides a structure similar to a triacylglycerol (TAG), is stored in lipid droplets (13). In the Golgi apparatus, Cer is also metabolized to sphingolipids of higher complexity, such as glycosphingolipids (GSL) and sphingomyelins (SM) (14). GSL and SM are there after shuttled to the plasma membrane, where they are used as structural components (14). The degradation of GSL, catalyzed by subsequent reactions of specific hydrolases, yields glucosylceramide and galactosylceramide, which are in turn hydrolyzed by distinct β-glucosidases and β-galactosidases to produce Cer (15).
Figure 1 shows schematically the metabolism of sphingolipids, due to the complexity of the processes, only the main molecules and enzymes are shown.
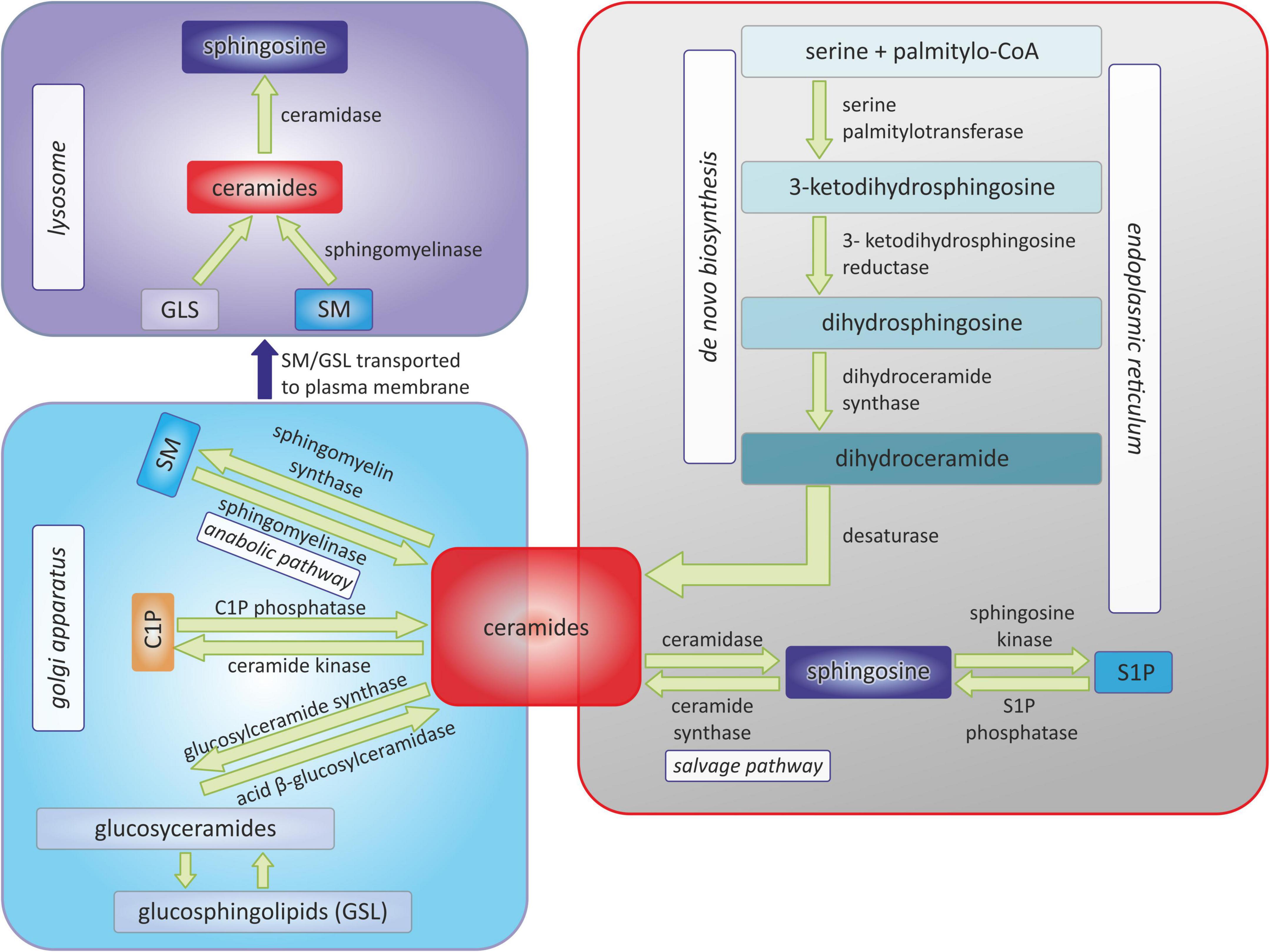
Figure 1. Pathways of sphingolipids metabolism. C1P, ceramide-1-phosphate; SM, sphingomyelin; S1P, sphingosine-1-phosphate.
Sphingolipids in the regulation of the cellular function
Sphingolipids play a major role in the regulation of the cellular function and influence almost all of the primary aspects of cell biology, including growth, the cell cycle, cell death, cell senescence, apoptosis, inflammation, the immune response, cell adhesion and migration, angiogenesis, nutrient uptake, metabolism, the response to stress stimuli, and autophagy (8).
Due to the fact that the article deals with the metabolism and signaling of sphingolipids in cardiovascular diseases, and most of the chapters are devoted to their involvement in heart diseases, this chapter will also discuss the role of sphingolipids in the regulation of cellular functions on the example of the heart muscle.
It has been demonstrated that the sphingolipid family is widely distributed throughout the heart muscle (1). In addition, sphingolipids perform many important physiological functions in the heart, among others, Cer and S1P were shown to directly regulate cardiomyocyte contractility. S1P has been proven to exert a negative inotropic effect on isolated mouse cardiomyocytes in the way of two intracellular pathways. First, works via Gi mediated which reduce L-type calcium channel current, blunt calcium-induced calcium release, and a decrease in the intracellular Ca2+ concentration. Second, acts via Gi decrease in activation of inwardly rectifying K+ current, consequently shortens the action potential duration (APD). A shorter APD leads to reduced calcium influx, decreased intracellular Ca2+ concentration, and therefore exerts negative inotropic effect (16). In turn, the addition of a cell-permeable Cer analog (C2-ceramide) to isolated rat cardiomyocytes has been shown to exert a positive inotropic effect due to enhanced sarcoplasmic reticulum calcium release and reabsorption as well as increased calcium sensitivity of the cardiomyocytes (17, 18). Moreover, treatment of isolated rat cardiomyocytes with C6-ceramide, which mimics endogenous ceramide generation and is considered to be more relevant to physiological conditions, decreased the peak amplitude of cell shortening without any influence on the peak Ca2+ transient. The observed negative inotropic response was mediated by the activation of the protein kinase C isoform ε, which is calcium-independent, and by the subsequent phosphorylation of cardiac troponin I (TnI) and cardiac myosin binding protein-C (cMyBP-C) (19). Based on the above data, it can be concluded that the influence of S1P and Cer on the force of heart contraction is opposite. While S1P weakens the force of muscle contraction (negative inotropic effect), Cer increases it (positive ionotropic effect). These data appear to be of particular interest for cardiovascular pharmacotherapy.
Sphingolipids are also involved in the pathways of myocardial fibrosis, which is one of the cornerstones of myocardial dysfunction in heart failure (HF). Pre-treatment of primary ventricular rat neonatal cardiac fibroblasts with dihydrosphingosine reduced transforming growth factor β (TGF-β)-induced collagen synthesis and increased the de novo synthesis pathway of dihydrosphingosine-1-phosphate, which on the other hand reduced the expression of the sphingosine-1-phosphate receptor 1 (S1PR1) (20). Administration of S1P to isolated cardiac fibroblasts resulted in increased α-smooth muscle actin (a marker of the transformation into collagen-producing myofibroblasts) and collagen expression in a sphingosine-1-phosphate receptor 2 (S1PR2)- and Rho kinase-dependent mechanism. Moreover, the authors have proven that TGF-β increases the expression and activity of SPHK1, whereas the blockage of SPHK1 and S1PR2 and the administration of anti-S1P monoclonal antibody completely blocks TGF-β-stimulated collagen production (21). Transgenic mice with overexpression of SPHK1 developed interstitial and perivascular myocardial fibrosis as well as sporadic calcification within the fibrotic areas, which was not observed in wild type mice. Transgenic mice also showed increased expression levels of collagen type I α1 and α2 proteins when compared with wild type animals. Sphingosine-1-phosphate receptor 3 (S1PR3) deletion partially inhibited cardiac fibrosis in transgenic mice with overexpression of SPHK1, therefore the authors concluded that SPHK1-dependent myocardial fibrosis involved the S1PR3-Rho family small G protein signaling pathway, and the release of reactive oxygen species and the activation of TGF-β (22). Moreover, S1PR1 transgenic mice showed hyperplastic fibroblasts and/or myofibroblasts, ventricular hypertrophy, and diffuse interstitial fibrosis without any hemodynamic stress. The above studies indicate that S1P promote myocardial fibrosis due to an activating effect on cardiac fibroblasts and myofibroblasts, which synthesize and release collagen into the extracellular matrix. This leads directly to interstitial fibrosis, cardiac hypertrophy and stiffness and my consequently cause HF.
Sphingolipids are also thought to mediate cardiomyocyte hypertrophy, although the results are ambiguous. Robert et al. reported that incubation of rat neonatal cardiomyocytes with S1P results in a concentration-dependent cardiomyocyte hypertrophy mediated by S1PR1 (23). On the other hand, Sekiguchi et al. revealed that S1P does not induce hypertrophy of neonatal rat cardiomyocytes, which was induced by sphingosylphosphorylcholine (SPC), however, SPC is not an agonist of S1P receptor, therefore the exact mechanism responsible for this observation is not yet fully understood (24).
Sphingolipids in cardiovascular diseases
Hypertension
According to the ESC/ESH Guidelines for the management of arterial hypertension from 2018, hypertension is defined as systolic blood pressure (SBP) ≥140 mmHg and/or diastolic blood pressure (DBP) ≥90 mmHg measured in a doctor’s office. Hypertension is a significant clinical problem. It is predicted that by 2025 the number of people with hypertension will increase to 1.5 billion (25).
The available data show the pivotal role of sphingolipids in the regulation of vascular tone and endothelial cells dysfunction (26–28). Therefore, it can be hypothesized that sphingolipids represent a new pathophysiological mechanism involved in the development of hypertension.
The above hypothesis seems to be confirmed by in vitro studies. In isolated bovine small coronary arteries and in human endothelial cells, Cer induced the production of reactive oxygen species (ROS) causing endothelial cell dysfunction and reducing bioactive nitric oxide (NO) (29, 30). In addition, in isolated carotid arteries from spontaneously hypertensive (SHR) rats shifting the Cer/S1P ratio toward ceramide dominance by the administration a sphingosine kinase inhibitor (dimethylsphingosine), and neutral sphingomyelinase contributed to endothelial-dependent vasoconstriction in which phospholipase A2, cyclooxygenase-1, and thromboxane synthase which was not observed in normotensive Wistar Kyoto rats (31). Additionally, it has been shown that Cer induced vasoconstriction in isolated canine cerebral arterial smooth muscle by increasing intracellular calcium concentration (32).
Moreover, in vivo studies seem to support the role of sphingolipids in the pathophysiology of hypertension. Namely, SHR rats were shown to have increased total ceramide (mainly C16:0, C22:0, C24:1, and C24:0) and sphingosine plasma concentrations (31). Based on the available data, it can be assumed that the plasma concentration of ceramides may be a potential marker of the early diagnosis of hypertension. It has been shown that endothelial deletion of SPTLC2 (long chain subunit 2 of serine palmitoyltransferase–the first enzyme of the de novo pathway) in mice revealed an important role of endothelial ceramides de novo biosynthesis in vascular and blood pressure homeostasis, and demonstrated that endothelium was a key source of plasma ceramides (33). In turn, Siedlinski et al. revealed that angiotensin II (AngII)-induced hypertension is associated with increased plasma concentrations of S1P in wild type (C57BL/6J) mice and chronic infusion of S1P caused a moderate, increase in systolic blood pressure, endothelial dysfunction, and vascular contractility in the mesenteric arteries (34). Meissner et al. showed that AngII increased the blood pressure in wild type mice, whereas in SPHK1 knockout (SPHK1–/–) mice the blood pressure response to AngII was significantly lower or even absent in the SPHK2 knockout (SPHK2–/–) mice (35). Similarly, Siedlinski et al. showed in SPHK1–/–mice that AngII infusion caused less advanced hypertension in comparison with wild type mice (34). Moreover, administration of SPHK2 antagonists, but not the SPHK1 antagonist, significantly reduced blood pressure in wild type mice injected with AngII (35). It was also shown that mice lacking Nogo-B (an endoplasmic reticulum membrane protein which inhibits serine palmitoyltransferase, present with hypotension) are resistant to AngII-induced hypertension and have preserved endothelial function and NO release (36).
While most of the data support a significant role of ceramide in the generation of hypertension, information regarding S1P in the pathogenesis of hypertension is inconclusive. Exogenous S1P appears to act as an activator of endothelial nitric oxide synthase (eNOS) through its receptors S1PR1 and S1PR3 (37, 38). S1P is also secreted from the endothelial cells and regulates flow-mediated vasodilation through the S1P-S1PR1 autocrine signaling axis (39). However, in greater concentrations, exogenous S1P was shown to induce vasoconstriction in resistance arteries mediated predominantly by S1PR3 and S1PR2 receptors (40, 41). Nevertheless, it has been suggested that activation of eNOS through S1PR1 and S1PR3 neutralizes S1PR2 and S1PR3-mediated vasoconstriction in vascular smooth muscle cells (26). In addition, Hu et al. (42) revealed that mice with S1PR1 knockout specifically in renal collecting ducts have reduced urinary sodium excretion, impaired pressure natriuresis, and developed more severe hypertension after 10-day deoxycorticosterone acetate (DOCA) salt treatment in comparison with wild type mice. Therefore, the authors concluded that collecting duct-S1PR1 signaling can play a significant role as an antihypertensive pathway by the induction of sodium excretion and they suggested that the dysfunction of the renal medullary S1PR1 could be a novel mechanism for salt-sensitive hypertension (42). It has also been shown that mice lacking endothelial S1PR1 had higher systolic blood pressure than the controls, had reduced S1P- and flow-mediated vasodilation, had reduced basal and acetylcholine (Ach)-induced nitric oxide production in the mesenteric arteries, and had lower plasma levels of nitrite. Therefore, the S1P-S1PR1-NO axis played an important role in the flow-mediated regulation of vascular tone and therefore the regulation of blood pressure (43).
Clinical studies seem to confirm the possibility of using sphingolipids as markers of arterial hypertension. Hypertensive patients also had significantly higher total plasma levels of Cer (mainly C24:1 and C24:0) and S1P than normotensive controls (31). Jujic et al. analyzed the family based cohort of Malmö Offspring Study and revealed that patients who had systolic blood pressure ≥140 mmHg had significantly higher plasma concentrations of S1P in comparison with patients with systolic blood pressure below 120 mmHg (44). Moreover, it has been shown that plasma concentrations of S1P were significantly correlated with impaired endothelial function assessed by the Reactive Hyperemia index and a higher aortic systolic pressure (34). In addition, Zheng et al. showed that plasma SM concentrations in patients with hypertension but without a physiological nocturnal reduction of ≥10% of blood pressure (non-dippers) was significantly higher in comparison with the dipper group and plasma SM levels were negatively correlated with the degree of systolic and diastolic blood pressure decrease at night (45).
Interestingly, both experimental and clinical studies have shown that hypertensive treatment may interact with sphingolipid metabolism. Administration of fingolimod (FTY720), which is a non-selective S1PR1, 3, 4, 5 agonist, increased the blood pressure in normotensive C57BL/6 mice and exacerbates hypertension in AngII mouse model, underlining the antihypertensive function of S1PR1 signaling (35, 43). Acute administration two differentially selective S1P agonists, FTY720 and BAF312 (more selective S1P1,5 agonist) to Sprague Dawley rats resulted in S1P1-mediated bradycardia. However, administration of FTY720, but not BAF312, resulted in caused dose-dependent hypertension mediated by S1PR3 (46). In addition, oral administration of FTY720 in SHR rats, but not in Wistar Kyoto rats, increased the mean arterial pressure and induced large contractions in isolated carotid arteries, most probably due to the inhibition of sphingosine kinase (47). Spijkers et al. (48) have shown that treatment of SHR rats with losartan (Ang II type 1 receptor antagonist) or hydralazine (the non-selective vasodilator) not only reduced blood pressure, but also decreased vascular Cer concentration and improved endothelial-dependent vasorelaxation of isolated carotid arteries. Moreover, only losartan reduced sphingomyelinase-induced contractions of isolated carotid arteries, which may result from the reduction in the endothelial expression of calcium-independent phospholipase A2 (48). Furthermore, S1P has been associated with a higher baseline plasma renin activity and a reduced blood pressure response to hydrochlorothiazide (thiazide diuretic) in hypertensive patients (49).
Coronary artery disease
Atherosclerosis and chronic coronary syndromes
Atherosclerosis is a pathophysiological mechanism underlying coronary artery disease (CAD). Atherosclerotic plaques are formed within the regions of disrupted endothelium and/or the arterial wall. Within the bloodstream, lipids attach to water-soluble lipoproteins named apolipoproteins. Lipoproteins containing apo-B, which are the major protein component of very low-density lipoprotein (VLDL), intermediate density lipoprotein (IDL), and low-density lipoprotein (LDL), pass through the disrupted endothelium and are endocytosed by macrophages within the intimal layer of the arteries. Therefore, lipoproteins are oxidized, which induces leukocyte accumulation and the formation of foam cells. It has been shown that LDL aggregation promotes the accumulation of cholesteryl ester-rich lipid droplets in the human primary monocyte-derived macrophages, induces matrix metalloproteinase-7 secretion from macrophage foam cells, and activates T-cells in vitro (50). LDL aggregation susceptibility depends on the particle lipid composition, including higher concentrations of sphingomyelins and ceramides and may be decreased by diet or proprotein convertase subtilisin/kexin 9 (PCSK9) inhibition (50). Moreover, blood flow disturbance within atherosclerosis-susceptible regions promotes the rearrangement of the microenvironment of the arterial wall intima, with vascular smooth muscle cell migration, extracellular matrix remodeling, plaque growth, fibrous cap formation, and necrotic core formation with calcifications (51, 52). Interestingly, LDL aggregation susceptibility prognosticates cardiovascular death in patients with CAD, therefore it has been concluded that intimal LDL aggregation facilitates atherogenesis, inflammation, and plaque rupture by inducing foam cell formation and the secretion of matrix metalloproteinase-7, and by activating T-cells, which increases the risk of death (50). Therefore, the reduction of sphingolipid content in aggregation-prone LDL with diet or treatment with a PCSK9 inhibitor could be beneficial in reducing the risk of CAD (50). Another important predisposing factor for the development of atherosclerosis is the deposition of the mineral hydroxyapatite in the extracellular matrix and in the arterial smooth muscle cells, leading to arterial calcification, which in turn is associated with plaque rupture, and an increased risk of heart disease and stroke (53). The calcification process can affect both the inner (arterial intimal calcification; AIC) and the middle (arterial medial calcification; AMC) walls of the arteries. AIC is associated with the obstruction of the arteries and the rupture of the atherosclerotic plaque (54), whereas, AMC predisposes to vessel stiffness and the development of systolic hypertension and increased pulse wave velocity, consequently leading to diastolic dysfunction and heart failure (55). Yuan et al. (56) showed that endothelium-specific AC gene knockout mic (Asah1fl/fl/ECcre) with streptozotocin-induced diabetes significantly augmented the production and activation of NOD-like receptor pyrin domain 3 (NLRP3) inflammasomes in the coronary artery endothelial cells, as well as significantly higher expression of exosome markers auch as CD63 and alkaline phosphatase (ALP) in the coronary arterial wall and interleukin 1 beta (IL-1β) release of exosomes in comparison with wild-type (WT/WT) littermates. Moreover, Asah1fl/fl/ECcre mice trated with streptozotocin revealed significant thickening of the coronary arterial wall and lower expression of tight junction protein compared to WT/WT littermates (56). Additionally, it has been shown that smooth muscle-specific acid ceramidase (Ac) gene knockout mice (Asah1fl/fl/SMCre) during hypercalcemia caused by high doses of vitamin D had more severe AMC in both the aorta and the coronary arteries as well as upregulation of osteopontin and RUNX2 (osteogenic markers) in the aortic and coronary arterial medial wall, and CD63, AnX2 (sEV markers) and ALP expression (mineralization marker) in the coronary arterial media in comparison with Asah1fl/fl/SMwt and WT/WT mice (57). In addition, the same researchers showed that high doses of vitamin D-induced hypercalcemia in Smpd1trg/SMcre mice with overexpressing the acid lysosomal sphingomyelinase (SMPD1) gene specific for aortic and coronary smooth muscle cells contributed to higher aortic and coronary AMC as well as increased expression of RUNX2 and osteopontin in the coronary and aortic media (58).
On the basis of the available studies, it can be hypothesized that sphingolipids play an important role in endothelial dysfunction and thus may promote the atherosclerotic processes.
S1P appears to have a twofold effect on the development of atherosclerosis depending on the receptors through which it acts (59) (Figure 2). The interaction of S1P with S1PR1 may have antiatherosclerotic effects by inhibiting macrophage apoptosis and endothelial inflammation via intracellular pathway dependent on the phosphatidylinositol 3-kinase (PI3K) and protein kinase B (PI3K/Akt) (60). On the other hand, S1PR1 may have a positive effect on the development of atherosclerosis by activating pro-inflammatory endothelial factors such as: intercellular adhesion molecule (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), as well as by facilitating the formation of neointima formation by increasing the level of interleukin 6 (IL-6), which stimulates the proliferation and migration of vascular smooth muscle cells (VSMCs) (61, 62). S1P interacting with S1PR2 may also have a pro-atherosclerotic effect by promoting the inflammatory response of endothelial cells due to the increased expression of pro-inflammatory factors such as tumor necrosis factor α (TNF-α), interleukin 1 (IL-1), stimulating endothelial dysfunction (63). However, the interaction of S1P with S1PR2 may also have antiatherosclerotic effects by inhibiting the proliferation and migration of smooth muscle cells via the Rac protein and delaying the neointima formation (64, 65). Skoura et al. (66) have shown that ApoE and S1PR2-deficient Apoe-/-S1pr2-/-mice show reduced development of western diet-induced atherosclerosis, present decreased fibrous tissue, collagen and lipid content, as well as decreased necrotic core within the atherosclerotic plaque and a reduced number of macrophages within the vessel wall in comparison with the Apoe-/-S1pr2 + / + controls. Therefore, the authors suggested that S1PR2 signaling regulates macrophage infiltration and inflammatory cytokine secretion, thereby promoting atherosclerosis (66). On the other hand, Guanbataar et al. showed that ApoE-deficient mice receiving a S1PR2 antagonist and fed on a western-type diet resulted in the reduction of atherosclerotic plaque development, and alleviated endothelial dysfunction as well as resulting in significantly decreased lipid deposition, macrophage accumulation, and the expression of inflammatory proteins within the aorta (67). Similarly, S1P by activating S1PR3 can also promote the development of atherosclerosis, as does S1PR1 by activating endothelial ICAM-1 and VCAM-1 and causing its dysfunction (68). But S1P/S1PR3 signaling may also have antiatherosclerotic effects by inhibiting neointima formation (69). This same authors shown that S1PR3 genetic deficiency significantly increased neointima formation in the mouse carotid artery ligation model (69).
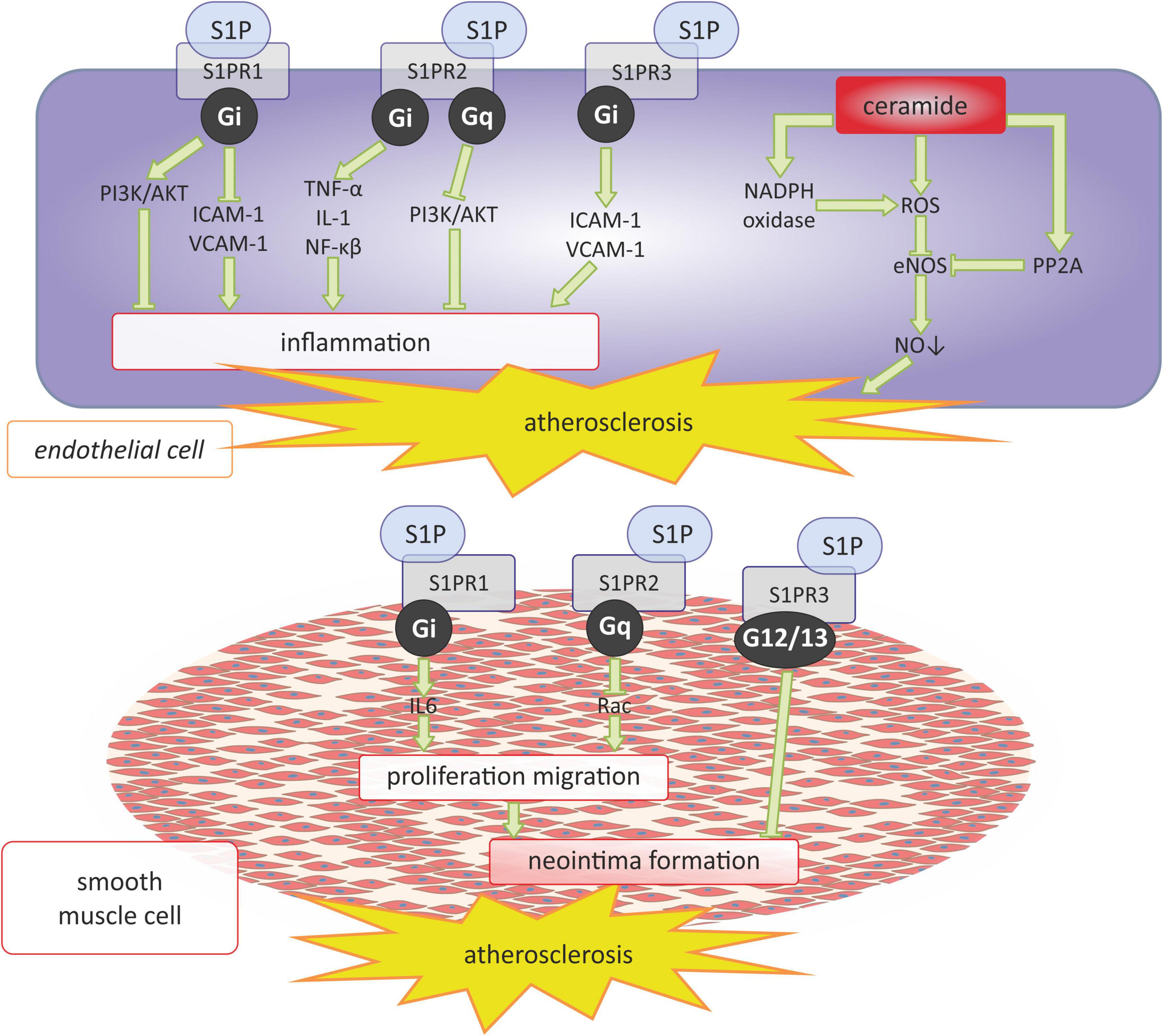
Figure 2. Sphingosine 1-phosphate and ceramide signaling in atherosclerosis. AKT, serine/threonine protein kinase B; eNOS, endothelial nitric oxide synthase; Gi, Gi protein alpha subunit; Gq, Gq protein alpha subunit; ICAM-1 -intercellular adhesion molecule 1; IL-1, interleukin 1; IL-6, interleukin 6; NADPH oxidase, nicotinamide adenine dinucleotide phosphate oxidase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NO, nitric oxide; PI3K, phosphoinositide 3-kinase; PP2, protein phosphatase 2, Rac, family small GTPase; ROS, reactive oxygen species; S1P, sphingosine-1-phosphate; S1PR1, sphingosine-1-phosphate receptor 1; S1PR2, sphingosine-1-phosphate receptor 2; S1PR3, sphingosine-1-phosphate receptor 3; TNF-α, tumor necrosis factor α; VCAM-1, vascular cell adhesion molecule 1.
Interestingly, the available data indicate that not only the interaction of S1P with its receptors, but also the enzymes leading to S1P synthesis, may play an important role in the pathogenesis of atherosclerosis. Ishimaru et al. revealed that SPHK2, but not SPHK1-deficient mice, developed larger atherosclerotic plaques in comparison with the control mice, with no influence on the plasma lipid profile (70). Moreover, the authors found that SPHK2 is responsible for autophagosome- and lysosome-mediated catabolism of the intracellular lipids, therefore SPHK2 has been suggested to be a novel target for the treatment of atherosclerosis (70). The above observations seem to be corroborated by Feuerborn et al. who showed that LDL receptor-deficient mice transplanted with bone marrow lacking SPKH2 have an increased concentration of S1P in the erythrocytes, plasma, and high-density lipoprotein (HDL), and a reduced development of atherosclerotic plaques with smaller necrotic cores within lesions (71). Potì et al. revealed that, in LDL receptor-deficient mice on a western high-cholesterol diet, lowering plasma S1P levels by treatment with SKI-II, a SPHK1 inhibitor, increased the development of atherosclerotic lesions, and increased the levels of TNF-α and the expression of endothelial adhesion molecules (soluble intercellular adhesion molecule-1, sICAM-1; soluble vascular cell adhesion molecule-1, sVCAM-1) (72).
Similar to the enzymes that influence S1P synthesis, sphingomyelinases, which play a role in the anabolic pathways leading to the formation of ceramides from sphingomyelin, appear to play an important role in the pathogenesis of atherosclerosis. For example, it was shown that neutral as well as acid sphingomyelinase, which are expressed in the intima of atherosclerotic arteries, promotes foam cell formation by the production of Cer in lipoproteins and the enhancement of LDL uptake and has the ability to induce cholesteryl ester accumulation in macrophages (73–75). Moreover, sphingomyelinase induces aggregation and fusion of LDL, which can be a potential mechanism responsible for the focal retention of extracellular lipids in the arterial wall (76, 77). Moreover, ceramides themselves exert a pro-atherosclerotic effect by inducing endothelial dysfunction. It has been shown that ceramides in endothelial cells generate the ROS production and activating protein phosphatase 2A (PP2A), and consequently reduce the activity of eNOS, thus impairing the production and bioavailability of NO (28) (Figure 2).
The activity of sphingomyelin synthases also plays an important role in the pathogenesis of atherosclerosis. In ApoE-deficient mice, the administration of an adenovirus containing sphingomyelin synthase 2 resulted in the increased development of atherosclerosis, and its overexpression is associated with an increased aortic inflammatory response, contributing to endothelial dysfunction and plaque instability (78, 79). In addition, the overexpression of sphingomyelin synthases 1 and 2 significantly decreased (high-density lipoprotein) HDL-sphingomyelin and HDL-cholesterol, and increased non-HDL-sphingomyelin, non-HDL-cholesterol, and apolipoprotein B concentrations, thus exerting a proatherogenic effect (80).
Moreover, the latest research also indicates the important role of sphingolipids in the process of arterial calcification, which is one of the main mechanisms leading to the development of atherosclerosis (53) (Figure 3). The research of Koka et al. conducted both in the primary cultures of the mouse carotid arterial endothelial cells as well as in the mouse model of hypercholesterolemia showed that acid sphingomyelinase and ceramidases related to the formation of the membrane raft were necessary for the activation of NRLP3 inflammasomes in the endothelial cells and further increased atherosclerosis associated with hypercholesterolemia (81).
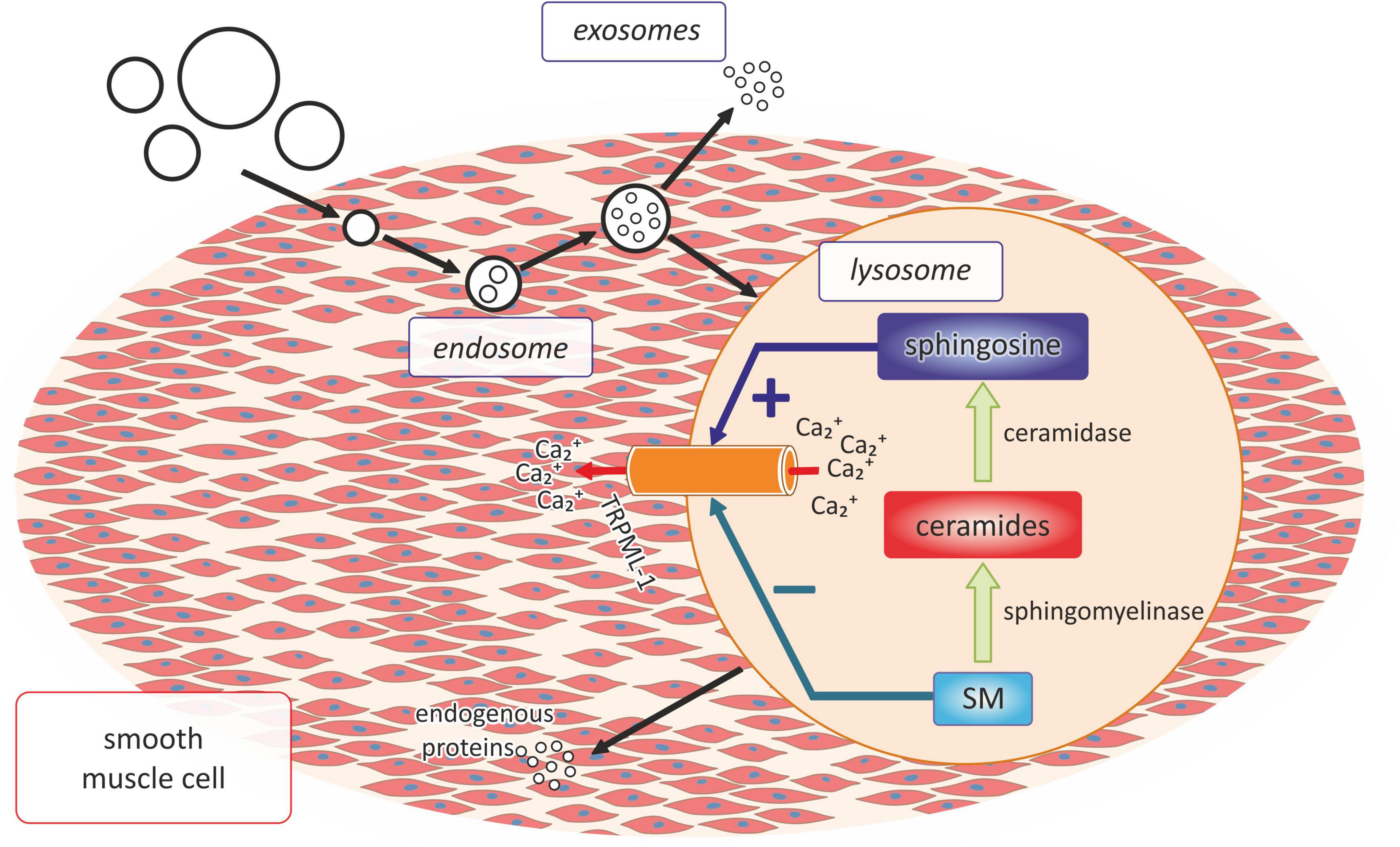
Figure 3. The role of lysosomal sphingolipid in the process of vascular calcification. SM, sphingomyelin; Ca2+, calcium ions.
Numerous experimental data indicate the beneficial effect of modulating sphingolipid system in the pharmacotherapy of atherosclerosis. Exogenous administration of fingolimod decreased the development of atherosclerosis in LDL receptor-deficient mice and ApoE-deficient mice (82, 83). Administration of KRP-203, a S1PR1 agonist, reduced the development of atherosclerosis in LDL receptor-deficient mice on a cholesterol-rich diet by modulating the lymphocyte and macrophage function and improving the endothelial barrier (84). In ApoE-deficient mice on a chow or high fat diet, inhibition of de novo ceramide synthesis by the administration of myriocin, an inhibitor of SPT, resulted in significantly decreased plasma concentration of sphingomyelin, Cer, and S1P, and a significant increase in plasma phosphatidylcholine levels with no significant change in the cholesterol and triglyceride plasma concentrations. Most interestingly, both in ApoE-deficient mice fed on a chow or high fat diet, the administration of myriocin resulted in a significantly reduced atherosclerotic lesion area (85). In other studies on ApoE-knockout mice fed on a western diet, treatment with myriocin decreased plasma concentrations of sphingomyelin, sphinganine, cholesterol, and triglyceride with a concomitant decrease in beta-very-low-density lipoprotein (beta-VLDL) and LDL cholesterol, an increase in HDL cholesterol, a significant reduction of arterial atherosclerosis development, a reduction in the lesion and macrophage area, and the inhibition of necrotic core formation (86, 87).
Clinical studies seem to confirm the important role of sphingolipids in the atherogenic process and in the pathogenesis of CAD. However, data on the concentration of non-HDL-bound S1P in patients with CAD were inconclusive, but they were consistent with the decreased concentration of HDL-bound S1P in this group of patients, suggesting that CAD is associated with an uptake in the disturbance of HDL for plasma S1P (88–90). Moreover, HDL-associated S1P concentrations showed negative correlation with the severity of CAD and distinguishes one-vessel disease from multi-vessel disease, and a low concentration of HDL-associated S1P was predictive for CAD advancement (89). The serum concentration of S1P has been shown to be more predictive of obstructive CAD in comparison with traditional risk factors, such as age, sex, a family history of CAD, diabetes mellitus, lipid profile, and hypertension (90). Nevertheless, patients with stable CAD have increased plasma concentrations of sphingolipids, for example: ceramides, sphingomyelins, sphinganine, and sphingosine (91, 92). In a study by Poss et al., the total C24:1-containing sphingolipids and/or total dihydroceramides, independent of the chain length, were most strongly associated with the occurrence of CAD (91). Higher levels of plasma Cer(d18:1/20:0), Cer(d18:1/22:0), and Cer(d18:1/24:0) were significantly associated with the presence of left anterior descending coronary artery (LAD) stenosis ≥50% (93). Also, a high ratio of Cer(d18:1/24:1) to Cer(d18:1/24:0) was shown to be associated with more severe coronary artery stenosis (94). Finally, Meeusen et al. revealed that increased plasma concentrations of Cer(16:0), Cer(18:0), and Cer(24:1) are independently associated with major adverse cardiovascular events in patients with and without CAD (95). Moreover, a recent meta-analysis has shown that higher plasma concentrations of Cer(d18:1/16:0), Cer(d18:1/18:0), and Cer(d18:1/24:1) were associated with major adverse cardiovascular events (96). Therefore, novel CAD predictive ceramide risk scores were proposed, which show superior performance in comparison with traditional cardiovascular risk markers (91, 93, 97–100). Hilvo et al. improved the previously proposed ceramide risk score (CERT1) and established CERT2, which is a ceramide-based and phospholipid-based risk score for predicting the residual CVD event risk, especially cardiovascular death, in patients with CAD (97). CERT2 has been shown to have superior prognostic efficacy over troponin T (TnT) and LDL cholesterol and is associated with biomarkers of inflammation, myocardial necrosis, myocardial dysfunction, renal dysfunction, and dyslipidemia (97, 101).
In addition, based on available clinical studies, it appears that Cer composition seems to influence the morphology and vulnerability of atherosclerotic plaque. In the ATHEROREMO-IVUS study, Cheng et al. (102) revealed that higher plasma concentrations of Cer(d18:1/16:0) and Cer(d18:1/24:0) were associated with higher necrotic core fractions and lactosylceramide (d18:1/18:0), and with a higher coronary lipid core burden index (LCBI) on near-infrared spectroscopy (NIRS). A higher Cer(d18:1/20:0)/Cer(d18:1/24:0) ratio was associated with a higher plaque burden and the presence of dense calcium (102). Pan et al. (103) performed Optical Coherence Tomography in patients with STEMI and correlated culprit lesion characteristics with plasma Cer concentrations. The authors revealed that lipid-rich plaque, plaque rupture, and thin-cap fibroatheroma were more prevalent, while calcification occurred less often in patients with a high plasma Cer concentration in comparison with individuals with lower plasma Cer levels (103). Patients with a high plasma Cer concentration had a longer lipid core length, a larger mean lipid arc, and a bigger lipid volume index, which positively correlated with plasma Cer(d18:1/16:0), Cer(d18:1/18:0), and Cer(d18:1/24:1), and a thinner fibrous cap thickness (negatively correlated with the abovementioned Cer species) when compared with the low plasma Cer group. Moreover, four Cer species which have previously been shown to have a strong predictive value for major adverse cardiovascular events [Cer(d18:1/16:0), Cer(d18:1/18:0), Cer(d18:1/24:1), and Cer(d18:1/24:0)] were also predictors for thin-cap fibroatheroma (103).
Myocardial ischemia/reperfusion injury
The current treatment of choice for acute myocardial ischemia is based on the immediate restoration of blood flow using primary coronary intervention (PCI) or thrombolytic therapy. Paradoxically, such early and fast restoration of blood flow further increases the ischemia-induced myocardial injury, which is called myocardial ischemia/reperfusion injury (IRI) (104–106).
Numerous studies have revealed that ischemic hearts show an increase in Cer and S1P levels, therefore it can be concluded that sphingolipids are involved in the pathophysiology of IRI. However, considering the important role of ceramides in apoptosis of cardiomyocytes, researchers most often attribute an unfavorable effect on the myocardium to them, while S1P is considered a cardioprotective effect.
As mentioned above, Cer mediates the ischemia/reperfusion-induced apoptosis of cardiomyocytes (107, 108). In vitro studies on isolated rat hearts subjected to ischemia/reperfusion (I/R) have an increased myocardial level of Cer and S1P, and decreased levels of sphingomyelin and sphingosine (109–111). Moreover, it was found that in isolated rat hearts subjected to I/R, Cer was accumulated specifically in the mitochondria, but not in the microsomal fraction (112). The I/R group also had increased mitochondrial activity of neutral sphingomyelinase, which was decreased in rats treated with a sphingomyelinase inhibitor (D609), with no changes in the microsomal fraction in any group. Moreover, in isolated rat hearts subjected to I/R, the injection of D609 significantly reduced infarct size, myocardial ceramide concentration, and the ratio of active caspase 3/pro-caspase 3, as a marker of active apoptosis and ROS production (112). Additionally, ischemic pre-conditioning seems to partially prevent a reperfusion-induced increase in myocardial Cer concentrations (113).
Moreover, in vivo studies have shown that increased myocardial concentrations of ceramide due to I/R are probably associated with the reduced activity of ceramidase (111). A rat model of I/R revealed that the ischemic myocardium as well as the ischemic and reperfused myocardium have an increased concentration of Cer(16:0, 18:0, 18:1, 18:2, and 20:4) (108, 113). Additionally, it was revealed that I/R in mice increased the myocardial concentration of ceramide both in the infarct area and in the area at risk, with a concomitant increased expression of two subunits of the SPTLC1 and SPTLC2 in the area at risk, suggesting that I/R enhances the accumulation of ceramide via the activation of the de novo synthesis pathway (114). In turn, intraventricular administration of nanocarriers loaded with the SPT inhibitor myriocin at reperfusion significantly reduced the infarct size, decreased the inflammatory response in the area at risk, and reduced the myocardial reactive oxygen species production (114). Since myriocin also regulates cardiac metabolism and inflammatory responses after I/R injury, it has been proposed that myriocin might serve as a post-conditioning therapeutic tool (115).
Whereas Cer has a rather an adverse effect on the heart muscle, S1P protected the heart from I/R myocardial injury and is released together with adenosine during ischemic pre-conditioning and post-conditioning (116–118). Pre-conditioning or post-conditioning with S1P protected the isolated rat heart against I/R-induced myocardial injury (119–121). This is probably done via controlling the mitochondrial function and Akt/glycogen synthase kinase β (Gsk3β) phosphorylation (121). Moreover, S1P played a cardioprotective role in I/R also through the activation of the S1PR3- Ras homolog family member A/protein kinase D (RhoA/PKD) signaling cascade in S1P3 KO mouse cardiomyocytes (122). Moreover, pharmacological inhibition of S1P lyase prior to I/R in isolated murine hearts increased the myocardial levels of S1P, reduced the infarct size and enhanced hemodynamic recovery Moreover, pharmacological inhibition of S1P lyase prior to I/R in isolated murine hearts increased the myocardial levels of S1P, reduced the infarct size and enhanced hemodynamic recovery (123). On the other hand, studies on isolated mouse and rat hearts showed that ischemia decreased the myocardial activity of sphingosine kinase and activates S1P lyase, which leads to reduced myocardial levels of S1P (123, 124).
S1P can affect the myocardium during I/R through its receptors. Studies carried out on both mouse and rat hearts have shown that exogenously administered S1P and stimulation of the S1PR1 receptor, as well as activation of SPHK1, protect against hypoxia- and oxygen-glucose deprivation/reoxygenation-induced myocardial cell death, respectively (125–128). Furthermore, in mouse model of I/R, S1P reduced the myocardial infarction size, reduced inflammatory cell recruitment and apoptosis of cardiomyocytes, thus exerting cardioprotective effects against I/R injury, through the activation of S1PR3-mediated and nitric-oxide dependent pathways (129). Additionally, Means et al. showed that activation of both S1PR2 and S1PR3 receptors are involved in cardioprotection against I/R injury, since the knockout of either S1PR2 or S1PR3 alone does not influence the in vivo response to I/R injury, while the knockout of both receptors significantly increases the I/R injury-related infarct size (130). Recently, Zhang et al. (131) in the rat model of I/R showed that blockage of the S1PR2/S1PR3 receptors with specific antagonists increased IR-induced mortality and infarction size, induced conduction abnormalities, and decreased the fractional shortening and ejection fraction of LV. While pre-treatment with S1PR2 and S1PR3 agonists at least partially reversed those changes (131). Afterward, the interaction of S1P with its receptors activates intracellular pathways also having a cardioprotective effect against ischemia. It have been shown that two signaling pathways to be involved in the above process: the Survivor Activating Factor Enhancement (SAFE) and the Reperfusion Injury Salvage Kinase (RISK) pathways. The SAFE pathway is activated by ischemic conditioning and/or by reperfusion and promotes cell survival through the activation of TNF-α which therefore activates its receptor TNF-R2, leading to the Janus kinase/signal transducer and activator of transcription 3 pathway (JAK/STAT-3) (132). The RISK pathway is activated by reperfusion and has a cardioprotective effect through the activation of two independent cascades, i.e., the PI3K-Akt or mitogen-activated extracellular signal-regulated kinase (MAPK, MEK1/ERK1/ERK2) (132). Somers et al. (133) revealed that S1P administered at the time of reperfusion to isolated rat hearts subjected to I/R reduced the myocardial infarct size in wild type animals, but not in STAT-3 or TNF-deficient mice. Moreover, after reperfusion in wild type mice, the authors revealed increased nuclear levels of phosphorylated STAT-3, Akt, and FOXO1, which is a downstream target of the RISK pathway (133). Moreover, it has been shown that activation of STAT-3 is required for the S1P-induced reduction of the infarction area in I/R (134).
Although human studies regarding sphingolipid involvement in I/R are scarce, it has been shown that, in patients undergoing elective percutaneous coronary intervention, coronary sinus levels of S1P, sphingosine, and sphinganine were increased as soon as 1 min following transient ischemia and reperfusion (135).
Acute coronary syndromes
Regarding the currently used nomenclature, acute coronary syndromes (ACS) include ST-segment elevation ACS. Most patients will develop ST-segment elevation myocardial infarction (STEMI) or non-ST-segment elevation ACS (NSTE-ACS), including myocardial ischemia without cardiomyocyte necrosis (unstable angina) or non-ST-segment elevation myocardial infarction (NSTEMI) (106). From the pathophysiological point of view, myocardial infarction (MI) mostly results from atherosclerotic plaque rupture or erosion and in the vast majority of cases depends on the composition and vulnerability of the atherosclerotic plaque (51).
Based on the available data, it can be concluded that sphingolipids also play an important role in the pathogenesis of acute coronary syndrome and myocardial infarction. Moreover, the studies presented below reveal a rather unfavorable effect of Cer on the heart muscle and a definitely cardioprotective effect of S1P.
It has been shown that ceramides by their activity in mitochondrial membranes caused alter cellular energetics, generating ROS formation and initiating cytochrome C release to promote apoptosis through caspase 3 pathways (Figure 4) (28). Cer also play a profibrotic role, because they can active of cyclic adenosine monophosphate responsive element binding protein 3 like 1 (CREB3L1) pathway to promote collagen deposition consequently leading to the development of heart failure with reduced ejection fraction (HFrEF) as well as heart failure with preserved ejection fraction (HFpEF) (Figure 4) (28).
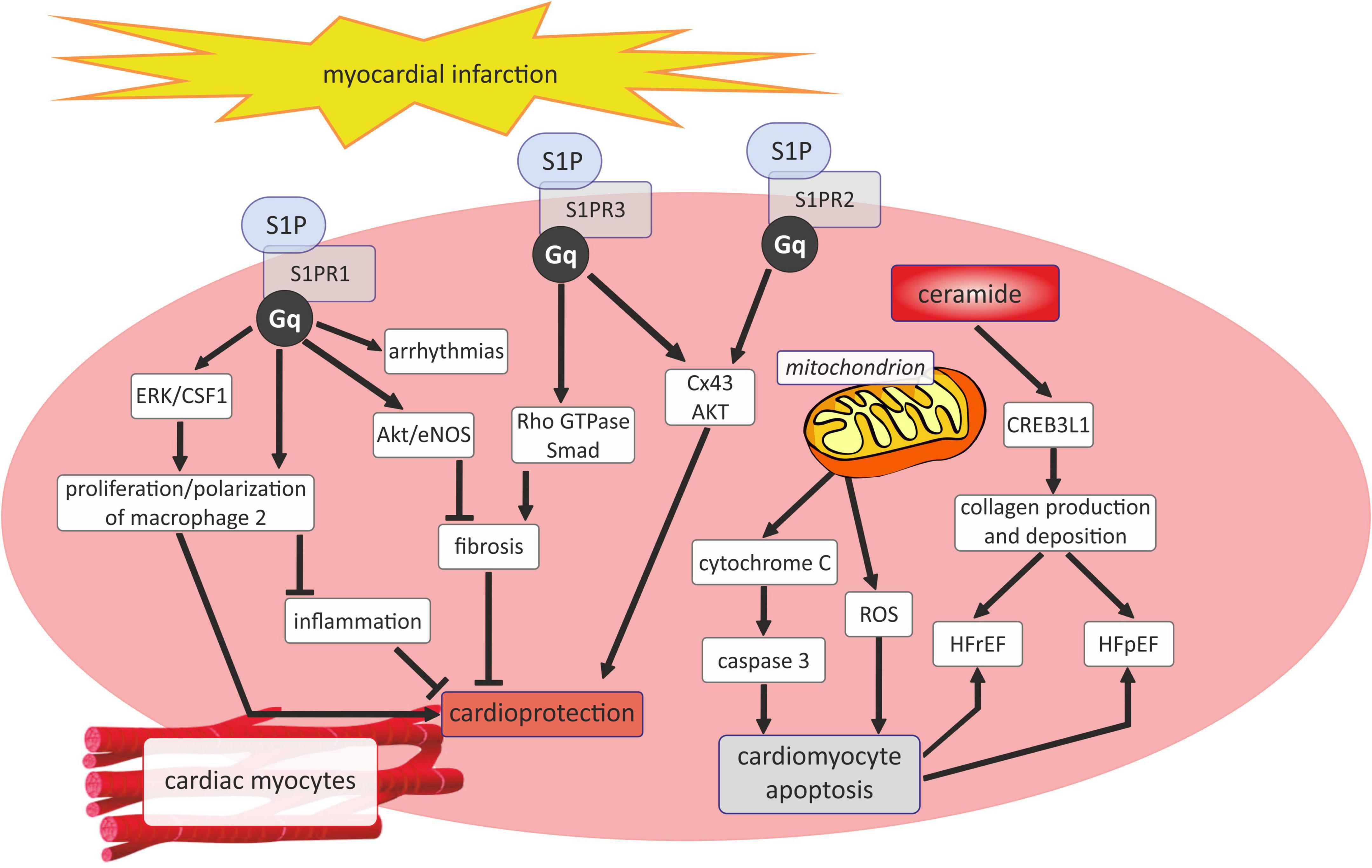
Figure 4. Sphingosine 1-phosphate and ceramide signaling in myocardial infarction. AKT, serine/threonine protein kinase B; CREB3L1, cyclic adenosine monophosphate responsive element binding protein 3 like 1; CSF1, colony stimulating factor 1; Cx43, connexin43; ERK, extracellular signal-regulated kinases; Gq, Gq protein alpha subunit; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure or heart failure with reduced ejection fraction; Rho GTPase, family small GTPase; ROS, reactive oxygen species; SMAD -suppressor of mothers against decapentaplegic; S1P, sphingosine-1-phosphate; S1PR1, sphingosine-1-phosphate receptor 1; S1PR2, sphingosine-1-phosphate receptor 2; S1PR3, sphingosine-1-phosphate receptor 3.
The adverse effects of Cer on the heart were reflected in in vivo experimental studies. Hadas et al. (136) have proven that, in Swiss Webster (CFW) mice 24 h after ligation of LAD, several genes involved in de novo synthesis of Cer were upregulated and myocardial concentrations of C16, C20, C20:1, and C24 ceramides were increased. Moreover, treatment with modified mRNA (modRNA) enhanced the activity of acid ceramidase and reduced hypoxia-induced cell death and resulted in a reduced inflammatory response, in improved cardiac function, in a smaller scar area, and in longer survival post infarction (136). In the reparative phase post MI induced by LAD ligation, C57BL/6 mice have significantly higher myocardial concentrations of Cer16:0, Cer24:1, ceramide-1-phosphates (Cer1P; 14:0, 18:1, 18:2, 18:3), dihydroceramide-1-phosphates (14:0, 16:0, 18:2, 20:4, 20:5), monohexosylceramides (20:0, 22:0, 24:0, and 24:1), dihexosylceramides (22:0, 24:1, 24:0), and lower myocardial concentrations of two dihydro sphingomyelins (38:4 and 40:0) in comparison with the sham-operated control group (137). Moreover, the MI group had higher expressions of Cer kinase (Cerk), as well as higher ratios of the Cer1P/Cer, which suggests that in the reparative phase post MI, the upregulated expression of Cerk may lead to enhanced conversion of Cer to Cer1P (137). In addition, myocardial concentrations of Cer(d18:1/16:0), Cer(d18:1/18:0), Cer(d18:1/20:0), Cer(d18:1/22:0), and Cer(d18:1/24:0), as well as the expression of serine palmitoyl transferase-2 (the gene encoding a base subunit of SPT), ceramide synthase 6 (CerS6), and neutral sphingomyelinase (nSMase) were found to be significantly higher in Wistar rats with acute MI induced by LAD occlusion in comparison with the sham-operated control group (138).
As mentioned before, unlike Cer, S1P is assigned a rather cardioprotective role in the course of acut coronary syndrome and myocardial infarction. It has been shown that the beneficial effects of S1P on the heart can be exerted by its receptors, of which S1PR1 and S1PR3 are widespread, while S1PR2 expression is low in the myocardium (Figure 4). S1PR1 has been disclosed to act as a protector in ischemic myocardial injury since soothes inflammatory responses and stimulates the proliferation of repair macrophages as well as inhibits myocardial fibrosis via Akt/eNOS dependent pathway (59). Nevertheless, S1PR1 also affect negatively on the heart by aggravating arrhythmias (59). In turn, the interaction of S1P with S1PR2 and S1PR3 exerts a cardioprotective effect, mainly by maintaining the conduction of myocyte action potential by inducing connexin 43 (Cx43) phosphorylation and phosphorylation of serine/threonine protein kinase B (AKT) (59). Similarly, the available experimental studies show a beneficial effect of S1P on the heart muscle in the course of MI. In C57BL/6 mice, pharmacological elevation of circulating S1P attracted hematopoietic stem cells and promoted tissue regeneration within the infarct zone, thus leading to cardiac structural and functional improvement after myocardial infarction (139). The studies of Yang et al. revealed that S1P induced the autophagy of cardiomyocytes after MI through the inhibition of the mammalian target of rapamycin (mTOR) signaling pathway, preventing adverse remodeling following MI in Sprague-Dawley rats (140). Moreover, in rats, myocardial infarction reduced the plasma concentrations of S1P, dihydrosphingosine-1-phosphate, sphingosine, and dihydrosphingosine but increased the concentration of total Cer, with concomitant alteration in the sphingolipid levels in the erythrocytes and platelets (141).
Based on the available data, it can be concluded that clinical trials seem to confirm the experimental results. Recently Yao et al. has shown that the Cer(d18:1/24:1(15Z)/Cer(d18:1/24:0) ratio, Cer(d18:1/14:0), and Cer(d18:1/22:0) can be independent predictors of acute coronary syndrome in patients with chest pain and added to high-sensitive troponin and traditional factors to improve the diagnostics of ACS (142). Patients with acute coronary syndrome, including unstable angina (UA) and AMI, had significantly higher concentrations of ceramide and secretory acid sphingomyelinase in comparison with healthy controls and individuals with stable angina (103, 143). Interestingly, increased plasma concentrations of Cer was sustained until a day after percutaneous coronary intervention in UA or day 7 in AMI patients (143). In another study, patients with ACS have been shown to have higher plasma concentrations of Cer(d18:1/16:0), Cer(d18:1/24:0), lactosylceramide (d18:1/18:0), and the Cer(d18:1/16:0)/Cer(d18:1/24:0) ratio in comparison with stable CAD (102). Recently, Burrello et al. revealed that patients with STEMI have significantly increased extracellular vesicle total content of Cer, dihydroceramides, and sphingomyelins in comparison with the controls and the prognostic ability of their levels was not inferior to troponin as a biomarker for AMI (144). The sphingolipid concentration within the extracellular vesicles (EV) was correlated to the peak level of high sensitive troponin and decreased 24 h post coronary artery reperfusion (144). Karagiannidis et al. evaluated patients with STEMI subjected to primary percutaneous coronary intervention and thrombus aspiration and revealed that higher ceramide C16:0 plasma concentrations were significantly correlated with larger aspirated thrombus volume, larger intracoronary thrombus burden, and poorer pre- and post-procedural thrombolysis in myocardial infarction (TIMI) flow; ceramides C24:0 and C24:1 were also associated with a larger intracoronary thrombus burden (145). Furthermore, plasma concentration of sphingomyelin, ceramide and glucosylceramide has been shown to correlate positively with high-sensitivity C-reactive protein, as a marker of inflammation in patients with myocardial infarction (146). Ceramides has also been proposed as a promising prognostic biomarker in patients with myocardial infarction. De Carvalho et al. proposed a 12-ceramide plasma signature which predicted cardiovascular death, MI, and stroke at 1 year in patients with acute MI (138). Cer (d18:0/16:0) and Cer (t18:0/12:0) and sphinganine have been shown to have high prognostic sensitivity and specificity for major adverse cardiovascular events for patients under 45 years of age with STEMI (147).
In addition, patients with STEMI have decreased plasma concentrations of S1P and sphinganine-1-phosphate, while the erythrocyte concentrations of S1P, sphingosine, sphinganine, sphinganine-1-phosphate were significantly increased on hospital admission and some of them were sustained for 2 years following infarction (148, 149). Moreover, in patients with myocardial infarction, the uninfected area of the myocardium presents a reduced S1P/Cer ratio (141). It has been shown that increased plasma concentrations of S1P correlated significantly with the occurrence of pre-infarction angina (150). Recently, Polzin et al. suggested that S1P may be responsible for the “Ang II receptor blockers (ARB)-myocardial infarction” paradox, which is a phenomenon in which ARB do not reduce the risk of myocardial infarction, despite effective blood pressure control (151). The authors revealed that patients treated for 3 months with ARB have a significantly lower plasma concentration of S1P in comparison with the patients treated with angiotensin converting enzyme inhibitors (ACEI) (151).
Heart failure
Heart failure is a highly occurring disease, whose prevalence is considered to be 1–2% of the adult population in developed countries, and more than 10% among people over 70 years of age (152). The current terminology distinguishes three groups of patients with HF in regard to the left ventricular (LV) ejection fraction (LVEF): HF with preserved ejection fraction (EF) (HFpEF; LVEF ≥50%), HF with mildly reduced EF (HFmrEF; LVEF 41–49%), and HF with reduced EF (HFrEF; LVEF ≤40%). There are substantial differences in the pathophysiology of HFpEF and HFrEF. HFpEF is considered to derive from the compound of several risk factors and comorbidities, such as older age, female sex, obesity, hypertension, diabetes mellitus, renal dysfunction, anemia, iron deficiency, sleep disorders, and chronic obstructive pulmonary disease (153). The main underlying pathological mechanisms leading to HFpEF are impaired cardiac relaxation and/or filling, increased chamber stiffness, and higher filling pressure with ventricular pressure overload resulting from cardiomyocyte hypertrophy and impaired energetic metabolism, interstitial fibrosis, inflammation, oxidative stress, endothelial and microvascular dysfunction (153, 154). On the other hand, HFrEF is more commonly related to CAD, including myocardial infarction, valvular disease, and uncontrolled hypertension. HFrEF may result mainly from cardiomyocyte injury due to ischemic insult such as myocardial infarction, myocarditis, valvular disease with cell death due to overload and a genetic mutation. Those processes result in systolic dysfunction, extensive myocardial fibrosis, and eccentric remodeling with ventricular dilation (152–154). Sphingolipids are considered to be involved in the cellular processes which underlie the development of both HFpEF and HFrEF.
Based on the available experimental studies, it seems that in the course of HF there is an increase in the level of Cer and a decrease in S1P in the myocardium. This likely had direct implications for the metabolism and function of the failing heart.
A mouse model of post-myocardial infarction HF induced by ligation of the left coronary artery revealed increased levels of C16, C24:1, and C24 ceramides in a failing myocardium after 10 weeks following MI in comparison with the sham operated controls (155). Recently, Hoffman et al. (156) revealed that genetically engineered mice with the overexpression of cardiomyocyte-specific Krüppel-like factor 5 (KLF5; a transcription factor which regulates the metabolism of myocardial fatty acids), have increased SPTLC1 and SPTLC2 expression, and higher cardiac levels of Cer and systolic dysfunction. Based on the above data, it can be concluded that KLF5 is a transcriptional regulator of SPTLCs which contributes to de novo ceramide synthesis and promotes myocardial dilation in ischemic cardiomyopathy, therefore its inhibition may be a therapeutic target for ischemic HF and myocardial remodeling (156).
Nevertheless, male C57BL/6 mice with post-MI HF showed reduced myocardial levels of sphinganine and sphingosine with increased levels of S1P at a chronic phase of HF (56 days post MI). Similarly, mRNA expression of S1P1R in the LV increased 1 day post MI and then decreased 5 days post MI, while in chronic HF the expression significantly increased, which suggests that myocardial S1P/S1PR1 signaling is enhanced during the chronic phase of HF (157). It is known that HF is associated with persistently increased activity of the sympathetic nervous system and, in experimental studies, chronic administration of β-adrenoceptor agonists is an established animal model of HF. Cannavo et al. have also proven that in C57BL/6 mice with HF, ISO resulted in a S1PR1 downregulation at the level of the plasma membrane, while chronic administration of a S1PR1-selective agonist resulted in the downregulation of β1AR cardiac plasma membrane levels; both agonists significantly increased the heart-to-body weight (HW/BW) ratio (158). Whereas, long-term overexpression of S1PR1 as a result of gene therapy in chronic HF post-MI in Sprague Dawley rats resulted in significantly smaller LV internal diastolic diameter, lower LV end diastolic pressure, and higher systolic and diastolic function, which reduced the infiltration of immune cells and restored the total plasma membrane β-adrenergic receptor (βAR) density in comparison with the HF controls with physiologically lower myocardial expression of S1PR1. Therefore, the authors concluded that treatment aimed at restoring cardiomyocyte membrane expression of S1PR1 exerts beneficial effects counterbalancing the detrimental overstimulation of β1AR present in HF (158).
A few experimental studies indicate the possibility of using sphingolipids in HF. It was shown that treatment with myriocin, a SPT-inhibitor, prevented LV dilation, improved LV systolic function, reduced the extent of myocardial fibrosis and macrophage content in C57B/L6 mice with MI-induced HF (155). Moreover, in Long Evans rats with hypotensive acute HF followed by a recovery phase, exogenous administration of S1P during the recovery phase improved the heart rate, which was accompanied by the activation of the signal transducer and activator of transcription 3 (STAT3) pathway (159).
The available clinical studies confirm changes in the expression of sphingolipids in the myocardium in the course of HF. Moreover, they indicate the possibility of using some substances from the great family of sphingolipids as potential prognostic markers in patients with HF. Knapp et al. reported decreased plasma concentrations of free sphingosine and sphinganine in patients with chronic HF, both ischemic and idiopathic dilated cardiomyopathy, with no significant differences in the levels of S1P, sphinganine-1-phosphate, and ceramide in comparison with the healthy controls (160). However, later research showed that a higher plasma concentration of the C16:0/C24:0 ceramide ratio may be associated with the increased risk of HF and was associated with a lower left ventricular ejection fraction, worse global circumferential strain, higher left atrial end-systolic volume, and lower left atrial emptying fraction (161). Recently, Wittenbecher et al. analyzed the relationship of single lipid metabolites and the lipidomic networks with the risk of developing HF and highlighted that two single lipid metabolites, CER 16:0 and phosphatidylcholine 32:0, apart from several other lipidomic patterns, were significantly associated with HF risk (162). Post-mortem heart tissue samples of patients with ischemic HF showed a significant increase in sphingosine and sphinganine concentration and an almost 10-fold decrease in the concentration of S1P within the ischemic myocardium when compared with healthy controls, with no significant differences in S1PR1 expression (157). However, Pérez-Carrillo et al. (163) observed the accumulation of Cer and S1P in the HF myocardial tissue, with an increased ceramide/S1P ratio by 57% in HF hearts. They also found that 12 genes involved in sphingolipid metabolism (especially genes participating in the de novo and salvage pathways) were differentially expressed in HF compared with the control patients (163).
Plasma ceramide levels have been found to be increased proportionally to the New York Heart Association (NYHA) functional class and were an independent risk factor of mortality in patients with chronic HF with reduced ejection fraction (164). Whereas, in patients with ischemic HF, plasma S1P and sphingomyelin concentrations were negatively associated with LV ejection fraction and NYHA class (165). Moreover, in HFpEF patients, increased plasma concentrations of Cer 16:0 and Cer 18:0 were associated with an increased risk of death or HF admission, with no such relationship in regard to Cer 24:0 (166). Targher et al. analyzed outpatients with chronic HF irrespective of LVEF enrolled in the Gruppo Italiano per lo Studio della Sopravvivenza nell’ Insufficienza Cardiaca-Heart Failure (GISSI-HF) trial and revealed a plasma ratio of Cer(d18:1/16:0), Cer(d18:1/18:0), Cer(d18:1/20:0), Cer(d18:1/22:0),Cer(d18:1/24:1) to Cer(d18:1/24:0) were significantly related to a higher risk of cardiovascular mortality, however after adjustment for established cardiovascular risk factors, medication use, and plasma concentrations of N-terminal (NT)-pro hormone BNP (NT-proBNP), these associations were impaired (167).
Arrhythmias and conduction disorders
Arrhythmias are a common heart dysfunction manifested as disruption of the appropriate periodicity and regularity of electromechanical activity (168). Conduction disorders, also known as heart blocks, are dysfunctions in which electrical signal production or its propagation through the myocardium is abnormal (169).
Considering the fact that sphingolipids are components of cell membranes, it can be hypothesized that sphingolipids also regulate cardiac repolarization, and therefore may play an important role in the pathophysiology of arrhythmia and conduction disorders.
The above hypothesis seems to be confirmed by experimental studies. Based on the available data, it can be assumed that particularly ceramides have the ability to regulate cardiac repolarization. Whole-cell patch clamp studies carried out on the human embryonic kidney cell line HEK293, which had been incubated for several hours with Cer, reacted with the inhibition of the human Ether-à-go-go-Related Gene (hERG) potassium channel current (a major component of rapid delayed rectifier K + current/IKr). Such an inhibition may prolong the action potential duration and QT interval, predisposing to ventricular arrhythmias and sudden cardiac death (170–172). Similarly, Huang et al. revealed that, in isolated rabbit pulmonary vein tissues, C2-ceramide regulates its electrophysiological properties, therefore it may be involved in the pathogenesis of pulmonary vein-induced arrhythmogenesis (173). Furthermore, in the rat model of lethal ventricular tachyarrhythmia (LVTA)-sudden cardiac death (SCD) (Sprague Dawley rats injected with aconitine into the tail vein), Wu et al. showed disruption of the plasma concentration of Cer, sphingomyelin, phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine after an LVTA-SCD event (174, 175). Tachycardia in Wistar rats has also been shown to change the metabolism of bioactive sphingolipids differently in each ventricle, in LV increasing the myocardial concentration of sphingosine and reducing concentrations of ceramide, S1P, and sphinganine-1-phosphate, while in RV increasing the concentration of sphingosine and sphinganine, with a reduction of S1P, sphinganine-1-phosphate, and ceramide (176).
In turn, S1P is involved in particular in the regulation of the intracellular potassium current in the myocardium. It was demonstrated that in isolated human atrial cardiomyocytes, S1P-S1PR3 signaling is responsible for the activation of the muscarinic receptor-activated inward rectifier potassium current, which has been involved in the vagally mediated regulation of the heart rate (177). Additionally, Ochi et al. (178) have proven that S1P activates a weakly inwardly rectifying K+ current in guinea pig atrial myocytes, which significantly shortens the action potential duration and shortens the effective refractory period (ERP). A shortening of the ERP may predispose to the development of reentry arrhythmias, such as atrial fibrillation (178). Moreover, in Sprague Dawley rats, pharmacological inhibition of S1P lyase caused bradycardia (179).
Several clinical studies appear to confirm the role of sphingolipids in the regulation of cardiac polarization. Recently, an analysis of the Cardiovascular Health Study population without a history of atrial fibrillation revealed that plasma ceramides and sphingomyelins with very long chain saturated fatty acids were associated with a reduced risk of incident atrial fibrillation, whereas ceramides and sphingomyelins with palmitic acid were associated with an increased atrial fibrillation risk (180). Moreover, in clinical settings, treatment with S1P receptor modulators was associated with side effects, such as bradycardia, atrioventricular blocks, and also probably ventricular tachycardia, which were suggested to be mainly S1PR1/S1PR3-dependent (46, 181–184). In addition, patients with atrial fibrillation had significantly lower plasma concentration of S1P in comparison with the controls with sinus rhythm, however, the exact role of S1P in atrial fibrillation needs to be further investigated (185). Furthermore, in patients with rheumatoid arthritis, psoriasis, and other inflammatory conditions who frequently had prolonged QT, administration of fingolimod prolonged the QT interval and inhibited the hERG current (186).
Stroke
Vascular diseases of the brain are one of the most frequent causes of death and disability, and the number of cases in many countries around the world is steadily increasing. Stroke is the acute neurologic injury that occurs as a result of brain ischemia or brain hemorrhage, both of which present with different clinical manifestations and outcomes (187). Sphingolipids have been shown to play a pivotal role in the pathophysiology of ischemic stroke.
Based on the literature data, it can be concluded that sphingolipids are widely represented in the central nervous system (CNS), for example neurons and astrocytes have the ability to synthesize and release S1P (2). Moreover, expression of all the S1P receptors has been found within the central nervous system: oligodendrocytes mainly express S1PR1 and S1PR5, astrocytes: S1PR1 and S1PR3, while microglia express S1PR1-3 (2). Due to the common occurrence of S1P and its receptors in brain tissue, it can be assumed that they may also play a role in the pathophysiology central nervous system diseases, including stroke. Especially that both experimental and clinical studies suggest the important role of S1P as a potentially mediator in ischemic stroke. Ischemia increases the concentration of S1P in the brain tissue and has been suggested to be a microglial-dependent process (188–190). Exogenous administration of S1P to the healthy mouse brain activates microglia and astrocytes, inducing a local neuroinflammatory response (191). S1P may play a role in stroke through its receptors (59). Overall, all S1P receptors (SPR1-3) accelerate the development of ischemic stroke (59). It was revealed that the interaction of S1P with S1PR1 and S1PR2 play a pivotal role in the disruption of the cerebrovascular integrity thereby promoting the destruction of the blood-brain barrier (BBB) (59). Damage to the BBB throughout S1PR1 is mediated by ICAM-1, while S1PR2 may increase the activity of metalloproteinase 9 (MMP-9) and enhance expression of p38 MAPK and ERK1/2 proteins consequently activating the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) directly leading to an increase the permeability of cerebral vessels (192–194) (Figure 5). S1PR1, 2, and 3 have been shown to mediate the activation of microglial cells and the inflammatory response in male ICR mice, which may play an important role in the pathogenesis of ischemic stroke (192, 195). Inhibitions of S1PR1 and S1PR3 activity in the mouse model of transient middle cerebral artery occlusion/reperfusion (tMCAO) significantly reduced the brain infarct volume, improved the neurological deficit, and decreased the number of the activated microglia and expression of proinflammatory cytokines (192, 195, 196). Moreover, inhibition of S1PR2 attenuates microglial activation and microglial proliferation in the mouse post-ischemic brain after tMCAO (197). S1PR2 has also been found to play a pivotal role in the disruption of the cerebrovascular integrity in the experimental model of stroke (198). The role of S1PR3 in promoting macrophage proliferation is perhaps the best explained. It has been reported that the mechanisms of action of S1PR3 were associated with ERK1/2 and p38 MAPK activation and with Akt inactivation (195) (Figure 5). However, it has been shown that the interaction of S1P with S1PR1 can also protect the brain after ischemic stroke by inhibiting the inflammatory response and neuronal apoptosis, as well as promoting angiogenesis (59) (Figure 5).
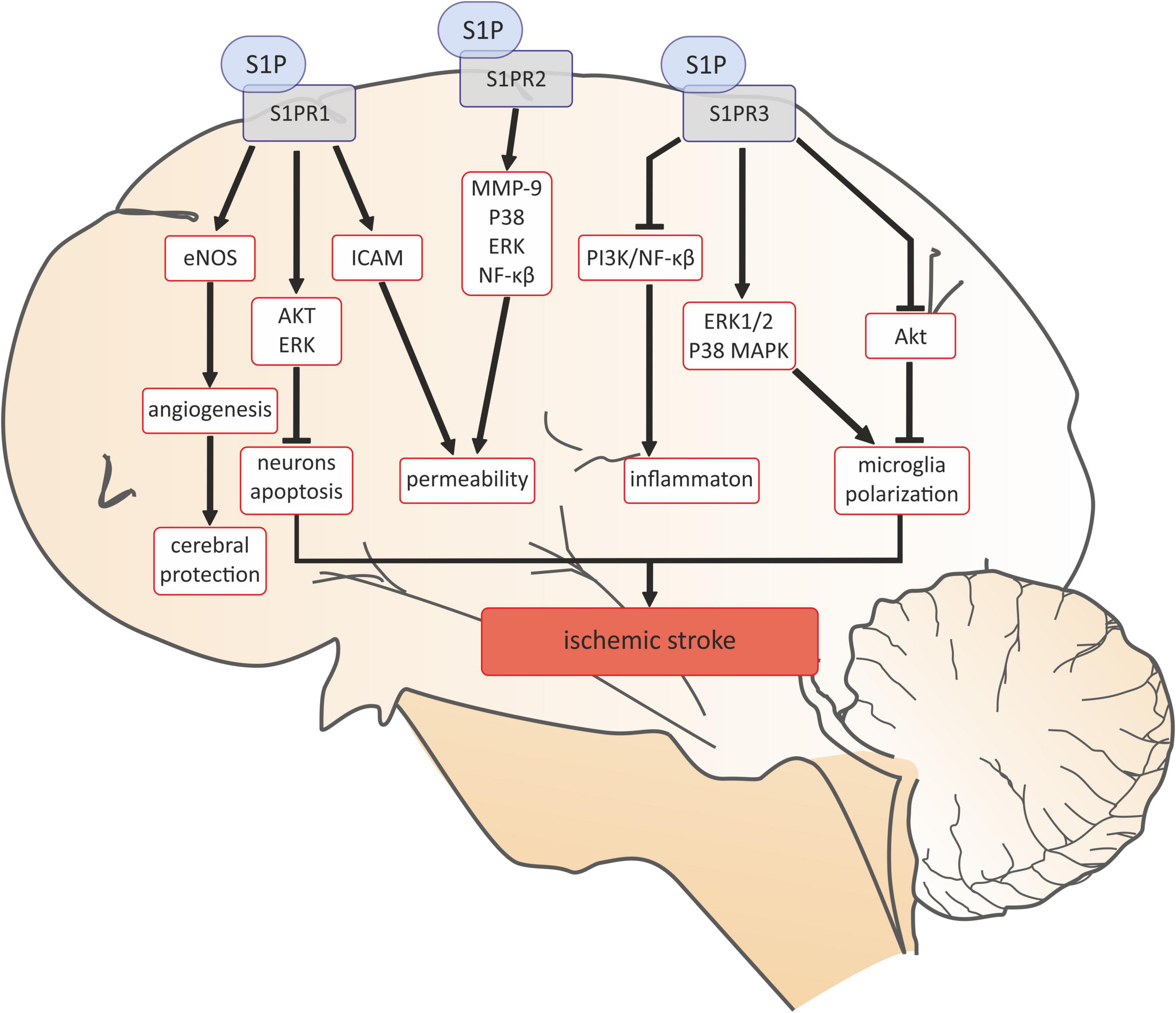
Figure 5. Sphingosine 1-phosphate signaling in ischemic stroke. AKT, serine/threonine protein kinase B; eNOS, endothelial nitric oxide synthase; ERK, extracellular signal-regulated kinases; ERK1/2, Ras-dependent extracellular signal-regulated kinase; ICAM-1 -intercellular adhesion molecule 1; MMP-9, matrix metalloproteinase-9; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; p38, p38 mitogen-activated protein kinases; PI3K, phosphoinositide 3-kinase; S1P, sphingosine-1-phosphate; S1PR1, sphingosine-1-phosphate receptor 1; S1PR2, sphingosine-1-phosphate receptor 2; S1PR3, sphingosine-1-phosphate receptor 3.
In addition, the activity of enzymes leading to the formation of S1P, i.e., SPHK1 and SPHK2 play a role in the ischemia-induced brain injury, however, their activity is opposite. In vitro studies have revealed that neurons subjected to glucose-oxygen deprivation have an increased expression of SPHK1 and pro-inflammatory mediators in the primary microglia, while the inhibition of SPHK1 attenuates the microglial induction of the proinflammatory mediators by ischemic neurons (199, 200). Zheng et al. in a mouse model of stroke induced by middle cerebral artery occlusion (MCAO) noticed an increase in the expression of SPHK1 predominantly in the microglia, and a reduction in the expression of inflammatory mediators in the cortical penumbra (199). Additionally, pharmacological inhibition of SPHK1in the same animal model decreased the size of infarction, preserves the integrity of the blood-brain barrier, and improves the neurological deficits after reperfusion (199, 201). Su et al. have shown that SPHK1/S1P mediates neuroinflammation and neuronal apoptosis via tumor necrosis factor receptor-associated factor 2 (TRAF2) and NF-κB in the activated microglia in the rat model of cerebral I/R (202). Furthermore, inhibition of SPHK1 by siRNA reduced the production of interleukin-17A (IL-17A) and decreased the degree of microglia subjected to oxygen-glucose deprivation, suggesting that SPHK1/S1P mediates the expression of IL-17A in the activated microglia, thereby inducing neuronal apoptosis in cerebral I/R (200). On the other hand, SPHK2 seems to exert beneficial neuroprotective effects in cerebral ischemia. Yung et al. have shown that C57BL/J mice exposed to pre-conditioning with isoflurane or hypoxia before tMCAO have upregulated cerebral SPHK2 and the protective effects of pre-conditioning, including reduced infarct volumes and improved neurological outcomes are attenuated in animals lacking SPHK2 (SPK2–/–) (203). Wacker et al. also showed that SPHK2 inhibition by dimethylsphingosine in Swiss-Webster ND4 mice, subjected to hypoxic pre-conditioning and tMCAO, reduced infarct volume, neurological deficits, and ipsilateral edema (204). Moreover, in mice subjected to tMCAO, the knockout of SPHK2, but not SPHK1, increased the ischemic lesion size and worsened the neurological function (205).
In addition, tMCAO, as well as lethal ischemia, increased brain tissue Cer concentration in rodents (206, 207). Yu et al. (208) reported that in wildtype mice tMCAO increases the activity of acid sphingomyelinase, increases the ischemic cortical tissue concentration of Cer, and increases the production of reactive oxygen species, which was absent in mice lacking acid sphingomyelinase. Moreover, mice lacking acid sphingomyelinase had a decreased degree of apoptosis and their neurological deficits were improved in comparison with wildtype animals (208). In the rat model of chronic cerebral ischemia induced by the clipping of the common carotid arteries, Ohtani et al. revealed that after 14 days there is a significant increase in brain tissue activity of acid sphingomyelinase and Cer concentration, specifically in astroglia (209).
Clinical trials confirm the important role of sphingolipids in the pathogenesis of stroke. Namely, the Cer signaling pathway appears to exert a detrimental effect in the pathophysiology of stroke. In vitro studies on the human neuroblastoma cell line SHEP subjected to experimental ischemia (serum/oxygen deprivation) have increased the activity of acid sphingomyelinase and ceramide production, which reached a maximum of 30 min following reoxygenation (206). Moreover, in patients after acute ischemic stroke, Lee et al. (210) have proven that the plasma concentration of S1P and very long chain ceramides are significantly lower in comparison with the non-stroke controls. A group of patients who suffered from ischemic stroke also had higher concentrations of long chain ceramides. Moreover, greater levels of Cer(d18:1/18:0), Cer(d18:1/20:0), and Cer(d18:1/22:0) measured at 48–72 h following the onset of stroke were predictors of poorer functional outcomes (210). Gui et al. (211) have shown that plasma levels of Cer(d18:1/16:0), Cer(d18:1/22:0), and Cer(d18:1/24:0) were significantly higher in stroke patients than in patients from the control group. Higher plasma levels of C16:0, C22:0, and C24:0 were associated with the clinical severity of stroke, and patients with minor stroke (defined on the National Institutes of Health Stroke Scale (NIHSS) <6) had lower ceramide serum levels that those with moderate-to-high clinical severity (211). Fiedorowicz et al. found two sphingolipid ratios (Sph-1-P/Cer-C24:1and Cer-C24:0/Cer-C24:1) to be strongly characteristic and both had diagnostic potential in ischemic stroke (212). Furthermore, patients with post-stroke depression (PSD) have significantly higher plasma concentrations of Cer16:0, Cer18:0, Cer24:0, and Cer24:1 in comparison with non-post-stroke depression (non-PSD) patients. Moreover, the specific ceramides C16:0 (vs. major depression; MD), C18:0 (vs. MD), and C16:0 (vs. Non-PSD, P = 0.002) can be used as predictive risk factors for diagnosis (213). Similar results were shown by an open-label, evaluator-blinded, parallel-group clinical pilot trial, where patients, with an anterior cerebral circulation occlusion and onset of stroke that exceeded 4.5 h and who had received standard management with additional fingolimod orally for3 consecutive days, had lower circulating lymphocyte counts, milder neurological deficits, better recovery of neurological functions, and less enlargement of lesion size between baseline and day 7 (214), which indicates the possibility of using sphingolipids in the pharmacotherapy of strokes in the future.
Conclusion
Sphingolipids are not only an integral part of the cell membrane, but also play an important role in the pathophysiology of cardiovascular diseases, such as hypertension, coronary artery disease, heart failure, arrhythmias, and stroke. Moreover, recent experimental and clinical studies indicate the possibility of using sphingolipids in the pharmacotherapy of the above diseases. However, more research is needed to better understand the role of sphingolipids in cardiovascular disease.
Author contributions
SB-J, AC-J, and KC: conceptualization. SB-J, PJ, WŁ, and KC: literature review. SB-J, PJ, WŁ, AC-J, and KC: writing—original draft preparation and writing—review and editing. KC and AC-J: supervision. KC: project administration. All authors have read and agreed to the published version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Borodzicz S, Czarzasta K, Kuch M, Cudnoch-Jedrzejewska A. Sphingolipids in cardiovascular diseases and metabolic disorders. Lipids Health Dis. (2015) 14:55. doi: 10.1186/s12944-015-0053-y
2. Soliven B, Miron V, Chun J. The neurobiology of sphingosine 1-phosphate signaling and sphingosine 1-phosphate receptor modulators. Neurology. (2011) 76(Suppl. 3):S9–14. doi: 10.1212/WNL.0b013e31820d9507
3. Merrill AH Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. (2011) 111:6387–422. doi: 10.1021/cr2002917
4. Kovilakath A, Jamil M, Cowart LA. Sphingolipids in the heart: From cradle to grave. Front Endocrinol (Lausanne). (2020) 11:652. doi: 10.3389/fendo.2020.00652
5. Bartke N, Hannun YA. Bioactive sphingolipids: Metabolism and function. J Lipid Res. (2009) 50(Suppl.):S91–6. doi: 10.1194/jlr.R800080-JLR200
6. Abed Rabbo M, Khodour Y, Kaguni LS, Stiban J. Sphingolipid lysosomal storage diseases: From bench to bedside. Lipids Health Dis. (2021) 20:44. doi: 10.1186/s12944-021-01466-0
7. Grassi S, Chiricozzi E, Mauri L, Sonnino S, Prinetti A. Sphingolipids and neuronal degeneration in lysosomal storage disorders. J Neurochem. (2019) 148:600–11. doi: 10.1111/jnc.14540
8. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. (2018) 19:175–91. doi: 10.1038/nrm.2017.107
9. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat Rev Mol Cell Biol. (2008) 9:139–50. doi: 10.1038/nrm2329
10. Shinghal R, Scheller RH, Bajjalieh SM. Ceramide 1-phosphate phosphatase activity in brain. J Neurochem. (1993) 61:2279–85. doi: 10.1111/j.1471-4159.1993.tb07470.x
11. Dawoody Nejad L, Stumpe M, Rauch M, Hemphill A, Schneiter R, Bütikofer P, et al. Mitochondrial sphingosine-1-phosphate lyase is essential for phosphatidylethanolamine synthesis and survival of Trypanosoma brucei. Sci Rep. (2020) 10:8268. doi: 10.1038/s41598-020-65248-x
12. Ichikawa S, Hirabayashi Y. Glucosylceramide synthase and glycosphingolipid synthesis. Trends Cell Biol. (1998) 8:198–202. doi: 10.1016/S0962-8924(98)01249-5
13. Senkal CE, Salama MF, Snider AJ, Allopenna JJ, Rana NA, Koller A, et al. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab. (2017) 25:686–97. doi: 10.1016/j.cmet.2017.02.010
14. Aguilar A, Saba JD. Truth and consequences of sphingosine-1-phosphate lyase. Adv Biol Regul. (2012) 52:17–30. doi: 10.1016/j.advenzreg.2011.09.015
15. Tettamanti G. Ganglioside/glycosphingolipid turnover: New concepts. Glycoconj J. (2004) 20:301–17. doi: 10.1023/B:GLYC.0000033627.02765.cc
16. Landeen LK, Dederko DA, Kondo CS, Hu BS, Aroonsakool N, Haga JH, et al. Mechanisms of the negative inotropic effects of sphingosine-1-phosphate on adult mouse ventricular myocytes. Am J Physiol Heart Circ Physiol. (2008) 294:H736–49. doi: 10.1152/ajpheart.00316.2007
17. Liu SJ, Kennedy RH. Positive inotropic effect of ceramide in adult ventricular myocytes: Mechanisms dissociated from its reduction in Ca2+ influx. Am J Physiol Heart Circ Physiol. (2003) 285:H735–44. doi: 10.1152/ajpheart.01098.2002
18. Relling DP, Hintz KK, Ren J. Acute exposure of ceramide enhances cardiac contractile function in isolated ventricular myocytes. Br J Pharmacol. (2003) 140:1163–8. doi: 10.1038/sj.bjp.0705510
19. Simon JN, Chowdhury SA, Warren CM, Sadayappan S, Wieczorek DF, Solaro RJ, et al. Ceramide-mediated depression in cardiomyocyte contractility through PKC activation and modulation of myofilament protein phosphorylation. Basic Res Cardiol. (2014) 109:445. doi: 10.1007/s00395-014-0445-6
20. Magaye RR, Savira F, Xiong X, Huynh K, Meikle PJ, Reid C, et al. Dihydrosphingosine driven enrichment of sphingolipids attenuates TGFβ induced collagen synthesis in cardiac fibroblasts. Int J Cardiol Heart Vasc. (2021) 35:100837. doi: 10.1016/j.ijcha.2021.100837
21. Gellings Lowe N, Swaney JS, Moreno KM, Sabbadini RA. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc Res. (2009) 82:303–12. doi: 10.1093/cvr/cvp056
22. Takuwa N, Ohkura S, Takashima S, Ohtani K, Okamoto Y, Tanaka T, et al. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc Res. (2010) 85:484–93. doi: 10.1093/cvr/cvp312
23. Robert P, Tsui P, Laville MP, Livi GP, Sarau HM, Bril A, et al. EDG1 receptor stimulation leads to cardiac hypertrophy in rat neonatal myocytes. J Mol Cell Cardiol. (2001) 33:1589–606. doi: 10.1006/jmcc.2001.1433
24. Sekiguchi K, Yokoyama T, Kurabayashi M, Okajima F, Nagai R. Sphingosylphosphorylcholine induces a hypertrophic growth response through the mitogen-activated protein kinase signaling cascade in rat neonatal cardiac myocytes. Circ Res. (1999) 85:1000–8. doi: 10.1161/01.RES.85.11.1000
25. Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. (2018) 39:3021–104. doi: 10.1097/HJH.0000000000001940
26. Cantalupo A, Di Lorenzo A. S1P signaling and de novo biosynthesis in blood pressure homeostasis. J Pharmacol Exp Ther. (2016) 358:359–70. doi: 10.1124/jpet.116.233205
27. Boini KM, Xia M, Koka S, Gehr TW, Li PL. Sphingolipids in obesity and related complications. Front Biosci (Landmark Ed). (2017) 22:96–116. doi: 10.2741/4474
28. Choi RH, Tatum SM, Symons JD, Summers SA, Holland WL. Ceramides and other sphingolipids as drivers of cardiovascular disease. Nat Rev Cardiol. (2021) 18:701–11. doi: 10.1038/s41569-021-00536-1
29. Zhang DX, Zou AP, Li PL. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am J Physiol Heart Circ Physiol. (2003) 284:H605–12. doi: 10.1152/ajpheart.00697.2002
30. Li H, Junk P, Huwiler A, Burkhardt C, Wallerath T, Pfeilschifter J, et al. Dual effect of ceramide on human endothelial cells: Induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation. (2002) 106:2250–6. doi: 10.1161/01.CIR.0000035650.05921.50
31. Spijkers LJ, van den Akker RF, Janssen BJ, Debets JJ, De Mey JG, Stroes ES, et al. Hypertension is associated with marked alterations in sphingolipid biology: A potential role for ceramide. PLoS One. (2011) 6:e21817. doi: 10.1371/journal.pone.0021817
32. Zheng T, Li W, Wang J, Altura BT, Altura BM. Sphingomyelinase and ceramide analogs induce contraction and rises in [Ca(2+)](i) in canine cerebral vascular muscle. Am J Physiol Heart Circ Physiol. (2000) 278:H1421–8. doi: 10.1152/ajpheart.2000.278.5.H1421
33. Cantalupo A, Sasset L, Gargiulo A, Rubinelli L, Del Gaudio I, Benvenuto D, et al. Endothelial sphingolipid de novo synthesis controls blood pressure by regulating signal transduction and NO via ceramide. Hypertension. (2020) 75:1279–88. doi: 10.1161/HYPERTENSIONAHA.119.14507
34. Siedlinski M, Nosalski R, Szczepaniak P, Ludwig-Gałȩzowska AH, Mikołajczyk T, Filip M, et al. Vascular transcriptome profiling identifies Sphingosine kinase 1 as a modulator of angiotensin II-induced vascular dysfunction. Sci Rep. (2017) 7:44131. doi: 10.1038/srep44131
35. Meissner A, Miro F, Jiménez-Altayo F, Jurado A, Vila E, Planas AM. Sphingosine-1-phosphate signalling-a key player in the pathogenesis of Angiotensin II-induced hypertension. Cardiovasc Res. (2017) 113:123–33. doi: 10.1093/cvr/cvw256
36. Cantalupo A, Zhang Y, Kothiya M, Galvani S, Obinata H, Bucci M, et al. Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat Med. (2015) 21:1028–37. doi: 10.1038/nm.3934
37. Igarashi J, Bernier SG, Michel T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J Biol Chem. (2001) 276:12420–6. doi: 10.1074/jbc.M008375200
38. Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck Lipinski K, Baba HA, et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest. (2004) 113:569–81. doi: 10.1172/JCI200418004
39. Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell. (2012) 23:600–10. doi: 10.1016/j.devcel.2012.07.015
40. Salomone S, Potts EM, Tyndall S, Ip PC, Chun J, Brinkmann V, et al. Analysis of sphingosine 1-phosphate receptors involved in constriction of isolated cerebral arteries with receptor null mice and pharmacological tools. Br J Pharmacol. (2008) 153:140–7. doi: 10.1038/sj.bjp.0707581
41. Kamiya T, Nagaoka T, Omae T, Yoshioka T, Ono S, Tanano I, et al. Role of Ca2+ -dependent and Ca2+ -sensitive mechanisms in sphingosine 1-phosphate-induced constriction of isolated porcine retinal arterioles in vitro. Exp Eye Res. (2014) 121:94–101. doi: 10.1016/j.exer.2014.01.011
42. Hu G, Zhu Q, Wang W, Xie D, Chen C, Li PL, et al. Collecting duct-specific knockout of sphingosine-1-phosphate receptor 1 aggravates DOCA-salt hypertension in mice. J Hypertens. (2021) 39:1559–66. doi: 10.1097/HJH.0000000000002809
43. Cantalupo A, Gargiulo A, Dautaj E, Liu C, Zhang Y, Hla T, et al. S1PR1 (Sphingosine-1-Phosphate Receptor 1) signaling regulates blood flow and pressure. Hypertension. (2017) 70:426–34. doi: 10.1161/HYPERTENSIONAHA.117.09088
44. Jujic A, Matthes F, Vanherle L, Petzka H, Orho-Melander M, Nilsson PM, et al. Plasma S1P (Sphingosine-1-Phosphate) links to hypertension and biomarkers of inflammation and cardiovascular disease: Findings from a translational investigation. Hypertension. (2021) 78:195–209. doi: 10.1161/HYPERTENSIONAHA.120.17379
45. Zheng H, Xie X, Xie N, Xu H, Huang J, Luo M. Sphingomyelin levels in nondipper and dipper hypertensive patients. Exp Ther Med. (2014) 7:599–603. doi: 10.3892/etm.2013.1455
46. Fryer RM, Muthukumarana A, Harrison PC, Nodop Mazurek S, Chen RR, Harrington KE, et al. The clinically-tested S1P receptor agonists, FTY720 and BAF312, demonstrate subtype-specific bradycardia (S1P1) and hypertension (S1P3) in rat. PLoS One. (2012) 7:e52985. doi: 10.1371/journal.pone.0052985
47. Spijkers LJ, Alewijnse AE, Peters SL. FTY720 (fingolimod) increases vascular tone and blood pressure in spontaneously hypertensive rats via inhibition of sphingosine kinase. Br J Pharmacol. (2012) 166:1411–8. doi: 10.1111/j.1476-5381.2012.01865.x
48. Spijkers LJ, Janssen BJ, Nelissen J, Meens MJ, Wijesinghe D, Chalfant CE, et al. Antihypertensive treatment differentially affects vascular sphingolipid biology in spontaneously hypertensive rats. PLoS One. (2011) 6:e29222. doi: 10.1371/journal.pone.0029222
49. Mehanna M, McDonough CW, Smith SM, Gong Y, Gums JG, Chapman AB, et al. Metabolomics signature of plasma renin activity and linkage with blood pressure response to beta blockers and thiazide diuretics in hypertensive European American patients. Metabolites. (2021) 11:645. doi: 10.3390/metabo11090645
50. Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. (2018) 39:2562–73. doi: 10.1093/eurheartj/ehy319
51. Kowara M, Cudnoch-Jedrzejewska A. Different approaches in therapy aiming to stabilize an unstable atherosclerotic plaque. Int J Mol Sci. (2021) 22:4354. doi: 10.3390/ijms22094354
52. Malakar AK, Choudhury D, Halder B, Paul P, Uddin A, Chakraborty S. A review on coronary artery disease, its risk factors, and therapeutics. J Cell Physiol. (2019) 234:16812–23. doi: 10.1002/jcp.28350
53. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
54. Nicoll R, Henein MY. The predictive value of arterial and valvular calcification for mortality and cardiovascular events. Int J Cardiol Heart Vessel. (2014) 3:1–5. doi: 10.1016/j.ijchv.2014.02.001
55. Chow B, Rabkin SW. The relationship between arterial stiffness and heart failure with preserved ejection fraction: A systemic meta-analysis. Heart Fail Rev. (2015) 20:291–303. doi: 10.1007/s10741-015-9471-1
56. Yuan X, Bhat OM, Lohner H, Zhang Y, Li PL. Endothelial acid ceramidase in exosome-mediated release of NLRP3 inflammasome products during hyperglycemia: Evidence from endothelium-specific deletion of Asah1 gene. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:158532. doi: 10.1016/j.bbalip.2019.158532
57. Bhat OM, Li G, Yuan X, Huang D, Gulbins E, Kukreja RC, et al. Arterial medial calcification through enhanced small extracellular vesicle release in smooth muscle-specific Asah1 gene knockout mice. Sci Rep. (2020) 10:1645. doi: 10.1038/s41598-020-58568-5
58. Bhat OM, Yuan X, Cain C, Salloum FN, Li PL. Medial calcification in the arterial wall of smooth muscle cell-specific Smpd1 transgenic mice: A ceramide-mediated vasculopathy. J Cell Mol Med. (2020) 24:539–53. doi: 10.1111/jcmm.14761
59. Lu S, She M, Zeng Q, Yi G, Zhang J. Sphingosine 1-phosphate and its receptors in ischemia. Clin Chim Acta. (2021) 521:25–33. doi: 10.1016/j.cca.2021.06.020
60. Al-Jarallah A, Chen X, González L, Trigatti BL. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists. PLoS One. (2014) 9:e106487. doi: 10.1371/journal.pone.0106487
61. Galvani S, Sanson M, Blaho VA, Swendeman SL, Obinata H, Conger H, et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal. (2015) 8:ra79. doi: 10.1126/scisignal.aaa2581
62. Kitano T, Usui S, Takashima SI, Inoue O, Goten C, Nomura A, et al. Sphigosine-1-phosphate receptor 1 promotes neointimal hyperplasia in a mouse model of carotid artery injury. Biochem Biophys Res Commun. (2019) 511:179–84. doi: 10.1016/j.bbrc.2019.02.047
63. Ren K, Lu YJ, Mo ZC, Liu X, Tang ZL, Jiang Y, et al. ApoA-I/SR-BI modulates S1P/S1PR2-mediated inflammation through the PI3K/Akt signaling pathway in HUVECs. J Physiol Biochem. (2017) 73:287–96. doi: 10.1007/s13105-017-0553-5
64. Fan JL, Zhang L, Bo XH. MiR-126 on mice with coronary artery disease by targeting S1PR2. Eur Rev Med Pharmacol Sci. (2020) 24:893–904.
65. Shimizu T, Nakazawa T, Cho A, Dastvan F, Shilling D, Daum G, et al. Sphingosine 1-phosphate receptor 2 negatively regulates neointimal formation in mouse arteries. Circ Res. (2007) 101:995–1000. doi: 10.1161/CIRCRESAHA.107.159228
66. Skoura A, Michaud J, Im DS, Thangada S, Xiong Y, Smith JD, et al. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol. (2011) 31:81–5. doi: 10.1161/ATVBAHA.110.213496
67. Ganbaatar B, Fukuda D, Shinohara M, Yagi S, Kusunose K, Yamada H, et al. Inhibition of S1P receptor 2 attenuates endothelial dysfunction and inhibits atherogenesis in apolipoprotein E-deficient mice. J Atheroscler Thromb. (2021) 28:630–42. doi: 10.5551/jat.54916
68. Kimura T, Tomura H, Mogi C, Kuwabara A, Ishiwara M, Shibasawa K, et al. Sphingosine 1-phosphate receptors mediate stimulatory and inhibitory signalings for expression of adhesion molecules in endothelial cells. Cell Signal. (2006) 18:841–50. doi: 10.1016/j.cellsig.2005.07.011
69. Keul P, Lucke S, von Wnuck K, Lipinski C. Bode, Gräler M, Heusch G, et al. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ Res. (2011) 108:314–23. doi: 10.1161/CIRCRESAHA.110.235028
70. Ishimaru K, Yoshioka K, Kano K, Kurano M, Saigusa D, Aoki J, et al. Sphingosine kinase-2 prevents macrophage cholesterol accumulation and atherosclerosis by stimulating autophagic lipid degradation. Sci Rep. (2019) 9:18329. doi: 10.1038/s41598-019-54877-6
71. Feuerborn R, Besser M, Poti F, Burkhardt R, Weißen-Plenz G, Ceglarek U, et al. Elevating Endogenous Sphingosine-1-Phosphate (S1P) levels improves endothelial function and ameliorates atherosclerosis in low density lipoprotein receptor-deficient (LDL-R-/-) mice. Thromb Haemost. (2018) 118:1470–80. doi: 10.1055/s-0038-1666870
72. Potì F, Ceglarek U, Burkhardt R, Simoni M, Nofer JR. SKI-II–a sphingosine kinase 1 inhibitor–exacerbates atherosclerosis in low-density lipoprotein receptor-deficient (LDL-R-/-) mice on high cholesterol diet. Atherosclerosis. (2015) 240:212–5. doi: 10.1016/j.atherosclerosis.2015.03.020
73. Morita SY, Kawabe M, Sakurai A, Okuhira K, Vertut-Doi A, Nakano M, et al. Ceramide in lipid particles enhances heparan sulfate proteoglycan and low density lipoprotein receptor-related protein-mediated uptake by macrophages. J Biol Chem. (2004) 279:24355–61. doi: 10.1074/jbc.M402035200
74. Xu XX, Tabas I. Sphingomyelinase enhances low density lipoprotein uptake and ability to induce cholesteryl ester accumulation in macrophages. J Biol Chem. (1991) 266:24849–58. doi: 10.1016/S0021-9258(18)54306-4
75. Marathe S, Kuriakose G, Williams KJ, Tabas I. Sphingomyelinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler Thromb Vasc Biol. (1999) 19:2648–58. doi: 10.1161/01.ATV.19.11.2648
76. Oörni K, Hakala JK, Annila A, Ala-Korpela M, Kovanen PT. Sphingomyelinase induces aggregation and fusion, but phospholipase A2 only aggregation, of low density lipoprotein (LDL) particles. Two distinct mechanisms leading to increased binding strength of LDL to human aortic proteoglycans. J Biol Chem. (1998) 273:29127–34. doi: 10.1074/jbc.273.44.29127
77. Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J Clin Invest. (1996) 98:1455–64. doi: 10.1172/JCI118934
78. Wang X, Dong J, Zhao Y, Li Y, Wu M. Adenovirus-mediated sphingomyelin synthase 2 increases atherosclerotic lesions in ApoE KO mice. Lipids Health Dis. (2011) 10:7. doi: 10.1186/1476-511X-10-7
79. Zhao YR, Dong JB, Li Y, Wu MP. Sphingomyelin synthase 2 over-expression induces expression of aortic inflammatory biomarkers and decreases circulating EPCs in ApoE KO mice. Life Sci. (2012) 90:867–73. doi: 10.1016/j.lfs.2012.04.003
80. Dong J, Liu J, Lou B, Li Z, Ye X, Wu M, et al. Adenovirus-mediated overexpression of sphingomyelin synthases 1 and 2 increases the atherogenic potential in mice. J Lipid Res. (2006) 47:1307–14. doi: 10.1194/jlr.M600040-JLR200
81. Koka S, Xia M, Chen Y, Bhat OM, Yuan X, Boini KM, et al. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. (2017) 13:336–44. doi: 10.1016/j.redox.2017.06.004
82. Keul P, Tolle M, Lucke S, von Wnuck Lipinski K, Heusch G, Schuchardt M, et al. The sphingosine-1-phosphate analogue FTY720 reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2007) 27:607–13. doi: 10.1161/01.ATV.0000254679.42583.88
83. Nofer JR, Bot M, Brodde M, Taylor PJ, Salm P, Brinkmann V, et al. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. (2007) 115:501–8. doi: 10.1161/CIRCULATIONAHA.106.641407
84. Potì F, Gualtieri F, Sacchi S, Weißen-Plenz G, Varga G, Brodde M, et al. KRP-203, sphingosine 1-phosphate receptor type 1 agonist, ameliorates atherosclerosis in LDL-R-/- mice. Arterioscler Thromb Vasc Biol. (2013) 33:1505–12. doi: 10.1161/ATVBAHA.113.301347
85. Hojjati MR, Li Z, Zhou H, Tang S, Huan C, Ooi E, et al. Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J Biol Chem. (2005) 280:10284–9. doi: 10.1074/jbc.M412348200
86. Park TS, Panek RL, Mueller SB, Hanselman JC, Rosebury WS, Robertson AW, et al. Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. (2004) 110:3465–71. doi: 10.1161/01.CIR.0000148370.60535.22
87. Park TS, Panek RL, Rekhter MD, Mueller SB, Rosebury WS, Robertson A, et al. Modulation of lipoprotein metabolism by inhibition of sphingomyelin synthesis in ApoE knockout mice. Atherosclerosis. (2006) 189:264–72. doi: 10.1016/j.atherosclerosis.2005.12.029
88. Sattler KJ, Elbasan S, Keul P, Elter-Schulz M, Bode C, Gräler MH, et al. Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res Cardiol. (2010) 105:821–32. doi: 10.1007/s00395-010-0112-5
89. Sattler K, Lehmann I, Gräler M, Bröcker-Preuss M, Erbel R, Heusch G, et al. HDL-bound sphingosine 1-phosphate (S1P) predicts the severity of coronary artery atherosclerosis. Cell Physiol Biochem. (2014) 34:172–84. doi: 10.1159/000362993
90. Deutschman DH, Carstens JS, Klepper RL, Smith WS, Page MT, Young TR, et al. Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am Heart J. (2003) 146:62–8. doi: 10.1016/S0002-8703(03)00118-2
91. Poss AM, Maschek JA, Cox JE, Hauner BJ, Hopkins PN, Hunt SC, et al. Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. J Clin Invest. (2020) 130:1363–76. doi: 10.1172/JCI131838
92. Jiang XC, Paultre F, Pearson TA, Reed RG, Francis CK, Lin M, et al. Plasma sphingomyelin level as a risk factor for coronary artery disease. Arterioscler Thromb Vasc Biol. (2000) 20:2614–8. doi: 10.1161/01.ATV.20.12.2614
93. Mantovani A, Bonapace S, Lunardi G, Canali G, Dugo C, Vinco G, et al. Associations between specific plasma ceramides and severity of coronary-artery stenosis assessed by coronary angiography. Diabetes Metab. (2020) 46:150–7. doi: 10.1016/j.diabet.2019.07.006
94. Tu C, Xie L, Wang Z, Zhang L, Wu H, Ni W, et al. Association between ceramides and coronary artery stenosis in patients with coronary artery disease. Lipids Health Dis. (2020) 19:151. doi: 10.1186/s12944-020-01329-0
95. Meeusen JW, Donato LJ, Bryant SC, Baudhuin LM, Berger PB, Jaffe AS. Plasma ceramides. Arterioscler Thromb Vasc Biol. (2018) 38:1933–9. doi: 10.1161/ATVBAHA.118.311199
96. Mantovani A, Dugo C. Ceramides and risk of major adverse cardiovascular events: A meta-analysis of longitudinal studies. J Clin Lipidol. (2020) 14:176–85. doi: 10.1016/j.jacl.2020.01.005
97. Hilvo M, Meikle PJ, Pedersen ER, Tell GS, Dhar I, Brenner H, et al. Development and validation of a ceramide- and phospholipid-based cardiovascular risk estimation score for coronary artery disease patients. Eur Heart J. (2020) 41:371–80. doi: 10.1093/eurheartj/ehz387
98. Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J. (2016) 37:1967–76. doi: 10.1093/eurheartj/ehw148
99. Tarasov K, Ekroos K, Suoniemi M, Kauhanen D, Sylvänne T, Hurme R, et al. Molecular lipids identify cardiovascular risk and are efficiently lowered by simvastatin and PCSK9 deficiency. J Clin Endocrinol Metab. (2014) 99:E45–52. doi: 10.1210/jc.2013-2559
100. Mundra PA, Barlow CK, Nestel PJ, Barnes EH, Kirby A, Thompson P, et al. Large-scale plasma lipidomic profiling identifies lipids that predict cardiovascular events in secondary prevention. JCI Insight. (2018) 3:e121326. doi: 10.1172/jci.insight.121326
101. Hilvo M, Wallentin L, Ghukasyan Lakic T, Held C, Kauhanen D, Jylha A, et al. Prediction of residual risk by ceramide-phospholipid score in patients with stable coronary heart disease on optimal medical therapy. J Am Heart Assoc. (2020) 9:e015258. doi: 10.1161/JAHA.119.015258
102. Cheng JM, Suoniemi M, Kardys I, Vihervaara T, de Boer SP, Akkerhuis KM, et al. Plasma concentrations of molecular lipid species in relation to coronary plaque characteristics and cardiovascular outcome: Results of the ATHEROREMO-IVUS study. Atherosclerosis. (2015) 243:560–6. doi: 10.1016/j.atherosclerosis.2015.10.022
103. Pan W, Dong H, Sun R, Zhao L, Sun M, Li L, et al. Plasma ceramides in relation to coronary plaque characterization determined by optical coherence tomography. J Cardiovasc Transl Res. (2021) 14:140–9. doi: 10.1007/s12265-020-09978-3
104. Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. (2012) 16:123–32. doi: 10.1177/1089253211436350
105. Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. (2018) 39:119–77.
106. Collet JP, Thiele H, Barbato E, Barthélémy O, Bauersachs J, Bhatt DL, et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. (2021) 42:1289–367. doi: 10.1093/eurheartj/ehaa909
107. Umansky SR, Shapiro JP, Cuenco GM, Foehr MW I, Bathurst C, Tomei LD. Prevention of rat neonatal cardiomyocyte apoptosis induced by simulated in vitro ischemia and reperfusion. Cell Death Differ. (1997) 4:608–16. doi: 10.1038/sj.cdd.4400282
108. Bielawska AE, Shapiro JP, Jiang L, Melkonyan HS, Piot C, Wolfe CL, et al. Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am J Pathol. (1997) 151:1257–63.
109. Cordis GA, Yoshida T, Das DK. HPTLC analysis of sphingomylein, ceramide and sphingosine in ischemic/reperfused rat heart. J Pharm Biomed Anal. (1998) 16:1189–93. doi: 10.1016/S0731-7085(97)00260-4
110. Cui J, Engelman RM, Maulik N, Das DK. Role of ceramide in ischemic preconditioning. J Am Coll Surg. (2004) 198:770–7. doi: 10.1016/j.jamcollsurg.2003.12.016
111. Zhang DX, Fryer RM, Hsu AK, Zou AP, Gross GJ, Campbell WB, et al. Production and metabolism of ceramide in normal and ischemic-reperfused myocardium of rats. Basic Res Cardiol. (2001) 96:267–74. doi: 10.1007/s003950170057
112. Ramírez-Camacho I, Bautista-Pérez R, Correa F, Buelna-Chontal M, Román-Anguiano NG, Medel-Franco M, et al. Role of sphingomyelinase in mitochondrial ceramide accumulation during reperfusion. Biochim Biophys Acta. (2016) 1862:1955–63. doi: 10.1016/j.bbadis.2016.07.021
113. Beresewicz A, Dobrzyn A, Górski J. Accumulation of specific ceramides in ischemic/reperfused rat heart; effect of ischemic preconditioning. J Physiol Pharmacol. (2002) 53:371–82.
114. Reforgiato MR, Milano G, Fabriàs G, Casas J, Gasco P, Paroni R, et al. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res Cardiol. (2016) 111:12. doi: 10.1007/s00395-016-0533-x
115. Bonezzi F, Piccoli M, Dei Cas M, Paroni R, Mingione A, Monasky MM, et al. Sphingolipid synthesis inhibition by myriocin administration enhances lipid consumption and ameliorates lipid response to myocardial ischemia reperfusion injury. Front Physiol. (2019) 10:986. doi: 10.3389/fphys.2019.00986
116. Vessey DA, Li L, Kelley M, Zhang J, Karliner JS. Sphingosine can pre- and post-condition heart and utilizes a different mechanism from sphingosine 1-phosphate. J Biochem Mol Toxicol. (2008) 22:113–8. doi: 10.1002/jbt.20227
117. Vessey DA, Li L, Honbo N, Karliner JS. Sphingosine 1-phosphate is an important endogenous cardioprotectant released by ischemic pre- and postconditioning. Am J Physiol Heart Circ Physiol. (2009) 297:H1429–35. doi: 10.1152/ajpheart.00358.2009
118. Egom EE, Mohamed TM, Mamas MA, Shi Y, Liu W, Chirico D, et al. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am J Physiol Heart Circ Physiol. (2011) 301:H1487–95. doi: 10.1152/ajpheart.01003.2010
119. Araibi H, van der Merwe E, Gwanyanya A, Kelly-Laubscher R. The effect of sphingosine-1-phosphate on the endothelial glycocalyx during ischemia-reperfusion injury in the isolated rat heart. Microcirculation. (2020) 27:e12612. doi: 10.1111/micc.12612
120. Morel S, Christoffersen C, Axelsen LN, Montecucco F, Rochemont V, Frias MA, et al. Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc Res. (2016) 109:385–96. doi: 10.1093/cvr/cvw004
121. Fang R, Zhang LL, Zhang LZ, Li W, Li M, Wen K. Sphingosine 1-phosphate postconditioning protects against myocardial ischemia/reperfusion injury in rats via mitochondrial signaling and Akt-Gsk3β phosphorylation. Arch Med Res. (2017) 48:147–55. doi: 10.1016/j.arcmed.2017.03.013
122. Yung BS, Brand CS, Xiang SY, Gray CB, Means CK, Rosen H, et al. Selective coupling of the S1P(3) receptor subtype to S1P-mediated RhoA activation and cardioprotection. J Mol Cell Cardiol. (2017) 103:1–10. doi: 10.1016/j.yjmcc.2016.12.008
123. Bandhuvula P, Honbo N, Wang GY, Jin ZQ, Fyrst H, Zhang M, et al. S1P lyase: A novel therapeutic target for ischemia-reperfusion injury of the heart. Am J Physiol Heart Circ Physiol. (2011) 300:H1753–61. doi: 10.1152/ajpheart.00946.2010
124. Vessey DA, Kelley M, Li L, Huang Y, Zhou HZ, Zhu BQ, et al. Role of sphingosine kinase activity in protection of heart against ischemia reperfusion injury. Med Sci Monit. (2006) 12:Br318–24.
125. Shao JJ, Peng Y, Wang LM, Wang JK, Chen X. Activation of SphK1 by K6PC-5 inhibits oxygen-glucose deprivation/reoxygenation-induced myocardial cell death. DNA Cell Biol. (2015) 34:669–76. doi: 10.1089/dna.2015.2959
126. Zhang J, Honbo N, Goetzl EJ, Chatterjee K, Karliner JS, Gray MO. Signals from type 1 sphingosine 1-phosphate receptors enhance adult mouse cardiac myocyte survival during hypoxia. Am J Physiol Heart Circ Physiol. (2007) 293:H3150–8. doi: 10.1152/ajpheart.00587.2006
127. Karliner JS, Honbo N, Summers K, Gray MO, Goetzl EJ. The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J Mol Cell Cardiol. (2001) 33:1713–7. doi: 10.1006/jmcc.2001.1429
128. Tao R, Zhang J, Vessey DA, Honbo N, Karliner JS. Deletion of the sphingosine kinase-1 gene influences cell fate during hypoxia and glucose deprivation in adult mouse cardiomyocytes. Cardiovasc Res. (2007) 74:56–63. doi: 10.1016/j.cardiores.2007.01.015
129. Theilmeier G, Schmidt C, Herrmann J, Keul P, Schäfers M, Herrgott I, et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. (2006) 114:1403–9. doi: 10.1161/CIRCULATIONAHA.105.607135
130. Means CK, Xiao CY, Li Z, Zhang T, Omens JH, Ishii I, et al. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. (2007) 292:H2944–51. doi: 10.1152/ajpheart.01331.2006
131. Zhang X, Chen D, Wang J, Liu J, Guo H, Zhang G. Involvement of sphingosine-1-phosphate receptors 2/3 in IR-induced sudden cardiac death. Heart Vessels. (2019) 34:1052–63. doi: 10.1007/s00380-018-01323-8
132. Soares ROS, Losada DM, Jordani MC, Évora P, Castro-E-Silva O. Ischemia/Reperfusion injury revisited: An overview of the latest pharmacological strategies. Int J Mol Sci. (2019) 20:5034. doi: 10.3390/ijms20205034
133. Somers SJ, Frias M, Lacerda L, Opie LH, Lecour S. Interplay between SAFE and RISK pathways in sphingosine-1-phosphate-induced cardioprotection. Cardiovasc Drugs Ther. (2012) 26:227–37. doi: 10.1007/s10557-012-6376-2
134. Kelly-Laubscher RF, King JC, Hacking D, Somers S, Hastie S, Stewart T, et al. Cardiac preconditioning with sphingosine-1-phosphate requires activation of signal transducer and activator of transcription-3. Cardiovasc J Afr. (2014) 25:118–23. doi: 10.5830/CVJA-2014-016
135. Egom EE, Mamas MA, Chacko S, Stringer SE, Charlton-Menys V, El-Omar M, et al. Serum sphingolipids level as a novel potential marker for early detection of human myocardial ischaemic injury. Front Physiol. (2013) 4:130. doi: 10.3389/fphys.2013.00130
136. Hadas Y, Vincek AS, Youssef E, Żak MM, Chepurko E, Sultana N, et al. Altering sphingolipid metabolism attenuates cell death and inflammatory response after myocardial infarction. Circulation. (2020) 141:916–30. doi: 10.1161/CIRCULATIONAHA.119.041882
137. Hua T, Bao Q, He X, Cai W, He J. Lipidomics revealed alteration of sphingolipid metabolism during the reparative phase after myocardial infarction injury. Front Physiol. (2021) 12:663480. doi: 10.3389/fphys.2021.663480
138. de Carvalho LP, Tan SH, Ow GS, Tang Z, Ching J, Kovalik JP, et al. Plasma ceramides as prognostic biomarkers and their arterial and myocardial tissue correlates in acute myocardial infarction. JACC Basic Transl Sci. (2018) 3:163–75. doi: 10.1016/j.jacbts.2017.12.005
139. Klyachkin YM, Nagareddy PR, Ye S, Wysoczynski M, Asfour A, Gao E, et al. Pharmacological elevation of circulating bioactive phosphosphingolipids enhances myocardial recovery after acute infarction. Stem Cells Transl Med. (2015) 4:1333–43. doi: 10.5966/sctm.2014-0273
140. Yang LG, Wang AL, Li L, Yang H, Jie X, Zhu ZF, et al. Sphingosine-1-phosphate induces myocyte autophagy after myocardial infarction through mTOR inhibition. Eur J Pharmacol. (2021) 907:174260. doi: 10.1016/j.ejphar.2021.174260
141. Knapp M, Zendzian-Piotrowska M, Błachnio-Zabielska A, Zabielski P, Kurek K, Górski J. Myocardial infarction differentially alters sphingolipid levels in plasma, erythrocytes and platelets of the rat. Basic Res Cardiol. (2012) 107:294. doi: 10.1007/s00395-012-0294-0
142. Yao K, Wang Y, Xu D, Liu X, Shen C, Hu W, et al. Effect of combined testing of ceramides with high-sensitive troponin T on the detection of acute coronary syndrome in patients with chest pain in China: A prospective observational study. BMJ Open. (2019) 9:e028211. doi: 10.1136/bmjopen-2018-028211
143. Pan W, Yu J, Shi R, Yan L, Yang T, Li Y, et al. Elevation of ceramide and activation of secretory acid sphingomyelinase in patients with acute coronary syndromes. Coron Artery Dis. (2014) 25:230–5. doi: 10.1097/MCA.0000000000000079
144. Burrello J, Biemmi V, Dei Cas M, Amongero M, Bolis S, Lazzarini E, et al. Sphingolipid composition of circulating extracellular vesicles after myocardial ischemia. Sci Rep. (2020) 10:16182. doi: 10.1038/s41598-020-73411-7
145. Karagiannidis E, Papazoglou AS, Stalikas N, Deda O, Panteris E, Begou O, et al. Serum ceramides as prognostic biomarkers of large thrombus burden in patients with STEMI: A micro-computed tomography study. J Pers Med. (2021) 11:89. doi: 10.3390/jpm11020089
146. Park JY, Lee SH, Shin MJ, Hwang GS. Alteration in metabolic signature and lipid metabolism in patients with angina pectoris and myocardial infarction. PLoS One. (2015) 10:e0135228. doi: 10.1371/journal.pone.0135228
147. Huang L, Li T, Liu YW, Zhang L, Dong ZH, Liu SY, et al. Plasma metabolic profile determination in young ST-segment elevation myocardial infarction patients with ischemia and reperfusion: Ultra-performance liquid chromatography and mass spectrometry for pathway analysis. Chin Med J (Engl). (2016) 129:1078–86. doi: 10.4103/0366-6999.180527
148. Knapp M, Baranowski M, Czarnowski D, Lisowska A, Zabielski P, Górski J, et al. Plasma sphingosine-1-phosphate concentration is reduced in patients with myocardial infarction. Med Sci Monit. (2009) 15:Cr490–3.
149. Knapp M, Lisowska A, Zabielski P, Musial W, Baranowski M. Sustained decrease in plasma sphingosine-1-phosphate concentration and its accumulation in blood cells in acute myocardial infarction. Prostaglandins Other Lipid Mediat. (2013) 106:53–61. doi: 10.1016/j.prostaglandins.2013.10.001
150. Kiziltunc E, Abaci A, Ozkan S, Alsancak Y, Unlu S, Elbeg S, et al. The relationship between pre-infarction angina and serum sphingosine-1-phosphate levels. Acta Cardiol Sin. (2014) 30:546–52. doi: 10.1093/eurheartj/eht307.P477
151. Polzin A, Helten C, Dannenberg L, Müller T, Gräler M, Kelm M, et al. Sphingosine-1-phosphate: A mediator of the ARB-MI paradox? Int J Cardiol. (2021) 333:40–2. doi: 10.1016/j.ijcard.2021.02.082
152. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. (2016) 37:2129–200. doi: 10.1093/eurheartj/ehw128
153. Pieske B, Tschöpe C, de Boer RA, Fraser AG, Anker SD, Donal E, et al. How to diagnose heart failure with preserved ejection fraction: The HFA-PEFF diagnostic algorithm: A consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur Heart J. (2019) 40:3297–317. doi: 10.1093/eurheartj/ehz641
154. Schwinger RHG. Pathophysiology of heart failure. Cardiovasc Diagn Ther. (2021) 11:263–76. doi: 10.21037/cdt-20-302
155. Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight. (2017) 2:e82922. doi: 10.1172/jci.insight.82922
156. Hoffman M, Palioura D I, Kyriazis D, Cimini M, Badolia R, Rajan S, et al. Cardiomyocyte krüppel-like factor 5 promotes de novo ceramide biosynthesis and contributes to eccentric remodeling in ischemic cardiomyopathy. Circulation. (2021) 143:1139–56. doi: 10.1161/CIRCULATIONAHA.120.047420
157. Gowda SB, Gowda D, Kain V, Chiba H, Hui SP, Chalfant CE, et al. Sphingosine-1-phosphate interactions in the spleen and heart reflect extent of cardiac repair in mice and failing human hearts. Am J Physiol Heart Circ Physiol. (2021) 321:H599–611. doi: 10.1152/ajpheart.00314.2021
158. Cannavo A, Rengo G, Liccardo D, Pagano G, Zincarelli C, De Angelis MC, et al. β1-adrenergic receptor and sphingosine-1-phosphate receptor 1 (S1PR1) reciprocal downregulation influences cardiac hypertrophic response and progression to heart failure: Protective role of S1PR1 cardiac gene therapy. Circulation. (2013) 128:1612–22. doi: 10.1161/CIRCULATIONAHA.113.002659
159. Deshpande GP, Imamdin A, Lecour S, Opie LH. Sphingosine-1-phosphate (S1P) activates STAT3 to protect against de novo acute heart failure (AHF). Life Sci. (2018) 196:127–32. doi: 10.1016/j.lfs.2018.01.023
160. Knapp M, Baranowski M, Lisowska A, Musial W. Decreased free sphingoid base concentration in the plasma of patients with chronic systolic heart failure. Adv Med Sci. (2012) 57:100–5. doi: 10.2478/v10039-011-0057-4
161. Nwabuo CC, Duncan M, Xanthakis V, Peterson LR, Mitchell GF, McManus D, et al. Association of circulating ceramides with cardiac structure and function in the community: The Framingham heart study. J Am Heart Assoc. (2019) 8:e013050. doi: 10.1161/JAHA.119.013050
162. Wittenbecher C, Eichelmann F, Toledo E, Guasch-Ferre M, Ruiz-Canela M, Li J, et al. Lipid profiles and heart failure risk: Results from two prospective studies. Circ Res. (2021) 128:309–20. doi: 10.1161/CIRCRESAHA.120.317883
163. Pérez-Carrillo L, Giménez-Escamilla I, Martínez-Dolz L I, ánchez-Lázaro JS, Portolés M, Roselló-Lleti E, et al. Implication of sphingolipid metabolism gene dysregulation and cardiac sphingosine-1-phosphate accumulation in heart failure. Biomedicines. (2022) 10:135. doi: 10.3390/biomedicines10010135
164. Yin W, Li F, Tan X, Wang H, Jiang W, Wang X, et al. Plasma ceramides and cardiovascular events in hypertensive patients at high cardiovascular risk. Am J Hypertens. (2021):hab105. [Online ahead of print]. doi: 10.1093/ajh/hpab105
165. Polzin A, Piayda K, Keul P, Dannenberg L, Mohring A, Gräler M, et al. Plasma sphingosine-1-phosphate concentrations are associated with systolic heart failure in patients with ischemic heart disease. J Mol Cell Cardiol. (2017) 110:35–7. doi: 10.1016/j.yjmcc.2017.07.004
166. Javaheri A, Allegood JC, Cowart LA, Chirinos JA. Circulating ceramide 16:0 in heart failure with preserved ejection fraction. J Am Coll Cardiol. (2020) 75:2273–5. doi: 10.1016/j.jacc.2020.02.062
167. Targher G, Lunardi G, Mantovani A, Meessen J, Bonapace S, Temporelli PL, et al. Relation between plasma ceramides and cardiovascular death in chronic heart failure: A subset analysis of the GISSI-HF trial. ESC Heart Fail. (2020) 7:3288–97. doi: 10.1002/ehf2.12885
168. Brugada J, Katritsis DG, Arbelo E, Arribas F, Bax JJ, Blomström-Lundqvist C, et al. 2019 ESC Guidelines for the management of patients with supraventricular tachycardiaThe Task Force for the management of patients with supraventricular tachycardia of the European Society of Cardiology (ESC). Eur Heart J. (2020) 41:655–720.
169. Glikson M, Nielsen JC, Kronborg MB, Michowitz Y, Auricchio A I, Barbash M, et al. 2021 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy. Eur Heart J. (2021) 42:3427–520. doi: 10.1093/eurheartj/ehab699
170. Bai Y, Wang J, Shan H, Lu Y, Zhang Y, Luo X, et al. Sphingolipid metabolite ceramide causes metabolic perturbation contributing to HERG K+ channel dysfunction. Cell Physiol Biochem. (2007) 20:429–40. doi: 10.1159/000107527
171. Ganapathi SB, Fox TE, Kester M, Elmslie KS. Ceramide modulates HERG potassium channel gating by translocation into lipid rafts. Am J Physiol Cell Physiol. (2010) 299:C74–86. doi: 10.1152/ajpcell.00462.2009
172. Chapman H, Ramström C, Korhonen L, Laine M, Wann KT, Lindholm D, et al. Downregulation of the HERG (KCNH2) K(+) channel by ceramide: Evidence for ubiquitin-mediated lysosomal degradation. J Cell Sci. (2005) 118(Pt 22):5325–34. doi: 10.1242/jcs.02635
173. Huang SY, Lu YY, Lin YK, Chen YC, Chen YA, Chung CC, et al. Ceramide modulates electrophysiological characteristics and oxidative stress of pulmonary vein cardiomyocytes. Eur J Clin Invest. (2022) 52:e13690. doi: 10.1111/eci.13690
174. Wu J, Wu Q, Dai W, Kong J, Lv J, Yu X, et al. Serum lipid feature and potential biomarkers of lethal ventricular tachyarrhythmia (LVTA) induced by myocardial ion channel diseases: A rat model study. Int J Legal Med. (2018) 132:439–48. doi: 10.1007/s00414-017-1710-7
175. Wu J, Zhang Y, Wu Q, Xie D, Dai W, Zhang X, et al. Integrative analyses of myocardial lipidome and proteome implicate mitochondrial dysfunction in lethal ventricular tachyarrhythmia (LVTA) induced by acute myocardial ischemia (AMI). J Proteomics. (2019) 197:14–22. doi: 10.1016/j.jprot.2019.01.021
176. Wojcik B, Baranowski M, Chabowski A, Gorski J. Effect of atrial pacing on the level of bioactive sphingolipids in the heart ventricles of the rat. J Physiol Pharmacol. (2015) 66:385–9.
177. Himmel HM, Meyer Zu Heringdorf D, Graf E, Dobrev D, Kortner A, Schüler S, et al. Evidence for Edg-3 receptor-mediated activation of I(K.ACh) by sphingosine-1-phosphate in human atrial cardiomyocytes. Mol Pharmacol. (2000) 58:449–54. doi: 10.1124/mol.58.2.449
178. Ochi R, Momose Y, Oyama K, Giles WR. Sphingosine-1-phosphate effects on guinea pig atrial myocytes: Alterations in action potentials and K+ currents. Cardiovasc Res. (2006) 70:88–96. doi: 10.1016/j.cardiores.2006.01.010
179. Harris CM, Mittelstadt S, Banfor P, Bousquet P, Duignan DB, Gintant G, et al. Sphingosine-1-Phosphate (S1P) lyase inhibition causes increased cardiac S1P levels and bradycardia in rats. J Pharmacol Exp Ther. (2016) 359:151–8. doi: 10.1124/jpet.116.235002
180. Jensen PN, Fretts AM, Hoofnagle AN, Sitlani CM, McKnight B I, King B, et al. Plasma ceramides and sphingomyelins in relation to atrial fibrillation risk: The cardiovascular health study. J Am Heart Assoc. (2020) 9:e012853. doi: 10.1161/JAHA.119.012853
181. Mori M. Lethal arrhythmia due to fingolimod, a S1P receptor modulator: Are we overestimating or underestimating? J Neurol Neurosurg Psychiatry. (2015) 86:823. doi: 10.1136/jnnp-2015-310451
182. Sanna MG, Vincent KP, Repetto E, Nguyen N, Brown SJ, Abgaryan L, et al. Bitopic Sphingosine 1-Phosphate Receptor 3 (S1P3) antagonist rescue from complete heart block: Pharmacological and genetic evidence for direct S1P3 regulation of mouse cardiac conduction. Mol Pharmacol. (2016) 89:176–86. doi: 10.1124/mol.115.100222
183. Pournajaf S, Dargahi L, Javan M, Pourgholami MH. Molecular pharmacology and novel potential therapeutic applications of fingolimod. Front Pharmacol. (2022) 13:807639. doi: 10.3389/fphar.2022.807639
184. Lasa JS, Olivera PA, Bonovas S, Danese S, Peyrin-Biroulet L. Safety of S1P modulators in patients with immune-mediated diseases: A systematic review and meta-analysis. Drug Saf. (2021) 44:645–60. doi: 10.1007/s40264-021-01057-z
185. Holzwirth E, Fischer-Schaepmann T, Obradovic D, von Lucadou M, Schwedhelm E, Daum G, et al. Anti-inflammatory HDL effects are impaired in atrial fibrillation. Heart Vessels. (2022) 37:161–71. doi: 10.1007/s00380-021-01908-w
186. Sordillo PP, Sordillo DC, Helson L. Review: The prolonged QT interval: Role of Pro-inflammatory cytokines, reactive oxygen species and the ceramide and sphingosine-1 phosphate pathways. In Vivo. (2015) 29:619–36.
187. Thrift AG, Thayabaranathan T, Howard G, Howard VJ, Rothwell PM, Feigin VL, et al. Global stroke statistics. Int J Stroke. (2017) 12:13–32. doi: 10.1177/1747493016676285
188. Kimura A, Ohmori T, Kashiwakura Y, Ohkawa R, Madoiwa S, Mimuro J, et al. Antagonism of sphingosine 1-phosphate receptor-2 enhances migration of neural progenitor cells toward an area of brain. Stroke. (2008) 39:3411–7. doi: 10.1161/STROKEAHA.108.514612
189. Salas-Perdomo A, Miró-Mur F, Gallizioli M, Brait VH, Justicia C, Meissner A, et al. Role of the S1P pathway and inhibition by fingolimod in preventing hemorrhagic transformation after stroke. Sci Rep. (2019) 9:8309. doi: 10.1038/s41598-019-44845-5
190. Brunkhorst R, Kanaan N, Koch A, Ferreirós N, Mirceska A, Zeiner P, et al. FTY720 treatment in the convalescence period improves functional recovery and reduces reactive astrogliosis in photothrombotic stroke. PLoS One. (2013) 8:e70124. doi: 10.1371/journal.pone.0070124
191. Moon E, Han JE, Jeon S, Ryu JH, Choi JW, Chun J. Exogenous S1P exposure potentiates ischemic stroke damage that is reduced possibly by inhibiting S1P receptor signaling. Mediators Inflamm. (2015) 2015:492659. doi: 10.1155/2015/492659
192. Gaire BP, Lee CH, Sapkota A, Lee SY, Chun J, Cho HJ, et al. Identification of Sphingosine 1-phosphate receptor subtype 1 (S1P(1)) as a pathogenic factor in transient focal cerebral ischemia. Mol Neurobiol. (2018) 55:2320–32. doi: 10.1007/s12035-017-0468-8
193. Cao C, Dai L, Mu J, Wang X, Hong Y, Zhu C, et al. S1PR2 antagonist alleviates oxidative stress-enhanced brain endothelial permeability by attenuating p38 and Erk1/2-dependent cPLA(2) phosphorylation. Cell Signal. (2019) 53:151–61. doi: 10.1016/j.cellsig.2018.09.019
194. Wan Y, Jin HJ, Zhu YY, Fang Z, Mao L, He Q, et al. MicroRNA-149-5p regulates blood-brain barrier permeability after transient middle cerebral artery occlusion in rats by targeting S1PR2 of pericytes. Faseb J. (2018) 32:3133–48. doi: 10.1096/fj.201701121R
195. Gaire BP, Song MR, Choi JW. Sphingosine 1-phosphate receptor subtype 3 (S1P(3)) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J Neuroinflammation. (2018) 15:284. doi: 10.1186/s12974-018-1323-1
196. Brait VH, Tarrasón G, Gavalda A, Godessart N, Planas AM. Selective Sphingosine 1-phosphate receptor 1 agonist is protective against ischemia/reperfusion in mice. Stroke. (2016) 47:3053–6. doi: 10.1161/STROKEAHA.116.015371
197. Sapkota A, Gaire BP, Kang MG, Choi JW. S1P(2) contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci Rep. (2019) 9:12106. doi: 10.1038/s41598-019-48609-z
198. Kim GS, Yang L, Zhang G, Zhao H, Selim M, McCullough LD, et al. Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat Commun. (2015) 6:7893. doi: 10.1038/ncomms8893
199. Zheng S, Wei S, Wang X, Xu Y, Xiao Y, Liu H, et al. Sphingosine kinase 1 mediates neuroinflammation following cerebral ischemia. Exp Neurol. (2015) 272:160–9. doi: 10.1016/j.expneurol.2015.03.012
200. Lv M, Zhang D, Dai D, Zhang W, Zhang L. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm Res. (2016) 65:551–62. doi: 10.1007/s00011-016-0939-9
201. Nakagawa S, Aruga J. Sphingosine 1-phosphate signaling is involved in impaired blood-brain barrier function in ischemia-reperfusion injury. Mol Neurobiol. (2020) 57:1594–606. doi: 10.1007/s12035-019-01844-x
202. Su D, Cheng Y, Li S, Dai D, Zhang W, Lv M. Sphk1 mediates neuroinflammation and neuronal injury via TRAF2/NF-κB pathways in activated microglia in cerebral ischemia reperfusion. J Neuroimmunol. (2017) 305:35–41. doi: 10.1016/j.jneuroim.2017.01.015
203. Yung LM, Wei Y, Qin T, Wang Y, Smith CD, Waeber C. Sphingosine kinase 2 mediates cerebral preconditioning and protects the mouse brain against ischemic injury. Stroke. (2012) 43:199–204. doi: 10.1161/STROKEAHA.111.626911
204. Wacker BK, Park TS, Gidday JM. Hypoxic preconditioning-induced cerebral ischemic tolerance: Role of microvascular sphingosine kinase 2. Stroke. (2009) 40:3342–8. doi: 10.1161/STROKEAHA.109.560714
205. Pfeilschifter W, Czech-Zechmeister B, Sujak M, Mirceska A, Koch A, Rami A, et al. Activation of sphingosine kinase 2 is an endogenous protective mechanism in cerebral ischemia. Biochem Biophys Res Commun. (2011) 413:212–7. doi: 10.1016/j.bbrc.2011.08.070
206. Herr I, Martin-Villalba A, Kurz E, Roncaioli P, Schenkel J, Cifone MG, et al. FK506 prevents stroke-induced generation of ceramide and apoptosis signaling. Brain Res. (1999) 826:210–9. doi: 10.1016/S0006-8993(99)01288-3
207. Nakane M, Kubota M, Nakagomi T, Tamura A, Hisaki H, Shimasaki H, et al. Lethal forebrain ischemia stimulates sphingomyelin hydrolysis and ceramide generation in the gerbil hippocampus. Neurosci Lett. (2000) 296:89–92. doi: 10.1016/S0304-3940(00)01655-4
208. Yu ZF, Nikolova-Karakashian M, Zhou D, Cheng G, Schuchman EH, Mattson MP. Pivotal role for acidic sphingomyelinase in cerebral ischemia-induced ceramide and cytokine production, and neuronal apoptosis. J Mol Neurosci. (2000) 15:85–97. doi: 10.1385/JMN:15:2:85
209. Ohtani R, Tomimoto H, Kondo T, Wakita H, Akiguchi I, Shibasaki H, et al. Upregulation of ceramide and its regulating mechanism in a rat model of chronic cerebral ischemia. Brain Res. (2004) 1023:31–40. doi: 10.1016/j.brainres.2004.07.024
210. Lee TH, Cheng CN, Chao HC, Lee CH, Kuo CH, Tang SC, et al. Plasma ceramides are associated with outcomes in acute ischemic stroke patients. J Formos Med Assoc. (2022) 121(Pt 1):43–50. doi: 10.1016/j.jfma.2021.01.006
211. Gui YK, Li Q, Liu L, Zeng P, Ren RF, Guo ZF, et al. Plasma levels of ceramides relate to ischemic stroke risk and clinical severity. Brain Res Bull. (2020) 158:122–7. doi: 10.1016/j.brainresbull.2020.03.009
212. Fiedorowicz A, Kozak-Sykała A, Bobak L, Kałas W, Strza̧dała L. Ceramides and sphingosine-1-phosphate as potential markers in diagnosis of ischaemic stroke. Neurol Neurochir Pol. (2019) 53:484–91. doi: 10.5603/PJNNS.a2019.0063
213. Hong L, Hongmei W, Leijie X, Dandan Z, Peng L, Zhifei H, et al. Serum ceramide concentrations are associated with depression in patients after ischemic stroke-A two-center case-controlled study. Clin Chim Acta. (2021) 518:110–5. doi: 10.1016/j.cca.2021.03.014
Keywords: sphingolipids, heart failure, hypertension, coronary artery disease, ceramides, sphingosine-1 phosphate, stroke, cardiovascular disease
Citation: Borodzicz-Jażdżyk S, Jażdżyk P, Łysik W, Cudnoch-Jȩdrzejewska A and Czarzasta K (2022) Sphingolipid metabolism and signaling in cardiovascular diseases. Front. Cardiovasc. Med. 9:915961. doi: 10.3389/fcvm.2022.915961
Received: 08 April 2022; Accepted: 05 August 2022;
Published: 31 August 2022.
Edited by:
Marco Piccoli, IRCCS San Donato Polyclinic, ItalyReviewed by:
Sara Grassi, University of Milan, ItalyOwais Bhat, Virginia Commonwealth University, United States
Paola Signorelli, University of Milan, Italy
Copyright © 2022 Borodzicz-Jażdżyk, Jażdżyk, Łysik, Cudnoch-Jȩdrzejewska and Czarzasta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarzyna Czarzasta, katarzyna.czarzasta@wum.edu.pl