
Abstract
Type III secretion system (T3SS) effector proteins are primarily recognized for binding host proteins to subvert host immune response during infection. Besides their known host target proteins, several T3SS effectors also interact with endogenous bacterial proteins. Here we demonstrate that the Salmonella T3SS effector glycosyltransferase SseK1 glycosylates the bacterial two-component response regulator OmpR on two arginine residues, R15 and R122. Arg-glycosylation of OmpR results in reduced expression of ompF, a major outer membrane porin gene. Glycosylated OmpR has reduced affinity to the ompF promoter region, as compared to the unglycosylated form of OmpR. Additionally, the Salmonella ΔsseK1 mutant strain had higher bile salt resistance and increased capacity to form biofilms, as compared to WT Salmonella, thus linking OmpR glycosylation to several important aspects of bacterial physiology.
Similar content being viewed by others


Introduction
Salmonella is responsible for ~ 1.35 million infections in the USA each year1. Identifying virulence factor mechanisms involved in pathogenesis and environmental persistence is essential to finding better approaches to reduce Salmonella disease burden. Salmonella uses a specialized secretion system named Type Three Secretion System (T3SS) to inject effector proteins into host cells2,3,4. Many of these effector proteins inhibit host immune responses. The Salmonella T3S effector SseK and its ortholog NleB target host cell immune response pathways to reduce host inflammatory responses5. SseK and NleB are glycosyltransferases that glycosylates several host proteins and on specific arginine residues and interfere with their physiological function6,7,8. For example, SseK1 glycosylates the death domain containing protein TRADD (Tumor necrosis factor Receptor type 1-Associated DEATH Domain protein), and Tubulin Folding Cofactor TBCB7,9,10,11. SseK2 glycosylates FADD (FAS-Associated Death Domain protein)7. NleB from EHEC is known to target FADD, TRADD, RIPK1 (Receptor Interacting Protein Kinase 1), TNFR1 (Tumor Necrosis Factor Receptor superfamily 1), and HIF-1α protein12,13. These modifications ultimately interfere with proper physiological functions of the target proteins. Besides death domain containing proteins, NleB1 glycosylates arginine residues of GAPDH13. Arginine glycosylation of GAPDH prevents its interaction with TRAF2 and the subsequent induction of NF-kB signaling. Deletion of any one of these glycosyltransferase effectors is correlated with reduced bacterial virulence in a mouse model8,9.
In addition to their known host targets, our group and others have recently demonstrated that NleB/SseK orthologs also glycosylate bacterial proteins. For example, Citrobacter rodentium effector NleB Arg-glycosylates the glutathione synthase GshB, leading to enhanced glutathione synthase activity and consequently increased resistance to oxidative stress14. Salmonella T3SS effector SseK1 also plays a significant role in methylglyoxal detoxification by glycosylating the GloA, GloB, GloC, and YajL proteins in this pathway15. Additionally, our latest study on SseK1 intrabacterial activity demonstrates that SseK1 upregulates UDP-GlcNAc synthesis by glycosylating NagC and GlmR16.
Two-component response regulators are used by bacteria to sense and respond accordingly to the surrounding environment17,18,19. Two-component systems are comprised of a membrane-bound kinase and a corresponding response regulator that exerts the effect of the external stimuli typically by regulating transcription of target genes20. It was recently described that SseK3-mediated Arg-glycosylation plays an important role in modulating the DNA-binding activity of Salmonella PhoP, a two-component response regulator21. Another critical two component response regulator of Salmonella is the EnvZ-OmpR system. The EnvZ-OmpR system is known for its regulatory effects on the major outer membrane porins OmpF and OmpC in response to extracellular pH and osmolarity change22,23,24. With increasing osmolarity, EnvZ phosphorylates the response regulator protein OmpR. Once phosphorylated, the binding affinity of OmpR to target gene promoters increases, resulting in their transcriptional upregulation25,26. Additionally, OmpR can also non-canonically regulate transcription of its target genes under its non-phosphorylated state23. Besides regulating several stress related genes27,28,29,30,31,32, OmpR also regulates the expression of effector genes in Salmonella Pathogenicity Island 2 (SPI2) which is especially relevant to the intracellular adaptation of Salmonella26,33,34,35,36,37.
In a previous study, pull down experiments combined with mass spectrometry sugar analysis were performed to identify novel host targets of effector glycosyltransferase, wherein OmpR was unexpectedly detected as SseK1 target. Here we found that OmpR is glycosylated by SseK1. Glycosylation of OmpR leads to decreased expression of its target gene ompF, presumably by reducing the binding affinity of OmpR to the ompF promoter region. We also found that whereas a Salmonella ΔompR mutant has a significant growth defect in the presence of bile salts and a reduced capacity to form biofilms, a Salmonella ΔsseK1 mutant has the opposite phenotype, indicating an overall repression of OmpR transcriptional activity through SseK1 mediated-glycosylation.
Results
SseK1 glycosylates OmpR
Salmonella enterica serover Typhimurium encodes three SseK glycosyltransferase paralogs named SseK1,SseK2, and SseK3. We expressed OmpR in wild type, single, double, and triple sseK mutants. We used an anti R-GlcNac monoclonal antibody to conduct western blot analysis of cell lysates expressing recombinant His tagged OmpR to investigate which SseK paralog glycosylates OmpR. Only SseK1 glycosylated OmpR (Fig. 1A). Additionally, another two component response regulator, QseF, was not glycosylated by any of the SseK paraologs, serving as a negative control for the assay (Fig. 1A). We conducted an in vitro glycosylation assay with purified SseK1, a catalytically inactive mutant of SseK1 (SseK1 HEN mutant), and OmpR (Fig. 1B). In vitro glycosylation assays demonstrated that OmpR is glycosylated by wild-type SseK1 whereas SseK1 HEN failed to glycosylate OmpR with GlcNAc (Fig. 1C).

SseK1 Arg-glycosylates OmpR. (A) Western blot analysis of intra-bacterial glycosylation of OmpR and QseF in different Salmonella sseK mutant strains; (B) SDS-PAGE image of the enzymes and substrates used for in vitro glycosylation assays; (C) Western blot analysis of in vitro glycosylation of OmpR in the presence of active or inactive (HEN) forms of SseK1.
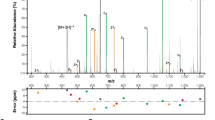
After confirming that OmpR is a bacterial SseK1 target, we wanted to identify the specific OmpR glycosylation sites. We detected, using mass spectrometry suger analysis, two potential OmpR arginine residues, R15 and R122 (Fig. 2A,B). We validated the mass spectrometry data by creating recombinant OmpR proteins with R15A, R122A, and R15,R122A point mutations. We expressed the recombinant OmpR point mutants in WT Salmonella. Western blot analysis of cell lysates indicated that OmpR R12A is glycosylated, albeit at a reduced level. The R122A and R15,R122A point mutants were not glycosylated, indicating that R122 is the primary SseK1 target residue (Fig. 2C).

Identification of OmpR glycosylation sites. (A) HCD spectra of the in vivo glycosylated OmpR tryptic peptides containing glycosylated R15; (B) R122; (C) Western blot verification of Arg-glycosylation of WT and R-to-A point mutations of OmpR.
Glycosylation of OmpR results in altered expression of ompF
OmpR is a transcriptional regulator. To understand the consequence of OmpR glycosylation by SseK1, we measured the transcription of an important OmpR-regulated gene, ompF. OmpF is a key outer membrane porin protein that is essential for Salmonella adaptability to pH and osmolarity stress30,38,39. We used a mRFP (monomeric red fluorescent protein) transcriptional fusion assay wherein the mRFP gene was fused with the upstream promoter region of ompF and the RFP levels were measured in either WT, ΔsseK1, ΔompR, or ΔsseK1/ompR double mutant strains. Our mRFP transcriptional reporter assay data showed a decreased mRFP signal for ompF promoter fusions in WT Salmonella as compared to the ΔsseK1 strain (Fig. 3A). As expected, ΔompR and the ΔsseK1/ompR double mutant showed significantly reduced activity of the ompF promoter (Fig. 3A). There was no significant growth difference among the strains (Fig. 3B).

ompF::mrfp transcriptional reporter assay. (A) Measurement of mRFP expression levels of ompF::rfp transcriptional fusions in WT Salmonella enterica and its ΔsseK1 or ΔompR or ΔsseK1/ΔompR derivatives. mRFP levels are expressed as RFU (relative fluorescence units)/OD600 ratio; (B) Growth rates of Salmonella strains used in the mRFP fusion assay.
To further validate our findings, we complemented ΔsseK1 Salmonella with either sseK1 or sseK1 HEN (inactive) and repeated the mRFP transcriptional assay. Our assay showed a significant reduction of ompF promoter activity in the SseK1-complemented strain, as compared to the SseK1 HEN (inactive)-complemented strain (Fig. 4A). As expected, Salmonella ΔompR and ΔsseK1/ompR mutant strains failed to induce any significant mRFP signal (Figs. 3A and 4A). There was no significant growth difference among the strains (Fig. 4B).

ompF::mrfp transcriptional reporter assay of ΔsseK1 complemented strains. (A) Measurement of mRFP expression levels of ompF::rfp transcriptional fusions in ΔsseK1 or ΔsseK1/ompR Salmonella complemented with either sseK1 or sseK1 HEN. mRFP levels are expressed as RFU (relative fluorescence units)/OD600 ratio; (B) Growth rates of Salmonella strains used in the mRFP fusion assay.
To understand the molecular mechanism of SseK1-mediated reduced promoter activity of OmpR target genes, we conducted an EMSA assay in which purified native or Arg-glycosylated OmpR proteins (Fig. 5A) were incubated with fluorescently labelled ompF promoter region DNA. We observed reduced affinity of glycosylated OmpR to its target DNA as compared to unglycosylated OmpR (Fig. 5B), consistent with the ompF promoter activity assay data.

Glycosylation reduces OmpR DNA binding affinity. (A) Purification of native and Arg-glycosylated OmpR combined with validation of glycosylation by Western blotting; (B) EMSAs comparing the DNA-binding activity of OmpR and OmpR-GlcNAc towards ompF promoter DNA.
Salmonella ΔsseK1 strain has increased bile tolerance and biofilm formation capacity
OmpR modulates several critical Salmonella pathways34,35,40,41, one of which is bile salt tolerance38. Bile salts are natural antimicrobial compounds produced by the host to reduce pathogen proliferation42. OmpR is a positive regulator of bile salt tolerance43 and thus we wanted to investigate whether glycosylation of OmpR by SseK1 has any effect on bile salt tolerance. We measured the growth of WT, ΔsseK1, ΔompR, and ΔsseK1/ompR double mutant Salmonella strains for their capacity to grow in the presence of bile salts. Our data shows that in the presence of 0.6% sodium deoxycholate, ΔsseK1 Salmonella strains grew faster than WT Salmonella (Fig. 6A,B). In the presence of 0.3% sodium deoxycholate, the difference in growth was reduced (Fig. 6A). In contrast, the Salmonella ΔompR mutant grew poorly in bile salts, as expected (Fig. 6B). The ΔsseK1/ompR double mutant also grew poorly in the presence of bile salts, indicating that the observed increased bile salt tolerance of ΔsseK1 mutant Salmonella is exerted through OmpR (Fig. 6B). No significant growth difference between these strains was observed when they were grown in the absence of sodium deoxycholate (Fig. 6A).

OmpR glycosylation results in altered bile salt tolerance and biofilm formation capacity of Salmonella. (A) Growth curve analysis of WT Salmonella enterica and its ΔsseK1 or ΔompR or ΔsseK1/ompR derivatives in no bile salt, 0.3% bile salt (sodium deoxycholate), 0.6% bile salt; (B) CFU counts of Salmonella strains after 16 h growth in 0.6% bile salts in LB; (C) Biofilm formation capacity of different Salmonella enterica mutant and complemented strains. Mutant strains contain empty vector. Complemented strains harbors plasmids expressing either WT sseK1 or sseK1 HEN point mutant. Biofilms were also classified as described by Christensen et al.73 into the following categories: non-adherent (0), weakly (+), moderately (++), or strongly (+++) adherent, based upon the ODs of bacterial biofilm.
Another key virulence aspect of Salmonella is the biofilm formation process44,45,46. OmpR is a positive regulator of biofilm formation in several pathogens including E. coli, Klebsiella, and Salmonella43,47,48. We hypothesized that OmpR glycosylation might have an impact on Salmonella biofilm formation capacity. We compared the biofilm formation ability of Salmonella WT, ΔsseK1, ΔompR and ΔsseK1/ompR. ΔsseK1 produced significantly more biofilm as compared to WT Salmonella (Fig. 6C). As expected, the ΔompR strain formed significantly less biofilm as compared to the WT strain (Fig. 6C). Additionally, we observed no statistically significant difference in biofilm production between ΔompR and the ΔompR/sseK1 double mutant, indicating an epistatic effect of the ompR mutation on sseK1 for this particular phenotype (Fig. 6C). This observation was further supported by the complementation studies where expression of sseK1 did not complement the biofilm formation defect seen in ΔompR/sseK1 double mutant (Fig. 6C).
Discussion
Salmonella harbors two Pathogenicity Islands—SPI1 and SPI2. Although encoded by a gene located outside SPI1 and SPI2, SseK1 was shown to be secreted by both T3SS1 and T3SS2 secretion systems49. Once translocated, SseK1 glycosylates host proteins to reduce host immune responses during infection10. SseK1 and its ortholog NleB also glycosylate several bacterial proteins. Here we demonstrate that among the SseK paralogs, only SseK1 targets the two-component response regulator OmpR indicating a highly specific interaction, the effect of which was observed through the differential expression of an OmpR target gene—ompF.
The regulation of ompF by OmpR is a complex multiunit affair with several other transcription factors working in concert to either up- or down-regulate ompF expression24,50. Although the total amount of outer membrane porins (OMPs) of a cell generally does not change significantly, the combination of different OMPs is altered in response to different environmental stimuli. These changes are orchestrated largely by the ratio of phosphorylated and unphosphorylated OmpR along with several other regulators of OMP expression48,51,52,53. While conducting the first set of mRFP fusion assays we used either WT or mutant Salmonella strains to assess the effect of endogenously expressed sseK1 on OmpR transcriptional activity. A significant differential expression level of ompF promoter activity was detected under these conditions, indicating that OmpR glycosylation by SseK1 is likely to occur as an evolutionary bacterial adaptation to specific stress conditions.
Since ompF is part of the OmpR regulon54, we investigated whether OmpR glycosylation alters its DNA binding affinity of ompF promoter. Our results showed a reduction in DNA binding capacity of glycosylated OmpR as compared to the unglycosylated OmpR. Interestingly, the OmpR glycosylation sites detected by mass spectrometry analysis and validated by site directed mutagenesis are located outside the OmpR DNA-binding motif55,56, rather than in the N-terminal response regulatory region. Moreover, the OmpR glycosylation sites R15 and R122 are not in vicinity of the D55 phosphorylation target site22,51,57. One potential explanation of these data is that glycosylation could alter the conformation of the OmpR DNA binding domain to prevent its binding to target promoters. The silenced affinity of OmpR to its target promoters upon glycosylation by SseK1 possibly evolved as a gene modulatory mechanism to reverse the enhanced affinity of this transcriptional factor to DNA upon phosphorylation by EnvZ at the residue D55. Thus, this study illustrates an original example of bacterial transcription gene regulation where the same transcription factor can undergo two different post-transcriptional modifications (phosphorylation and glycosylation) with opposite effects on target gene expression.
Two-component response regulators are key players in bacterial adaptation to dynamic environmental conditions18,19,58. OmpR is vital for Salmonella adaptation in changing environments. As such, we investigated the significance of OmpR glycosylation by SseK1. Even though our finding that Salmonella ΔsseK1 strain has increased bile salt and biofilm production capacity seems counterintuitive at the first glance, we believe it can be rationally explained. Our experiments were performed in laboratory conditions with rich media which are not always ideally representative of the natural pathogen lifestyle. In Salmonella, SseK1 is transcribed from SPI-2, which is upregulated during its intracellular lifestyle4,33,34,35,59,60,61. While inside the host cell, increased SseK1 expression could lead to increased glycosylation of OmpR, leading to downregulation of pathways that are of less significance in the intracellular environment, such as bile salt tolerance and biofilm formation. Conversely, outside of host cells, SPI-2 is not induced62,63,64,65, leading to less SseK1 which then leads to lower inhibition of OmpR activity. This would allow WT Salmonella to exhibit more of a ΔsseK1 mutant-like phenotype during its extracellular lifestyle, with increased bile salt tolerance and surface adherence.
In contrast to our recent work on NagC glycosylation leading to increased DNA binding affinity16, here we observed decreased DNA binding affinity of glycosylated OmpR. One significant difference between glycosylated NagC and glycosylated OmpR is that the glycosylated Arg residues of NagC reside within the HTH DNA binding motif. Combined with work on PhoP glycosylation leading to altered DNA affinity, this work reaffirms the phenomenon of T3SS effector glycosyltransferases altering transcription factors to modulate gene expression. In addition to identifying additional glycosyltransferase targets, understanding the biochemical fundamentals of how glycosylation of different transcription factors can alter their DNA binding capacity and how it could differ among transcription factors merits additional research.
Materials and methods
Plasmids, strains, and cloning
The plasmids and strains used in this study are listed in Tables 1 and 2, respectively. Wild type sseK1 (Salmonella enterica) and its derivative H244A E255A N256A, were cloned into pET42a using ABC cloning66. ompR and qseF were cloned in pTac using ABC cloning66. ompR and sseK1/ompR deletions were constructed using lambda red recombination with the pKD3 and pKD119 plasmids67. Mutants were screened on LB medium supplemented with 10 µg/mL chloramphenicol and mutations were confirmed by PCR and DNA sequencing. Protein purification was performed as described previously14. For the purification of glycosylated OmpR, His-tagged OmpR was co-expressed (or not) with FLAG-tagged SseK1 and purified against the His-epitope, as described previously15.
Glycosyltransferase assay
In vitro glycosylation assays were conducted as previously described7. 200 nM SseK1 or SseK1 HEN were incubated in 50 mM Tris–HCl buffer pH 7.4, 1 mM UDP-GlcNAc, 10 mM MnCl2, and 1 mM DTT with 1 mM OmpR. After a 2-h incubation period at room temperature, samples were blotted with anti-R-GlcNAc and anti-His tag monoclonal antibodies (Abcam, Cambridge, MA, USA). Western blot images were captured in a LI-COR (LI-COR Biosciences, Lincoln, NY, USA) imager.
Digest of gel-separated proteins
Affinity-purified proteins were separated by SDS-PAGE, fixed, and then visualized with Coomassie staining. Bands of interest were excised and Coomassie staining removed by destaining with 50 mM NH4HCO3, 50% ethanol for 20 min at room temperature with shaking at 750 rpm. Destained samples then dehydrated with 100% ethanol, before being reduced by being rehydrated with 10 mM DTT in 50 mM NH4HCO3. Samples were reduced for 1 h at 56 °C with shaking and then washed twice in 100% ethanol for 10 min to remove DTT. Reduced dehydrated gel bands were then rehydrated with 55 mM iodoacetamide in 50 mM NH4HCO3 and allowed to alkylate in the dark for 45 min at room temperature. Alkylation buffer was removed, and the gel samples washed with 50 mM NH4HCO3, followed by two rounds of 100% ethanol before being vacuum dried. Alkylated samples were then rehydrated with 20 ng/µL of trypsin (Promega) in 40 mM NH4HCO3 at 4 °C for 1 h. Excess protease was removed, gel pieces were covered in 40 mM NH4HCO3 and incubated overnight at 37 °C. Peptides were collected, desalted using homemade R3/C18 stage tips as previously described68 before analysis by LC–MS (Supplementary Fig. 1).
Reverse phase LC–MS/MS
Peptide samples were resuspended in Buffer A* (2% MeCN, 0.1% TFA) and separated using a two-column chromatography set on a Dionex Ultimate 3000 UHPLC (Thermo Fisher Scientific). Samples were first concentrated on a PepMap100 C18 20 mm × 75 μm trap at 5 μl/min for 5 min with Buffer A (0.1% formic acid, 2% DMSO) and then separated on a PepMap C18 500 mm × 75 μm analytical column (Thermo Fisher Scientific). Separated peptide were infused into a Orbitrap Eclipse Mass Spectrometer (Thermo Fisher Scientific) at 300 nL/min for 65-min by altering the buffer composition from 2% Buffer B (0.1% formic acid, 77.9% acetonitrile, 2% DMSO) to 28% B over 35 min, then from 28% B to 4% B over 10 min, then from 40% B to 80% B over 5 min. The composition was held at 100% B for 5 min, and then dropped to 2% B over 1 min before being held at 2% B for another 9 min. The Eclipse Mass Spectrometer was operated in a data-dependent mode, acquiring one full precursor scan (resolution 120,000; 375–2000 m/z, AGC target of 1 × 106) followed by up to 3 s of data-dependent HCD MS-MS events (using three collision energies of 25, 30, and 35; resolution 15k AGC target of 250% with a maximum injection time of 22 ms).
Mass spectrometry data analysis
Identification of Arg-glycosylation events was accomplished using MaxQuant (v1.6.17.0)69. The predicted amino acid sequences for OmpR were combined into a database with the Escherichia coli K12 proteome (Uniprot accession: UP000000625) the Salmonella Typhimurium SL1344 OmpR-his sequence and searched, allowing carbamidomethylation of cysteine set as a fixed modification and the variable modifications of oxidation of methionine and Arg-GlcNAcylation (H13C8NO5; 203.0793 Da to Arginine). Searches were performed with Trypsin cleavage specificity, allowing 2 miscleavage events with a maximum false discovery rate (FDR) of 1.0% set for protein and peptide identifications. The resulting modified peptide output was processed within the Perseus (v1.4.0.6)70 analysis environment to remove reverse matches and common protein contaminants. To ensure high quality data, assigned glycopeptides were manually assessed and the HCD spectra assigned to each unique glycopeptide annotated with the Interactive Peptide Spectral Annotator71 (http://www.interactivepeptidespectralannotator.com/PeptideAnnotator.html).
mRFP reporter assay
A low-copy number plasmid (pHG165) carrying ompF promoter transcriptional fusions to mRFP (monomeric red fluorescent protein) was electroporated into Salmonella. 200 µL of LB media with Cb was used to grow the transformed bacteria in 96 well clear bottom black walled assay plates. mRFP expression levels were measured every 20 min of growth by a synergy H1 microplate reader. OD600 values were measured concurrently and mRFP data were presented as an average of RFU (Relative Fluorescence Units)/OD600 ratio.
Electrophoretic mobility shift assay (EMSA)
A 5′ Alexa-fluor labelled DNA corresponding to the Salmonella ompF promoter region was amplified by PCR from Salmonella gDNA using the oligonucleotides: 5′ Alexa-fluor-tttttacgtcacactcaaggccagctatgctg-3′ and 5′-ttattaccctcattggtttttttatatgac-3′ as forward and reverse primers respectively. Two nmoles of purified PCR product were incubated for 10 min at room temperature in the presence of either OmpR or OmpR-GlcNAc in 10 μL buffer containing 50 mM HEPES, 100 mM K glutamate (pH 8.0), and 0.5 mg/mL BSA. Samples (10 µL) were loaded on 0.5% agarose gels and subjected to electrophoresis in 0.5 × TBE buffer. DNA–protein complexes were visualized by using a Li-COR Odyssey.
Bile salt resistance and biofilm assays
Overnight cultures of Salmonella strains were diluted 1:100 to start a growth assay in LB with 0.6% or 0.3% Sodium deoxycholate in a 96 well plates with OD600 values measured every 3 h. For biofilm assays, overnight cultures of Salmonella strains were inoculated at 1:100 dilution into LB without sodium chloride into 96 well polystyrene plates. The plate was incubated at 30 °C without agitation. After 36 h of growth, the planktonic cells were removed, and wells were washed 3 times with PBS. Biofilm was fixed by adding 200 μL of methanol to the wells and incubating for 20 min at room temperature. 150 μL of 1% (w/v) crystal violet solution was added to the wells and incubated for 15 min. Wells were rinsed with PBS and air-dried. 150 μL of 30% (v/v) acetic acid was added to the wells and the plate was shaken gently to solubilize the crystal violet. The OD570 was measured to quantify biofilm.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE72 partner repository with the dataset identifier PXD039412.
References
Salmonella Homepage | CDC. Published February 2, 2022. https://www.cdc.gov/salmonella/index.html. Accessed Feb 13, 2022.
Büttner, D. Protein export according to schedule: Architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 76(2), 262–310. https://doi.org/10.1128/MMBR.05017-11 (2012).
Deng, W. et al. Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 15(6), 323–337. https://doi.org/10.1038/nrmicro.2017.20 (2017).
DeSouzaSantos, M. & Orth, K. The role of the type III secretion system in the intracellular lifestyle of enteric pathogens. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.BAI-0008-2019 (2019).
Araujo-Garrido, J. L., Bernal-Bayard, J. & Ramos-Morales, F. Type III secretion effectors with arginine N-glycosyltransferase activity. Microorganisms 8(3), 357. https://doi.org/10.3390/microorganisms8030357 (2020).
Newton, H. J. et al. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-κB p65. PLoS Pathog. 6, 5. https://doi.org/10.1371/journal.ppat.1000898 (2010).
El Qaidi, S. et al. NleB/SseK effectors from Citrobacter rodentium, Escherichia coli, and Salmonella enterica display distinct differences in host substrate specificity. J. Biol. Chem. 292(27), 11423–11430. https://doi.org/10.1074/jbc.M117.790675 (2017).
Kelly, M. et al. Essential role of the type III secretion system effector NleB in colonization of mice by Citrobacter rodentium. Infect. Immun. 74(4), 2328–2337. https://doi.org/10.1128/IAI.74.4.2328-2337.2006 (2006).
Xue, J. et al. Arg-GlcNAcylation on TRADD by NleB and SseK1 is crucial for bacterial pathogenesis. Front. Cell Dev. Biol. 8, 641. https://doi.org/10.3389/fcell.2020.00641 (2020).
Günster, R. A., Matthews, S. A., Holden, D. W. & Thurston, T. L. M. SseK1 and SseK3 type III secretion system effectors inhibit NF-κB signaling and necroptotic cell death in Salmonella-infected macrophages. Infect. Immun. https://doi.org/10.1128/IAI.00010-17 (2017).
Araujo-Garrido, J. L., Baisón-Olmo, F., Bernal-Bayard, J., Romero, F. & Ramos-Morales, F. Tubulin folding cofactor TBCB is a target of the Salmonella effector protein SseK1. Int. J. Mol. Sci. 21(9), 3193. https://doi.org/10.3390/ijms21093193 (2020).
Li, S. et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 501(7466), 242–246. https://doi.org/10.1038/nature12436 (2013).
Gao, X. et al. NleB, a bacterial effector with glycosyltransferase activity targets GADPH function to inhibit NF-κB activation. Cell Host Microbe 13(1), 87–99. https://doi.org/10.1016/j.chom.2012.11.010 (2013).
El Qaidi, S. et al. An intra-bacterial activity for a T3SS effector. Sci. Rep. 10(1), 1073. https://doi.org/10.1038/s41598-020-58062-y (2020).
El Qaidi, S., Scott, N. E. & Hardwidge, P. R. Arginine glycosylation enhances methylglyoxal detoxification. Sci. Rep. 11(1), 3834. https://doi.org/10.1038/s41598-021-83437-0 (2021).
El Qaidi, S., Scott, N. E., Hays, M. P. & Hardwidge, P. R. Arginine glycosylation regulates UDP-GlcNAc biosynthesis in Salmonella enterica. Sci. Rep. 12(1), 5293. https://doi.org/10.1038/s41598-022-09276-9 (2022).
Stock, A. M., Robinson, V. L. & Goudreau, P. N. Two-component signal transduction. Annu. Rev. Biochem. 69(1), 183–215. https://doi.org/10.1146/annurev.biochem.69.1.183 (2000).
Capra, E. J. & Laub, M. T. Evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 66(1), 325–347. https://doi.org/10.1146/annurev-micro-092611-150039 (2012).
Two-component signal transduction in Bacillus subtilis: How one organism sees its world. J. Bacteriol. https://doi.org/10.1128/JB.181.7.1975-1983.1999. Accessed Jan 30, 2023.
Feng, L. et al. Elucidation of a complete mechanical signaling and virulence activation pathway in enterohemorrhagic Escherichia coli. Cell Rep. 39(1), 110614. https://doi.org/10.1016/j.celrep.2022.110614 (2022).
Xue, J. et al. Arginine GlcNAcylation and activity regulation of PhoP by a type III secretion system effector in Salmonella. Front. Microbiol. 12, 825743. https://doi.org/10.3389/fmicb.2021.825743 (2021).
Forst, S., Delgado, J. & Inouye, M. Phosphorylation of OmpR by the osmosensor EnvZ modulates expression of the ompF and ompC genes in Escherichia coli. Proc. Natl. Acad. Sci. 86(16), 6052–6056. https://doi.org/10.1073/pnas.86.16.6052 (1989).
Non-canonical activation of OmpR drives acid and osmotic stress responses in single bacterial cells. Nat. Commun. https://www.nature.com/articles/s41467-017-02030-0. Accessed Jan 22, 2023.
Transcription regulation of ompF and ompC by a single transcription factor, OmpR*. J. Biol. Chem. https://www.jbc.org/article/S0021-9258(20)55738-4/fulltext. Accessed Jan 30, 2023.
Barbieri, C. M., Wu, T. & Stock, A. M. Comprehensive analysis of OmpR phosphorylation, dimerization, and DNA binding supports a canonical model for activation. J. Mol. Biol. 425(10), 1612–1626. https://doi.org/10.1016/j.jmb.2013.02.003 (2013).
Feng, X., Oropeza, R. & Kenney, L. J. Dual regulation by phospho-OmpR of ssrA/B gene expression in Salmonella pathogenicity island 2. Mol. Microbiol. 48(4), 1131–1143. https://doi.org/10.1046/j.1365-2958.2003.03502.x (2003).
Batchelor, E., Walthers, D., Kenney, L. J. & Goulian, M. The Escherichia coli CpxA-CpxR envelope stress response system regulates expression of the porins OmpF and OmpC. J. Bacteriol. 187(16), 5723–5731. https://doi.org/10.1128/JB.187.16.5723-5731.2005 (2005).
Chou, J. H., Greenberg, J. T. & Demple, B. Posttranscriptional repression of Escherichia coli OmpF protein in response to redox stress: Positive control of the micF antisense RNA by the soxRS locus. J. Bacteriol. 175(4), 1026–1031. https://doi.org/10.1128/jb.175.4.1026-1031.1993 (1993).
Foo, Y. H., Spahn, C., Zhang, H., Heilemann, M. & Kenney, L. J. Single cell super-resolution imaging of E. coli OmpR during environmental stress. Integr. Biol. 7(10), 1297–1308. https://doi.org/10.1039/c5ib00077g (2015).
Kaeriyama, M. et al. OmpC and OmpF are required for growth under hyperosmotic stress above pH 8 in Escherichia coli. Lett. Appl. Microbiol. 42(3), 195–201. https://doi.org/10.1111/j.1472-765X.2006.01845.x (2006).
Quinn, H. J., Cameron, A. D. S. & Dorman, C. J. Bacterial regulon evolution: Distinct responses and roles for the identical OmpR proteins of Salmonella Typhimurium and Escherichia coli in the acid stress response. PLoS Genet. 10(3), e1004215. https://doi.org/10.1371/journal.pgen.1004215 (2014).
Eydallin, G. et al. Genome-wide screening of genes affecting glycogen metabolism in Escherichia coli K-12. FEBS Lett. 581(16), 2947–2953. https://doi.org/10.1016/j.febslet.2007.05.044 (2007).
Bustamante, V. H. et al. HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc. Natl. Acad. Sci. 105(38), 14591–14596. https://doi.org/10.1073/pnas.0801205105 (2008).
Cameron, A. D. S. & Dorman, C. J. A fundamental regulatory mechanism operating through OmpR and DNA topology controls expression of Salmonella pathogenicity islands SPI-1 and SPI-2. PLoS Genet. 8(3), e1002615. https://doi.org/10.1371/journal.pgen.1002615 (2012).
Garmendia, J., Beuzón, C. R., Ruiz-Albert, J. & Holden, D. W. The roles of SsrA-SsrB and OmpR-EnvZ in the regulation of genes encoding the Salmonella typhimurium SPI-2 type III secretion system. Microbiology (Reading) 149(Pt 9), 2385–2396. https://doi.org/10.1099/mic.0.26397-0 (2003).
Lee, A. K., Detweiler, C. S. & Falkow, S. OmpR regulates the two-component system SsrA-SsrB in Salmonella pathogenicity island 2. J. Bacteriol. 182(3), 771–781 (2000).
Xu, X. & Hensel, M. Systematic analysis of the SsrAB virulon of Salmonella enterica. Infect. Immun. 78(1), 49–58. https://doi.org/10.1128/IAI.00931-09 (2010).
Villarreal, J. M. et al. The Salmonella enterica serovar Typhi ltrR-ompR-ompC-ompF genes are involved in resistance to the bile salt sodium deoxycholate and in bacterial transformation. Mol. Microbiol. 92(5), 1005–1024. https://doi.org/10.1111/mmi.12610 (2014).
Pratt, L. A., Hsing, W., Gibson, K. E. & Silhavy, T. J. From acids to osmZ: Multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Mol. Microbiol. 20(5), 911–917. https://doi.org/10.1111/j.1365-2958.1996.tb02532.x (1996).
Chakraborty, S. & Kenney, L. J. A new role of OmpR in acid and osmotic stress in Salmonella and E. coli. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.02656 (2018).
OmpR regulates the stationary-phase acid tolerance response of Salmonella enterica serovar Typhimurium. J. Bacteriol. https://doi.org/10.1128/JB.182.8.2245-2252.2000. Accessed Jan 30, 2023.
Urdaneta, V. & Casadesús, J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front. Med. https://doi.org/10.3389/fmed.2017.00163 (2017).
Lucchini, V. et al. The role of OmpR in bile tolerance and pathogenesis of adherent-invasive Escherichia coli. Front. Microbiol. https://doi.org/10.3389/fmicb.2021.684473 (2021).
Harrell, J. E. et al. Salmonella biofilm formation, chronic infection, and immunity within the intestine and hepatobiliary tract. Front. Cell. Infect. Microbiol. https://doi.org/10.3389/fcimb.2020.624622 (2021).
Beshiru, A., Igbinosa, I. H. & Igbinosa, E. O. Biofilm formation and potential virulence factors of Salmonella strains isolated from ready-to-eat shrimps. PLoS One 13(9), e0204345. https://doi.org/10.1371/journal.pone.0204345 (2018).
Steenackers, H., Hermans, K., Vanderleyden, J. & De Keersmaecker, S. C. J. Salmonella biofilms: An overview on occurrence, structure, regulation and eradication. Food Res. Int. 45(2), 502–531. https://doi.org/10.1016/j.foodres.2011.01.038 (2012).
Biofilm Formation of Salmonella | IntechOpen. https://www.intechopen.com/chapters/50456. Accessed Jan 30, 2023.
Lin, T. H. et al. Phosphorylated OmpR is required for type 3 fimbriae expression in Klebsiella pneumoniae under hypertonic conditions. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.02405 (2018).
Baisón-Olmo, F., Galindo-Moreno, M. & Ramos-Morales, F. Host cell type-dependent translocation and PhoP-mediated positive regulation of the effector SseK1 of Salmonella enterica. Front. Microbiol. https://doi.org/10.3389/fmicb.2015.00396 (2015).
Chetri, S. et al. Transcriptional response of OmpC and OmpF in Escherichia coli against differential gradient of carbapenem stress. BMC Res. Notes 12(1), 138. https://doi.org/10.1186/s13104-019-4177-4 (2019).
Mattison, K., Oropeza, R., Byers, N. & Kenney, L. J. A phosphorylation site mutant of OmpR reveals different binding conformations at ompF and ompC11Edited by R. Ebright. J. Mol. Biol. 315(4), 497–511. https://doi.org/10.1006/jmbi.2001.5222 (2002).
Mattison, K. & Kenney, L. J. Phosphorylation alters the interaction of the response regulator OmpR with its sensor kinase EnvZ*. J. Biol. Chem. 277(13), 11143–11148. https://doi.org/10.1074/jbc.M111128200 (2002).
Batchelor, E. & Goulian, M. Robustness and the cycle of phosphorylation and dephosphorylation in a two-component regulatory system. Proc. Natl. Acad. Sci. U.S.A. 100(2), 691–696. https://doi.org/10.1073/pnas.0234782100 (2003).
Seo, S. W. et al. Revealing genome-scale transcriptional regulatory landscape of OmpR highlights its expanded regulatory roles under osmotic stress in Escherichia coli K-12 MG1655. Sci. Rep. 7(1), 2181. https://doi.org/10.1038/s41598-017-02110-7 (2017).
Martínez-Hackert, E. & Stock, A. M. The DNA-binding domain of OmpR: Crystal structures of a winged helix transcription factor. Structure 5(1), 109–124. https://doi.org/10.1016/s0969-2126(97)00170-6 (1997).
Itou, H. & Tanaka, I. The OmpR-family of proteins: Insight into the tertiary structure and functions of two-component regulator proteins. J. Biochem. 129(3), 343–350. https://doi.org/10.1093/oxfordjournals.jbchem.a002863 (2001).
Delgado, J., Forst, S., Harlocker, S. & Inouye, M. Identification of a phosphorylation site and functional analysis of conserved aspartic acid residues of OmpR, a transcriptional activator for ompF and ompC in Escherichia coli. Mol. Microbiol. 10(5), 1037–1047. https://doi.org/10.1111/j.1365-2958.1993.tb00974.x (1993).
The two-component regulatory system TCS08 is involved in cellobiose metabolism of Streptococcus pneumoniae R6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1797370/. Accessed May 23, 2021.
Chakravortty, D., Hansen-Wester, I. & Hensel, M. Salmonella pathogenicity island 2 mediates protection of intracellular Salmonella from reactive nitrogen intermediates. J. Exp. Med. 195(9), 1155–1166. https://doi.org/10.1084/jem.20011547 (2002).
Dandekar, T., Fieselmann, A., Popp, J. & Hensel, M. Salmonella enterica: A surprisingly well-adapted intracellular lifestyle. Front. Microbiol. https://doi.org/10.3389/fmicb.2012.00164 (2012).
Diacovich, L., Lorenzi, L., Tomassetti, M., Méresse, S. & Gramajo, H. The infectious intracellular lifestyle of Salmonella enterica relies on the adaptation to nutritional conditions within the Salmonella-containing vacuole. Virulence 8(6), 975–992. https://doi.org/10.1080/21505594.2016.1270493 (2017).
Coombes, B. K., Wickham, M. E., Lowden, M. J., Brown, N. F. & Finlay, B. B. Negative regulation of Salmonella pathogenicity island 2 is required for contextual control of virulence during typhoid. Proc. Natl. Acad. Sci. 102(48), 17460–17465. https://doi.org/10.1073/pnas.0505401102 (2005).
Choi, J. et al. Salmonella pathogenicity island 2 expression negatively controlled by EIIANtr–SsrB interaction is required for Salmonella virulence. Proc. Natl. Acad. Sci. U.S.A. 107(47), 20506–20511. https://doi.org/10.1073/pnas.1000759107 (2010).
Walthers, D. et al. The response regulator SsrB activates expression of diverse Salmonella pathogenicity island 2 promoters and counters silencing by the nucleoid-associated protein H-NS. Mol. Microbiol. 65(2), 477–493. https://doi.org/10.1111/j.1365-2958.2007.05800.x (2007).
Silphaduang, U., Mascarenhas, M., Karmali, M. & Coombes, B. K. Repression of intracellular virulence factors in Salmonella by the Hha and YdgT nucleoid-associated proteins. J. Bacteriol. 189(9), 3669–3673. https://doi.org/10.1128/JB.00002-07 (2007).
Qaidi, S. E. & Hardwidge, P. R. ABC cloning: An efficient, simple, and rapid restriction/ligase-free method. MethodsX 6, 316–321. https://doi.org/10.1016/j.mex.2019.02.007 (2019).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. PNAS 97(12), 6640–6645. https://doi.org/10.1073/pnas.120163297 (2000).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2(8), 1896–1906. https://doi.org/10.1038/nprot.2007.261 (2007).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26(12), 1367–1372. https://doi.org/10.1038/nbt.1511 (2008).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11(12), 2301–2319. https://doi.org/10.1038/nprot.2016.136 (2016).
Brademan, D. R., Riley, N. M., Kwiecien, N. W. & Coon, J. J. Interactive peptide spectral annotator: A versatile web-based tool for proteomic applications. Mol. Cell. Proteom. 18(8 suppl 1), S193–S201. https://doi.org/10.1074/mcp.TIR118.001209 (2019).
Csordas, A. et al. PRIDE: Quality control in a proteomics data repository. Database (Oxford) 2012, bas004. https://doi.org/10.1093/database/bas004 (2012).
Christensen, G. D. et al. Adherence of coagulase-negative staphylococci to plastic tissue culture plates: A quantitative model for the adherence of staphylococci to medical devices. J. Clin. Microbiol. 22(6), 996–1006. https://doi.org/10.1128/jcm.22.6.996-1006.1985 (1985).
Park, J. B. et al. Structural basis for arginine glycosylation of host substrates by bacterial effector proteins. Nat. Commun. 9(1), 4283. https://doi.org/10.1038/s41467-018-06680-6 (2018).
El Qaidi, S. et al. High-throughput screening for bacterial glycosyltransferase inhibitors. Front. Cell. Infect. Microbiol. 8, 1. https://doi.org/10.3389/fcimb.2018.00435 (2018).
Acknowledgements
The project described was supported by Grant Numbers AI127973 and AI153202 from the National Institute of Allergy and Infectious Diseases (NIAID), by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number P20GM130448 (PRH), and by a pilot project (SE) from P20GM130448. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. N.E.S is supported by an Australian Research Council Future Fellowship (FT200100270) and an ARC Discovery Project Grant (DP210100362). We thank the Melbourne Mass Spectrometry and Proteomics Facility of The Bio21 Molecular Science and Biotechnology Institute for access to MS instrumentation.
Funding
This study was funded by National Institute of General Medical Sciences (NIGMS, No. P20GM130448 (PRH)), National Health and Medical Research Council of Australia (NHMRC) (No. APP1100164) and Center on Emerging and Zoonotic Infectious Diseases (No. GVDM503011).
Author information
Authors and Affiliations
Contributions
N.E.S. Performed the mass spectrometry experiments, S.E. conceived of the project, Md.K.H. performed the other experiments and wrote the manuscript. S.E. provided technical assistance, project resources, and coordinated the project. All authors reviewed the data and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hasan, M.K., Scott, N.E., Hays, M.P. et al. Salmonella T3SS effector SseK1 arginine-glycosylates the two-component response regulator OmpR to alter bile salt resistance. Sci Rep 13, 9018 (2023). https://doi.org/10.1038/s41598-023-36057-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-36057-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
