
Abstract
Most of the knowledge about genetic variants at the sequence level in cattle is for Bos primigenius taurus populations. Here, we presented a complete genomic characterization of 52 Nellore (Bos primigenius indicus) bulls, revealing specific zebu DNA variants with putative impact in tropical adaptation and productive traits. Single nucleotide polymorphisms (SNPs) and insertion/deletion (INDELs) mutations were identified using the newest bovine reference genome ARS_UCD1.2, and variant functional consequences were predicted using the Ensembl VEP software. A total of 35,753,707 SNPs and 4,492,636 INDELs were detected and annotated to their functional effects. We identified 400 genes that comprised both, a SNP and an INDEL, of high functional impact on proteins (i.e. variants that cause protein truncation, loss of function or triggering nonsense-mediated decay). Among these, we highlight the following genes: BoLA, associated with cattle immune response to infections and reproduction aspects; HSPA8, DNAJC27, and DNAJC28, involved with thermoregulatory protective mechanisms in mammals; and many olfactory signaling pathway related genes that are important genetic factors in the evolution of mammalian species. All these functional aspects are directly related to cattle adaptability to tropical environments.
Similar content being viewed by others


Introduction
Cattle have played important roles in human societies for a long time, supplying products such as milk, meat, leather, and power. The current two major domesticated cattle, taurine (Bos primigenius taurus) and indicine or zebu (Bos primigenius indicus), descend from the extinct wild aurochs (Bos primigenius) that have diverged, between 250,000 and 330,000 years ago, into two distinct lineages1. Taurine cattle originated from the domestication of the Bos primigenius primigenius at, approximately, 10,000 years ago in the Fertile Crescent, while the indicine cattle descendant of the Bos primigenius nomadicus, domesticated about 8,000 years ago in the Indus Valley1,2,3,4,5.
Natural selection, followed by the post-domestication selection driven by humans modified cattle genotypes, leading to distinct genetic and phenotypes profiles between taurine and indicine cattle. In general, these two groups have been selected and are adapted to temperate and tropical environments, respectively2,4. Since the taurine cattle genome sequencing6, an extensive use of re-sequenced animals has allowed the identification of a considerable number of genetic variants segregating in different populations, essentially represented by taurine breeds7,8,9,10,11,12,13. To this point, a few studies have explored sequence information of indicine cattle breeds14,15,16.
Nellore figures between the most important domestic indicine cattle with a great impact on the global beef industry. It is a tropically adapted breed and the main responsible for transforming Brazil in one of the largest beef producers and exporters of the world17,18. The Brazilian Nellore originated from Ongole cattle which were brought to Brazil from India between 1868 and 196319. Crosses with local taurine cattle increased the population size and subsequently backcrosses with the original imported Ongole lineages recovered indicine adaptive and productive traits20,21. Indeed, the currently available Brazilian Nellore genetic resources present low levels of taurine introgression22. Since the 1950s, well organized Nellore breeding programs have been established in Brazil, in order to explore and improve the adaptability and performance of this breed under tropical environmental conditions23,24.
The objectives of the present study were to: 1) characterize the Nellore genomic variability by mapping single nucleotide variants and insertion/deletion mutations based on a complete genome scan of influential Nellore bulls; and 2) unravel specific zebu DNA variants with functional impact in tropical adaptation- and/or production-related traits.
Results and discussion
The sequencing generated an average of 360 million reads per individual with 97.9% of them being properly paired in the alignment process. The average individual sequence coverage was 18.50-folds (13.08 to 26.26-folds). These results, similar to other re-sequenced reports, which varied from 9 to 30-folds14,16,25, indicate the quality of raw sequence data and alignment process as well as that the depth of coverage was adequate for detecting variants with high confidence. The 1000 bull genomes project, for example, requires animals sequenced at a minimum of 10-folds coverage (http://www.1000bullgenomes.com/).
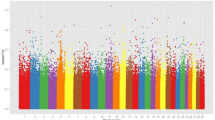
The distribution of the 35,753,707 SNPs and 4,492,636 INDELs across the genome is presented in the Figs. 1 and 2, respectively. Among INDELs, 51.6% and 48.4% were characterized as deletions and insertions, respectively. Most of them (80.0% and 96.7%) correspond respectively to deletions and insertions of less than 3 bp. The prevalence of small INDELs has also been reported in studies involving different breeds8,14,16. Here, 13.6% of SNPs and 42.1% of INDELs correspond to novel variants (Ensembl release 96). This high proportion of novel variants, mainly INDELs, suggests Nellore as a unique and significant source of cattle-specific genomic variation, which is in accordance to the known genetic and phenotypic differentiation of this breed in comparison with taurine or even other indicine breeds4,26,27,28. In addition, considering the bovine genome length 2715.85 Mb (https://www.ncbi.nlm.nih.gov/genome/82), on average, there were identified approximately 15 polymorphisms per kb. A similar rate of polymorphism was reported by25, who, working with sequencing data of 46 Brahman bulls, reported the identification of about 36 million SNPs and 4.7 million INDELs. All these results are indicative of the magnitude and genetic diversity captured in this group of influential Nellore bulls.

Single nucleotide polymorphism distribution by chromosome.

Insertion/deletion distribution by chromosome.
The majority of SNPs and INDELs were intergenic (59.9% and 58,7%, respectively) and intronic (32.3% and 33.4%, respectively) (Table 1). Only 0.3% of the total variants were classified as high or moderate functional impact. It includes the splice acceptor and splice donor, start and stop lost, stop gained, missense, frameshift, inframe insertion or deletion, protein altering variant and transcript ablation variants (Table 1). Even though the small proportion of variants with high or moderate impact on proteins (0.3%), these variants overlapped 17,675 genes (approximately 64% of the total bovine genome annotated genes). As the variants were identified based on a taurine genome reference, this result indicates how expressive is the genetic differentiation with effective impact on phenotypes between Nellore and taurine cattle. To properly address it, our results have to be compared to a database with sequenced animals of various taurine breeds.
Here, we identified 400 genes that comprise both, a SNP and an INDEL, of high functional impact on proteins (see Supplementary Table S1). The gene interaction network of this set of genes (Fig. 3 and Supplementary Table S2) showed pathways closely related to the immune response to infections and also impacting reproduction.

Gene interaction network of genes marked by variants (SNPs and INDELs) with high functional impact.
The Bovine Major Histocompatibility Complex (MHC), also known as The Bovine Lymphocyte Antigen (BoLA), is the primary genetic component of cattle immune system29. The main function of BoLA class I (MHC-I) is to present peptides to CD8 + T-lymphocytes, which kill virus-infected and neoplastic cells30. Bovine lymphocyte antigen restricts cytotoxic cells generated during Theileria infections31, evidencing the associations between BoLA class l antigens and tick resistance27. BoLA genes have been flagged as Nellore selection signatures32. In addition, non-classical MHC-I genes, such as the BOLA-NC1, plays a role in cattle reproduction by regulating maternal immunity to the fetus, which is essential for pregnancy establishment and maintenance33, showing that MHC-I play a role in embryo maternal interactions34.
As the immune response to infections and reproduction aspects, heat tolerance is one of the main indicatives of adaptability to harsh environments. It is well-documented that zebu animals, such as Nellore, present a more efficient thermoregulatory system to deal with heat stress than taurine breeds26,35. This efficiency is expected to be related to several genetically controlled physical and physiological parameters. In this sense, among the genes identified here as potential candidates to tropical cattle adaptability, we could highlight the HSPA8, DNAJC24, and DNAJC28. HSPA8 plays role in thermoregulatory protective mechanisms in cattle and buffalo under tropical environments36, and it is also associated with cellular response to heat stress in goats37. DNAJC24 and DNAJC28 are type III hsp40 genes that belong to the heat shock protein family38. In general, the expression of HSP genes is induced by high temperature, hypoxia, infection and a number of other stresses30,39. Thus, zebu-specific variations affecting HSP-mediated response to environmental stressors would explain part of tropical cattle adaptability.
Another important result regarding the putative genes related to adaptability in Nellore is the number of olfactory signaling pathway related genes that have been identified (OR4S1, OR4F15, OR2G2, OR9A4, OR2AP1, OR51B4, OR5R1, OR10AD1, OR2AJ1, OR8U1, and OR1P1). The multigene family of olfactory receptor (OR) is an important genetic factor in the evolution of mammalian species40. Variations in cattle OR repertoire could be related to evolution associated with environmental changes41. ORs are expressed on millions of olfactory sensory neurons within the olfactory epithelium, but also in organs outside the nasal cavity where they bind to molecules such as nutrients and metabolites42. Physiological responses mediated by these chemoreceptors exert a crucial role in animal’s appetite and energy balance regulation, which could affect the feed intake, weight gain, and animal body composition42. Olfactory receptors also affect the reproduction in mammals by playing a role in production of germ cells, which are the precursors of gametes43. According to44, the OR2AP1 gene, identified in this study, is associated with fertility and semen quality of zebu cattle under heat stress conditions. Thus, genetic variations of OR genes could be directly linked to cattle adaptations under extreme environmental conditions.
We have also evaluated the distribution and annotation of the variants fixed for the alternative allele.25 investigated the fixed non-reference alleles in the Brahman genome. According to these authors, such variants could represent genomic regions strongly selected in indicine cattle for providing adaptive advantages in tropical environments. Indeed,25 identified some genes with missense mutations fixed in Brahman for the alternative allele related to immunity. Here, the MAF histogram (Fig. 4A) showed a number of fixed variants in Nellore across the genome (Fig. 4B). We have used our Nellore reference population of about 10,000 animals to verify whether these high impact variants fixed for the alternative allele are not really segregating in Nellore. As we can observe in the Supplementary Figure S2, these variants are really rare in our Nellore reference population.

Minor allele frequency (MAF) distribution (A) and the number of fixed variants by chromosome (B).
Among the Nellore fixed variants, 2,935 have a high or a moderate impact on proteins and they overlap a total of 1,672 annotated genes. As shown in Fig. 5 and Supplementary Table S3, many of these genes play a role in immune system pathways. This result complements our findings discussed before for the list of genes identified as potential candidates to tropical cattle adaptability based on the presence of a simultaneously high impact SNP and INDEL variants. As shown in Fig. 5, this second list of genes (linked to fixed Nellore alleles) also illustrate the possible effective major impact of Bovine Lymphocyte Antigen (BoLA) related genes in zebu cattle adaptation. In addition, a search for known QTLs affecting traits related to adaptation in the QTL database45 showed that the region between 15 and 40 Mb on chromosome 23 harbors putative QTLs affecting cattle Cell and antibody-mediated immune response, tick resistance, heat tolerance, and respiratory rate. Nineteen genes associated with missense mutations fixed in Nellore (see Supplementary Table S1), including the genes BOLA-DQA5, BOLA-NC1, CNPY3, JSP.1, TRIM10, TRIM15, and UBD presented at the Fig. 5, are located in this QTL region.

Gene interaction network of genes marked by variants fixed in Nellore with high or moderate functional impact.
At Fig. 5, it could be also highlighted a group of cluster differentiation (CD) genes (CD5, CD27, CD48, and CD59), which are expressed on leukocytes and other cells of the immune system46, and a group of Interleukin (IL) superfamily members (IL6ST, IL20, IL18RAP, IL18R1, IL36B, IL36RN), which are also important for the immune system regulation. Both CD and IL family genes act on the immune system activation in response to environmental stress, being, in this way, important candidate genes to affect tropical adaptation47. Evaluating gene expression patterns in cattle selected for resistance or susceptibility to intestinal nematodes,48 found that the CD27, CD45, and IL18 genes were highly expressed in resistant animals while the CD59 and IL6 were highly expressed in susceptible animals.
Interestingly, as in the list of candidate genes identified based on the presence of a simultaneously high impact SNP and INDEL variants, members of the multigene family of olfactory receptor (OR) genes (OR11H7, OR11L1, OR1L1, OR2T11, OR4D5, OR4K14, OR51B4, OR52M1, OR6V1, and OR6Y1) have also been found in the list of genes affected by high or moderate impact fixed Nellore alleles (see Supplementary Table S1). As discussed previously, it has been documented that OR genes are important genetic factors influencing evolution and adaptation of mammalian species40.
Beef cattle production in tropical conditions is dominated by Zebu cattle on grass-fed systems, in which the animals are exposed to natural infestations of parasites and have also to tolerate high temperatures and humidity24. Remarkably, animal adaptation to the tropics is directly related to its ability to survive, grow and reproduce in the presence of endemic environmental stressors49. Our findings have evidenced the polygenic nature of climatic adaptation in the Zebu breed, Nellore, but also suggested the Bovine Major Histocompatibility Complex (BoLA) gene family as one of the main responsible for Nellore adaptation, together with the cluster of differentiation (CD) and Interleukin (IL) superfamily members, and the olfactory receptor (OR) multigene family. The mediated physiological responses by these genes are of paramount importance for the survival and reproduction of animals in challenging environments.
Conclusions
A genomic characterization of Nellore was done at a sequence level, and new insights into genetic basis of zebu cattle adaptation to harsh tropical environments were provided. We identified various single nucleotide variants, including those that are fixed in this Nellore reference population, and insertion/deletion mutations with high impact in product of genes with functions directly related to cattle adaptability such as disease resistance, heat tolerance, and reproduction.
Methods
Animal ethics statement
The animal DNA samples used in this study were extracted from commercially collected semen straws purchased from AI (Artificial insemination) companies or donated to the project.
Genome sequence information
The studied animals were chosen based on their contributions to the genetic diversity of the Brazilian Nellore population, evaluated through a pedigree file contained 2,688,124 individuals in total, 9,811 (6,040 founders) sires and 915,371 dams. For the sequencing, there were prioritized the founders less genetically related to each other, with high number of progenies, and also taking into account their genetic contributions to our genotyping database, which includes more than 10,000 genotyped animals from five Brazilian Nellore breeding programs (DeltaGen http://deltagen.com.br/, Nelore Qualitas https://qualitas.agr.br/, Cia do Melhoramento, CRV PAINT https://www2.crvlagoa.com.br/paint, and Instituto de Zootecnia http://www.iz.sp.gov.br/). Supplementary Fig. S1 shows the genetic structure of the sequenced sires and how they represent the genetic diversity of the aforementioned Nellore reference population.
The whole-genome sequencing of 52 key Nellore bulls was performed using the Illumina HiSeq X™ Ten platform. Quality control, alignment, and variant calling processes were carried out according to the guidelines suggested by the 1000 bull Genomes Project, available at http://www.1000bullgenomes.com/doco/1000bullsGATK3.8pipelineSpecifications_Run8_Revision_20191101.docx. Initially, the FastQC program50 was used to check the raw sequence quality. Trimmomatic software51 was used to trim paired and single-reads of the adapter and low-quality bases at their extremities, then to filter out reads that were left with less length than 35 bp or mean qscore lower than 20. The reads were then aligned to the ARS-UCD1.2 reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCA_002263795.2) using the Burrows-Wheeler Aligner - BWA-MEM52. Further, BAM files were sorted using SAMtools53, and PCR duplicates were removed using the Picard tools (http://broadinstitute.github.io/picard/).
Variant calling and annotation process
Single nucleotide variants (SNPs) and insertion and deletion mutations (INDELs) were identified using the HaplotypeCaller tool, implemented by the Genome Analysis Toolkit software – GATK54. SNPs and INDELs were filtered for quality purposes considering the following exclusion criteria25: quality by depth - QD < 2.0; Fisher Strand test - FS > 60.0; root mean square of the mapping quality score - MQ < 40.0; ranked sum test for the distance of alleles from the end of the reads - ReadPosRankSum < −8.0; mapping qualities of reads - MQRankSum < −12.5; and SOR > 3.0. A total of 35,753,707 SNPs and 4,492,636 INDELs of autosomal chromosomes and X were left after this quality control procedure. The Ensembl VEP platform55 was then used for variant annotation.
In silico functional analysis of genes comprising variants of high impact on proteins
The Ensembl VEP software classified SNPs and INDELS according to their functional consequences on transcripts. Variants were classified as high when they caused premature stop codons, loss of function or trigger nonsense-mediated decay, and as moderate if lead to non-disruptive variants that might change protein effectiveness. Genes comprising high impact markers were split into two lists: 1) genes containing at least a SNP and an INDEL, both with high functional impact on proteins; and 2) genes marked by high or moderate functional impact variants that are fixed in the sequenced sires. Each list of genes was independently submitted to the ClueGO56, a Cytoscape57 plug-in that integrates Gene Ontology and KEGG pathways to create an organized GO/pathway annotation network56, to verify whether these genes could be statistically (p-value <0.01) related to pathways associated with cattle adaptability in the tropics.
Data availability
The data used in this study were obtained under license and so cannot be publicly available. Data are however available from the authors upon reasonable request, and with authorization of Nellore breeding programs.
Change history
26 January 2021
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Pitt, D. et al. Domestication of cattle: Two or three events? Evol. Appl. 12, 123–136 (2019).
MacHugh, D. E. et al. variation and the evolution, domestication and phylogeography of taurine and zebu cattle (Bos taurus and Bos indicus). Genetics 146, 1071–1086 (1997).
Murray, C., Huerta-Sanchez, E., Casey, F. & Bradley, D. G. Cattle demographic history modelled from autosomal sequence variation. Philos. Trans. R. Soc. B Biol. Sci. 365, 2531–2539 (2010).
Porto-Neto, L. R. et al. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics 14 (2013).
Park, S. D. E. et al. Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol. 16 (2015).
Elsik, C. G. et al. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science (80-.). https://doi.org/10.1126/science.1169588. (2009)
Eck, S. H. et al. Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biol. 10 (2009).
Kawahara-Miki, R. et al. Whole-genome resequencing shows numerous genes with nonsynonymous SNPs in the Japanese native cattle Kuchinoshima-Ushi. BMC Genomics 12 (2011).
Stothard, P. et al. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics 12 (2011).
Jansen, S. et al. Assessment of the genomic variation in a cattle population by re-sequencing of key animals at low to medium coverage. BMC Genomics 14 (2013).
Tsuda, K. et al. Abundant sequence divergence in the native Japanese cattle Mishima-Ushi (Bos taurus) detected using whole-genome sequencing. Genomics 102, 372–378 (2013).
Daetwyler, H. D. et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 46, 858–865 (2014).
Choi, J. W. et al. Whole-genome resequencing analysis of hanwoo and yanbian cattle to identify genome-wide SNPs and signatures of selection. Mol. Cells 38, 466–473 (2015).
Stafuzza, N. B. et al. Single nucleotide variants and InDels identified from whole-genome re-sequencing of Guzerat, Gyr, Girolando and Holstein cattle breeds. PLoS One 12 (2017).
Hayes, B. J. & Daetwyler, H. D. 1000 Bull Genomes Project to Map Simple and Complex Genetic Traits in Cattle: Applications and Outcomes. Annu. Rev. Anim. Biosci. 7, 89–102 (2019).
Iqbal, N. et al. Genomic variants identified from wholegenome resequencing of indicine cattle breeds from Pakistan. PLoS One 14 (2019).
USDA. Livestock and poultry: world markets and trade. United States Department of Agriculture and Foreign Agricultural Service 31 http://apps.fas.usda.gov/psdonline/circulars/livestock_poultry.PDFhttps://doi.org/10.1016/S1097-8690(11)70006-3 (2019).
ABIEC. Brazilian Livestock Profile Contents. (2016).
Vozzi, P. A. et al. Structure and genetic variability in Nellore (Bos indicus) cattle by pedigree analysis. Genet. Mol. Biol. 29, 482–485 (2006).
Oliveira, J. H. F., Magnabosco, C. U. & Borges, A. M. S. M. Nelore: Base Genética e Evolução Seletiva no Brasil. Embrapa Cerrados 50 (2002).
Dani, M. A. C., Heinneman, M. B. & Dani, S. U. Brazilian Nelore cattle: A melting pot unfolded by molecular genetics. Genet. Mol. Res. 7, 1127–1137 (2008).
O’Brien, A. M. P. et al. Low levels of taurine introgression in the current Brazilian Nelore and Gir indicine cattle populations. Genet. Sel. Evol. 47 (2015).
Carvalheiro, R. Genomic Selection in Nelore Cattle in Brazil. in 10th World Congress of Genetics Applied to Livestock Production (2014).
Albuquerque, L. G., Fernandes Júnior, G. A. & Roberto, C. Beef Cattle Genomic Selection in Tropical Environments. in Proc. Assoc. Advmt. Anim. Breed. Genet 22, 255–263 (2017).
Koufariotis, L. et al. Sequencing the mosaic genome of Brahman cattle identifies historic and recent introgression including polled. Sci. Rep. https://doi.org/10.1038/s41598-018-35698-5 (2018).
McManus, C. et al. Use of multivariate analyses for determining heat tolerance in Brazilian cattle. Trop. Anim. Health Prod. 43, 623–630 (2011).
Porto Neto, L. R., Jonsson, N. N., D’Occhio, M. J. & Barendse, W. Molecular genetic approaches for identifying the basis of variation in resistance to tick infestation in cattle. Vet. Parasitol. 180, 165–172 (2011).
Porto-Neto, L. R. et al. The genetic architecture of climatic adaptation of tropical cattle. PLoS One 9 (2014).
Amills, M., Ramiya, V., Norimine, J. & Lewin, H. A. The major histocompatibility complex of ruminants. OIE Rev. Sci. Tech. 17, 108–120 (1998).
Song, L; et al. & Liu, Z. Genome-Wide Identification of Hsp40 Genes in Channel Catfish and Their Regulated Expression after Bacterial Infection. PLoS One https://doi.org/10.1371/journal.pone. 0115752 (2014).
Preston, P. M., Brown, C. G. D. & Spooner, R. L. Cell-mediated cytotoxicity in Theileria annulata infection of cattle with evidence for BoLA restriction. Clin. Exp. Immunol. 53, 88–100 (1983).
Cardoso, D. F. et al. Genome-wide scan reveals population stratification and footprints of recent selection in Nelore cattle. Genet. Sel. Evol. 50 (2018).
Shu, L. et al. Non-classical major histocompatibility complex class makes a crucial contribution to reproduction in the dairy cow. J. Reprod. Dev. 58, 569–575 (2012).
Fair, T. Embryo maternal immune interactions in cattle. Anim. Reprod. 13, 346–354 (2016).
Cardoso, C. C. et al. Physiological and thermographic response to heat stress in zebu cattle. Livest. Sci. 182, 83–92 (2015).
Kumar, A. et al. Expression profiling of major heat shock protein genes during different seasons in cattle (Bos indicus) and buffalo (Bubalus bubalis) under tropical climatic condition. J. Therm. Biol. 51, 55–64 (2015).
Jagan Mohanarao, G. et al. HSP70 family genes and HSP27 expression in response to heat and cold stress in vitro in peripheral blood mononuclear cells of goat (Capra hircus). Small Rumin. Res. 116, 94–99 (2014).
Kampinga, H. H. et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14, 105–111 (2009).
Feder, M. E. & Hofmann, G. E. Heat-Shock Proteins, Molecular Chaperones, and the Stress Response: Evolutionary and Ecological Physiology. Annu. Rev. Physiol. 61, 243–282 (1999).
Niimura, Y. & Nei, M. Extensive gains and losses of olfactory receptor genes in mammalian evolution. PLoS One 2 (2007).
Lee, K. et al. Analysis of cattle olfactory subgenome: The first detail study on the characteristics of the complete olfactory receptor repertoire of a ruminant. BMC Genomics 14 (2013).
Connor, E. E., Zhou, Y. & Liu, G. E. The essence of appetite: Does olfactory receptor variation play a role? J. Anim. Sci. 96, 1551–1558 (2018).
Diedrichs, F. et al. Comparative molecular portraits of human unfertilized oocytes and primordial germ cells at 10 weeks of gestation. Int. J. Dev. Biol. 56, 789–797 (2012).
H., B. et al. Signatures of positive selection in East African Shorthorn Zebu: A genome-wide single nucleotide polymorphism analysis. Sci. Rep. (2015).
Hu, Z. L., Park, C. A. & Reecy, J. M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 47 (2019).
Engel, P. et al. CD Nomenclature 2015: Human Leukocyte Differentiation Antigen Workshops as a Driving Force in Immunology. J. Immunol. 195, 4555–4563 (2015).
Chan, E. K. F., Nagaraj, S. H. & Reverter, A. The evolution of tropical adaptation: Comparing taurine and zebu cattle. Anim. Genet. 41, 467–477 (2010).
Araujo, R. N. et al. Use of a candidate gene array to delineate gene expression patterns in cattle selected for resistance or susceptibility to intestinal nematodes. Vet. Parasitol. 162, 106–115 (2009).
Burrow, H. M. Importance of adaptation and genotype × environment interactions in tropical beef breeding systems. Animal 6, 729–740 (2012).
Andrews, S. FASTQC A Quality Control tool for High Throughput Sequence Data. Babraham Inst. (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
H, L. & R, D. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17 (2016).
Bindea, G. et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009).
Shannon, P. et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Acknowledgements
This work was supported by Sao Paulo Research Foundation (FAPESP grants: #2009/16118–5, #2017/10630–2, #2018/10109–3, #2018/20026–8, and #2019/12434–1). H.N.O., R.C., and L.G.A. acknowledge the National Council for Science and Technological Development (CNPq) for financial support. We also thank the Coordination for the Improvement of Higher Education Personnel (CAPES; financial code 001).
Author information
Authors and Affiliations
Contributions
L.G.A. conceived and led the coordination of the study. L.F.S.F. participated in the collection and preparation of the samples. G.A.F.J., H.N.O., R.C., D.F.C. and R.V.V. participated in the statistical analysis. G.A.F.J. draft the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernandes Júnior, G.A., de Oliveira, H.N., Carvalheiro, R. et al. Whole-genome sequencing provides new insights into genetic mechanisms of tropical adaptation in Nellore (Bos primigenius indicus). Sci Rep 10, 9412 (2020). https://doi.org/10.1038/s41598-020-66272-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66272-7
This article is cited by
-
Local ancestry and selection in admixed Sanjiang cattle
Stress Biology (2023)
-
Genome sequencing and de novo and reference-based genome assemblies of Bos indicus breeds
Genes & Genomics (2023)
-
Identification of eQTLs and differential gene expression associated with fetal programming in beef cattle
Journal of Applied Genetics (2022)
-
Effect of acute heat shock on stress gene expression and DNA methylation in zebu (Bos indicus) and crossbred (Bos indicus × Bos taurus) dairy cattle
International Journal of Biometeorology (2022)
-
Imputation accuracy to whole-genome sequence in Nellore cattle
Genetics Selection Evolution (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}