
Abstract
Simultaneous, enantiomer-specific identification of chiral molecules in multi-component mixtures is extremely challenging. Many established techniques for single-component analysis fail to provide selectivity in multi-component mixtures and lack sensitivity for dilute samples. Here we show how enantiomers may be differentiated by mass-selected photoelectron circular dichroism using an electron–ion coincidence imaging spectrometer. As proof of concept, vapours containing ∼1% of two chiral monoterpene molecules, limonene and camphor, are irradiated by a circularly polarized femtosecond laser, resulting in multiphoton near-threshold ionization with little molecular fragmentation. Large chiral asymmetries (2–4%) are observed in the mass-tagged photoelectron angular distributions. These asymmetries switch sign according to the handedness (R- or S-) of the enantiomer in the mixture and scale with enantiomeric excess of a component. The results demonstrate that mass spectrometric identification of mixtures of chiral molecules and quantitative determination of enantiomeric excess can be achieved in a table-top instrument.
Similar content being viewed by others
Introduction
There is an increasing awareness of the benefits stemming from enantiomer-specific analysis of mixtures of volatile organics (VOCs). These advantages have long been apparent in the analysis of food odours where hundreds of chiral odorant pairs having different perceived smells are known1. Recent work has highlighted the role for enantiomer-specific detection of, for example, the monoterpene limonene in analysing biogenic emissions of VOCs2,3. In a related manner, detection of enantiomeric metabolites in clinical breath analysis may play an increasing role in the understanding and development of chiral pharmaceuticals. Much progress has been made in the analysis of mixtures of VOCs in such areas using direct injection mass spectrometry4,5, where ‘soft’ ionization techniques can obviate the need for prior chromatographic separation, a particular advantage for field and in vivo studies. Yet mass spectrometry, traditionally a ‘chirally blind’ technique, does not directly address the identification of enantiomers in these mixtures, providing a stimulus for the development of enhanced approaches that can provide such capabilities.
Mass spectrometric techniques for determination of single-component enantiomeric excess (e.e.) relevant to pharmaceutical development have, however, developed greatly over the past decade6,7 and rely on interactions with a known chiral reference reagent to discriminate enantiomers. These reference compounds have to be carefully chosen in advance according to the specific target species, and have themselves to be sourced in enantiopure form. Likely interference from contaminants in the analysed sample stream has also to be considered, although hybrid tandem mass spectrometric techniques can be used to reduce such interferences.
As an alternative to chemical discriminants, a fully generic chiral reference is provided by circularly polarized light (CPL), readily prepared and characterized by well-established optical techniques. Chiroptical methods, such as electronic circular dichroism and vibrational optical activity (VOA) and vibrational circular dichroism (VCD) rely on typically rather weak chiral asymmetries (0.001–0.01%) in light–matter interaction, but are widely used techniques for the detection and structural determination of solvated chiral molecules8,9.
Recently, circular dichroism in ion yields generated by CPL multiphoton laser ionization of chiral molecules has been shown to be capable of discriminating enantiomers in solvent free conditions10,11. An alternative, gas phase approach for the analysis of chiral isomer mixtures, based on phase-sensitive microwave spectroscopy, has also been demonstrated12,13. By eliminating the often indeterminate contributions from solvation14, gas phase studies of biomolecules can be of considerable value for developing fundamental understanding15 and permit the weak interactions responsible for chiral molecular recognition to be directly examined16,17.
In this report we describe a proof of concept demonstration of a direct, generic method for the simultaneous determination of enantiomeric components of a gaseous mixture by laser-based mass spectrometric detection, coupled with coincident electron imaging detection in a single table-top instrument. The detection of mass-selected parent ions in coincidence with energy dispersed electrons allows the electron spectra to be tagged by the precursor neutral. Recent work with synchrotron radiation has demonstrated the analytical potential of recording correlated ion and photoelectron ion spectra in this fashion18. Here, we add the convenience and additional capabilities of a laboratory-based ultrafast laser ionization source for optimized parent ion production with, crucially, the use of circular polarizations and energy- and angle-resolved imaging detection of the coincident electron. The latter provides the capability of enantiomeric differentiation by photoelectron circular dichroism (PECD)19,20,21. PECD derives from chiral asymmetries in the photoelectron angular distribution that, for single vacuum ultraviolet (VUV) photon ionization, routinely fall in the range 1–10%. These expectations are maintained in the recent emergence of multiphoton PECD where, again, asymmetries of up to 10% have been reported from single pure enantiomers22,23,24,25,26,27. Such large asymmetries provide more than sufficient sensitivity to identify enantiomers of even minor (that is, few %) components in a mass spectrum28. Hence, electron–ion coincidence imaging29 is here shown to create the capability to record fully multiplexed mass-tagged photoelectron circular dichroism spectra of chiral molecules in multi-component mixtures.
Results
Coincidence detection of electrons and ions
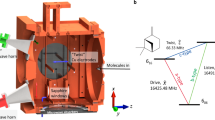
Key features of the mass-selective photoelectron circular dichroism (MS-PECD) experiment are provided in Fig. 1. Briefly the electron and ion from an individual molecular ionization by a laser pulse are projected to respective detectors by electric fields. The three-dimensional momentum distribution of the electron is recovered from its position and time-of-flight (ToF) at the detector, while the mass of the ion can be determined from its ToF. Coincident detection of the pair establishes their common origin, and allows the electron data to be ‘tagged’ according to the parent molecular mass. The accumulated electron data from many such tagged coincidence detections can be used to create the species-specific photoelectron energy and angular distributions required for PECD analysis.

(a) Following an ionizing laser pulse that crosses a molecular sample beam, ions and electrons are extracted in opposing directions by electric fields to reach their respective detectors. Electrons are focussed using a velocity map imaging arrangement of electrodes to yield the complete (px,py,pz) momentum from the time- and position-sensitive imaging detector33. For the present experiment mass analysis is achieved from measuring just the ion ToF. Detection of a mass-selected ion in delayed coincidence with an electron identifies particles from a single molecular ionization event and these electron–ion correlations can be used to ‘tag’ the accumulated electron data by their respective parent molecular masses. Panel (b) shows how the mass-tagged electron angular distributions recorded with circular polarized light display a forward–backward asymmetry that can distinguish between enantiomers, while the radius maps the electron kinetic energy.
Mixture analysis
As a demonstration we sample prepared mixtures containing <1% of two enantiomeric monoterpenes, camphor (C10H16O) and limonene (C10H16), in an inert carrier gas (Ne). Both terpenes occur as biogenic VOCs with stereoiosomerism of limonene, in particular, being of significant interest in odour analysis and monitoring terrestrial plant emissions2. Very recently, PECD asymmetries of ∼8% have been demonstrated for pure camphor enantiomers in multiphoton ionization using femtosecond (fs) laser pulses around 400 nm21,22,23,24, while multiphoton PECD asymmetries of ∼4% have been reported for limonene using fs laser pulses centred at 420 nm27. Fig. 2 shows schematically the simultaneously excited two-photon-resonant, three-photon ionization scheme for this mixture.

Panel (a) shows schematically the ‘soft’, two photon-resonant, three-photon-ionization producing parent molecular ions and coincident electrons (e1,M1+) and (e2,M2+). Panel (b) indicates how the laser may be tuned to excite via specific resonant neutral states. The different photoelectron energy distributions (spectra) obtained from the mass-tagged coincident electrons e1 and e2 are shown schematically and may be used to help chemical identification.
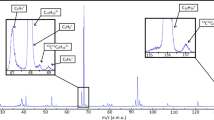
In Fig. 3, the ion ToF mass spectra for two different mixtures of enantiomeric vapours, Mix-RR (R-limonene/R-camphor) and Mix-RS (R-limonene/S-camphor), are shown after ionization with circular polarized 150 fs duration laser pulses with wavelength centred at 392.3 nm. The bandwidth of these fs laser pulses simultaneously encompasses two-photon resonant excitations to excited 3 s Rydberg states of either molecular species24,30, followed by absorption of a third photon taking each system above its ionization limit (Fig. 2a). Ion ToF-analysis produces similar mass spectra from both RS-(Fig. 3a) and RR-(Fig. 3b) mixtures. Only a small change in relative peak heights of the camphor versus limonene is observed between the Mix-RS and Mix-RR samples, due to slightly different experimental conditions, of no relevance here. The insets in Fig. 3 show the mass spectra in the region of 136–152 a.m.u., where the peaks corresponding to the parent ions of limonene (136 a.m.u.) and camphor (152 a.m.u.) can be clearly observed. From the width of the peaks in the mass spectra we can extract a mass resolution of m/Δm∼1,100 in this mass region. In this study we aimed for soft ionization with as low as possible fragmentation. The intensity and focus position of the femtosecond laser were adjusted such that the peaks due to fragmentation of the parent ion were only minor components in the mass spectra. As was reported before for ionization of pure camphor samples near 400 nm24, parent ion fragmentation can be easily induced by increasing the laser fluence although, noting the invariance of mass-selected photoelectron spectra (PES) of various ionic fragments, it was concluded that fragmentation was induced after photoionization of the neutral molecule. Hence, in principle, the laser fluence could be adjusted to obtain further structural identification from the fragmentation fingerprint31,32, and furthermore, the wavelength could be varied to achieve more selective ionization of preferred mixture components.

In panel (a) the ToF spectrum of a mixture containing R-limonene and S-camphor is shown, while in panel (b) the ToF spectrum of a mixture containing R-limonene and R-camphor is shown. The mixtures were seeded in neon and expanded through a nozzle as a molecular beam. The parent mass region is expanded in the insets. The small peaks at masses 137 and 153 are parent molecule isotopomers with 13C at natural abundance.
In Fig. 4, we show the PES obtained after ionization at 392 nm with CPL. The electron kinetic energy distributions are extracted from their full three-dimensional momentum distributions recorded by position and time-of-arrival encoding of individual electrons at our imaging delay line detector33. Furthermore, the coincidence between ion and electron events allows each electron to be associated with formation of an ion of given mass. For each mixture PES are shown for ionization with both right and left circular polarized (RCP and LCP) light. In Fig. 4a the PES obtained from the arrival of all electrons without any correlation of the electron with the mass of the coincident ion are shown; in Fig. 4b the PES of electrons coincident with an ion mass 136 (limonene) are shown, and in Fig. 4c the spectra of electrons coincident with an ion mass 152 (camphor) are presented. As expected the PES for the mixtures Mix-RR and Mix-RS and the two light helicities are all the same for a given choice of electron mass-tagging. However, we do see clear differences between the mass-tagged electron spectra coincident with camphor or limonene parent ions within a single mixture. A prominent peak around 0.6 eV is observed in the camphor tagged PES. Using the ionization energy of camphor of 8.7 eV and a laser photon energy of 3.16 eV the electron peak at 0.6 eV is assigned to three-photon ionization of the highest occupied molecular orbital (MO)24, leaving only 0.18 eV of internal vibrational excitation in the camphor parent ion. The limonene tagged PES shows somewhat broader structures with a dominant peak around 0.3 eV. This peak is assigned to three-photon ionization of the second highest occupied MO30.

The mixtures Mix-RR and Mix-RS were irradiated with a femtosecond laser beam at 392 nm with either LCP or RCP. In panel (a) the kinetic energy of all detected electrons is shown; below that are shown the kinetic energy of coincident mass-tagged electrons at m/z=136 (limonene—panel (b)) and m/z=152 (camphor—panel (c)). The mass-tagged electrons in the shaded areas around the main peaks, (0.25±0.25) eV for limonene and (0.6±0.2) eV for camphor, were used to extract the three-dimensional angular distribution from which the PECD asymmetry was calculated, see Fig. 5.
Multiphoton PECD measurements can be used to characterize the selected enantiomer by means of a simple asymmetry factor, defined for a given CPL as:24

where Nf and Nb are, respectively, the number of electrons emitted into forward and backward hemispheres (that is, 0≤θ<π/2 and π/2≤θ<π relative to the light propagation direction) and are derived from the accumulated electron distribution data after filtering by coincident ion mass and the preferred energy range. In practice, measurements are made by alternating LCP and RCP light and the two counts, Nf and Nb, obtained by combining the two polarization measurements in a manner that cancels any residual instrumental forward–backward asymmetry, as detailed below in Methods and section II of ref. 24).
In Fig. 5 the mass-tagged PECD asymmetries for 392 nm excitation, determined across the respective PES peak ranges identified in Fig. 4, are plotted for Mix-RR and Mix-RS. It is observed that PECD is different from zero (absolute PECD values ∼2% and ∼4%), indicating that the compounds in this gaseous mixture at both the mass 136 and mass 152 are chiral. Furthermore, when changing from Mix-RR to Mix-RS the mass 136 (limonene) tagged PECD does not change (within experimental error, see Table 1), while for mass 152 (camphor) PECD reverses sign when changing the mixture. This latter sign reversal is anticipated and clearly correlates with the change of the camphor enantiomer in the mixture. Conversely, the absence of change in the mass 136 tagged PECD indicates that the enantiomer composition of limonene has not changed between the two mixtures. This is exactly in agreement with the composition of the two mixtures that we prepared in this experiment.

Mass-selected PECD asymmetries, G, measured by multiphoton MS-PECD for two mixtures of enantiomerically pure limonene and camphor. Between Mix-RR (R-limonene/R-camphor) and Mix-RS (R-limonene/S-camphor) the camphor MS- PECD asymmetry flips sign, as anticipated for a change from R- to S-camphor, while for limonene the MS-PECD asymmetry remains the same. See also Table 1 for explicit values with uncertainties.
Analysis for enantiomeric excess
As a final demonstration recordings were made with Mix-S[Rs], a combination of S-limonene and a 3:1 mix of R:S camphor vapours (50% enantiomeric excess (e.e.)). The results are compared with those for the previous two mixtures in Table 1. The S-limonene PECD retains the same magnitude but, as would be predicted, opposite sign to the previous R-limonene mixtures while the camphor PECD is reduced in magnitude, reflecting the reduction in enantiomeric purity.
For any component the observed PECD can be written as G=fRGR+fSGS=(2fR−1)GR, where fX is the fractional abundance of enantiomer X and GX its PECD asymmetry. But since e.e.=(fR−fS) × 100% it is straightforward to show that e.e.=G/GR × 100%. We can obtain a value for camphor GR under the conditions of these measurements, assuming 100% e.e. of our commercial R- and S- camphor products, from the mean magnitude measured for Mix-RR and Mix-RS, which from Table 1 gives GR= 0.041(2) where the uncertainty in the last digit is the sample s.d. Hence the measured camphor PECD for Mix-R[Rs] (Table 1) suggests the result e.e.=0.5±0.2, in excellent agreement with the prepared composition of this sample.
Discussion
It is evident from the preceding results that the PECD asymmetry factor, G (equations (1) and (5)), evaluated simply from the electron emission yields in forward and backward directions (relative to the laser beam direction) provides a convenient, quantitative characterization of enantiomeric composition. These are identical to asymmetry factors obtained from the full analysis of the photoelectron angular distribution (see Methods section and Supplementary Table 1) allowing us to conclude that a full angular resolving electron detector is not a prerequisite for MS-PECD enantiomeric analysis; in principle a bi-detector, capable of discriminating just forward and backward electron scattering directions would be sufficient. Table 1 includes another comparison, this of results obtained with energy selection but without mass-tagging of the electrons. Even the partial overlapping of the PES regions assigned to camphor and limonene in Fig. 4 leads to a complete scrambling of the non-tagged PECD G values which no longer display an intuitively discernable pattern. Interpretation of such data therefore poses at best a considerably greater challenge and would require far greater a priori knowledge concerning the mixture under examination. The correlation of electron and ion data by coincidence detection methods therefore is capable of yielding a level of insight additional to either the electron- or ion mass-spectroscopy alone.
Many (chiral) molecules have two-photon absorptions accessible at single photon wavelengths ∼400 nm and ionization energies in a corresponding three-photon region around 9 eV. Combined with the large spectral bandwidth of an ultrafast laser pulse, the simultaneous ionization of multiple molecular components in a mixture can be expected to be relatively facile. Conversely, at longer wavelengths more selective ionization via selected chromophores becomes feasible. PECD itself provides a very rich signature of the ionized species, with asymmetries that vary depending on initial orbital, photon energy and molecular conformation20,21. Variation of the resonant laser ionization scheme would allow for effective exploitation of these properties, perhaps by employing more selective excitation through different resonant states (see Fig. 2) or by establishing conditions that optimize the magnitude of detectable PECD asymmetry—and hence further enhance sensitivity. (In this context it may be noted that despite the large excess of neon carrier gas (>99%) ionization of Ne is completely suppressed precisely because there is no accessible resonantly enhanced pathway with 392 nm photons, and a non-resonant mechanism would demand much higher laser intensity.) Furthermore, with picosecond lasers one can selectively excite individual vibrational bands within the electronically excited intermediate. This may provide opportunities for conformer specific resonant ionization34 and also open the door to possible rich vibronic phenomena in PECD capable of providing further structural insights35,36.
The precision of our e.e. determination is comparable to that achieved, with comparable data acquisition times, using a polarized microwave double resonance technique12,13,37 with detected phase providing enantiomer discrimination; for example, an e.e. for (-)-menthone was measured as 0.625±0.24238. Shorter acquisition times have been reported using a variation of this technique, permitting an e.e. of 0.025±0.005 to be measured with a 1,2-propanediol mixture with a known prepared 0.02 enantiomeric excess39. The asymmetry factors reported here are determined as a relatively simple function of two effective counters recording the number of electrons emitted into forward and backward hemispheres. These counts are assumed to be subject to Poisson statistics, which will determine the precision of the reported asymmetries, and especially the e.e. ratio. The reproducibility of repeated MS-PECD determinations, in both single and multi-component samples is, as shown in Table 1, very encouraging. Nevertheless, it is clear that an increase in data acquisition rate would provide benefits of reduced uncertainty associated with measurements and/or reduced measurement times with correspondingly improved convenience and sample consumption.
One obvious, and (in a limited sense) trivial to implement improvement would be an increase in laser repetition rate. A hundred-fold increase from the current 3 kHz, would yield a 10-fold improvement in statistical uncertainty, reducing that in the e.e. determination reported here to the level of a few %. Coincidence measurements with high (MHz) repetition rate sources are well established using synchrotron sources operating in their ‘few bunch’ temporal modes; indeed the first electron–ion coincidence imaging PECD measurements were obtained in such conditions40. More recently, electron–ion coincidence experiments with a 400 kHz short pulse laser source have been reported41 and indeed suitable high rep-rate fibre lasers providing ps–fs pulse durations are now commercially available. An alternative, and not mutually exclusive, possibility would be a relaxation of the severe ionization rate limitation, here set at less than 0.08 electrons per laser shot, imposed to minimize false coincidences. The adoption of covariance mapping techniques to replace electron–ion coincidence detection42, may offer one route to accommodate significantly enhanced count rates without loss of the fundamental capabilities we have demonstrated. But this limitation can be eliminated completely if mass-tagged electron detection were not required for discriminating mixture components; for example if wavelength selective excitation/ionization techniques could instead be employed, or for e.e. determinations on single components. It is clear in such cases that, subject to detector dynamic range, ionization rates increased by three or more orders of magnitude could then be utilized. A combination of higher laser repetition rate and relaxation of the limiting ionization rate per laser shot can feasibly be expected to yield an order of magnitude improvement in both precision and acquisition times.
There is an alternative approach to enantioselective laser ionization mass spectrometry, LIMS-CD, that measures CD in the overall ion yield. To date, most reported experiments have utilized low-order (n+n′) resonance enhanced excitations by ns UV lasers11,43,44, although recently Weitzel and co-workers have demonstrated fs laser LIMS-CD, including non-resonant 800 nm excitation10. LIMS-CD and MS-PECD experiments with an ultrafast laser can thus share the potential capabilities of this ionization technique, as detailed above. Beyond that, while the (multiphoton) PECD effect develops in the pure electric-dipole approximation, LIMS-CD appears to depend on higher order multipole interactions, and in some way conflates CD contributions from various linear and nonlinear photon absorption steps. This can be evidenced by the observation of different CD from nascent parent ion and more photon hungry fragmentation channels10,45. In MS-PECD any post-ionization absorption should not contribute differently to the measured asymmetry.
Both LIMS-CD and MS-PECD require steps to reduce instrumental asymmetry. In LIMS-CD intensity differences in the LCP and RCP beams, and pulse-to-pulse fluctuations of the temporal and spatial distribution of the laser pulse energy are most effectively overcome for a single chiral component by concurrent measurement of an achiral reference which is thus required to have a similar absorption spectra and laser intensity dependence10,44,46—ideally for both parent and daughter ion channels. In some circumstances this might be quite demanding to arrange in a multi-component simultaneous measurement. In contrast, our MS-PECD data acquisition methodology (outlined in Methods, below) deliberately accumulates equal counts from the two polarizations such that when forming the dichroism as a difference between the two polarizations any residual, polarization-independent instrumental asymmetry in the angular distributions, for example, resulting from detector spatial inhomogeneities, gets fully cancelled. This precaution is trivially implemented for all species, but so doing actively precludes observation of any CD that may be present in the ionization yields.
A laboratory scale ultrafast laser imaging mass spectrometer capable of simultaneously detecting multiple enantiomeric species in gaseous mixtures without requiring prior separation techniques or reactions with complexing reagents, such as we have considered here, will open new analytical applications for the sensitive and selective detection of chiral molecules. As the measurements are performed on mass-selected species we can envisage extending the technique to molecular complexes like chiral dimers, or microsolvated chiral molecules28. Furthermore, the combination of MS-PECD with laser desorption of chiral molecules from site-selected regions of surface samples15, will extend the MS-PECD technique to sample chiral molecules directly from spatially selected regions on a two-dimensional surface47. A complementary approach using a table-top laser source of elliptically polarized VUV for single photon PECD measurements has also been recently described48.
Finally we note that theoretical modeling of single photon PECD is generally of sufficient quality to identify unambiguously absolute molecular configuration from the experimental observations20,49. The current theoretical modelling for multiphoton PECD is less developed24,50 although work such as that presented here should help stimulate further theoretical efforts to model and quantify multiphoton PECD.
Methods
Experimental setup
The experimental setup used in the present work has been described in detail before33 and is schematically shown in Fig. 1. The expanding gas mixture generates a continuous molecular beam in the source chamber which is doubly skimmed downstream of the nozzle as it passes through the buffer chamber into the imaging spectrometer chamber. In the imaging chamber the molecular beam intersects the laser beam at 90°. After ionization of a molecule, the electron and ion are detected in coincidence on two opposing time- and position-sensitive delay line detectors. In our experiment we operated velocity map imaging voltages for electron detection and optimized voltages for optimal mass resolution (and not necessarily optimal voltages for velocity map imaging of ions) for ion detection.
Camphor and limonene enantiomers—R and S forms—of high purity (>96%) were purchased from Sigma Aldrich. The liquid limonene samples were held in an external stainless steel reservoir maintained at room temperature. A stream of pure neon gas at 0.6 bar backing pressure sweeps the vapour of limonene through a stainless steel transfer line and through a further sample reservoir, containing camphor solids, also at room temperature. A continuous seeded molecular beam is created by expanding the gas mixture through a nozzle into the first vacuum chamber from where it is doubly skimmed before entering the ionization region. To prepare Mix-S[Rs] a 3:1 mixture of R:S camphor (e.e.=50%) was accurately weighed out. It was then ground together to ensure a homogeneous sample of the enantiomers before being placed in the reservoir.
A commercial femtosecond laser system manufactured by Spectra Physics was used. The laser had a pulse duration of about 150 fs with 3 kHz repetition rate, and the frequency doubled output at 392 nm was attenuated to provide pulse energies of ∼10–12 μJ (Mix-RR and Mix-S[Rs]) and 13–16 μJ (Mix-RS). It was loosely focused into the molecular beam with a lens of about 30 cm focal length. The corresponding intensity in the ionization volume is estimated to be 1011–1012 W cm−2. Further details are provided in Supplementary Figs 1–4 and Supplementary Methods.
Data acquisition and analysis
For each mixture data were accumulated for ∼2 × 108 laser shots, during which the circular polarization (LCP/RCP) was switched every 500 s in order to reduce the effect of any experimental drift. The average number of electron events per laser shot, independent of coincident ion was limited to about 0.08. This count rate ensures that the probability for false coincidences is less than 0.3% assuming Poisson statistics.
The photoelectron angular distribution, Ip(θ), arising from an n-photon ionization of a chiral molecule with circular polarized light is given by24

where p is the polarization (p=+1 and p=−1 correspond to LCP and RCP radiation, respectively). Here, θ is the angle between the electron momentum and the propagation direction of the laser beam, Pi (cosθ) are Legendre polynomials and  are the corresponding coefficients. Because the odd
are the corresponding coefficients. Because the odd  coefficients are anti-symmetric with respect to circular polarization switching (
coefficients are anti-symmetric with respect to circular polarization switching ( ), whereas the even coefficients are unaltered21,22,24 the chiral (odd) coefficients can be isolated by forming the dichroism or difference angular distribution:
), whereas the even coefficients are unaltered21,22,24 the chiral (odd) coefficients can be isolated by forming the dichroism or difference angular distribution:

Forward–backward asymmetry arises from these odd polynomials in cos(θ). A useful measure to quantify the enantiomeric sensitivity of multiphoton PECD in a single asymmetry parameter has been suggested as

(truncated as shown for a three-photon process) and this can be trivially evaluated from considering, for each polarization p, the numbers of electrons emitted in the forward and backward hemispheres (respectively Np,f and Np,b ):21,22,24

This form of combining polarization measurements has the experimental advantage of cancelling any polarization-independent residual instrumental asymmetry that might result, for example, from detector gain inhomogeneity. To ensure optimum cancellation it is arranged that we achieve the same total count for each polarization such that (N+1,f+N+1,b)=(N−1,f+N−1,b). Assuming Poisson counting statistics there is an uncertainty  in each count and so the error in G is obtained by standard error propagation.
in each count and so the error in G is obtained by standard error propagation.
Mass-tagged and photoelectron-energy selected coincidence events were filtered from the data for the various polarization settings, and PECD asymmetry factors calculated using equation (5) are presented in Table 1, with errors estimated by error propagation assuming Poisson counting statistics.
Further experimental details are provided in Supplementary Methods and we also compare G factors derived from the full electron angular distribution analysis (equation (4)) with those from application of equation (5) in Supplementary Table 1.
Additional information
How to cite this article: Fanood, M. M. R. et al. Enantiomer-specific analysis of multi-component mixtures by correlated electron imaging–ion mass spectrometry. Nat. Commun. 6:7511 doi: 10.1038/ncomms8511 (2015).
References
Brenna, E., Fuganti, C. & Serra, S. Enantioselective perception of chiral odorants. Tetrahedron-Asymmetry 14, 1–42 (2003).
Song, W., Staudt, M., Bourgeois, I. & Williams, J. Laboratory and field measurements of enantiomeric monoterpene emissions as a function of chemotype, light and temperature. Biogeosciences 11, 1435–1447 (2014).
Eerdekens, G. et al. VOC measurements within a boreal forest during spring 2005: on the occurrence of elevated monoterpene concentrations during night time intense particle concentration events. Atmos. Chem. Phys. 9, 8331–8350 (2009).
Biasioli, F., Yeretzian, C., Mark, T. D., Dewulf, J. & Van Langenhove, H. Direct-injection mass spectrometry adds the time dimension to (B)VOC analysis. Trac-Trends Anal. Chem 30, 1003–1017 (2011).
Deleris, I. et al. Comparison of direct mass spectrometry methods for the on-line analysis of volatile compounds in foods. J. Mass Spectrom. 48, 594–607 (2013).
Tao, W. A., Gozzo, F. C. & Cooks, R. G. Mass spectrometric quantitation of chiral drugs by the kinetic method. Anal. Chem. 73, 1692–1698 (2001).
Wu, L. & Vogt, F. G. A review of recent advances in mass spectrometric methods for gas-phase chiral analysis of pharmaceutical and biological compounds. J. Pharm. Biomed. Anal. 69, 133–147 (2012).
Berova, N., Nakanishi, K. & Woody, R. Circular dichroism principles and applications 2nd edn Wiley-VCH (2000).
Berova, N., Polavarapu, P., Nakanishi, K. & Woody, R. Comprehensive chiroptical spectroscopy John Wiley & Sons (2012).
Horsch, P., Urbasch, G. & Weitzel, K. M. Analysis of chirality by femtosecond laser ionization mass spectrometry. Chirality 24, 684–690 (2012).
Boesl, U., Bornschlegl, A., Loge, C. & Titze, K. Resonance-enhanced multiphoton ionization with circularly polarized light: chiral carbonyls. Anal. Bioanal. Chem. 405, 6913–6924 (2013).
Patterson, D., Schnell, M. & Doyle, J. M. Enantiomer-specific detection of chiral molecules via microwave spectroscopy. Nature 497, 475 (2013).
Shubert, V. A., Schmitz, D., Patterson, D., Doyle, J. M. & Schnell, M. Identifying enantiomers in mixtures of chiral molecules with broadband microwave spectroscopy. Angew. Chem. Int. Ed 53, 1152–1155 (2014).
Mukhopadhyay, P., Zuber, G., Wipf, P. & Beratan, D. N. Contribution of a solute’s chiral solvent imprint to optical rotation. Angew. Chem. Int. Ed 46, 6450–6452 (2007).
de Vries, M. S. & Hobza, P. Gas-phase spectroscopy of biomolecular building blocks, theory and experiment. Annu. Rev. Phys. Chem. 58, 585–612 (2007).
Zehnacker, A. & Suhm, M. A. Chirality recognition between neutral molecules in the gas phase. Angew. Chem. Int. Ed 47, 6970–6992 (2008).
Zehnacker, A. Chiral recognition in the gas phase Taylor & Francis (2010).
Bodi, A., Hemberger, P., Osborn, D. L. & Sztaray, B. Mass-resolved isomer-selective chemical analysis with imaging photoelectron photoion coincidence spectroscopy. J. Phys. Chem. Lett. 4, 2948–2952 (2013).
Ritchie, B. Theory of the angular distribution of photoelectrons ejected from optically active molecules and molecular negative ions. Phys. Rev. A 13, 1411–1415 (1976).
Powis, I. in Adv. Chem. Phys. ed. Light J. C. Vol. 138, 267–329Wiley (2008).
Janssen, M. H. M. & Powis, I. Detecting chirality in molecules by imaging photoelectron circular dichroism. Phys. Chem. Chem. Phys. 16, 856–871 (2014).
Ram, N. B., Lehmann, C. S. & Janssen, M. H. M. Probing chirality with a femtosecond reaction microscope. EPJ Web Conf. 41, 02029 (2013).
Lux, C. et al. Circular dichroism in the photoelectron angular distributions of camphor and fenchone from multiphoton ionization with femtosecond laser pulses. Angew. Chem. Int. Ed. 51, 5001–5005 (2012).
Lehmann, C. S., Ram, N. B., Powis, I. & Janssen, M. H. M. Imaging photoelectron circular dichroism of chiral molecules by femtosecond multiphoton coincidence detection. J. Chem. Phys. 139, 234307 (2013).
Rafiee Fanood, M. M., Powis, I. & Janssen, M. H. M. Chiral asymmetry in the multiphoton ionization of methyloxirane using femtosecond electron-ion coincidence imaging. J. Phys. Chem. A 118, 11541–11546 (2014).
Lux, C., Wollenhaupt, M., Sarpe, C. & Baumert, T. Photoelectron circular dichroism of bicyclic ketones from multiphoton ionization with femtosecond laser pulses. Chemphyschem. 16, 115–137 (2015).
Rafiee Fanood, M. M., Janssen, M. H. M. & Powis, I. Enantioselective femtosecond laser photoionization spectrometry of limonene using photoelectron circular dichroism. Phys. Chem. Chem. Phys. 17, 8614–8617 (2015).
Powis, I. et al. A photoionization investigation of small, homochiral clusters of glycidol using circularly polarized radiation and velocity map electron-ion coincidence imaging. Phys. Chem. Chem. Phys. 16, 467–476 (2014).
Vredenborg, A., Lehmann, C. S., Irimia, D., Roeterdink, W. G. & Janssen, M. H. M. The reaction microscope: imaging and pulse shaping control in photodynamics. Chemphyschem. 12, 1459–1473 (2011).
Smialek, M. A. et al. Limonene: electronic state spectroscopy by high-resolution vacuum ultraviolet photoabsorption, electron scattering, He(I) photoelectron spectroscopy and ab initio calculations. Phys. Chem. Chem. Phys. 14, 2056–2064 (2012).
Lozovoy, V. V. et al. Control of molecular fragmentation using shaped femtosecond pulses. J. Phys. Chem. A 112, 3789–3812 (2008).
Duffy, M. J. et al. Fragmentation of neutral amino acids and small peptides by intense, femtosecond laser pulses. J. Am. Soc. Mass Spectrom. 24, 1366–1375 (2013).
Vredenborg, A., Roeterdink, W. G. & Janssen, M. H. M. A photoelectron-photoion coincidence imaging apparatus for femtosecond time-resolved molecular dynamics with electron time-of-flight resolution of σ=18ps and energy resolution ΔE/E=3.5%. Rev. Sci. Inst 79, 063108 (2008).
Karaminkov, R., Chervenkov, S., Delchev, V. & Neusser, H. J. High-resolution mass-selective uv spectroscopy of pseudoephedrine: evidence for conformer-specific fragmentation. J. Phys. Chem. A 115, 9704–9713 (2011).
Powis, I. The influence of vibrational parity in chiral photoionization dynamics. J. Chem. Phys. 140, 111103 (2014).
Garcia, G. A., Nahon, L., Daly, S. & Powis, I. Vibrationally induced inversion of photoelectron forward-backward asymmetry in chiral molecule photoionization by circularly polarized light. Nat. Commun 4, 2132 (2013).
Patterson, D. & Schnell, M. New studies on molecular chirality in the gas phase: enantiomer differentiation and determination of enantiomeric excess. Phys. Chem. Chem. Phys. 16, 11114–11123 (2014).
Shubert, V. A., Schmitz, D. & Schnell, M. Enantiomer-sensitive spectroscopy and mixture analysis of chiral molecules containing two stereogenic centers—microwave three-wave mixing of menthone. J. Mol. Spectrosc 300, 31–36 (2014).
Patterson, D. & Doyle, J. M. Sensitive chiral analysis via microwave three-wave mixing. Phys. Rev. Lett. 111, 023008 (2013).
Garcia, G. A. et al. Circular dichroism in the photoelectron angular distribution from randomly oriented enantiomers of camphor. J. Chem. Phys. 119, 8781–8784 (2003).
Furch, F. J. et al. Carrier-envelope phase stable few-cycle pulses at 400 kHz for electron-ion coincidence experiments. Opt. Express 21, 22671–22682 (2013).
Mikosch, J. & Patchkovskii, S. Coincidence and covariance data acquisition in photoelectron and -ion spectroscopy. II. Analysis and applications. J. Mod. Opt 60, 1439–1451 (2013).
Li, R., Sullivan, R., Al-Basheer, W., Pagni, R. M. & Compton, R. N. Linear and nonlinear circular dichroism of R-(+)-3-methylcyclopentanone. J. Chem. Phys. 125, 144304 (2006).
Loge, C. & Boesl, U. Multiphoton ionization and circular dichroism: new experimental approach and application to natural products. Chemphyschem. 12, 1940–1947 (2011).
Loge, C. & Boesl, U. Laser mass spectrometry with circularly polarized light: circular dichroism of molecular ions. Chemphyschem. 13, 4218–4223 (2012).
Titze, K., Zollitsch, T., Heiz, U. & Boesl, U. Laser mass spectrometry with circularly polarized light: circular dichroism of cold molecules in a supersonic gas beam. Chemphyschem. 15, 2762–2767 (2014).
Jungmann, J. H. & Heeren, R. M. A. Emerging technologies in mass spectrometry imaging. J. Proteomics 75, 5077–5092 (2012).
Ferré, A. et al. A table-top ultrashort light source in the extreme-ultraviolet for circular dichroism experiments. Nature Photon. 9, 93–98 (2015).
Powis, I. in Comprehensive chiroptical spectroscopy eds Berova N., Polavarapu P., Nakanishi K., Woody R. Vol. 1, pp 407–431John Wiley & Sons (2012).
Dreissigacker, I. & Lein, M. Photoelectron circular dichroism of chiral molecules studied with a continuum-state-corrected strong-field approximation. Phys. Rev. A 89, 053406 (2014).
Acknowledgements
This research has been financially supported by the division of Chemical Sciences of the Netherlands Organisation for Scientific Research (NWO-CW VICI and CW-BAZIS). It has further received funding from LASERLAB-EUROPE (grant agreement no. 284464, EC’s Seventh Framework Programme) and the Marie Curie Initial Training Network ICONIC. We are very grateful for the discussions on chiral mass spectrometry with Prof. N.N.M. Nibbering who passed away in 2014, and we dedicate this paper to his memory. We also gratefully acknowledge the collaboration with Prof. W.J. Buma on REMPI spectroscopy of cold chiral molecules. The authors thank Dr. H. Alvaro Galue for assistance in some of the data acquisition. Furthermore, the authors gratefully acknowledge the excellent technical support by Mr R. Kortekaas in the construction of the imaging mass spectrometer and supporting experimental apparatus.
Author information
Authors and Affiliations
Contributions
The experiment was first conceived by M.H.M.J., and designed by M.H.M.J. and I.P. Experimental data recording was carried out by M.M.R.F. with some assistance from I.P. Data analysis was done by M.M.R.F. C.S.L. and N.B.R. contributed to development of the instrument and data handling methodology. The paper was written by I.P. and M.H.M.J. with contributions from the other authors.
Corresponding author
Ethics declarations
Competing interests
M.H.M.J. is the owner of SciTechAdvisors. All other authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-4, Supplementary Table 1, Supplementary Methods and Supplementary References (PDF 285 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fanood, M., Ram, N., Lehmann, C. et al. Enantiomer-specific analysis of multi-component mixtures by correlated electron imaging–ion mass spectrometry. Nat Commun 6, 7511 (2015). https://doi.org/10.1038/ncomms8511
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms8511
This article is cited by
-
Identification of the enantiomeric nature of 2-keto-3-deoxy-galactonate in the catabolic pathway of 3,6-anhydro-l-galactose
Applied Microbiology and Biotechnology (2023)
-
Full quantum control of enantiomer-selective state transfer in chiral molecules despite degeneracy
Communications Physics (2022)
-
Assignment-free chirality detection in unknown samples via microwave three-wave mixing
Communications Chemistry (2022)
-
Conformer-dependent vacuum ultraviolet photodynamics and chiral asymmetries in pure enantiomers of gas phase proline
Communications Chemistry (2021)
-
Chiral discrimination by recollision enhanced femtosecond laser mass spectrometry
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
