
Abstract
Originally described by the late evolutionary biologist Leigh Van Valen, the Red Queen hypothesis posits that the evolutionary arms race between hosts and their pathogens selects for discrete, genetically encoded events that lead to competitive advantages over the other species. Examples of immune evasion strategies are seen throughout the co-evolution of the mammalian immune system and pathogens, such as the enzymatic inactivation of nuclear factor-κB signaling or host translation by pathogen-encoded virulence factors. Such immunoevasive maneuvers would be expected to select for the evolution of innate immune counterstrategies. Recent advances in our understanding of host immunity and microbial pathogenesis have provided insight into a particular innate immune adaptation, termed bystander activation. Bystander activation occurs as a consequence of infected cells alerting and instructing neighboring uninfected cells to produce inflammatory mediators, either through direct cell contact or paracrine signals. Thus, bystander activation can allow the immune system to overcome the ability of pathogens to disarm immune signaling in directly infected cells. This review presents an overview of the general hallmarks of bystander activation and their emerging role in innate immunity to intracellular pathogens, as well as examples of recent mechanistic discoveries relating to the bystander activation during infection with specific pathogens relevant to human health and disease.
Similar content being viewed by others


Introduction
Pathogens have developed a number of strategies to evade recognition and clearance by the host immune system. One such strategy, which exists across the microbial taxonomic spectrum, is to invade and establish a unique intracellular niche permissive for microbial replication within host cells. To survive within the host intracellular environment, many pathogens employ virulence factors that manipulate host cell processes, such as protein translation and membrane trafficking, to allow for the acquisition of nutrients necessary for growth and replication. Conversely, the innate immune system (and eventual effector cells of the adaptive immune system) is capable of recognizing the presence of an intracellular microbial infection and mounting a variety of responses aimed at defeating the pathogen. Examples of recognition strategies include the ligation of host cytosolic pattern recognition receptors (PRRs) by evolutionarily conserved pathogen-associated molecular patterns (PAMPs) introduced into the host cytosol by intracellular pathogens, or the detection of ‘patterns of pathogenesis’, such as the recognition of viral-mediated downregulation of major histocompatibility class I protein expression on the cell surface by natural killer cells.1, 2, 3, 4 However, many pathogens have evolved to evade immune detection, as exemplified by the alteration or downregulation of bacterial flagellin expression to evade recognition by the innate immune receptors Toll-like receptor (TLR) 5 and NLR family, apoptosis inhibitory protein 5 (NAIP5).5, 6, 7 At the expense of additional energy and genomic utilization, the host and microbe competitively evolve to occupy spaces of increasingly superior evolutionary fitness.
It has long been known that one of the most potent downstream outcomes of innate immune recognition of intracellular infection involves the rapid production of pro-inflammatory cytokines, such as type I interferons (IFNs), interleukin 12 (IL-12) and tumor necrosis factor (TNF), which are important for antimicrobial defense.4 The prevailing dogma is that these cytokines are produced by directly infected cells in response to PRR ligation to alert neighboring, uninfected cells to the presence of a pathogen. However, it has also long been known that many intracellular pathogens deploy a number of strategies, such as blocking mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) signaling or host translation, to silence this wave of signals in the infected cell and prevent the subsequent propagation of cytokine-mediated immune responses.8, 9, 10, 11, 12 Thus, it remained unclear how, in the face of immune silencing, the host was capable of initiating and amplifying these signaling and cytokine cascades, as the recognition and response events were thought to take place in cis within an infected cell. However, with more recent technical advances, particularly those that allow for the study of host:pathogen interactions with single-cell resolution, scientists have begun to appreciate how and where these early cytokines are made. Observations in these studies have led to novel insight into the role of uninfected bystander cells in the primary immune activation events immediately following infection.
We would like to here define bystander cells in the context of innate immunity as uninfected, neighboring cells (although not necessarily adjacent to or in contact with the infected cell in three-dimensional organotypic space), which signal to the immune system, even in lieu of direct pathogen recognition, in a process known as bystander activation. In this model, bystander cells, which may or may not be of the same cell type as the infected cell, produce cytokines upon receiving indirect pathogen recognition signals or microbial-derived products from the infected cell, thus enabling bystander cells to bypass pathogen-mediated attenuation of innate immune signaling within the directly infected cell. Intercellular communication between infected and bystander cells can involve either direct cell–cell contact or soluble signals that act at a distance. The following sections provide examples of bystander activation in infection models of viral, bacterial and protozoan pathogens, and hosts ranging from Drosophila to humans. These diverse examples serve to support the concept of bystander activation as a critical evolutionary adaptation in metazoan innate immunity.
Viral pathogens
The innate immune system uses a variety of PRRs to detect viral infection. Many of these PRRs sense foreign nucleic acids and trigger the production of type I IFNs.13 However, many viruses have evolved virulence factors that antagonize type I IFN production by infected cells.11, 12 Thus, it is unclear how an effective type I IFN response can be generated during viral infection. A study utilized an IFN-sensitive response element–green fluorescent protein (GFP) reporter cell line that specifically reports activation of the transcription factor IFN regulatory factor (IRF) 3 rather than type I IFN signaling to probe IRF3-dependent responses at the single-cell level.14 Using fluorescence microscopy, this system revealed that the transfection of fluorescently labeled DNA complexes into cells induced IRF3-dependent reporter expression in both transfected and neighboring untransfected cells. Furthermore, clusters of transfected and untransfected bystander cells produced the majority of antiviral cytokines, such as TNF and IFNβ, following nucleic acid stimulation.14 Induction of antiviral responses in bystander cells required cellular contact via gap junctions, which are intercellular channels composed of oligomerized connexin proteins.14 The precise molecules communicated through gap junctions and responsible for bystander activation were not identified, in part, because the molecular mechanisms underlying immune sensing of cytosolic DNA were poorly understood at this time.
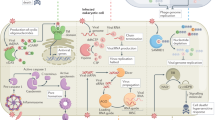
It is now known that cyclic GMP–AMP synthase (cGAS) is a key immune sensor critical for host detection of cytosolic DNA, both self and foreign.15, 16 Upon binding DNA, cGAS produces cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), which binds to the endoplasmic reticulum-resident adapter protein STING (stimulator of IFN genes), thus leading to IRF3 activation and subsequent induction of type I IFNs.16 Recently, cGAMP was shown to behave as a secondary messenger and be transmitted via gap junctions to activate bystander cells in an in vitro model of vaccinia virus infection (Figure 1).17 Fluorescence microscopy of cells infected with a GFP-tagged vaccinia strain revealed that the activation of STING by cGAMP took place not only in virally infected cells, but also in neighboring bystander cells.17 Therefore, the ligation of STING in uninfected cells by cGAMP produced in infected cells represents a means of bystander activation that circumnavigates the canonical, cell-intrinsic signaling cascade that is thought to occur upon PRR engagement. Activation of these bystander cells led to enhanced resistance to viral infection due to induction of antiviral genes.17 Thus, cell–cell transmission of cGAMP via gap junctions enables host cells to mount a successful antiviral response despite viral evasion strategies.

General mechanisms of bystander cell activation during infection. (a) An infected (green) cell transfers molecules, such as cGAMP, Ca2+ or other signals, via gap junctions to a neighboring bystander cell (blue). (b) Host-derived extracellular vesicles containing host signals or PAMPs, bacterial outer membrane vesicles containing PAMPs, or immune signaling complexes (inflammasomes) are released from infected cells and taken up by bystander cells. (c) Infected cells release soluble molecules, such as cytokines, that are detected by bystander cells. cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; PAMPs, pathogen-associated molecular patterns.
In addition to transmission of cGAMP via gap junctions, cGAMP can also be packaged into viral particles and extracellular vesicles (Figure 1).18, 19 These particles can deliver cGAMP and trigger STING-dependent signaling in newly infected cells. This represents yet another mechanism by which cGAMP produced by an infected cell can be transferred and propagate innate immune signaling in neighboring cells. As cGAS responds to many viruses, including retroviruses such as HIV,20 it would be expected that bystander activation caused by cGAMP transmitted through gap junctions or cGAMP packaged into viral particles could be a common immune strategy employed against a wide array of viruses. Furthermore, cGAS also produces cGAMP upon sensing of bacterial DNA, such as during Mycobacterium tuberculosis infection,21, 22 and many bacteria produce their own cyclic dinucleotides that are also recognized by STING and potently trigger the IRF3 signaling pathway.23, 24, 25, 26 One could envision that during bacterial infection, either cGAMP or bacterial cyclic dinucleotides could be transmitted from infected cells to neighboring bystander cells via gap junctions, thus ensuring and amplifying antibacterial immune responses as well.
Many viruses disrupt retinoic acid-inducible gene 1 (RIG-I)-like receptor and TLR-mediated signaling. For example, the positive-sense RNA virus, hepatitis C virus (HCV), encodes a viral protease, NS3/4A, which cleaves the RIG-I and melanoma differentiation-associated protein 5 (MDA-5) signaling adapter mitochondrial antiviral signaling protein (MAVS).27, 28, 29 Whether or how the host is capable of mounting a robust type I IFN response against HCV, in the face of immune evasion, has been unclear. Recent studies have indeed implicated bystander activation in mediating an effective type I IFN response as a result of cell–cell communication between HCV-infected hepatocytes and uninfected plasmacytoid dendritic cells (pDCs).30 By taking advantage of the inability of pDCs to support viral replication, it was shown through in vitro co-culture experiments that pDCs, not the infected hepatocytes, produced type I IFNs in response to infection. Further investigation revealed that hepatocyte-derived exosomes containing viral RNA were taken up by neighboring DCs, thereby triggering nucleic acid sensors in the neighboring pDCs (Figure 1).31 These exosomes are also detectable in the serum of HCV-infected patients, suggesting that they also mediate bystander activation during the natural course of HCV infection.32 Similarly, pDCs were activated in a TLR7-dependent manner, as a result of exosomal RNA released from hepatocytes in an in vitro model of negative-strand RNA lymphocytic choriomeningitis virus infection.33 Therefore, exosomal transfer of viral PAMPs to bystander immune cells may be a general mechanism employed by the immune system to circumvent viral inhibition of innate immune signaling.
This theme of viral nucleic acid transfer between host cells is not unique to mammals. Viral nucleic acid transfer to bystander cells, although exosome-independent, has been described in a Drosophila melanogaster model of Sindbis virus infection. In this case, infected host cells released unpackaged, viral double-stranded RNA (dsRNA). These nucleic acids were then taken up by uninfected cells, where they led to the induction of protective antiviral immunity throughout the fly via RNA interference mechanisms.34 Mutant flies deficient in the dsRNA uptake pathway were profoundly susceptible to infection with both Drosophila C virus and Sindbis virus. Thus, antiviral immunity in Drosophila relies on cell–cell propagation of innate immune signals as well.
In addition to bystander responses to viral nucleic acids, there is also evidence that bystander responses triggered by other viral PAMPs are critical for orchestrating both innate and adaptive responses. Many pathogens, including influenza virus, would be expected to disarm the function of DCs, a critical cell type that serves as a bridge between innate and adaptive immunity. During influenza infection, the NLR family pyrin domain containing 3 (NLRP3) is activated in response to the influenza M2 ion channel, leading to the formation of a multiprotein complex termed the inflammasome. The NLRP3 inflammasome then activates the host protease caspase-1, leading to processing and release of IL-1 family cytokines, including IL-1α and IL-1β. Subsequently, it was found that IL-1 receptor (IL-1R) signaling, rather than direct sensing of viral infection by the PRRs TLR7 or RIG-I, was required for DC activation during influenza infection (Figure 1). IL-1R signaling was both sufficient and necessary for DCs to upregulate expression of costimulatory molecules such as CD86, migrate to lymph nodes and prime a virus-specific CD8+ T-cell response.35 Why direct sensing of influenza virus by the PRRs TLR7 and RIG-I is not sufficient is unclear, but one possibility is that influenza, like many viral pathogens, may disarm signaling downstream of these innate immune sensors in directly infected DCs. Given that many viral infections induce inflammasome activation, release of IL-1 cytokines by infected cells and subsequent IL-1 signaling to uninfected DCs may provide a failsafe mechanism for ensuring a successful immune response to a wide variety of viruses.
Bacterial pathogens
Much like the gap junction-dependent transfer of cGAMP during vaccinia infection, infected host cells are capable of using gap junctions to directly transfer pathogen recognition signals to uninfected bystander cells in response to bacterial stimuli as well. In response to treatment with the bacterial PAMP lipopolysaccharide (LPS), immune cells and epithelial cells modulate expression of the gap junction protein connexin 43 (Cx43).36, 37 Furthermore, LPS-mediated activation of DCs requires Cx43-dependent gap junctions, suggesting that LPS signaling triggers molecular signals of an unknown nature that are then propagated via gap junctions to maximize immune activation (Figure 1).38 These early studies pointed to a role for gap junction-mediated intercellular communication in host responses to bacterial stimuli. Subsequent studies described below have since shown that cell–cell communication mediated by gap junctions indeed do have a role in propagating the host response to bacterial pathogens.
During infection with the extracellular Gram-negative pathogen Pseudomonas aeruginosa, TLR2 signaling induces a Ca2+ flux that stimulates NF-κB and MAPK signaling required for the recruitment and activation of neutrophils to the airway.39, 40 In response to TLR2 ligands or P. aeruginosa infection, Ca2+ fluxes could be transmitted from stimulated human airway cells to adjacent cells in a manner requiring Cx43-mediated gap junctions; this led to the increased production of the neutrophil-attracting chemokine CXCL8 by epithelial cells. Furthermore, treatment of P. aeruginosa-infected mice with pharmacological gap junction inhibitors significantly decreased neutrophil recruitment.40 These findings suggest that the gap junction-mediated activation of neighboring bystander cells is critical for promoting antibacterial host defense in vivo.
The intracellular Gram-negative pathogen Shigella flexneri uses a type III secretion system to translocate multiple bacterial effector proteins into the host cell cytosol to impair host cell signaling and production of cytokines and chemokines, such as the neutrophil chemoattractant IL-8.41 For example, the effector OspG inhibits host ubiquitylation, thus blocking IκB degradation and subsequent NF-κB activation, whereas OspF is a phosphothreonine lyase that irreversibly dephosphorylates p38 and ERK MAPKs.42, 43, 44 By employing microscopic analysis of S. flexneri-infected host cell monolayers at the single-cell level, it was found that infected cells were poor producers of IL-8.45 Instead, within minutes post infection, infected cells potentiated NF-κB, JNK and ERK signaling in neighboring uninfected cells, leading to IL-8 production by these bystander cells.45 Bystander IL-8 production was also observed during infection with the bacterial pathogens Listeria monocytogenes and Salmonella enterica serovar typhimurium.45 The induction of this bystander response required sensing of peptidoglycan-derived fragments by the intracellular receptor nucleotide-binding oligomerization domain-containing protein 1 (NOD1) in the infected cell, and then propagation of a signal to the neighboring cell via cell–cell contact through gap junctions. The nature of this transmitted signal is unknown, but could involve secondary messengers such as calcium or other host-derived signals.
Bystander activation in response to bacterial PAMPS or infection can also be mediated by soluble signals. It was found that in response to LPS, as well as viral PAMPs, a small number of early responder cells secrete IFNβ, which activates expression of antimicrobial genes in other cells to yield an efficient response at the population level.46, 47 Uncovering this response required single-cell analysis, either through fluorescence microscopic analysis of RNA expression or single-cell RNA-sequencing in conjunction with various techniques to chemically or physically block cell–cell communication. A study investigating the immune response mounted against L. monocytogenes infection employed single cell approaches, involving fluorescence microscopy and flow cytometric analysis of epithelial cells infected by GFP-expressing L. monocytogenes.48 Directly infected cells were impaired in their ability to produce the chemokines CXCL2 and CXCL5, suggesting that L. monocytogenes inhibits chemokine production. Instead, these chemokines were primarily produced in neighboring non-infected epithelial cells, and this cell–cell communication was independent of gap junctions or cytokine secretion. Instead, reactive oxygen intermediates, produced by NADPH oxidase (NOX) 4 in infected cells, mediated paracrine activation of neighboring bystander cells.48 How NOX4 is activated in response to L. monocytogenes infection is not entirely clear, but appears to involve detection of L. monocytogenes-derived ligands by NOD2 and other cytosolic innate immune receptors.
In another example of bystander activation, a limited repertoire of soluble inflammatory cytokines that is selectively synthesized by infected cells can activate bystander cells to produce cytokines and subsequently amplify and diversify the immune response. Legionella pneumophila utilizes a type IV secretion system (T4SS) to translocate bacterial effector proteins that aid in establishing a membrane-bound organelle that supports intracellular infection.49 This T4SS translocates several effectors, including Lgt1, Lgt2, Lgt3, SidI, SidL, Pkn5 and Lpg1489, which potently inhibit host protein synthesis, in part, by impairing host translational elongation.50, 51 Furthermore, L. pneumophila infection further inhibits host protein synthesis by suppressing translational initiation.52 Despite a >95% decrease in de novo protein synthesis, L. pneumophila-infected macrophages produce a robust cytokine response that paradoxically requires immune sensing of T4SS activity.53 A T4SS-dependent inflammatory response characterized by robust pro-inflammatory cytokine production and neutrophil recruitment to the lung is observed in mice during pulmonary infection as well.54 To investigate how pro-inflammatory cytokines are made despite the effector-driven block in host translation, two independent studies used flow cytometry to distinguish infected from uninfected macrophages within the same population.55, 56 In these studies, intracellular cytokine staining was performed following infection of macrophages with L. pneumophila reporter strains expressing either GFP or a T4SS effector translationally fused to the enzyme β-lactamase, which modifies a fluorescent dye loaded into host cells following T4SS-mediated translocation. By tracking directly infected macrophages, it was found that these cells still synthesized the cytokines IL-1α and IL-1β de novo.55, 56 Selective translation of IL-1α and IL-1β in infected macrophages required signaling through the TLR adapter MyD88 through an as-yet-unidentified molecular mechanism.55 Legionella-infected cells then go on to process and secrete mature IL-1 cytokines upon cytosolic sensing of bacterial ligands such as flagellin and LPS, and subsequent inflammasome activation.57, 58 Thus, inflammasome-dependent cytokines escape the effector-driven translational block, as they are still selectively translated and released by L. pneumophila-infected cells.
The translational block in L. pneumophila-infected macrophages, however, did hinder their ability to produce other important pro-inflammatory proteins, such as IL-6, IL-12, TNF and the costimulatory molecule CD86.56 These proteins were instead produced by bystander macrophages. Analysis of pulmonary infection similarly found that directly infected alveolar macrophages were also impaired in their ability to produce TNF, but still upregulated expression of IL-1α and IL-1β.56 TNF was produced primarily by uninfected alveolar macrophages, as well as a variety of other bystander immune cell types, including neutrophils, inflammatory monocytes and DCs.56 The costimulatory molecule CD86 was similarly upregulated by bystander DCs. Pro-inflammatory responses by these bystander populations required IL-1 signaling, as mice lacking the IL-1R exhibited a marked reduction in TNF and IL-12 production, and CD86 expression by bystander cells in vivo (Figure 1).56 How IL-1R signaling activates bystander cells during in vivo infection is not yet clear, in part, because it is unknown whether cell-intrinsic IL-1R signaling is required for bystander immune responses to L. pneumophila. Interestingly, IL-1 signaling was neither required nor sufficient to elicit expression of TNF and IL-6 in bone marrow-derived macrophages (BMDMs) in vitro, suggesting a role for other selectively translated signals in mediating bystander activation in BMDMs and potentially during pulmonary infection.56 Because inflammasomes respond to many bacterial infections, it would be expected that in addition to the key role of IL-1 signaling in driving neutrophil recruitment, IL-1 would similarly drive bystander production of pro-inflammatory cytokines in response to other bacterial pathogens as well.
The cytokine IL-12 has a major role in controlling many intracellular bacterial pathogens, including Mycobacterium spp. Infecting yet40 reporter mice expressing the Il12p40 gene linked via an internal ribosome entry site to YFP with Mycobacterium bovis bacillus Calmette-Guérin (BCG) expressing dsRed fluorescent protein enabled tracking of both the cellular source of IL-12p40 and the location of BCG bacteria.59 DCs were the major source of IL-12p40, and these cells were not only uninfected, but also did not co-localize with BCG. Further analysis indicated that DCs directly infected with BCG exhibited impaired IL-12p40 production, and that soluble mycobacterial components could initiate the IL-12p40 production in uninfected DCs during in vitro infection.59 Interestingly, macrophages infected with BCG or M. tuberculosis release exosomes containing bacterial lipoproteins and lipoaraminomannan, and these exosomes stimulate the cytokine production in naive macrophages (Figure 1).60, 61 Furthermore, exosomes isolated from the BALF and serum of BCG-infected mice can induce macrophages to produce cytokines.60, 62 In addition to exosomes, M. tuberculosis-infected macrophages actively release bacterial membrane vesicles containing bacterial lipoglycans and lipoproteins that induce TLR2 signaling and cytokine responses in uninfected macrophages during in vitro infection.63 Collectively, these studies suggest that bystander activation occurs during infection with various Mycobacterium spp., and that release of bacterial ligands by infected cells may provide another possible mechanism by which bacterial infection can be detected by uninfected bystanders.
In addition to cell–cell communication via cytokines or bacterial PAMPs, there is evidence that infected cells release innate immune signaling machinery that can be taken up by bystander cells. Two independent studies described that macrophages release active inflammasome complexes during in vitro stimulation.64, 65 These preformed inflammasome complexes were internalized and induced IL-1β maturation in neighboring bystander cells, without the need for additional inflammasome-triggering stimuli (Figure 1). This mechanism may be active in vivo, as extracellular inflammasome complexes were visualized in the lymph nodes of mice infected with P. aeruginosa, and injection of purified inflammasome complexes into mice led to IL-1-dependent recruitment of neutrophils.64 Thus, release of inflammasome complexes by infected cells represents yet another mechanism for propagating intercellular communication between infected and bystander cells.
Protozoan pathogens
There is evidence that bystander innate immune activation occurs in response to intracellular protozoan parasites as well. Macrophages infected with Leishmania amazonensis released extracellular vesicles that enhanced the production of pro-inflammatory cytokines, such as IL-12 and TNF, by uninfected macrophages in vitro (Figure 1).66 These extracellular vesicles appeared to be of host origin, but the nature of the immunostimulatory ligands contained within the vesicles and whether they are of host or parasite origin is unclear. Extracellular vesicles isolated from the plasma of Plasmodium berghei-infected mice or from Plasmodium falciparum-infected RBCs could also activate naïve host cells in vitro.67, 68 The extracellular vesicles released by P. falciparum-infected red blood cells (RBCs) were of host origin, and contained both host proteins and membrane-associated parasite antigens. During in vivo Toxoplasma gondii infection of mice, it was found that IL-12 was produced by bystander inflammatory monocytes and DCs.69 How these bystander cells produce IL-12 in response to T. gondii is unknown, but presumably involves immune sensing of a soluble host or parasite factor.
As the host immune system evolves bystander activation mechanisms to deal with a number of immune attenuation approaches by pathogenic microbes, one might suspect that pathogens, in turn, would evolve to overcome bystander activation—a further manifestation of the Red Queen hypothesis. Indeed, using T. gondii expressing an effector:Cre recombinase fusion protein that can be translocated into host cells, in conjunction with reporter mice that express the fluorescent protein ZsGreen following Cre-mediated recombination, it was revealed that T. gondii injects its effector proteins into both infected and uninfected cells.70 Effector injection into both infected and uninfected cells has biological consequences, as injection of the rhoptry protein, ROP16, leads to robust STAT6 activation in both infected and uninfected cells.70 STAT6 activation by T. gondii is thought to inhibit IL-12 production, a cytokine essential to parasite resistance. This ability to inject effectors into both infected and uninfected cells may critically enable T. gondii to attenuate innate immune activation in bystander cells. Thus, by injecting effectors into uninfected, as well as infected cells, the parasite can manipulate the host cytokine response to enable the parasite’s survival.
Conclusion
Lewis Carroll wrote in his 1871 novel, Through the Looking-Glass, from which the Red Queen hypothesis was derived, ‘Now, here, you see, it takes all the running you can do, to keep in the same place. If you want to get somewhere else, you must run at least twice as fast as that!’71 The observation that populations of hosts and pathogens are constantly under selective pressure to maintain a competitive advantage over one another embodies this idea. As pathogens have evolved numerous mechanisms to maintain intracellular niches within their hosts, host cells, in turn, have evolved ways to alert the immune system of the presence of an infection. Over time, host cells have developed means to communicate with neighboring, uninfected bystander cells in situations in which primary immune activation mechanisms have been silenced in the directly infected cells. This review synthesizes some of the most recent literature, regarding bystander activation across a diverse pathogenic microbial taxonomy, as it is important to the fields of microbial pathogenesis and immunology to understand some of these mechanisms and how they function to initiate and shape a variety of immune responses. As researchers continue to elucidate the complexities of the host–microbial relationship, we posit that bystander activation will be shown to be a critical component in many of these dynamics. We expect that there will be diverse mechanisms driving bystander activation, and that many pathogens will evade or manipulate mechanisms of bystander activation for their own advantage. Understanding the role for uninfected bystander cells in infectious pathologies is not only important for the advancement of our understanding of host:pathogen biology, but it also will continue to drive the forefronts of medicine, evolutionary biology and the vast study of infectious diseases.
References
Medzhitov R, Janeway CA Jr . Innate immunity: the virtues of a nonclonal system of recognition. Cell 1997; 91: 295–298.
Huard B, Fruh K . A role for MHC class I down-regulation in NK cell lysis of herpes virus-infected cells. Eur J Immunol 2000; 30: 509–515.
Vance RE, Isberg RR, Portnoy DA . Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 2009; 6: 10–21.
Iwasaki A, Medzhitov R . Control of adaptive immunity by the innate immune system. Nat Immunol 2015; 16: 343–353.
Leifer CA, McConkey C, Li S, Chassaing B, Gewirtz AT, Ley RE . Linking genetic variation in human Toll-like receptor 5 genes to the gut microbiome's potential to cause inflammation. Immunol Lett 2014; 162: 3–9.
Miao EA, Rajan JV . Salmonella and caspase-1: a complex interplay of detection and evasion. Front Microbiol 2011; 2: 85.
Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM et al. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci USA 2005; 102: 9247–9252.
Asrat S, Davis KM, Isberg RR . Modulation of the host innate immune and inflammatory response by translocated bacterial proteins. Cell Microbiol 2015; 17: 785–795.
Alto NM, Orth K . Subversion of cell signaling by pathogens. Cold Spring Harb Perspect Biol 2012; 4: a006114.
Brodsky IE, Medzhitov R . Targeting of immune signalling networks by bacterial pathogens. Nat Cell Biol 2009; 11: 521–526.
Hoffmann HH, Schneider WM, Rice CM . Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol 2015; 36: 124–138.
Orzalli MH, Knipe DM . Cellular sensing of viral DNA and viral evasion mechanisms. Annu Rev Microbiol 2014; 68: 477–492.
Sparrer KM, Gack MU . Intracellular detection of viral nucleic acids. Curr Opin Microbiol 2015; 26: 1–9.
Patel SJ, King KR, Casali M, Yarmush ML . DNA-triggered innate immune responses are propagated by gap junction communication. Proc Natl Acad Sci USA 2009; 106: 12867–12872.
Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci USA 2015; 112: E5699–E5705.
Sun L, Wu J, Du F, Chen X, Chen ZJ . Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339: 786–791.
Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013; 503: 530–534.
Bridgeman A, Maelfait J, Davenne T, Partridge T, Peng Y, Mayer A et al. Viruses transfer the antiviral second messenger cGAMP between cells. Science 2015; 349: 1228–1232.
Gentili M, Kowal J, Tkach M, Satoh T, Lahaye X, Conrad C et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 2015; 349: 1232–1236.
Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013; 341: 903–906.
Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015; 17: 820–828.
Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH et al. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med 2015; 21: 401–406.
Woodward JJ, Iavarone AT, Portnoy DA . c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 2010; 328: 1703–1705.
Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 2011; 79: 688–694.
Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011; 478: 515–518.
Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE et al. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio 2013; 4: e00018–13.
Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437: 1167–1172.
Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci USA 2006; 103: 6001–6006.
Cheng G, Zhong J, Chisari FV . Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc Natl Acad Sci USA 2006; 103: 8499–8504.
Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M et al. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci USA 2010; 107: 7431–7436.
Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C et al. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012; 12: 558–570.
Masciopinto F, Giovani C, Campagnoli S, Galli-Stampino L, Colombatto P, Brunetto M et al. Association of hepatitis C virus envelope proteins with exosomes. Eur J Immunol 2004; 34: 2834–2842.
Wieland SF, Takahashi K, Boyd B, Whitten-Bauer C, Ngo N, de la Torre JC et al. Human plasmacytoid dendritic cells sense lymphocytic choriomeningitis virus-infected cells in vitro. J Virol 2014; 88: 752–757.
Saleh MC, Tassetto M, van Rij RP, Goic B, Gausson V, Berry B et al. Antiviral immunity in Drosophila requires systemic RNA interference spread. Nature 2009; 458: 346–350.
Pang IK, Ichinohe T, Iwasaki A . IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol 2013; 14: 246–253.
Jara PI, Boric MP, Saez JC . Leukocytes express connexin 43 after activation with lipopolysaccharide and appear to form gap junctions with endothelial cells after ischemia-reperfusion. Proc Natl Acad Sci USA 1995; 92: 7011–7015.
Yeh TH, Hsu WC, Chen YS, Hsu CJ, Lee SY . Lipopolysaccharide decreases connexin 43 expression on nasal epithelial cells in vitro. Acta Otolaryngol 2005; 125: 1091–1096.
Matsue H, Yao J, Matsue K, Nagasaka A, Sugiyama H, Aoki R et al. Gap junction-mediated intercellular communication between dendritic cells (DCs) is required for effective activation of DCs. J Immunol 2006; 176: 181–190.
Soong G, Reddy B, Sokol S, Adamo R, Prince A . TLR2 is mobilized into an apical lipid raft receptor complex to signal infection in airway epithelial cells. J Clin Invest 2004; 113: 1482–1489.
Martin FJ, Prince AS . TLR2 regulates gap junction intercellular communication in airway cells. J Immunol 2008; 180: 4986–4993.
Ashida H, Mimuro H, Sasakawa C . Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front Immunol 2015; 6: 219.
Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C et al. An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol 2007; 8: 47–56.
Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C . The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci USA 2005; 102: 14046–14051.
Li H, Xu H, Zhou Y, Zhang J, Long C, Li S et al. The phosphothreonine lyase activity of a bacterial type III effector family. Science 2007; 315: 1000–1003.
Kasper CA, Sorg I, Schmutz C, Tschon T, Wischnewski H, Kim ML et al. Cell-cell propagation of NF-kappaB transcription factor and MAP kinase activation amplifies innate immunity against bacterial infection. Immunity 2010; 33: 804–816.
Patil S, Fribourg M, Ge Y, Batish M, Tyagi S, Hayot F et al. Single-cell analysis shows that paracrine signaling by first responder cells shapes the interferon-beta response to viral infection. Sci Signal 2015; 8: ra16.
Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014; 510: 363–369.
Dolowschiak T, Chassin C, Ben Mkaddem S, Fuchs TM, Weiss S, Vandewalle A et al. Potentiation of epithelial innate host responses by intercellular communication. PLoS Pathog 2010; 6: e1001194.
Finsel I, Hilbi H . Formation of a pathogen vacuole according to Legionella pneumophila: how to kill one bird with many stones. Cell Microbiol 2015; 17: 935–950.
Shen X, Banga S, Liu Y, Xu L, Gao P, Shamovsky I et al. Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell Microbiol 2009; 11: 911–926.
Fontana MF, Banga S, Barry KC, Shen X, Tan Y, Luo ZQ et al. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog 2011; 7: e1001289.
Ivanov SS, Roy CR . Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR. Nat Immunol 2013; 14: 1219–1228.
Shin S, Case CL, Archer KA, Nogueira CV, Kobayashi KS, Flavell RA et al. Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog 2008; 4: e1000220.
Sporri R, Joller N, Albers U, Hilbi H, Oxenius A . MyD88-dependent IFN-gamma production by NK cells is key for control of Legionella pneumophila infection. J Immunol 2006; 176: 6162–6171.
Asrat S, Dugan AS, Isberg RR . The frustrated host response to Legionella pneumophila is bypassed by MyD88-dependent translation of pro-inflammatory cytokines. PLoS Pathog 2014; 10: e1004229.
Copenhaver AM, Casson CN, Nguyen HT, Duda MM, Shin S . IL-1R signaling enables bystander cells to overcome bacterial blockade of host protein synthesis. Proc Natl Acad Sci USA 2015; 112: 7557–7562.
Lamkanfi M, Dixit VM . Mechanisms and functions of inflammasomes. Cell 2014; 157: 1013–1022.
Casson CN, Shin S . Inflammasome-mediated cell death in response to bacterial pathogens that access the host cell cytosol: lessons from legionella pneumophila. Front Cell Infect Microbiol 2013; 3: 111.
Rothfuchs AG, Egen JG, Feng CG, Antonelli LR, Bafica A, Winter N et al. In situ IL-12/23p40 production during mycobacterial infection is sustained by CD11bhigh dendritic cells localized in tissue sites distinct from those harboring bacilli. J Immunol 2009; 182: 6915–6925.
Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS . Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 2007; 110: 3234–3244.
Walters SB, Kieckbusch J, Nagalingam G, Swain A, Latham SL, Grau GE et al. Microparticles from mycobacteria-infected macrophages promote inflammation and cellular migration. J Immunol 2013; 190: 669–677.
Singh PP, Smith VL, Karakousis PC, Schorey JS . Exosomes isolated from mycobacteria-infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo. J Immunol 2012; 189: 777–785.
Athman JJ, Wang Y, McDonald DJ, Boom WH, Harding CV, Wearsch PA . Bacterial Membrane Vesicles Mediate the Release of Mycobacterium tuberculosis Lipoglycans and Lipoproteins from Infected Macrophages. J Immunol 2015; 195: 1044–1053.
Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G et al. The adaptor ASC has extracellular and 'prionoid' activities that propagate inflammation. Nat Immunol 2014; 15: 727–737.
Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 2014; 15: 738–748.
Cronemberger-Andrade A, Aragao-Franca L, de Araujo CF, Rocha VJ, Borges-Silva Mda C, Figueira CP et al. Extracellular vesicles from Leishmania-infected macrophages confer an anti-infection cytokine-production profile to naive macrophages. PLoS Negl Trop Dis 2014; 8: e3161.
Couper KN, Barnes T, Hafalla JC, Combes V, Ryffel B, Secher T et al. Parasite-derived plasma microparticles contribute significantly to malaria infection-induced inflammation through potent macrophage stimulation. PLoS Pathog 2010; 6: e1000744.
Mantel PY, Hoang AN, Goldowitz I, Potashnikova D, Hamza B, Vorobjev I et al. Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host Microbe 2013; 13: 521–534.
Christian DA, Koshy AA, Reuter MA, Betts MR, Boothroyd JC, Hunter CA . Use of transgenic parasites and host reporters to dissect events that promote interleukin-12 production during toxoplasmosis. Infect Immun 2014; 82: 4056–4067.
Koshy AA, Dietrich HK, Christian DA, Melehani JH, Shastri AJ, Hunter CA et al. Toxoplasma co-opts host cells it does not invade. PLoS Pathog 2012; 8: e1002825.
Van Valen L . A new evolutionary law. Evol Theory 1973; 1: 1–30.
Acknowledgements
We thank past and present members of the Shin laboratory and Igor Brodsky for insightful discussions on this topic, and critical reading of this manuscript. This work was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under grants R01AI118861 and R01AI121148 (to S.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Holmgren, A., McConkey, C. & Shin, S. Outrunning the Red Queen: bystander activation as a means of outpacing innate immune subversion by intracellular pathogens. Cell Mol Immunol 14, 14–21 (2017). https://doi.org/10.1038/cmi.2016.36
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2016.36
Keywords
This article is cited by
-
Reprogramming of microRNA expression via E2F1 downregulation promotes Salmonella infection both in infected and bystander cells
Nature Communications (2021)
-
Synthetic Host Defense Peptides Inhibit Venezuelan Equine Encephalitis Virus Replication and the Associated Inflammatory Response
Scientific Reports (2020)
-
Innate immunity and inflammation
Cellular & Molecular Immunology (2017)
