
Abstract
Glioblastoma is one of the deadliest tumors with barely over one-year median survival despite intensive efforts in defining its molecular characteristics and searching for innovative treatment strategies. While major progress has been made in cataloging cross-sectional genomic, transcriptomic and epigenomic features of the tumor, and inferring its main molecular pathways and niches for potential targeted intervention, we still do not have sufficient knowledge concerning evolutionary patterns and dynamics of molecular changes or the treatment-induced effects affecting glioblastoma biology. In this review, we summarize the results of recent longitudinal genomic, transcriptomic and epigenomic studies that brought us closer to a better understanding of this lethal disease. Evidence suggests that neuronal / glioma stem cells with accumulating mutations initiate glioblastoma development and recurrence, but the hypothetical models describing the courses that lead to established tumors have not been fully proven. Moving from the histopathological phenotype to the results of high resolution OMICS studies, we try to synthesize the currently available information from sequential glioblastoma analyses in order to highlight its multifaceted features and heterogenetity, as well as the expected complexity of potential treatment strategies that might once succeed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM), a grade IV glioma according to the classification by the World Health Organization [WHO], is one of the most common, and most malignant brain tumors. The majority, 90% of GBM, is primary tumor arising de novo, while the remaining subset is secondary that progresses from a WHO grade II diffuse astrocytoma or WHO grade III anaplastic astrocytoma [1].
Characteristically, GMB recurs usually within a year despite aggressive therapy [2]. The standart treatment of newly diagnosed GBM involves radiotherapy and temozolomide (TMZ)-based chemotherapy following gross total resection [3]. These interventions somewhat slow the progression of the tumor, but inevitably, a small residual population of cells escapes surgery and chemoradiation, and results in a typically fatal recurrence in about 7 months after diagnosis [4]. In contrast to the initially diagnosed tumors, there is no standard treatment protocol for the recurrent GBMs as yet. With the currently avaiblable medications, the median overall survival time is 12–15 months from initial diagnosis, although with the improvements of interventions and supportive care the figure may somewhat exceed 20 months at certain centers [5, 6]. Identification of the molecular drivers of tumor evolution is of major importance, since a better understandig of the etiology and recurrence might provide clues for the developement of efficient and targeted treatments [7]. The biggest step towards achieving this goal was the genome-wide analyses of chromosomal structural variations, single nucleotide variations (SNVs) and copy number variations (CNVs) in GBM by The Cancer Genome Atlas Network (TCGA) [8]. Subsequently, integrated analyses of genomic and transcriptomic data revealed that GBM may be subdiveded into molecular subtypes named proneural (PN), classical (CL), mesenchymal (MES) and neural (NE) (the latter category was later abandoned, since the pattern turned out to be due to normal cell contamination in the investigated sample) [9, 10]. Parallel, explorations of DNA CpG island methylation patterns genome-wide and correlations of these epigenomic patterns with the molecular GBM subtypes were reported [11]. Our group performed translational studies that reproduced separation of the three (PN, CL, MES) GBM molecular subgroups in formalin-fixed, paraffin-embedded (FFPE) specimens using immunohistochemistry (IHC) and pyrosequencing, and thereby demonstrated that the TCGA OMICS observations, may be applied, at least in part, in clinical practice [8, 9, 12]. The TCGA studies also revealed that the molecular abnormalities in GBM preferentially align in certain pathways including the activation of the epidermal growth factor receptor (EGFR) along with other tyrosin kinase receptors-initiated signaling pathways, the tumor protein 53 (TP53) and retinoblastoma (RB1) pathways [13,14,15,16,17]. These genetic alterations and pathways are known to promote tumor growth, while the roles of several novel or passanger mutations in GBM pathogenesis remain unclear [18]. Recent genomic and transcriptomic studies of sequential samples have also provided important observations for the elucidation of GBM development [4, 7, 10, 18,19,20,21]. Thus far, however, there has only been a few studies focused on epigenomic examination of GBM, and particularly, in sequential specimens [22,23,24,25]. Nevertheless, these few studies established that, in addition to alterations in the genome, epigenetic modifications, particularly CpG island methylation changes, underly the observed changes in the transcriptome and play an important role in defining disease progression. In depth mapping of genomic, transcriptomic and epigenomic alterations during the evolution of GBM is very important in order to gain reliable insights into tumorigenesis, progression and recurrence, as well as to assist the development of targeted treatments [26].
GBM is a notoriously heterogeneous tumor at both the histopathological and the molecular levels. Great degrees of variability may be observed among and within GBMs regarding grade as well as clonality. Intratumor heterogeneity (ITH) may reflect simultaneous presence of gliomas of various grades or GBM of various molecular subtypes, although the latter has been the subject of some debate. Similarly, whether GBM retains its original molecular subtype over time, or different transcriptional subtypes may be present at diagnosis and relapse has not been unequivocally established [8, 27, 28]. Evidence suggests that mutations accumulating in neuronal/glioma stem cells initiate the development and contribute to the recurrence of glioblastoma (for details see Tompa et al. [29]). Two common models have been proposed for capturing how heterogeneity arises and therapy resistance develops during the tumor’s course. The first model includes the cancer stem cell hypothesis or the ancestral cell origin model [30] assuming that a relatively small subset of cells with stem cell-like properties give rise to different tumor cell populations. In these cells, resistance develops through the acquisition of novel genetic alterations (GA). In contrast, the alternative clonal evolution model [31] suggests that all tumor cells independently acquire novel GA and later undergo natural selection. Every subclonal population has the potential to expand as enviromental or therapeutic conditions change [4, 18]. Which one of these two models describes tumor evolution more accurately remains to be determined. The aim of this survey is to provide a brief overview of genomic, transcriptomic and epigenomic data obtained from progressive GBM tumors in order to highlight main features and dynamics of tumor evolution.
Comparative Profiles of Initial and Recurrent GBMs (Table 1)
While reports on molecular characterization of GBM represent a significant proportion of the scientific literature, there are relatively few studies comparing the molecular profiles in primary and recurrent tumor pairs. There are two main reasons for this scarcity of information. First, only 25% of recurrent tumors are amenable to surgery, which leads to a very restricted number of suitable samples [5]. Second, even if the recurrent GBM tissue can be removed, the extent of necrosis, that is typically much more extensive in the recurrent tumor, often prohibits its use in further molecular studies [32]. However, because of the importance of information gained from such sequential profiling, we summarize below the available data, first presenting observations relying on selected markers, and then those relying on genome-wide analyses.
Protein Expression-Based Studies with Selected Markers
IHC is one of the most widely used techniques in the clinical setting and in translational research. It not only helps to establish the histological diagnosis, but also shows the tissue distribution and subcellular localization of expressed proteins. In addition, IHC is suitable for a comparative and quantitative determination of marker expression in various tissue regions. Two previous studies by Stark et al. [33, 34] worked with large numbers of paired samples using IHC, and focused on the expressions of pre-selected proteins with putative roles in DNA repair and tumor growth (MLH1, MSH2, MSH6, TP53 mutation, EGFR amplification). The authors found that expressions of MutL homolog 1 (MLH1), MutS homolog 2 (MSH2) and tumor protein 53 (TP53) (products of DNA repair and tumor suppressor genes) were significantly lower in recurrent tumors. Furthermore, expression levels of MLH1 and MSH2 (and of MSH6 only in initial tumors) were significantly associated with the Ki67 proliferation index in both initial and recurrent tumors, indicating a potential role of these proteins in GBM progression. Similar studies were performed by Shinsato et al. [35] using IHC analyses to compare MLH1, postmeiotic segregation increased 2 (PMS2) and O6-methylguanine DNA methyltransferase (MGMT) expression levels in primary and recurrent GBMs, the latter obtained from patients after TMZ treatment. This study also revealed a reduction in the MLH1 and PMS2 protein levels in TMZ-resistant cells. These observations suggest that a reduction in MLH1 protein expression leads to PMS2 protein instability, which in turn, confers TMZ resistance to GBM cells. Although the results were very exciting and the method of IHC could allow for addressing, the above studies did not analyze ITH. In one of our recent translational studies, we also included a small sequential cohort and found that molecular subgroups were largely retained in recurrent compared to primary GBM specimens, however, even with the few markers used in IHC signs of ITH and clonal evolution could be revealed in consensus with much more comprehensive analyses by the TCGA [8, 9, 12]. While these IHC-based studies provided some important observations and prognostic markers, altogether, the methodological approach involved technical limitations with low resolution of the gained information. Therefore, more comprehensive approaches were needed to advance further the field and to gain deeper insights into GBM pathogenesis.
Overview of Longitudinal Genome-Wide Studies
OMICS methods have been gaining ground since next generation sequencing (NGS) and microarray analyses became available [36]. These high resolution and high throughput methods have provided invaluable information about variations accumulating in the genomic DNA, the transcriptome and epigenome within tumors, including GBM. NGS analyses of genomic DNA define SNV, small insertions / deletions and CNVs genome-wide. It may also provide information concerning chromosomal rearrangements and even chromotripsis (see below). Whole-exome sequencing (WES) and targeted-exome sequencing (TES) reveal similar data, but restricted to the exomes or to a set of selected genes.
Transcriptome studies quantitatively define messenger (m)RNA expression genome-wide typically in bulk RNA of a selected tissue, while single cell RNA analyses are recently becoming more widely used [36]. As a transcriptome captures a snapshot of total transcripts present at a time, transcriptome analyses may allow us to define which cellular processes were active (which genes were expressed) and which were dormant (which genes were not expressed) at that moment in a given specimen. The active genes may align in certain pathways signifying certain biological processes. Mapping gene expression in a tissue (i.e. GBM vs. normal brain tissue) at different time points (at diagnoses and at relapse) provides information about how genes are regulated during the development of the disease.
Epigenetic mechanisms involving enzymatic modifications of DNA and associated histone proteins or altered expression of microRNAs regulate gene transcription and translation. Epigenomic alterations are increasingly recognized as a source of phenotypic variability [37]. DNA CpG island methylation is the most widely studied epigenetic mechanism in GBM, which results from the addition of a methyl group to cytosine to become 5-methylcytosine. This modification is generally observed at 5’-CpG-3′ dinucleotides in all mammals (occasionally observed at CpNpGs) [38], and is required for silencing of genes [39] and allele-specific imprinting [40]. The other important mechanism is the post-translational modification of N-terminal tails of histone proteins by acetylation, methylation, phosphorylation, and other biochemical modifications [41]. Expression changes in the microRNAs have recently been explored in the development and progression of several tumors. These small non-coding RNAs inhibit the translation and stability of mRNAs, and thereby are involved in different cellular processes such as cell cycle regulation, differentiation, apoptosis and migration [42].
GBMs are characterized by alterations affecting genes that control cell growth, apoptosis, angiogenesis and invasion. Genomic, transcriptomic and epigenomic alterations all contribute to these biological processes, and evolve during progression of GBM.
Genomic Analyses
Using WES and TES of 38 corresponding primary and recurrent GBM pairs, the study by Kim J. et al. [20] revealed that distally recurred tumors only shared a minority of initial tumor mutations (25%) indicative of divergent evolution, while most of the locally recurred tumors shared a majority (70%) of initial tumor mutations consistent with linear evolution. These results suggest that although GBMs may recur throughout the brain, recurrence at a distant cerebral location involves a high degree of clonal selection and consequent genomic divergence. The authors also speculate, that GBM clones that had diffusely invaded the brain parenchyma at early stages of tumor developement may wake up from their dormant state and repopulate distant locations with actively growing daughter cells [20]. To further investigate the nature of tumor evolution, another group of investigators carried out the largest-scale sequential study to date, with 114 patients’ samples. In 93 cases, DNA specimens from normal tissues (i.e. normal germ-line DNA sequences) were available in addition to those of primary and recurrent tumor samples. WES and transcriptome analyses showed that although 45% of the mutations are being shared between the primary and recurrent tumors, the dominant clone at diagnosis is generally not a linear ancestor of the dominant clone at relapse [19]. Another important finding in this study revealed that hypermutations preferentially target highly expressed genes, suggesting that the mutagenic mechanisms related to the alkylating effect of TMZ treatment affects most efficiently highly expressed regions of open chromatin. Wang J et al. [19] also stated that two-thirds of patients with primary GBM exhibit different transcriptional subtypes at diagnoses and relapse, in contrast to the conclusions of other studies suggesting that GBMs largely retain their initial transcriptional molecular subtypes [9, 10, 12].
A study by Martinez et al. [7] analyzed 20 paired GBM samples. Among primary GBMs, the authors observed 4 type 1 GBMs (secondary) which contained p53 mutation without EGFR amplification, 12 type 2 GBMs (de novo) which contained EGFR amplification in the absence of p53 mutation and 4 non-type 1-non-type 2 GBMs in which neither EGFR amplification nor p53 mutation was observed. Interestingly, all type 1 and type 2 GBMs conserved their p53 mutational status and EGFR amplification at relapse. This stability of the EGFR amplification status over time in the majority of the tumors was subsequently confirmed [17]. However, the study by Martinez et al [7] also showed that the four non-type 1-non-type 2 GBMs developed new EGFR amplifications, and thus acquired the type 2 GBM profile. These observations suggest that during relapses GBMs may accumulate additional molecular alterations and evolve along two distinct pathways acquiring type 1 GBM profile (harboring p53 mutation) or type 2 GBM profile (harboring EGFR amplification), depending on the profile of the original tumor [7]. The accumulation of new molecular alterations may explain, at least in part, the observed transcriptional subtype switching between initial and recurrent tumors.
A study by Sottoriva et al. [43] draws attention to that the residual disease, and not just the main tumor mass, must be investigated to understand how treatment-resistance develops. The investigators obtained multiple samples from multiple regions, including the main tumor mass, the infiltrating margin and the sub-ventricular zone of 11 patients along with their matching blood samples. The multi-regional WES analyses revealed extensive ITH as reflected by SNVs and CNVs, and involved EGFR amplification, the loss of chromosome 10 containing PTEN and homozygous deletions of CDKNA2. The phylogenetic trees built from WES data showed that residual disease subclones can arise early during tumor growth, and these infiltrative subclones may seed the growth of a recurrent tumor after treatment [43]. The genomic road map that leads to recurrence can be highly idiosyncratic, but may broadly be classified into two patterns: 1. linear recurrences share extensive genetic similarity with the primary tumor and can be directly traced to one of its specific sectors, and 2. divergent recurrences that share only few genetic alterations with the primary tumor and originate from cells that branched off early during tumorigenesis [4]. These authors analyzed 252 TCGA samples and 23 paired GBM samples with the goal to better elucidate the intratumoral clonal composition of primary GBM, and to reveal how GBM responds to therapeutic interventions. Two mutational clusters were detected, namely the clonal cluster with mutations from before sequential malignant transformation, which mutations were present in all tumor cells; and the subclonal cluster with mutations that occured later during the tumor expansion and branching evolution, which mutations were present only in a subset of the tumor cells. The distribution of the mutations was 67.9% clonal, 29.8% subclonal, and 2.3% could not be classified. The majority of TP53 and PI3KCA/PI3KR1 mutations (90.5%) were clonal. This study revealed that most affected genes and pathways affect cell cycle control, DNA damage response, cell death and differentation that may all underlie gliomagenesis. The TP53 mutant recurrent GBMs showed an increase in subclonal mutation frequency compared to wild-type TP53 recurrent GBMs, which suggests an association between TP53 mutation and subclonal tumor evolution [4]. This observation may be related to the known involvement of TP53 in tolerance to DNA damage or apoptosis suppression [44, 45]. Kim H. et al [4] also found a linear correlation between clonal mutations and age. However, the subclonal tumor group showed relatively more favorable event-free survival, than the clonal tumor group, which may be explained by the absence of a dominant aggressive clone [4].
Given the size of the human diploid genome and the vast potential of the acquisition of mutations suggest that clonal mutations might be acquired prior to gliomagenesis over the life span of an individual [46]. A recent paper describing a GBM mouse model, however, underscores that the more differentiated a cell is, the less susceptibility for new mutations it has [47]. This elegant study strongly supports the hypothesis that pathogenic mutations in neurogen stem cells or in lineage progenitors initiate gliomagenesis and growth.
Treatment-Induced Genomic Effects in Recurrent GBM
As mentioned above, the standard care for patients with newly diagnosed GBM includes surgical tumor removal and radio-chemotherapy, however, inevitably every tumor recurs [3, 48]. Recurrent tumors are less sensitive to therapy than the original tumors, and in most cases, invade functional brain areas, preventing a second surgical resection [5]. At present, it is unknown whether the primary reason for recurrence is lingering malignant cells, de novo clonal expansions or clonal selection under pressure from adjuvant radiation and chemotherapeutic treatments [49, 50]. There have been a few studies focusing on treatment effects in GBM where observations are available through sequential recurrences from the diagnosis to the patient’s death. Nickel et al. [51] analyzed a patient’s longitudinal tumor DNA samples by NGS, focusing on intratumor heterogeneity and the mutational differences between the primary tumor and recurrences. The patient was a 69 years old male. His treatment for the primary tumor involved surgical resection followed by radiotherapy and TMZ chemotherapy. At the first recurrence, he received the same treatment supplemented with thalidomine and bevacizumab, while at the second recurrence only surgical resection was applied. Prior to treatment, a PTEN mutation was noted in approximately half of the tumor cells, which likely acted as a driver mutation in the primary tumor. A small subset of cells also harbored PIK3CA mutation. The initial round of surgery, chemotherapy, and radiation did not eradicate the cells with the PTEN driver mutation, yet it was absent from the tumor at the second recurrence. In addition, the PIK3CA mutation harboring subclonal population acquired a „hypermutator” phenotype [51]. This observation is consistent with the results of some subsequent studies suggesting a potential hypermutator effect of TMZ-chemotherapy in low-grade gliomas and GBMs. However, following TMZ treatment hypermutation rarely develops in isocitrate dehydrogenase 1 (IDH1)-wild-type primary GBMs, indicating low risk for TMZ-induced hypermutation in these tumors under standard treatment. In contrast, the common IDH1 (R132H) mutation is known to induce a hypermethylation phenotype in gliomas by silencing MGMT (and other mismatch repair [MMR] genes) thus sensitizing the tumor to TMZ-induced mutagenesis [4, 16, 20, 52].
In another study, genomic DNA of 21 patients with primary and recurrent GBM were analyzed [21]. Three patients underwent only surgery without chemo- or radiation therapy. Analyzing the dynamic mutational profiles in the paired samples revealed evidence for therapy-mediated selection pressure in treated patients. These evidence included 1) decreased variant burden in recurrent tumors, 2) the neutral evolutionary pattern in untreated tumors shifted to non-neutral pattern in recurrent tumors after treatment, and 3) one recurrent tumor (out of the 18 TMZ-treated patients / tumors) showed a TMZ-induced hypermutator phenotype [21]. This latter observation suggesting a rarity of TMZ-induced hypermutation at recurrence is consistent with previous studies [4].
Erson-Omay et al. [53] treated and analyzed a single patient from diagnosis through double recurrences, and assessed the frequency of genomic events detected in this patient and in 110 exome- and whole-genome-sequenced specimens in the Yale-Glioma cohort. In the initial index patient’s tumor, WES analysis revealed amplification of chromosome 7 and deletion of chromosome 10 and focal deletion of CDKN2A locus on chromosome 9 along with an activating ectodomain EGFR A289V mutation, suggesting that the tumor cells had undergone chromothripsis. Chromothripsis is a sudden event with complex genomic rearrangements catastrophic for the harboring cell [54]. Based on the Yale-Glioma cohort, chromotripsis may be a frequent event in GBM. The patient in the Erson-Omay et al study [53] was enrolled in a clinical trial with a receptor tyrosine kinase inhibitor, vandetanib, beside the standard TMZ treatment. Despite all efforts, the disease progressed and a second gross total resection was carried out. WES analysis of the recurrent tumor showed a loss of tumor cells harboring the activating EGFR A289V mutation, most likely due to the targeted anti-EGFR therapy. However, this treatment had no impact on the high EGFR ploidy. After the second recurrence, DNA analyses showed double-minutes known to be resistant to targeted therapies [55, 56]. However, this tumor also harbored a hypermutator phenotype, involving the MSH6 gene and the MMR mechanism, allowing to design a new therapeutic strategy. Numerous reports pointed out that hypermutated tumors including endometrial, gastric, colorectal and small bowel cancers are susceptible to immune checkpoint inhibitors [57]. Based on these observations, the above patient with GBM was started on hydroxyurea and immune checkpoint inhibitor, pembrolizumab, and he lived for 5 years after the diagnosis [53]. While immune checkpoint inhibitors are now part of the standard treatment protocols in several solid tumors (e.g. melanoma, non-small cell cancer lung cancer, etc.…), systematic testing of these agents in GBM was only recently reported. The Ivy Foundation Early Phase Clinical Trials Consortium study patients were randomized to neoadjuvant pembrolizumab (a programmed cell death protein 1 [PD-1] - specific monoclonal antibody) before surgery and pembrolizumab adjuvant therapy after surgery. Patients who received neoadjuvant pembrolizumab before surgery and continued on pembrolizumab after surgery had longer overall survival than patients who received adjuvant, post-surgical PD-1 blockade alone. Neoadjuvant pemrolizumab was associated with upregulation of T cell– and interferon-γ-related gene expression, but downregulation of cell-cycle-related gene expression in the tumor. The neoadjuvant pembrolizumab therapy enhanced both the local and systemic antitumor immune response [58]. In the study by Zhao et al [59], genomic and transcriptomic analyses of the tumors revealed an enrichment of PTEN mutations associated with immunosuppressive gene profiles in nonresponders, whereas an enrichment of MAPK pathway alterations was detected in responders. Single-cell RNA sequencing of one PTEN-mutant tumor of a non-responder showed the association of the immunosuppressive signature (T regulatory cells, macrophages, microglia, neutrophils) with CD44 + tumor cells involved in invasion. Analyses of clonal evolution of mutations in a few responders and nonresponders suggested that neoantigenic mutations were lost, while genes associated with immunosuppression were enriched following therapy with PD-1 inhibitors. Schalper et al [60] tested a presurgical nivolumab (another PD-1 specific antibody) followed by postsurgical nivolumab until disease progression. Tumor tissues pre- and post-nivolumab dosing and from patients without nivolumab were analyzed for changes in the tumor immune microenvironment. Neoadjuvant nivolumab induced an increased expression of chemokines along with enhanced immune cell infiltration and diversity of tumor-infiltrating T lymphocyte clonality. While impressive effects were observed at the cellular and molecular levels, clinical benefits of these three studies were modest, and only a few individual patients showed longer survival.
Taken together, the above studies suggest that TMZ treatment can impact tumor heterogeneity by reducing the diversity of tumor subclones (allowing the resistant sublones to become dominant at relapse), shift the neutral mutational dynamics towards non-neutral patters and in rare cases, increases the mutation burden (hypermutator phenotype). Emerging evidence supports that GBMs undergo evolutionary changes even in the absence of any therapy. However, the selective pressure exerted by radio-chemotherapies and targeted therapies without doubt alter the molecular composition of these tumors [61]. Immune checkpoint inhibitors although initially beneficially impacted the molecular profiles of glioblastoma, the evolution of mutational profiles and the immune suppressive changes in the tumor microenvironment annulled the early benefits eventually leading to tumor recurrence.
Transcriptome Studies
Following the pivotal study of cross-sectional primary GBM specimens by Verhaak et al. [9], a number of elegant transcriptome analyses were published focusing on sequential GBM sampes. Li et al [62] investigated the transcriptome of 88 primary and 22 recurrent GBMs to define the distribution (heterogeneity) of molecular subtypes and gene signatures. Differences were detected in the distribution of the CL subtype in the recurrent (22%) and primary GBMs (36%), in the frequency of IDH1 mutations that were nearly twice as high in recurrent than in primary tumors, and in the frequency of TP53 mutations when PN recurrences (20%) and CL recurrences (80%) were compared. Furthermore, gene set enrichment analyses revealed that chromatine fracture, repair and remodeling gene sets were enriched in recurrent GBM coinciding with biological progression of the recurrent tumors [62]. Another interesting study focused on the interactions between the tumor and its microenvironment [10]. These authors compared the transcriptome profiles of 596 single glioma cells from 8 matching primary and recurrent gliomas, along with 124 additional glioma pairs from other datasets. Transcriptome profiling of tumor samples is a commonly used technique for interrogating pathway functionality and phenotype-based classification, however, the impact of tumor microenvironment may obscure the true transcriptional profile and the signaling activity in the tumor [63]. The study of Wang Q. et al [10] identified in their samples the TCGA-proposed transcriptional glioma subtypes, PN, CL and MES [9] however, the NE subtype was identified as normal cell contamination in the tumor sample. In the MES GBM subtype, the presence of tumor-associated glial and microglial cells was quite remarkable confirming that a macrophage/microglia-rich microenvironment shapes the MES glioma phenotype. The data also showed that neurofibromin (NF1) deactivation (characteristic of the MES phenotype) results in the attraction of macrophage/microglia, suggesting that there is a two-way interaction between tumor cells and microenvironment. The longitudinal transcriptome analysis showed that the gene expression-based subtype is retained in 55% of the cases [10], somewhat conflicting with another observation that two-thirds of primary GBMs exhibit different transcriptional subtypes at diagnoses and relapse [19]. Previously Verhaak et al. [9] suggested that three quarters of the tumors did not change class at recurrence [9], though this observation was based on a very small cohort (Murat dataset [64]). As both [10, 19] worked with large cohorts of specimens, the differences between their results indicate that transcriptome analyses may lead to dissimilar results depending on when and from where the samples are taken.
Finally, a few studies used single cell RNA-sequencing (scRNA-seq) to examine mutational differences between initial and recurrent GBMs [27, 28, 65]. Using scRNA-seq, Patel et al. [27] observed extensive ITH at transcriptional level. The authors also established GSC cultures to examine stemness signature, which subsequently applied to the single-cell transcriptional profiles revealed a stemness gradient in tumors. The expression of a number of transcription factors (NF1A, NF1B) previously implicated in tumor propagation and neural stem cell self-renewal, also significantly correlated with stemness gradient. In another study Chen et al [65] identified 3 relapse–specific homozygous missense mutations in three independent genes involved in RAS/GEF GTP-dependent signaling, which pathway is known to be involved in GBM pathogenesis [65, 66]. These studies highlighted that scRNA-seq is an approach suitable to gain a deeper insight into ITH and clonal evolution, while identifies defects in transcriptional programs related to oncogenic signaling and proliferation response.
GBM Epigenome Studies
The most widely studied epigenetic mechanism in GBM is DNA CpG methylation. CpG island hypermethylation and through that gene silencing is a hallmark of human cancers, but certain sets of genes may get hypomethylated and thus overexpressed. DNA CpG methylation profiles have been used as biomarkers for early detection of tumors in blood or body fluids, or predicting prognosis or treatment response and to monitor cancer recurrence [67]. As reviewed above, most research studies on tumor heterogeneity in GBM involved genomic and transcriptomic analyses [4, 8,9,10, 19, 20, 27, 43, 68]. DNA methylation microarrays and sequencing of bisulfite converted tumor DNA identified characteristic differences in the methylation profiles of cross-sectional and sequential GBM specimens, while also revealed ITH [11, 69,70,71].
The most extensively investigated epigenetic mark in GBM is the methylation of the MGMT gene promoter, which is an independent prognostic factor for favorable tumor biology and a beneficial predictor of response to TMZ and radiotherapy [22]. Another important achievment in GBM epigenomics is the identification of glioma-CpG island methylathor phenotype (G-CIMP), which has provided the basis for subsequent epigenomic studies [11, 24] focused on epigenetic differences to classify diffuse IDH mutant and IDH-wild-type gliomas into further molecular subgroups with characteristic patient outcome. Analyses of the DNA methylation profiles from 200 tumors of 77 patients were carried out to elucidate the epigenome-based signs of malignant transformation of initially lower grade gliomas. The initially G-CIMP high turned out to carry the worst prognosis, and the capability to recur as a more aggressive G-CIMP-low tumor, which can mimic an IDH-wild-type and stem cell-like GBM at recurrence. Since histopathological grading at first diagnosis is unable to predict phenotype changes, identification of this subgroup has crucial clinical implications for the assessment and therapeutic management of aggressive low grade gliomas at risk for malignant recurrences [24]. Others examined non-glioma CpG island methylator phenotype (non-G-CIMP) tumors, and found that a subset of gene promoter hypomethylation (for example TP73, TERT) leads to up-regulation of alternate transcripts with potential oncogenic consequences [23]. The most complex and forward-looking study recently carried out by Klughammer et al [25] highlights the importance and broad applicability of epigenetic studies. From a technical perspective, this study was facilitated by an optimized reduced representation bisulfite sequencing (RRBS) technique suitable to study FFPE specimens. The fact, that the authors were able to identify transcriptional subtypes based on RRBS data shows the utility of this approach for molecular classification. Therefore, the RRBS-based bisulfite sequencing may circumvent the dependence on high-quality RNA in transcriptional subtyping. This study also addressed whether or not the RRBS profiles (complemented with detailed histopathological characterization of tumors) capture relevant aspects of tumor microenvironment. This part of the study revealed significant differences in immune cell infiltration among the three transcripctional subtypes. The highest number of immune cells was found in tumors of MES subtype, consistent with previous data [10]. DNA methylation well differentiated between samples with high and low percentage of necrosis in histopathology, and with high versus low levels of specific immune cell infiltrates and CD8-positive cells, suggesting that DNA methylation data can be used to infer immune cell infiltration [25]. From the aspect of tumor evolution, patient-specific DNA methylation profiles were largely retained, but substantial inter- and intratumor heterogeneity was identified, although without clear trend for higher or lower heterogeneity in primary and recurrent samples. Biological pathway analyses showed during progression an enrichment for genes that gained methylation among those involved in neuronal development and apoptosis, while genes whose promoters lost methylation were enriched in the Wnt signaling pathway and T cell activation [25]. Aberrant activation of the Wnt signaling pathway has been linked to stemness, invasiveness, angiogenesis and therapeutic resistance in cancer [72] and is also known to play important roles in GBM [29]. The above epigenomic studies reflect advantages of the approach, most importantly representing a proxy to RNA-based analyses when using clinical FFPE specimens, but with the cost of somewhat reduced resolution. The information gained by these studies not only supplements data revealed by other OMICS methods, but also allows us to gain a more complex picture regarding molecular characteristics, dynamics and potential therapeutical targets in GBM.
Conclusions (Fig. 1)
The longitudinal studies briefly reviewed here have widened our understanding of GBM development and progression, however, several questions remained unanswered. We highlighted some important findings from IHC studies that revealed differences in protein expression patterns in primary and recurrent GBM, while also noted the limitations of such a low resolution, expression based evaluation method [33,34,35]. Subsequently, we surveyed the results of high-resolution genome-wide analyses in sequential GBM specimens, and presented the observed changes in the genomic, transcriptomic and epigenomic profiles over time as well as pointed to some treatment-induced effects on these profiles. These observations allow us to draw some conclusions on the evolution, dynamics and heterogeneity of the disease. The finding that the distally (in different microenvironment) recurred tumors share only a relatively low percentage of initial tumor mutations, in contrast to the locally (in relatively similar microenvironment) recurred ones would suggest that recurrence at a distant location involves clonal selection and genomic divergence, while local recurrence involves some degree of linearity [20]. However, the observation that a dominant clone in a recurrent tumor is generally not necessarily the linear descendant of the dominant clone of the initial tumor suggests that genomic alterations may independently arose in different founder cells [19]. It is also important to note that many tumors exhibit different transcriptional subtypes upon recurrence, and a given GBM specimen may also simultaneously harbor more than one molecular subgroup, even though detection of single molecular subgroups both in cross-sectional and longitudinal specimens have been noted [8, 27, 28]. Determination of the DNA CpG methylation patterns genome-wide allows us to infer the transcriptional subtypes of GBM even in FFPE specimens, and provides further insight into dynamics and pathways of molecular changes as well as of ITH [25]. Altogether, these observations suggest that the development of GBM over time is highly idiosyncratic, and follows different patterns and dynamics of molecular evolution. The accumulation of molecular changes may be influenced by prior mutations (e.g. IDH1, TP53, MMR genes) and gene expression profiles (DNA CpG hypomethylation and hypermethylation), the impact of microenvironmental changes (soluble molecules, cell-cell interactions, shifts in immune modulation) and treatment effects (traditional TMZ and irradiation, immune checkpoint inhibitors, and emerging other medications). Thus far, however, all approaches unequivocally establish that molecular profiles and dynamics of GBMs are extremely divers even though with some identifiable unifying features (e.g. existence of molecular subgroups). Therefore, success in treatment may only be expected from complex, likely individually adjusted therapeutic strategies, which need to be based on deep molecular profiling.

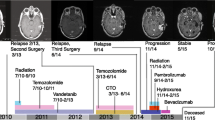
Molecular evolution of glioblastoma. The figure synthesizes main points of genomic, transcriptomic and epigenomic studies on longitudinal glioblastoma specimens and reflects the complexity of possible outcomes
References
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114(2):97–109. https://doi.org/10.1007/s00401-007-0278-6
Salcman M, Ebert PS (1991) In vitro response of human glioblastoma and canine glioma cells to hyperthermia, radiation, and chemotherapy. Neurosurgery 29(4):526–531. https://doi.org/10.1227/00006123-199110000-00007
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352(10):987–996. https://doi.org/10.1056/NEJMoa043330
Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ et al (2015) Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res 25(3):316–327
Weller M, Cloughesy T, Perry JR, Wick W (2012) Standards of care for treatment of recurrent glioblastoma—are we there yet? Neuro-Oncol 15(1):4–27. https://doi.org/10.1093/neuonc/nos273
Stupp R, Taillibert S, Kanner A, Read W, Steinberg DM, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K, di Meco F, Lieberman F, Zhu JJ, Stragliotto G, Tran DD, Brem S, Hottinger AF, Kirson ED, Lavy-Shahaf G, Weinberg U, Kim CY, Paek SH, Nicholas G, Bruna J, Hirte H, Weller M, Palti Y, Hegi ME, Ram Z (2017) Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318(23):2306–2316. https://doi.org/10.1001/jama.2017.18718
Martinez R, Rohde V, Schackert G (2010) Different molecular patterns in glioblastoma multiforme subtypes upon recurrence. J Neuro-Oncol 96(3):321–329. https://doi.org/10.1007/s11060-009-9967-4
Network CGAR (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216):1061–1068. https://doi.org/10.1038/nature07385
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, Cancer Genome Atlas Research Network (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17(1):98–110. https://doi.org/10.1016/j.ccr.2009.12.020
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, Barthel F, Cho HJ, Lin YH, Satani N, Martinez-Ledesma E, Zheng S, Chang E, Gabriel Sauvé CE, Olar A, Lan ZD, Finocchiaro G, Phillips JJ, Berger MS, Gabrusiewicz KR, Wang G, Eskilsson E, Hu J, Mikkelsen T, DePinho RA, Muller F, Heimberger AB, Sulman EP, Nam DH, Verhaak RGW (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32(1):42–56. https://doi.org/10.1016/j.ccell.2017.12.012
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, van den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K, Cancer Genome Atlas Research Network (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17(5):510–522. https://doi.org/10.1016/j.ccr.2010.03.017
Nagy Á, Garzuly F, Padányi G, Szűcs I, Feldmann Á, Murnyák B, Hortobágyi T, Kálmán B (2019) Molecular subgroups of glioblastoma–an assessment by immunohistochemical markers. Pathol Oncol Res 25(1):21–31. https://doi.org/10.1007/s12253-017-0311-6
Ueki K, Ono Y, Henson JW, Efird JT, von Deimling A, Louis DN (1996) CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res 56(1):150–153
Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y et al (2012). Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U.S.A 109(8), 3041–3046. https://doi.org/10.1073/pnas.1114033109
Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, Danussi C, Dolgalev I, Porrati P, Pellegatta S, Heguy A, Gupta G, Pisapia DJ, Canoll P, Bruce JN, McLendon RE, Yan H, Aldape K, Finocchiaro G, Mikkelsen T, Privé GG, Bigner DD, Lasorella A, Rabadan R, Iavarone A (2013) The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 45(10):1141–1149. https://doi.org/10.1038/ng.2734
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY et al (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343(6167):189–193. https://doi.org/10.1126/science.1239947
van den Bent MJ, Gao Y, Kerkhof M, Kros JM, Gorlia T, van Zwieten K, Prince J, van Duinen S, Sillevis Smitt PA, Taphoorn M, French PJ (2015) Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro-Oncol 17(7):935–941. https://doi.org/10.1093/neuonc/nov013
Neilsen BK, Sleightholm R, McComb R, Ramkissoon SH, Ross JS, Corona RJ, Miller VA, Cooke M, Aizenberg MR (2018) Comprehensive genetic alteration profiling in primary and recurrent glioblastoma. J Neuro-Oncol 142:1–8. https://doi.org/10.1007/s11060-018-03070-2
Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S et al (2016) Clonal evolution of glioblastoma under therapy. Nat Genet 48(7):768–776. https://doi.org/10.1038/ng.3590
Kim J, Lee IH, Cho HJ, Park CK, Jung YS, Kim Y, Nam SH, Kim BS, Johnson MD, Kong DS, Seol HJ, Lee JI, Joo KM, Yoon Y, Park WY, Lee J, Park PJ, Nam DH (2015) Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 28(3):318–328. https://doi.org/10.1016/j.ccell.2015.07.013
Muscat AM, Wong NC, Drummond KJ, Algar EM, Khasraw M, Verhaak R et al (2018) The evolutionary pattern of mutations in glioblastoma reveals therapy-mediated selection. Oncotarget 9(8):7844. https://doi.org/10.18632/oncotarget.23541
Hegi ME, Diserens AC, Gorlia T, Hamou MF, De Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352(10):997–1003. https://doi.org/10.1056/NEJMoa043331
Nagarajan RP, Zhang B, Bell RJ, Johnson BE, Olshen AB, Sundaram V et al (2014) Recurrent epimutations activate gene body promoters in primary glioblastoma. Genome Res 24(5):761–774 http://www.genome.org/cgi/doi/10.1101/gr.164707.113
de Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A, Laird PW, Wiznerowicz M, Iavarone A, Snyder J, deCarvalho A, Sanborn Z, McDonald KL, Friedman WA, Tirapelli D, Poisson L, Mikkelsen T, Carlotti CG Jr, Kalkanis S, Zenklusen J, Salama SR, Barnholtz-Sloan JS, Noushmehr H (2018) A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep 23(2):637–651. https://doi.org/10.1016/j.celrep.2018.03.107
Klughammer J, Kiesel B, Roetzer T, Fortelny N, Nemc A, Nenning KH, Furtner J, Sheffield NC, Datlinger P, Peter N, Nowosielski M, Augustin M, Mischkulnig M, Ströbel T, Alpar D, Ergüner B, Senekowitsch M, Moser P, Freyschlag CF, Kerschbaumer J, Thomé C, Grams AE, Stockhammer G, Kitzwoegerer M, Oberndorfer S, Marhold F, Weis S, Trenkler J, Buchroithner J, Pichler J, Haybaeck J, Krassnig S, Mahdy Ali K, von Campe G, Payer F, Sherif C, Preiser J, Hauser T, Winkler PA, Kleindienst W, Würtz F, Brandner-Kokalj T, Stultschnig M, Schweiger S, Dieckmann K, Preusser M, Langs G, Baumann B, Knosp E, Widhalm G, Marosi C, Hainfellner JA, Woehrer A, Bock C (2018) The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med 24(10):1611–1624. https://doi.org/10.1038/s41591-018-0156-x
Ferreira WAS, Pinheiro DDR, Costa Junior CAD, Rodrigues-Antunes S, Araujo MD, Leao Barros MB et al (2016) An update on the epigenetics of glioblastomas. Epigenomics 8(9):1289–1305. https://doi.org/10.2217/epi-2016-0040
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344(6190):1396–1401. https://doi.org/10.1126/science.1254257
Gill BJ, Pisapia DJ, Malone HR, Goldstein H, Lei L, Sonabend A et al (2014) MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc Natl Acad Sci U.S.A 111(34):12550–12555. https://doi.org/10.1073/pnas.1405839111
Tompa M, Kalovits F, Nagy A, Kalman B (2018) Contribution of the Wnt pathway to defining biology of glioblastoma. NeuroMolecular Med 20:1–15. https://doi.org/10.1007/s12017-018-8514-x
Birbrair A, Sattiraju A, Zhu D, Zulato G, Batista I, Nguyen VT, Messi ML, Solingapuram Sai KK, Marini FC, Delbono O, Mintz A (2017) Novel peripherally derived neural-like stem cells as therapeutic carriers for treating glioblastomas. Stem Cells Transl Med 6(2):471–481. https://doi.org/10.5966/sctm.2016-0007
Orzan F, De Bacco F, Crisafulli G, Pellegatta S, Mussolin B, Siravegna G et al (2017) Genetic evolution of glioblastoma stem-like cells from primary to recurrent tumor. Stem Cells 35(11):2218–2228. https://doi.org/10.1002/stem.2703
Marucci G, Fabbri PV, Morandi L, De Biase D, Di Oto E, Tallini G et al (2015) Pathological spectrum in recurrences of glioblastoma multiforme. Pathologica 107(1):1–8
Stark AM, Witzel P, Strege RJ, Hugo HH, Mehdorn HM (2003) p53, mdm2, EGFR, and msh2 expression in paired initial and recurrent glioblastoma multiforme. J Neurol Neurosurg Psychiatry 74(6):779–783. https://doi.org/10.1136/jnnp.74.6.779
Stark AM, Doukas A, Hugo HH, Mehdorn HM (2010) The expression of mismatch repair proteins MLH1, MSH2 and MSH6 correlates with the Ki67 proliferation index and survival in patients with recurrent glioblastoma. Neurol Res 32(8):816–820. https://doi.org/10.1179/016164110X12645013515052
Shinsato Y, Furukawa T, Yunoue S, Yonezawa H, Minami K, Nishizawa Y et al (2013) Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget 4(12):2261. https://doi.org/10.18632/oncotarget.1302
Horgan RP, Kenny LC (2011) ‘Omic’technologies: genomics, transcriptomics, proteomics and metabolomics. Obstet Gynecol 13(3):189–195. https://doi.org/10.1576/toag.13.3.189.27672
Nagarajan RP, Costello JF (2009, June) Epigenetic mechanisms in glioblastoma multiforme. In Seminars in cancer biology (Vol. 19, no. 3, pp. 188-197). Academic press. https://doi.org/10.1016/j.semcancer.2009.02.005
Clark SJ, Harrison J, Frommer M (1995) CpNpG methylation in mammalian cells. Nat Genet 10(1):20–27. https://doi.org/10.1038/ng0595-20
Okano M, Xie S, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19(3):219–220. https://doi.org/10.1038/890
Li E, Beard C, Jaenisch R (1993) Role for DNA methylation in genomic imprinting. Nature 366(6453):362–365. https://doi.org/10.1038/366362a0
Turner BM (2005) Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol 12(2):110–112. https://doi.org/10.1038/nsmb0205-110
Garzon R, Calin GA, Croce CM (2009) MicroRNAs in cancer. Annu Rev Med 60:167–179. https://doi.org/10.1146/annurev.med.59.053006.104707
Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC et al (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U.S.A 110(10):4009–4014. https://doi.org/10.1073/pnas.1219747110
Offer H, Erez N, Zurer I, Tang X, Milyavsky M, Goldfinger N, Rotter V (2002) The onset of p53-dependent DNA repair or apoptosis is determined by the level of accumulated damaged DNA. Carcinogenesis 23(6):1025–1032. https://doi.org/10.1093/carcin/23.6.1025
Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, Vassilev LT, Andreeff M (2005) MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood 106(9):3150–3159. https://doi.org/10.1182/blood-2005-02-0553
Lynch M (2010) Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U.S.A 107(3):961–968. https://doi.org/10.1073/pnas.0912629107
Llaguno SA, Sun D, Pedraza AM, Vera E, Wang Z, Burns DK, Parada LF (2019) Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat Neurosci 1:545–555. https://doi.org/10.1038/s41593-018-0333-8
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO, European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups., National Cancer Institute of Canada Clinical Trials Group (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet 10:459–466. https://doi.org/10.1016/S1470-2045(09)70025-7
Clarke J, Butowski N, Chang S (2010) Recent advances in therapy for glioblastoma. Arch Neurol 67(3):279–283. https://doi.org/10.1001/archneurol.2010.5
Hou LC, Veeravagu A, Hsu AR, Victor CK (2006) Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurg Focus 20(4):E3. https://doi.org/10.3171/foc.2006.20.4.2
Nickel GC, Barnholtz-Sloan J, Gould MP, McMahon S, Cohen A, Adams MD, Guda K, Cohen M, Sloan AE, LaFramboise T (2012) Characterizing mutational heterogeneity in a glioblastoma patient with double recurrence. PLoS One 7(4):e35262. https://doi.org/10.1371/journal.pone.0035262
Andor N, Harness JV, Mueller S, Mewes HW et al (2013) EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics 30(1):50–60. https://doi.org/10.1093/bioinformatics/btt622
Erson-Omay EZ, Henegariu O, Omay SB, Harmancı AS, Youngblood MW, Mishra-Gorur K, Li J, Özduman K, Carrión-Grant G, Clark VE, Çağlar C, Bakırcıoğlu M, Pamir MN, Tabar V, Vortmeyer AO, Bilguvar K, Yasuno K, DeAngelis LM, Baehring JM, Moliterno J, Günel M (2017) Longitudinal analysis of treatment-induced genomic alterations in gliomas. Genome Med 9(1):12. https://doi.org/10.1186/s13073-017-0401-9
Forment JV, Kaidi A, Jackson SP (2012) Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer 12(10):663–670. https://doi.org/10.1038/nrc3352
L'abbate A, Macchia G, D'addabbo P, Lonoce A, Tolomeo D, Trombetta D et al (2014) Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res 42(14):9131–9145. https://doi.org/10.1093/nar/gku590
Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, Paucar A, Yang H, Ohashi M, Zhu S, Wykosky J, Reed R, Nelson SF, Cloughesy TF, James CD, Rao PN, Kornblum HI, Heath JR, Cavenee WK, Furnari FB, Mischel PS (2014) Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343(6166):72–76. https://doi.org/10.1126/science.1241328
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD et al (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372(26):2509–2520. https://doi.org/10.1056/NEJMoa1500596
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, Kawaguchi ES, du L, Li G, Yong WH, Gaffey SC, Cohen AL, Mellinghoff IK, Lee EQ, Reardon DA, O’Brien BJ, Butowski NA, Nghiemphu PL, Clarke JL, Arrillaga-Romany IC, Colman H, Kaley TJ, de Groot JF, Liau LM, Wen PY, Prins RM (2019) Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 1:477–486. https://doi.org/10.1038/s41591-018-0337-7
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, Filip I, Orenbuch R, Goetz M, Yamaguchi JT, Cloney M, Horbinski C, Lukas RV, Raizer J, Rae AI, Yuan J, Canoll P, Bruce JN, Saenger YM, Sims P, Iwamoto FM, Sonabend AM, Rabadan R (2019) Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 1:462–469. https://doi.org/10.1038/s41591-019-0349-y
Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, Inogés S, de Andrea C, López-Diaz de Cerio A, Tejada S, Berraondo P, Villarroel-Espindola F, Choi J, Gúrpide A, Giraldez M, Goicoechea I, Gallego Perez-Larraya J, Sanmamed MF, Perez-Gracia JL, Melero I (2019) Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 1:470–476. https://doi.org/10.1038/s41591-018-0339-5
Campos B, Olsen LR, Urup T, Poulsen HS (2016) A comprehensive profile of recurrent glioblastoma. Oncogene 35(45):5819–5825. https://doi.org/10.1038/onc.2016.85
Li R, Chen X, You Y, Wang X, Liu Y, Hu Q, Yan W (2015) Comprehensive portrait of recurrent glioblastoma multiforme in molecular and clinical characteristics. Oncotarget 6(31):30968. https://doi.org/10.18632/oncotarget.5038
Kim H, Verhaak RG (2015) Transcriptional mimicry by tumor-associated stroma. Nat Genet 47(4):307–309. https://doi.org/10.1038/ng.3255
Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MCM, Hainfellner JA, Heppner FL, Dietrich PY, Zimmer Y, Cairncross JG, Janzer RC, Domany E, Delorenzi M, Stupp R, Hegi ME (2008) Stem cell–related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 26(18):3015–3024. https://doi.org/10.1200/JCO.2007.15.7164
Chen X, Wen Q, Stucky A, Zeng Y, Gao S, Loudon WG, Ho HW, Kabeer MH, Li SC, Zhang X, Zhong JF (2018) Relapse pathway of glioblastoma revealed by single-cell molecular analysis. Carcinogenesis 39(7):931–936. https://doi.org/10.1093/carcin/bgy052
Lee HK, Finniss S, Cazacu S, Xiang C, Poisson LM, Blumberg PM et al (2015) RasGRP3 regulates the migration of glioma cells via interaction with Arp3. Oncotarget 6:1850–1864. https://doi.org/10.18632/oncotarget.2575
Laird PW (2003) Early detection: the power and the promise of DNA methylation markers. Nat Rev Cancer 3(4):253–266. https://doi.org/10.1038/nrc1045
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155(2):462–477. https://doi.org/10.1016/j.cell.2013.09.034
Capper D, Jones DT, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555(7697):469–474. https://doi.org/10.1038/nature26000
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, Anjum S, Wang J, Manyam G, Zoppoli P, Ling S, Rao AA, Grifford M, Cherniack AD, Zhang H, Poisson L, Carlotti Jr CG, Tirapelli DP C, Rao A, Mikkelsen T, Lau CC, Yung WKA, Rabadan R, Huse J, Brat DJ, Lehman NL, Barnholtz-Sloan JS, Zheng S, Hess K, Rao G, Meyerson M, Beroukhim R, Cooper L, Akbani R, Wrensch M, Haussler D, Aldape KD, Laird PW, Gutmann DH, Noushmehr H, Iavarone A, Verhaak RGW, Anjum S, Arachchi H, Auman JT, Balasundaram M, Balu S, Barnett G, Baylin S, Bell S, Benz C, Bir N, Black KL, Bodenheimer T, Boice L, Bootwalla MS, Bowen J, Bristow CA, Butterfield YSN, Chen QR, Chin L, Cho J, Chuah E, Chudamani S, Coetzee SG, Cohen ML, Colman H, Couce M, D’Angelo F, Davidsen T, Davis A, Demchok JA, Devine K, Ding L, Duell R, Elder JB, Eschbacher JM, Fehrenbach A, Ferguson M, Frazer S, Fuller G, Fulop J, Gabriel SB, Garofano L, Gastier-Foster JM, Gehlenborg N, Gerken M, Getz G, Giannini C, Gibson WJ, Hadjipanayis A, Hayes DN, Heiman DI, Hermes B, Hilty J, Hoadley KA, Hoyle AP, Huang M, Jefferys SR, Jones CD, Jones SJM, Ju Z, Kastl A, Kendler A, Kim J, Kucherlapati R, Lai PH, Lawrence MS, Lee S, Leraas KM, Lichtenberg TM, Lin P, Liu Y, Liu J, Ljubimova JY, Lu Y, Ma Y, Maglinte DT, Mahadeshwar HS, Marra MA, McGraw M, McPherson C, Meng S, Mieczkowski PA, Miller CR, Mills GB, Moore RA, Mose LE, Mungall AJ, Naresh R, Naska T, Neder L, Noble MS, Noss A, O’Neill BP, Ostrom QT, Palmer C, Pantazi A, Parfenov M, Park PJ, Parker JS, Perou CM, Pierson CR, Pihl T, Protopopov A, Radenbaugh A,s Ramirez NC, Rathmell WK, Ren X, Roach J, Robertson AG, Saksena G, Schein JE, Schumacher SE, Seidman J, Senecal K, Seth S, Shen H, Shi Y, Shih J, Shimmel K, Sicotte H, Sifri S, Silva T, Simons JV, Singh R, Skelly T, Sloan AE, Sofia HJ, Soloway MG, Song X, Sougnez C, Souza C, Staugaitis SM, Sun H, Sun C, Tan D, Tang J, Tang Y, Thorne L, Trevisan FA, Triche T, van den Berg DJ, Veluvolu U, Voet D, Wan Y, Wang Z, Warnick R, Weinstein JN, Weisenberger DJ, Wilkerson MD, Williams F, Wise L, Wolinsky Y, Wu J, Xu AW, Yang L, Yang L, Zack TI, Zenklusen JC, Zhang J, Zhang W, Zhang J, Zmuda E (2016). Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164(3), 550–563. https://doi.org/10.1016/j.cell.2015.12.028
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22(4):425–437. https://doi.org/10.1016/j.ccr.2012.08.024
McCord M, Mukouyama YS, Gilbert MR, Jackson S (2017) Targeting WNT signaling for multifaceted glioblastoma therapy. Front Cell Neurosci 11:318. https://doi.org/10.3389/fncel.2017.00318
Acknowledgements
The authors’ works are supported by state funds through the University of Pecs, Graduate School in Neurosciences, the Institute of Laboratory Medicine and Szentagothai Research Center.
Funding
Open access funding provided by University of Pécs (PTE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kraboth, Z., Kalman, B. Longitudinal Characteristics of Glioblastoma in Genome-Wide Studies. Pathol. Oncol. Res. 26, 2035–2047 (2020). https://doi.org/10.1007/s12253-019-00705-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-019-00705-1