
Abstract
Thermoanaerobacter kivui is a thermophilic acetogen that can grow on carbon monoxide as sole carbon and energy source. To identify the gene(s) involved in CO oxidation, the genome sequence was analyzed. Two genes potentially encoding CO dehydrogenases were identified. One, cooS, potentially encodes a monofunctional CO dehydrogenase, whereas another, acsA, potentially encodes the CODH component of the CODH/ACS complex. Both genes were cloned, a His-tag encoding sequence was added, and the proteins were produced from a plasmid in T. kivui. His-AcsA copurified by affinity chromatography with AcsB, the acetyl-CoA synthase of the CO dehydrogenase/acetyl CoA synthase complex. His-CooS copurified with CooF1, a small iron-sulfur center containing protein likely involved in electron transport. Both protein complexes had CO:ferredoxin oxidoreductase as well as CO:methyl viologen oxidoreductase activity, but the activity of CooSF1 was 15-times and 231-times lower, respectively. To underline the importance of CooS, the gene was deleted in the CO-adapted strain. Interestingly, the ∆cooS deletion mutant did not grow on CO anymore. These experiments clearly demonstrated that CooS is essential for growth of T. kivui on CO. This is in line with the hypothesis that CooS is the CO-oxidizing enzyme in cells growing on CO.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carbon monoxide is an abundant atmospheric trace gas originating from biotic and abiotic sources (Stegenta-Dabrowska et al. 2019). The redox couple CO/CO2 has a rather negative redox potential (E0′[CO/CO2] = − 520 mV) (Thauer et al. 1977) and, thus, CO is an excellent electron donor for biological processes. Therefore, despite its high toxicity to many life forms, specialized bacteria and archaea are known to use carbon monoxide as electron and carbon source for growth (Henstra et al. 2007a; Sokolova et al. 2009; Robb and Techtmann 2018). These carboxydotrophs can be either aerobic or anaerobic but they share a common key enzyme, the CO dehydrogenase (CODH) (Ragsdale 2000; Dobbek et al. 2001). The enzyme is fundamentally different in aerobes and anaerobes (Doukov et al. 2002; Robb and Techtmann 2018). Aerobic bacteria have a molybdopterin as catalytic site for CO oxidation and channel the electrons into the quinone pool of a CO-insensitive electron transport chain (Wilcoxen et al. 2011; Meyer and Schlegel 1983). ATP is synthesized by electron transport phosphorylation and carbon is fixed by the Calvin cycle, allowing for a chemolithoautotrophic lifestyle (Meyer and Schlegel 1983). Anaerobic metabolism of CO is also chemolithoautotrophic, but the metabolism is different (Henstra et al. 2007b). The anaerobic CODH’s have a nickel atom as catalytic site and the enzymes involved can be either monofunctional (CooS) or bifunctional (AcsA) (Ragsdale 2000; Dobbek et al. 2001; Darnault et al. 2003). Monofunctional CODH’s are typically involved in CO oxidation, the direct electron acceptor is an iron sulfur center present in another protein (CooF), often encoded with cooS in an operon (Ensign and Ludden 1991; Kerby et al. 1992; Singer et al. 2006). The final electron acceptor may be a proton as in hydrogenotrophic carboxydotrophs (Henstra et al. 2007b), CO2 as in methanogenic archaea (Daniels et al. 1977; Rother and Metcalf 2004) or acetogenic bacteria (Diekert and Thauer 1978; Savage et al. 1987; Daniel et al. 1990; Diender et al. 2015) or sulfate as in sulfate reducing bacteria (Parshina et al. 2005) and archaea (Henstra et al. 2007a). Reduction of these electron acceptors lead to chemiosmotic energy conservation and the ATP gained is used to fix CO2, mainly by the Wood-Ljungdahl pathway (WLP) (Ragsdale and Wood 1985; Drake et al. 2008; Schuchmann and Müller 2014). In this two-branched, convergent pathway two molecules of CO2 are reduced to acetyl-CoA. The key enzyme of the WLP is the CODH/ACS that has a bifunctional CODH whose cellular function is to catalyze the reversal of the aforementioned reaction, the reduction of CO2 to CO, the precursor of the carboxyl group in acetyl-CoA (Diekert and Thauer 1978; Savage et al. 1987; Daniel et al. 1990; Diender et al. 2015; Schuchmann and Müller 2014) (Fig. 1). However, in vitro, the bifunctional CO dehydrogenase also catalyzes oxidation of CO (Carlson and Papoutsakis 2017).

A simplified pathway for acetogenesis from CO. The monofunctional CO dehydrogenase, CooS is hypothesized to oxidize CO. The physiological function of the bifunctional CODH/ACS (AcsAB) is to reduce CO2 to CO which is then condensed with a methyl group and CoA to give acetyl-CoA. During growth on H2 + CO2 or organic substrates, CO is generated from CO2 by AcsAB (grey)
Acetogenic bacteria are of outstanding biotechnological interest for a sustainable bioeconomy (Schiel-Bengelsdorf and Dürre 2012; Bertsch and Müller 2015; Müller 2019; Katsyv and Müller 2020), since they can convert carbon dioxide and molecular hydrogen or carbon monoxide or a combination thereof (synthesis gas; syngas) to valued-added chemicals (Demler and Weuster-Botz 2010; Dürre 2011; Köpke et al. 2011; Liew et al. 2016b; Wilkins and Atiyeh 2011). Some mesophilic acetogens are already used as biocatalysts for ethanol production from syngas (Najafpour and Younesi 2006; Maddipati et al. 2011; Bertsch and Müller 2015). In this process, carbon monoxide is oxidized alongside with molecular hydrogen. Recently, we have established that the thermophilic acetogen Thermoanaerobacter kivui, that had been described not to grow on CO (Daniel et al. 1990), can be adapted to grow on CO by serial transfers to media with increasing CO concentrations (Weghoff and Müller 2016). This bacterium also grows on CO or syngas in mineral media making it an ideal biocatalyst for CO or syngas-derived valued-added chemicals (Müller 2019). Recently, a procedure was established enabling T. kivui to convert carbon monoxide to formate, a very promising approach for CO conversion (Schwarz et al. 2020). The molecular basis of adaptation to CO as well as the enzyme involved in using CO as carbon and electron source remained elusive. Here, we have used a genetic approach to identify the CO dehydrogenase involved in growing on CO.
Materials and methods
Organisms and cultivation
T. kivui strains listed in Table 1 were routinely cultivated under anoxic conditions at 66 °C in complex or defined media described before (Weghoff and Müller 2016; Basen et al. 2018). All the growth experiments were performed in 120 ml serum bottles (Glasgerätebau Ochs GmbH, Bovenden-Lenglern, Germany) containing 20 or 50 ml media for growth on gases or sugars. For growth on CO, the gas phase was 100% CO (2 × 105 Pa), for growth on 25 mM glucose, the atmosphere was N2/CO2 (80/20 [v/v], 1 × 105 Pa). Growth was monitored by measuring the OD at 600 nm. Plating and cultivation method on solid media was the same as described in (Basen et al. 2018).
Production and purification of His-CooS and His-AcsA
Plasmid pMU131_His-cooS and pMU131_His-acsA were used for the expression of cooS (TKV_c08080) and acsA (TKV_c20100) in T. kivui. The plasmids are based on plasmid pMU131 (Shaw et al. 2010) which is replicating in T. kivui and confers resistance to kanamycin (Basen et al. 2018). The inserts His-cooS (size: 1954 bp) and His-acsA (size: 1927 bp) were amplified using the primers His_CooS_for (3) (5′- CAAGGAGGAGGATTGACTGTATGCACCATCATCATCACCATCATCATCATCATAGTGATAATTACATTTATTCTGCTG) and His_CooS_rev (4) (5′-TCC TGGATAAATTTAAAAAATTATATATTGAGTAGTTTGCGC) or His_AcsA_for (5) (5′- CAAGGAGGAGGATTGACTGTATGCACCATCATCATCACCATCATCATCATCATGAAGAGAAGAGAACTATTGACATTG) and His_AcsA_rev (6) (5′-TCCTGGATAAATTTA AAAAATTAAACACCCAAAGCCTTTC). The backbone pMU131 (size: 7192 bp) was amplified using the primers pMU131_for (1) (5′-TTTTTTAAATTTATCCAGGATAAAAGAGAAGACTC) and pMU131_rev (2) (5′-ACAGTCAATCCTCCTCCTTG), followed by the fusion of the PCR products via Gibson Assembly (Gibson Assembly Mastermix, NEB, Frankfurt/Main, Germany). T. kivui (DSM 2030) was transformed with the plasmids pMU131_His-cooS or pMU131_His-acsA as described (Basen et al. 2018). Cells were plated on agar medium containing 28 mM glucose as carbon source and 200 µg/ml kanamycin as selection marker. To verify the transformation, colonies were picked and the transformed plasmids were checked by using primer pairs seq1_for (7) (5′-TCTAACACAATTATATCATAAGGATTG ATA)/seq2_rev (8) (5′-AGTATTGTCAATATATTCAAGGCAA) binding on the pMU131 backbone and amplifying the complete His-cooS or His-acsA locus (Fig. S1).
For the purification of the His-tagged CooS or AcsA, T. kivui pMU131_His-cooS or pMU131_His-acsA cells were grown with 28 mM glucose as carbon and energy source and 200 µg/ml kanamycin. All purification steps were performed under strictly anoxic conditions at room temperature in an anoxic chamber (Coy Laboratory Products, Grass Lake, Michigan, USA) filled with 95–98% N2 and 2–5% H2. T. kivui cells were harvested and washed twice in buffer A (50 mM Tris/HCl, 150 mM NaCl, 20 mM MgSO4, 10 mM imidazole, 0.5 mM DTE, 4 µM resazurin, 20% [v/v] glycerol, pH 7.5). The cells were resuspended in 50 ml buffer A including 0.5 mM PMSF and 0.1 mg/ml DNAseI and passed one time through a French pressure cell (110 MPa). Cell debris was removed by centrifugation at 24,000×g for 20 min. Protein purification of the His-tagged proteins was carried out on a nickel nitrilotriacetic acid (Ni2+-NTA) resin (Qiagen, Hilden, Germany) using a gravity flow column under anoxic conditions as described previously (Katsyv et al. 2021). Fractions containing His-CooS or His-AcsA were collected, pooled, concentrated, using 50-kDa VIVASPIN tubes, and separated on a Superdex 200 10/300 GL increase prepacked column (GE Healthcare Life Sciences, Little Chalfont, UK). The sample was loaded on a Superdex 200 column equilibrated with buffer B (50 mM Tris/HCl, 150 mM NaCl, 20 mM MgSO4, 2 mM DTE, 4 µM resazurin, 20% [v/v] glycerol, pH 7.5) and eluted at a flow rate of 0.5 ml/min. CooS and AcsA activity eluted in a single peak with a maximum at 9.7 and 12.6 ml elution volume, respectively. Fractions containing His-CooS and His-AcsA were pooled and stored at 4 °C.
Preparation of cell free extract
Cells were cultivated in 1 L flasks (Müller-Krempel, Bülach, Switzerland) filled with 200 ml or 500 ml of complex media for growth on 100% CO (2 × 105 Pa) or 25 mM glucose, respectively. Mid exponential phase cells were harvested by centrifugation at 11,500 × g for 10 min at 4 °C and washed twice in buffer C (50 mM Tris/HCl, 20 mM MgSO4, 20% glycerol, 2 mM DTE, 4 μM resazurin, pH 7). After the washing steps, cells were resuspended in 20 ml buffer C with few crystals of DNAseI and 0.5 mM PMSF and disrupted by a passage through a French pressure cell (110 MPa). Cell debris was separated by centrifugation at 14,300 × g for 20 min and the supernatant was collected. The supernatant contained the cell free extract.
Measurement of CODH activity
Enzyme assays were routinely performed at 66 °C in 1.8 ml anoxic cuvettes (Glasgerätebau Ochs, Bovenden-Lenglern, Germany) sealed by rubber stoppers in a 100% CO atmosphere (2 × 105 Pa) with buffer D (50 mM Tris/HCl, 10 mM NaCl, 2 mM DTE, 4 μM resazurin, pH 7.5 or pH 7) or buffer E (100 mM HEPES/NaOH, 2 mM DTE, 2 μM resazurin, pH 7) at an overall liquid volume of 1 ml. His-CooSF1 or His-AcsAB activity was measured with methyl viologen (MV) or ferredoxin (Fd) as electron acceptor at 604 nm (ε = 13.9 mM−1 cm−1) or 430 nm (ε = 13.1 mM−1 cm−1), respectively. Fd was purified from Clostridium pasteurianum as described previously (Schönheit et al. 1978). CODH activity in cell free extracts was measured in buffer E using MV as an artificial electron acceptor. The assay was supplemented with crude extract or enriched His-CooSF1 or His-AcsAB preparation and the reaction was started with 10 mM MV or 30 μM Fd. For Km determination, the CO and Fd concentrations ranged between 0–300 µM and 0–200 µM, respectively. For the determination of the pH and temperature profile, the assay mixture including the protein was preincubated for 10 min at the pH or temperature indicated. The buffer (F) used to determine the pH optima was 50 mM MES, 50 mM CHES, 50 mM CAPS, 50 mM Bis–Tris, 50 mM Tris, 10 mM NaCl, 4 mM DTE, 4 μM resazurin.
Analytical methods
The concentration of proteins was measured according to Bradford (1976). The protein concentration in whole cells was measured according to Schmidt (Schmidt et al. 1963). The iron content of the purified enzymes was determined by colorimetric methods (Fish 1988). Proteins were separated in 12% polyacrylamide gels and stained with Coomassie brilliant blue G250. The molecular mass of the purified His-CooSF1 or His-AcsAB was determined using a calibrated Superdex 200 column, buffer B and defined size standards (ovalbumin: 43 kDa; albumin: 158 kDa; catalase: 232 kDa; ferritin: 440 kDa).
Generation of T. kivui ΔcooS strain
For the deletion of cooS (Tkv_c08080), plasmid pSJ006 (Fig. S2A) was constructed in E. coli DH5α. The plasmid was generated by inserting 1000 bp upstream flanking region (UFR) and downstream flanking region (DFR) of cooS into the backbone plasmid pMBTkv0012 (Jain et al. 2020). Primers used were ∆cooS_UFR_FP (5′- ACCCGGGGATCCGCAGGAAGATTGGAAGTCAT) & ∆cooS_UFR_RP (5′- CCCATATTTTTCAATTATTATCACAACTCCTTTT) for UFR and ∆cooS_DFR_FP (5′- GGAGTTGTGATAATAATTGAAAAATATGGGAGGAA) & ∆cooS_DFR_RP (5′- GCAGGTCGACTCTAGACTGGTCGGGGCAACAGGAT) for DFR amplification, followed by ligation into pMBTkv0012 using oligonucleotides ∆cooS_BB_FP (5′- GCCCCGACCAGTCTAGAGTCGACCTGCAGGCATG) & ∆cooS_BB_RP (5′- CAATCTTCCTGCGGATCCCCGGGTACCGAGCTCG).
T. kivui ΔpyrE (Basen et al. 2018) was transformed with the plasmid pSJ006. Since the plasmid contains pyrE gene as a selection marker, the first round of selection was performed on agar plates in defined media without uracil using 25 mM glucose as a substrate to select for transformants with the plasmid integration. Further, these transformants were subjected to second round of selection in media supplemented with 25 mM glucose, 50 µM uracil and 5 mM 5-fluoroorotate (5-FOA) to select the isolates with the loss of plasmid. The deletion of cooS was confirmed by PCR of isolated genomic DNA using forward primer (5′- GGGCTTTATAAAGCGAAATGGG) and reverse primer (5′- GCCTGTTGATAAGTCATAAAACCTGC) binding outside the cooS gene or using forward primer (5′- GCGTGATCCAAAATGTGGTTTCGG) and reverse primer (5′- CAAGCCATTGTGGTGCAGAAGC) binding inside the cooS gene in the genome.
For the requirement of ∆pyrE in the CO adapted strain, plasmid pMBTkv002b (Basen et al. 2018) was used. The CO adapted wild type (Weghoff and Müller 2016) was transformed with pMBTkv002b and subjected to 5-FOA selection with 50 µM uracil. The deletion of pyrE was verified by primers MB_IG_0005 and MB_IG_0006 (Basen et al. 2018). To delete the cooS in the generated CO adapted ∆pyrE, plasmid pSJ006 was used as described above. The deletion of cooS was confirmed by amplifying the flanking regions followed by DNA sequencing analysis (Sanger et al. 1977).
To integrate cooS gene back into the T. kivui ∆coos genome, plasmid pSJ008 was prepared (Fig. S2B). pJM006 originated from pMBTkv007 (Basen et al. 2018), was used as the backbone plasmid, which has pyrE gene under control of the promoter gyrase from Thermoanaerobacter sp. strain X514, and directly adjacent to the 3′-end, gene Teth514_0627 from Thermoanaerobacter sp. strain X514 under control of the promoter of the S-layer protein from T. kivui. pJM006 except for adhE was amplified by PCR using primers (5′- CTACTCAATATATAAAATTTAATTTAAAAATTTCACAGCAAGCAG) and (5′- TGTAATTATCACTCATACAGTCAATCCTCCTCCTTGTATTTG). The cooS gene was amplified by using forward primer (5′- GGAGGATTGACTGTATGAGTGATAATTACATTTATTCTGCTG) and reverse primer (5′- GTGAAATTTTTAAATTAAATTTTATATATTGAGTAGTTTGCGCC). The PCR products were then fused to generate the plasmid pSJ008 using Gibson assembly (Gibson Assembly Mastermix, NEB, Frankfurt/Main, Germany). T. kivui ∆cooS mutant was transformed with plasmid pSJ008. Selection for the transformants was performed by using defined media without uracil in the presence of 25 mM glucose.
Preparation of resting cells
Preparation of resting cells were performed under anoxic condition. Cells of T. kivui wild type and ∆cooS were grown on glucose or on glucose + 100% CO in the headspace, in 500 ml of complex media to mid exponential phase and were harvested by centrifugation (AvantiTMJ-25 and JA-10 Fixed-Angle Rotor; Beckman Coulter, Brea, CA, United States) at 11,500 × g, 4 °C for 10 min. The supernatant was discarded and the cells were washed three times with imidazole buffer (50 mM imidazole, 20 mM MgSO4, 20 mM KCl, 20 mM NaCl, 4 mM DTE, 4 μM resazurin, pH 7). After centrifugation, cells were resuspended in 5 ml of same buffer and kept in 16 ml gas tight Hungate tubes. The headspace of the Hungate tubes were changed to 100% N2. The protein concentration was measured according to (Schmidt et al. 1963).
Experiment with resting cells
For the experiment, 120 ml serum bottles (Glasgerätebau Ochs GmbH, Bovenden-Lenglern, Germany) under a N2/CO2 (80/20 [v/v], 1 × 105 Pa) atmosphere were filled with imidazole buffer (50 mM imidazole, 20 mM MgSO4, 20 mM KCl, 20 mM NaCl, 4 mM DTE, 4 μM resazurin, pH 7) in the presence of 50 mM of KHCO3. Cells were added to a protein concentration of 1 mg/ml, the final volume of the suspension was 10 ml. After the serum bottles were incubated at 66 °C for 10 min in pre-warmed water bath, the experiment was started by addition of H2 + CO2 (80/20 [v/v], 1 × 105 Pa).
Product analysis
Acetate production was measured by gas chromatography as described previously (Weghoff and Müller 2016).
Results
Identification and organization of genes involved in CO metabolism
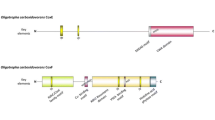
To identify genes that are involved in CO metabolism, the genome sequence of T. kivui was inspected. The genome harbors one gene encoding a potential monofunctional CODH, cooS (Tkv_c08080) (Hess et al. 2014). The downstream region is flanked by the gene cooF1 (Tkv_c08090), potentially involved in CO metabolism for transferring electrons to a membrane-bound hydrogenase (Fox et al. 1996; Schoelmerich and Müller 2020) (Fig. 2). CooF1 has a predicted molecular mass of 19 kDa and contains three 4Fe-4S cluster. Upstream of cooS is a hypothetical gene with unknown function. In silico analysis using the BlastP algorithm revealed 54, 32, 34, 53 and 52% identity of CooS of T. kivui to CooS of Caldicellulosiruptor hydrothermalis, Rhodospirillum rubrum, Desulfovibrio vulgaris, Clostridium carboxidivorans and Acetobacterium woodii, respectively. CooS has a predicted molecular mass of 68 kDa and contains a 4Fe-4S cluster and a Ni-4Fe-4S center where carbon monoxide oxidation occurs (Ragsdale and Kumar 1996). Additionally, the genome of T. kivui contains another putative CODH gene annotated as acsA (TKV_c20100) that together with acsB (TKV_c19820), encoding the acetyl-CoA synthase, forms the CODH/ACS complex. The gene acsA is flanked by a second copy of cooF (cooF2) (TKV_c20110), and a gene encoding potentially for nickel insertion, cooC2 (TKV_c20090) (Fig. 2), all transcribed in the same direction. AcsA is predicted to have a molecular mass of 67 kDa and has as well a 4Fe-4S cluster and Ni-4Fe-4S center, sharing 61 and 60% identity to Methanosarcina mazei and Thermoanaerobacter sp. YS13, respectively.

Genetic organization of CO dehydrogenase genes in T. kivui. Genes coding for potential monofunctional CODH in T. kivui, cooS, organized with cooF1 (upper panel), and a second CODH, acsA adjacent to cooF2 and cooC2 (lower panel)
Purification and characterization of His-CooS and His-AcsA
In order to establish that CooS and AcsA are indeed CO dehydrogenases, we took advantage of a plasmid-based production system in T. kivui (Katsyv et al. 2021). Therefore, we cloned cooS (TKV_c08080) or acsA (TKV_c20100) together with a DNA sequence coding for a 10 × histidine-tag into pMU131, which replicates in T. kivui (Fig. 3). Naturally competent cells of T. kivui (DSM 2030) were transformed with the plasmid and the encoded proteins containing a genetically engineered His-tag were purified. Therefore, crude extract of the genetically modified T. kivui strains was prepared as described in Material and Methods. The His-tagged CooS and AcsA were purified from the crude extract to apparent homogeneity by Ni2+-NTA-sepharose followed by a size exclusion chromatography on Superdex 200. Analyses of the purified His-CooS separated on a 12% SDS–polyacrylamide gel revealed two proteins with apparent molecular masses of ≈ 65 and ≈ 17 kDa (Fig. 4A). These molecular masses correspond well with the expected sizes for CooS (TKV_c08080, 68 kDa) and the downstream encoded CooF1 (TKV_c08090, 19 kDa) of T. kivui. Analytical size exclusion chromatography revealed a molecular mass of 537 kDa for the purified complex, which is consistent with His-CooSF1 being a hexamer. We measured 22.9 ± 0.6 mol of iron/mol of protein using the colorimetric assay to detect complexed iron (Fish 1988), which matches the prediction that His-CooSF1 contains four 4Fe-4S cluster and one Ni-4Fe-4S. When analyzing the enriched His-AcsA protein on a 12% SDS–polyacrylamide gel, two proteins with apparent molecular masses of ≈ 65 and ≈ 80 kDa were visible (Fig. 4B). These molecular masses correspond well with the expected sizes for AcsA (TKV_c20100, 67 kDa) and AcsB (TKV_c19820, 76 kDa) of T. kivui. Apparently, AcsA and AcsB form a stable complex that can be purified via affinity chromatography of AcsA. Stable complex formation of the CODH subunit (AcsA) with the acetyl-CoA synthase (AcsB) was shown before (Ragsdale et al. 1983; Ragsdale and Kumar 1996; Seravalli et al. 1997; Doukov et al. 2008). Analytical size exclusion chromatography revealed a molecular mass of 130.5 kDa for the purified complex, which is consistent with His-AcsAB being a monomer. We measured 9.0 ± 0.3 mol of iron/mol of protein, which matches the prediction that His-AcsAB contains one 4Fe-4S cluster and one Ni-4Fe-4S.

Cloning of pMU131_ His_cooS and pMU131_His_acsA. For the production of His-CooS and His-AcsA in T. kivui the constructs pMU131_ His-cooS and pMU131_His-acsA were cloned (A, E). Therefore, pMU131 backbone, including a S-layer-promoter, was amplified using corresponding primers via PCR (B, F). His-cooS (C) and His-acsA (G) were amplified from genomic DNA of T. kivui via PCR, using corresponding primers, containing an additional DNA sequence coding for a 10 × His-tag. Amplified His-cooS or His-acsA and pMU131 backbone were fused via Gibson Assembly and transformed in E. coli HB101. Afterwards, plasmids were isolated and digested with SacI (D, H). The resulting sizes for pMU131_ His-cooS were 7253 bp and 1826 bp and for pMU131_His-acsA 7280 bp and 1826 bp. M, Gene Ruler 1 kb DNA ladder

SDS-PAGE of the purified His-CooSF1 and His-AcsAB complex. His-CooSF1 (A) and His-AcsAB (B) of T. kivui, purified via affinity chromatography and separated on a gelfiltration column, were analyzed in a denaturating SDS-PAGE (12%). The proteins were stained with Coomassie Brilliant Blue G250. 10 µg of protein was applied to each lane. M, prestained page ruler
Both complexes exhibited CO:MV oxidoreductase as well as CO:Fd oxidoreductase activity (Table 2). The specific CO:MV oxidoreductase activity of His-AcsAB was 213.9 ± 7.6 U/mg, that of His-CooSF1 was 15-times lower with 13.9 ± 4.1 U/mg (Table 2). The bifunctional as well as the monofunctional CODH reduced Fd (isolated from C. pasteurianum) as electron acceptor with CO as electron donor. His-CooSF1 catalyzed CO:Fd oxidoreductase activity significantly lower with 0.5 ± 0.03 U/mg compared to His-AcsAB with 111.5 ± 15.4 U/mg (Table 2). Since this is the first purification of a monofunctional and bifunctional CODH from one organism, it is unknown whether the huge difference in activity is a unique feature of the enzymes for T. kivui.
We assessed key biochemical properties of the purified His-AcsAB and His-CooSF1, including temperature and pH stability, substrate affinities and cofactor dependence. To ensure an ideal reflection of the physiological conditions, we exclusively used the CO:Fd oxidoreductase assay. His-AcsAB was active at temperatures ranging from 22 to 85 °C with a maximal activity of 100.6 ± 7.8 U/mg at the optimal growth temperature of T. kivui (66 °C) (Fig. S3A). His-AcsAB activity decreased by 96% at 22 °C and by 65% at 40 °C (Fig. S3A). The pH range was relatively narrow with only 5% activity at pH 6 and 10 and an optimal activity of 48.5 ± 4.1 U/mg at pH 7 and 8 (Fig. S3B).
His-CooSF1 was active at temperatures ranging from 22 to 85 °C with a maximal activity of 0.5 ± 0.03 U/mg at the optimal growth temperature of T. kivui (66 °C) (Fig. S4A). His-AcsAB activity was decreased by 83% at 22 °C and by 62% at 40 °C (Fig. S4A). Compared to His-AcsAB, His-CooSF1 was almost fully active at temperatures higher than 66 °C (Fig. S4A). The pH range was relatively narrow with zero activity at pH 5 and 10. At pH 6 CO:Fd oxidoreductase activity was 0.2 ± 0.01 U/mg and reached an optimum of 0.3 ± 0.03 U/mg at pH 7 (Fig. S4B). All further analyses were subsequently carried out at pH 7.5 for His-AcsAB and pH 7 for His-CooSF1 and 66 °C, to ensure optimal enzyme activity.
Next, we assessed the Km values for all reaction partners of AcsAB and CooSF1. The dependence of the CO:Fd oxidoreductase reaction on CO and Fd was hyperbolic with saturation at ̴ 30 µM CO or 30 µM Fd for His-AcsAB and His-CooFS1 (Fig. S3C, D and Fig. S4C, D). The Km values of His-AcsAB for CO and Fd were 10.9 ± 3.6 µM and 15.9 ± 4.6 µM, respectively (Fig. S3C, D). The Km values of His-CooSF1 for CO and Fd were 5.0 ± 1.5 µM and 20.9 ± 6.0 µM, respectively (Fig. S4C, D). Unsurprisingly, the absence of any reaction partner led to a complete loss of activity for both CODHs.
Generation of cooS deletion in T. kivui
Since previous data revealed higher level of CooS compared to AcsA during growth on CO (Weghoff and Müller 2016), we aimed to delete cooS (Tkv_c08080) and study its involvement in growth on CO. Therefore, plasmid pSJ006 was generated containing approximately 1000 bp upstream and downstream of the cooS gene. The plasmid also contained the pyrE cassette as selectable marker that can be integrated into the pyrE-deficient uracil-auxotrophic strain TKV_MB002 (Basen et al. 2018) at one of the flanking regions. The subsequent disintegration was forced by the presence of 5-FOA since the plasmid contains a pyrE gene for production of a functional orotate phosphoribosyltransferase, leading to markerless deletion of cooS. Indeed, with glucose as carbon and energy source for growth of T. kivui, we were able to delete the cooS gene (data not shown). This strain is named as TKV_SJ001, to avoid confusion.
However, we could not adapt TKV_SJ001 strain to grow on CO, using the same procedure as described previously (Weghoff and Müller 2016). The failure to grow the mutant on CO could reflect the essentiality of CooS for CO metabolism or the inability to adapt the cells to the toxic gas. Therefore, we used a different approach and decided to delete cooS in a CO-adapted strain [∆cooS (CO)]. Thus, pyrE had to be deleted first; this was done essentially as described above, in the CO-adapted wild type T. kivui. In order to ease the mutant preparation on solid media, we made sure that the CO-adapted strain would start to grow on CO immediately when cultivated on glucose in between. Then, the cooS gene was deleted, essentially as described above using the same approach with cells grown on glucose. Again, we were able to delete the cooS gene, as exemplified by PCR with primers binding outside the cooS gene (Fig. S5A). Isolate 1 was additionally verified by PCR with the primers binding inside the cooS gene and as expected, an amplificate was not obtained whereas, with DNA from the parental strain ∆pyrE (CO) a DNA fragment of 1.5 kb was amplified (Fig S5B). All strains used in this work are listed in Table 1. To avoid confusion, from now on the ∆cooS strain in this work refers to the CO-adapted T. kivui ∆pyrE strain with a cooS deletion, if not otherwise specified.
The cooS mutant does not grow on carbon monoxide
To analyze the phenotype of the cooS mutant growth experiments were performed. The ∆cooS strain grew on 25 mM glucose, 25 mM mannitol, H2 + CO2 (2 × 105 Pa) or 100 mM formate, similar to the parent strain ∆pyrE (CO). The ∆pyrE (CO) strain grew on 100% CO with a rate of 0.012 (h−1) to an OD of 0.54 ± 0.04 (n = 3) after 5 days in complex media. In contrast, the ∆cooS mutant did not grow in the time frame observed. When the cooS gene was presented in trans on plasmid pSJ008 cells did grow on CO with rates and final yields were comparable to the ∆pyrE (CO) (Fig. 5A).

Growth of T. kivui ΔpyrE (CO) and ΔcooS on 100% CO. The cells were grown at 66 °C with 100% CO (2 × 105 Pa) in 120 ml serum bottle containing 20 ml of complex media (A) or mineral media with uracil (B). These experiments were performed in biological triplicates. Squares, T. kivui ΔpyrE (CO); circles, T. kivui ΔcooS; triangles, T. kivui ΔcooS plus re-introduced cooS in a different genome location
However, after 7 days the ∆cooS mutant started to grow with a rate of 0.0081 (h−1) to a final OD of 0.28 ± 0.06 (n = 3). In contrast, this was not observed in mineral media, indicating that cells of the ∆cooS mutant did not grow on CO but on a component of the complex media. When the cells were transferred from glucose-mineral medium to CO-mineral medium, slow growth was observed after 15 days leading to a final OD of 0.2, but after a second transfer, growth was no longer observed. In contrast, when ∆pyrE (CO) was transferred, it grew to an OD of 0.35 ± 0.06 (n = 3) in 5 days in mineral media (Fig. 5B).
To determine the CO dehydrogenase activity in ∆pyrE (CO) and the ∆cooS mutant, both were grown on CO in complex media (the ∆cooS for 7 days). Cells were harvested in late exponential growth phase, cell free extract was prepared and the CODH activity was determined with CO as electron donor and MV as electron acceptor (Table 3). The CO:MV oxidoreductase activity was 178.6 ± 12.8 U/mg in ∆pyrE (CO) but only 8% (14.9 ± 2.5 U/mg) in the ∆cooS mutant, clearly demonstrating that the majority of CODH activity is catalyzed by CooS. Complementation of the mutant restored CODH activity by 41.6% to 76.9 ± 6.9 U/mg. When the CO dehydrogenase activity was measured in glucose-grown cells, similar activities were observed in the ∆cooS mutant (50.6 ± 6.1 U/mg) and ∆pyrE (CO) (53.8 ± 11.4 U/mg).
Cell suspension experiments
The aforementioned experiments clearly revealed that CooS is essential for growth on CO. However, CO is also an intermediate in the WLP, produced by the CODH/ACS from CO2. Therefore, it was of interest to study the effect of the deletion of cooS on acetate formation from H2 + CO2. To this end, cells were grown on either glucose alone or on glucose under a CO atmosphere (100%), harvested in the mid exponential growth phase and resting cells were prepared. As can be seen in Fig. 6, acetate was produced from H2 + CO2 and cells pre-grown in the presence of CO (plus glucose) had a slightly higher activity. Interestingly, acetate formation was drastically stimulated by cooS deletion in any case, demonstrating a role of CooS also in acetogenesis from H2 + CO2.

Conversion of H2 + CO2 to acetate by resting cells of T. kivui wild type (CO) and ∆cooS. The cells were grown on 25 mM glucose (A) or 25 mM glucose with 100% CO in the headspace (B) to the mid exponential phase. The harvested and washed cells were resuspended in imidazole buffer (50 mM imidazole, 20 mM MgSO4, 20 mM KCl, 20 mM NaCl, 4 mM DTE, 4 μM resazurin, pH 7) to a final protein concentration of 1 mg/ml with the addition of 50 mM KHCO3 in anoxic serum bottles. The resting cells were incubated with H2 + CO2 (80/20 [v/v], 1 × 105 Pa) as a substrate in a shaking water bath at 66 °C. H2 + CO2 conversion to acetate was measured over the time. These experiments were performed in biological duplicates. Squares, T. kivui wild type (CO); circles, T. kivui ∆cooS
Discussion
T. kivui is a thermophilic acetogen with a high potential as catalyst in carbon capture and storage as well as utilization (Müller 2019). It grows in synthetic media with high rates on H2 + CO2 and, although it had originally been described to not use CO as sole carbon and energy source, it was adapted to CO by subsequent transfer to media with increasing CO concentrations since it finally grew at 100% CO (Weghoff and Müller 2016). The molecular basis for this adaptation still remains elusive but here we have identified the CO dehydrogenase gene that is essential for growth on CO.
T. kivui has a gene encoding a monofunctional CO dehydrogenase, cooS, as well as a gene encoding the CO dehydrogenase subunit of the bifunctional CODH/ACS complex. After homologous production and purification from T. kivui, CooS was co-purified with CooF1, a protein that is suspected to mediate the electron transfer to Fd, the natural electron acceptor of CO dehydrogenases. AcsA forms a stable complex with AcsB, the acetyl-CoA synthase. Both complexes, CooSF1 and AcsAB, reduced MV but also Fd with CO as electron donor, albeit the AcsAB complex had much higher activities than the CooSF1 complex with both electron acceptors. AcsAB of T. kivui was more active than the purified and characterized bifunctional CODHs (AcsAB) of A. woodii, Moorella thermoacetica or Clostridium formicoaceticum with MV (27, 20 or 14 U/mg) or Fd as electron acceptor (112, 60, 14 U/mg) (Ragsdale et al. 1983). Acetogenic, monofunctional CODHs have not been purified and characterized yet. The activities of present monomeric CODHs were only measured with viologens as electron acceptor. If comparing the specific CO:MV oxidoreductase activity of CooSF1 from T. kivui to known monofunctional CODHs e. g. from R. rubrum or D. vulgaris (660 or 160 U/mg) (Ensign and Ludden 1991; Hadj-Saïd et al. 2015) the enzyme of T. kivui is less active. However, one should keep in mind that the assays were done with artificial electron acceptors or ferredoxin isolated from a mesophile. Although His-AcsAB of T. kivui was more active than His-CooSF1 of T. kivui, deletion of cooS clearly demonstrated that CooS is essential for growing on CO. The physiological importance of AcsAB in CO metabolism could not be addressed since all attempts to generate a CODH/ACS deletion mutant failed so far. Deletion of AcsAB will lead to a non-functional WLP. Some acetogens may grow on syngas in the absence of WLP, for example by producing hydrogen, but T. kivui requires the WLP also for the heterotrophic growth (Jain et al., 2020). Under autotrophic conditions, the WLP is essential. Thus, a knockout of acsA in Clostridium autoethanogenum led to a complete loss of autotrophy, i.e. growth on H2 + CO2 or CO (Liew et al. 2016a). C. autoethanogenum has two additional cooS genes. Deletion of cooS2 had no significant effect on autotrophic growth, whereas deletion of cooS1 led to a long lag phase, slower growth, lower OD and a shift in the product spectrum from acetate to ethanol. Thus, in this acetogen the monofunctional CODH’s are dispensable for autotrophic growth (Liew et al. 2016a). The same was observed for the monofunctional CODH’s in Methanosarcina acetivorans (Matschiavelli et al. 2012; Rother et al. 2007). In sharp contrast, deletion of cooS in T. kivui led to the complete loss of growth on CO, demonstrating that AcsA cannot compensate for a loss of CooS, despite of its higher CO oxidizing activity. This may have to do with regulatory effects. The physiological function of CODH/ACS is to reduce CO2, not to oxidize CO, and this function may be downregulated in the cells.
Interestingly, and unexpectedly, acetate formation from H2 + CO2 was drastically increased in the cooS deletion strain. The precursor of acetate is acetyl-CoA which is also the central precursor for all the biosynthesis pathways in acetogens. Indeed, twice the amount of biomass was produced in a ∆cooS mutant of C. autoethanogenum. It was hypothesized that a deletion of cooS directs more CO2 to acetyl-CoA synthesis (Liew et al. 2016a). The same could be true for T. kivui, resulting in the observed enhanced acetate production. The molecular basis of this effect remains elusive but one scenario could be the following: during acetogenesis from CO2, one CO2 is reduced to CO by CODH/ACS. Since CO is toxic to cells, it is kept caged in a tunnel in the enzyme before it is bound to a Ni-4Fe-4S center of the enzyme. But CO may also escape from the enzyme and the CooS then acts as a safety guard to detoxify cytosolic CO by oxidizing it to CO2, leading to a futile cycle. Deletion of cooS would then direct more CO to the direction of acetyl CoA.
Abbreviations
- Fd:
-
Ferredoxin
- MV:
-
Methyl viologen
- CoA:
-
Coenzyme A
- WLP:
-
Wood–Ljungdahl pathway
- CODH/ACS:
-
Carbon monoxide dehydrogenase/acetyl-CoA synthase
- ATP:
-
Adenosine triphosphate
- 4Fe-4S:
-
Iron-sulfur cluster
- SDS:
-
Sodium dodecyl sulfate
- Ni-4Fe-4S:
-
Nickel–iron–sulfur center
- 5-FOA:
-
5-Fluoroorotate
- PMSF:
-
Phenylmethylsulfonyl fluoride
References
Basen M, Geiger I, Henke L, Müller V (2018) A genetic system for the thermophilic acetogenic bacterium Thermoanaerobacter kivui. Appl Environ Microbiol 84:e02210-02217. https://doi.org/10.1128/AEM.02210-17
Bertsch J, Müller V (2015) Bioenergetic constraints for conversion of syngas to biofuels in acetogenic bacteria. Biotechnol Biofuels 8:210. https://doi.org/10.1186/s13068-015-0393-x
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of proteine-dye-binding. Anal Biochem 72:248–254. https://doi.org/10.1006/abio.1976.9999
Carlson ED, Papoutsakis ET (2017) Heterologous expression of the Clostridium carboxidivorans CO dehydrogenase alone or together with the acetyl coenzyme A synthase enables both reduction of CO2 and oxidation of CO by Clostridium acetobutylicum. Appl Environ Microbiol 83:e00829-e917. https://doi.org/10.1128/AEM.00829-17
Daniel SL, Hsu T, Dean SI, Drake HL (1990) Characterization of the H2-dependent and CO-dependent chemolithotrophic potentials of the acetogens Clostridium thermoaceticum and Acetogenium kivui. J Bacteriol 172:4464–4471. https://doi.org/10.1128/jb.172.8.4464-4471.1990
Daniels L, Fuchs G, Thauer RK, Zeikus JG (1977) Carbon monoxide oxidation by methanogenic bacteria. J Bacteriol 132:118–126. https://doi.org/10.1128/JB.132.1.118-126.1977
Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC (2003) Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] clusters in closed and open subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat Struct Biol 10:271–279. https://doi.org/10.1038/nsb912
Demler M, Weuster-Botz D (2010) Reaction engineering analysis of hydrogenotrophic production of acetic acid by Acetobacterium woodii. Biotechnol Bioeng 108:470–474. https://doi.org/10.1002/bit.22935
Diekert GB, Thauer RK (1978) Carbon monoxide oxidation by Clostridium thermoaceticum and Clostridium formicoaceticum. J Bacteriol 136:597–606. https://doi.org/10.1128/JB.136.2.597-606.1978
Diender M, Stams AJ, Sousa DZ (2015) Pathways and bioenergetics of anaerobic carbon monoxide fermentation. Front Microbiol 6:1275. https://doi.org/10.3389/fmicb.2015.01275
Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O (2001) Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster. Science 293:1281–1285. https://doi.org/10.1126/science.1061500
Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL (2002) A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science 298:567–572. https://doi.org/10.1126/science.1075843
Doukov TI, Blasiak LC, Seravalli J, Ragsdale SW, Drennan CL (2008) Xenon in and at the end of the tunnel of bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Biochemistry 47:3474–3483. https://doi.org/10.1021/bi702386t
Drake HL, Gößner AS, Daniel SL (2008) Old acetogens, new light. Ann N Y Acad Sci 1125:100–128. https://doi.org/10.1196/annals.1419.016
Dürre P (2011) Fermentative production of butanol—the academic perspective. Curr Opin Biotechnol 22:331–336. https://doi.org/10.1016/j.copbio.2011.04.010
Ensign SA, Ludden PW (1991) Characterization of the CO oxidation/H2 evolution system of Rhodospirillum rubrum. Role of a 22-kDa iron-sulfur protein in mediating electron transfer between carbon monoxide dehydrogenase and hydrogenase. J Biol Chem 266:18395–18403. https://doi.org/10.1016/S0021-9258(18)55283-2
Fish WW (1988) Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol 158:357–364. https://doi.org/10.1016/0076-6879(88)58067-9
Fox JD, Kerby RL, Roberts GP, Ludden PW (1996) Characterization of the CO-induced, CO-tolerant hydrogenase from Rhodospirillum rubrum and the gene encoding the large subunit of the enzyme. J Bacteriol 178:1515–1524. https://doi.org/10.1128/jb.178.6.1515-1524.1996
Hadj-Saïd J, Pandelia ME, Léger C, Fourmond V, Dementin S (2015) The carbon monoxide dehydrogenase from Desulfovibrio vulgaris. Biochim Biophys Acta 1847:1574–1583. https://doi.org/10.1016/j.bbabio.2015.08.002
Henstra AM, Dijkema C, Stams AJ (2007a) Archaeoglobus fulgidus couples CO oxidation to sulfate reduction and acetogenesis with transient formate accumulation. Environ Microbiol 9:1836–1841. https://doi.org/10.1111/j.1462-2920.2007.01306.x
Henstra AM, Sipma J, Rinzema A, Stams AJ (2007b) Microbiology of synthesis gas fermentation for biofuel production. Curr Opin Biotechnol 18:200–206. https://doi.org/10.1016/j.copbio.2007.03.008
Hess V, Poehlein A, Weghoff MC, Daniel R, Müller V (2014) A genome-guided analysis of energy conservation in the thermophilic, cytochrome-free acetogenic bacterium Thermoanaerobacter kivui. BMC Genomics 15:1139. https://doi.org/10.1186/1471-2164-15-1139
Jain S, Dietrich HM, Müller V, Basen M (2020) Formate is required for growth of the thermophilic acetogenic bacterium Thermoanaerobacter kivui lacking hydrogen-dependent carbon dioxide reductase (HDCR). Front Microbiol 11:59. https://doi.org/10.3389/fmicb.2020.00059
Katsyv A, Müller V (2020) Overcoming energetic barriers in acetogenic C1 conversion. Front Bioeng Biotechnol 8:621166. https://doi.org/10.3389/fbioe.2020.621166
Katsyv A, Schoelmerich MC, Basen M, Muller V (2021) The pyruvate:ferredoxin oxidoreductase of the thermophilic acetogen, Thermoanaerobacter kivui. FEBS Open Bio 11:1332–1342. https://doi.org/10.1002/2211-5463.13136
Kerby RL, Hong SS, Ensign SA, Coppoc LJ, Ludden PW, Roberts GP (1992) Genetic and physiological characterization of the Rhodospirillum rubrum carbon monoxide dehydrogenase system. J Bacteriol 174:5284–5294. https://doi.org/10.1128/jb.174.16.5284-5294.1992
Köpke M, Mihalcea C, Liew F, Tizard JH, Ali MS, Conolly JJ, Sinawi-Al B, Simpson SD (2011) 2,3-butanediol production by acetogenic bacteria, an alternative route to chemical synthesis, using industrial waste gas. Appl Environ Microbiol 77:5467–5475. https://doi.org/10.1128/AEM.00355-11
Leigh JA, Mayer F, Wolfe RS (1981) Acetogenium kivui, a new thermophilic hydrogen-oxidizing, acetogenic bacterium. Arch Microbiol 129:275–280. https://doi.org/10.1007/BF00414697
Liew F, Henstra AM, Winzer K, Köpke M, Simpson SD, Minton NP (2016a) Insights into CO2 fixation pathway of Clostridium autoethanogenum by targeted mutagenesis. Mbio 7:e00427-00416. https://doi.org/10.1128/mBio.00427-16
Liew F, Martin ME, Tappel RC, Heijstra BD, Mihalcea C, Köpke M (2016b) Gas fermentation-a lexible platform for commercial scale production of low-carbon-fuels and chemicals from waste and renewable feedstocks. Front Microbiol 7:694. https://doi.org/10.3389/fmicb.2016.00694
Maddipati P, Atiyeh HK, Bellmer DD, Huhnke RL (2011) Ethanol production from syngas by Clostridium strain P11 using corn steep liquor as a nutrient replacement to yeast extract. Bioresour Technol 102:6494–6501. https://doi.org/10.1016/j.biortech.2011.03.047
Matschiavelli N, Oelgeschlager E, Cocchiararo B, Finke J, Rother M (2012) Function and regulation of isoforms of carbon monoxide dehydrogenase/acetyl coenzyme A synthase in Methanosarcina acetivorans. J Bacteriol 194:5377–5387. https://doi.org/10.1128/JB.00881-12
Meyer O, Schlegel HG (1983) Biology of aerobic carbon monoxide-oxidizing bacteria. Annu Rev Microbiol 37:277–310. https://doi.org/10.1146/annurev.mi.37.100183.001425
Müller V (2019) New horizons in acetogenic conversion of one-carbon substrates and biological hydrogen storage. Trends Biotechnol 37:1344–1354. https://doi.org/10.1016/j.tibtech.2019.05.008
Najafpour GD, Younesi H (2006) Ethanol and acetate synthesis from waste gas using batch culture of Clostridium ljungdahlii. Enzym Microb Technol 38:223–228. https://doi.org/10.1016/j.enzmictec.2005.06.008
Parshina SN, Sipma J, Nakashimada Y, Henstra AM, Smidt H, Lysenko AM, Lens PNL, Lettinga G, Stams AJM (2005) Desulfotomaculum carboxydivorans sp. nov., a novel sulfate-reducing bacterium capable of growth at 100% CO. Int J Syst Evol Microbiol 55:2159–2165. https://doi.org/10.1099/ijs.0.63780-0
Ragsdale SW (2000) Nickel containing CO dehydrogenases and hydrogenases. In: Holzenburg A, Scrutton NS (eds) Enzyme-catalyzed electron and radical transfer, vol 35. Springer, Boston, pp 487–518
Ragsdale SW, Kumar M (1996) Nickel-containing carbon monoxide dehydrogenase/acetyl-CoA synthase. Chem Rev 96:2515–2540. https://doi.org/10.1021/cr950058+
Ragsdale SW, Wood HG (1985) Acetate biosynthesis by acetogenic bacteria. Evidence that carbon monoxide dehydrogenase is the condensing enzyme that catalyzes the final steps in the synthesis. J Biol Chem 260:3970–3977
Ragsdale SW, Ljungdahl LG, DerVartanian DV (1983) Isolation of carbon monoxide dehydrogenase from Acetobacterium woodii and comparison of its properties with those of the Clostridium thermoaceticum enzyme. J Bacteriol 155:1224–1237. https://doi.org/10.1128/JB.155.3.1224-1237.1983
Robb FT, Techtmann SM (2018) Life on the fringe: microbial adaptation to growth on carbon monoxide. F1000Research 7:1981. https://doi.org/10.12688/f1000research.16059.1
Rother M, Metcalf WW (2004) Anaerobic growth of Methanosarcina acetivorans C2A on carbon monoxide: an unusual way of life for a methanogenic archaeon. Proc Natl Acad Sci USA 101:16929–16934. https://doi.org/10.1073/pnas.0407486101
Rother M, Oelgeschläger E, Metcalf WM (2007) Genetic and proteomic analyses of CO utilization by Methanosarcina acetivorans. Arch Microbiol 188:463–472. https://doi.org/10.1007/s00203-007-0266-1
Sanger FS, Nickelen F, Coulson AR (1977) DNA-sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467. https://doi.org/10.1073/pnas.0407486101
Savage MD, Wu ZG, Daniel SL, Lundie LL Jr, Drake HL (1987) Carbon monoxide-dependent chemolithotrophic growth of Clostridium thermoautotrophicum. Appl Environ Microbiol 53:1902–1906. https://doi.org/10.1128/AEM.53.8.1902-1906.1987
Schiel-Bengelsdorf B, Dürre P (2012) Pathway engineering and synthetic biology using acetogens. FEBS Lett 586:2191–2198. https://doi.org/10.1016/j.febslet.2012.04.043
Schmidt K, Jensen SL, Schlegel HG (1963) Die carotinoide der Thiorhodaceae. Arch Mikrobiol 46:117–126. https://doi.org/10.1007/BF00408204
Schoelmerich MC, Müller V (2020) Energy-converting hydrogenases: the link between H2 metabolism and energy conservation. Cell Mol Life Sci 77:1461–1481. https://doi.org/10.1007/s00018-019-03329-5
Schönheit P, Wäscher C, Thauer RK (1978) A rapid procedure for the purification of ferredoxin from Clostridia using polyethylenimine. FEBS Lett 89:219–222. https://doi.org/10.1016/0014-5793(78)80221-x
Schuchmann K, Müller V (2014) Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat Rev Microbiol 12:809–821. https://doi.org/10.1038/nrmicro3365
Schwarz FM, Ciurus S, Jain S, Baum C, Wiechmann A, Basen M, Müller V (2020) Revealing formate production from carbon monoxide in wild type and mutants of Rnf- and Ech-containing acetogens, Acetobacterium woodii and Thermoanaerobacter kivui. Microb Biotechnol 13:2044–2056. https://doi.org/10.1111/1751-7915.13663
Seravalli J, Kumar M, Lu WP, Ragsdale SW (1997) Mechanism of carbon monoxide oxidation by the carbon monoxide dehydrogenase/acetyl-CoA synthase from Clostridium thermoaceticum: kinetic characterization of the intermediates. Biochemistry 36:11241–11251. https://doi.org/10.1021/bi970590m
Shaw AJ, Hogsett DA, Lynd LR (2010) Natural competence in Thermoanaerobacter and Thermoanaerobacterium species. Appl Environ Microbiol 76:4713–4719. https://doi.org/10.1128/AEM.00402-10
Singer SW, Hirst MB, Ludden PW (2006) CO-dependent H2 evolution by Rhodospirillum rubrum: role of CODH:CooF complex. Biochim Biophys Acta 1757:1582–1591. https://doi.org/10.1016/j.bbabio.2006.10.003
Sokolova TG, Henstra AM, Sipma J, Parshina SN, Stams AJ, Lebedinsky AV (2009) Diversity and ecophysiological features of thermophilic carboxydotrophic anaerobes. FEMS Microbiol Ecol 68:131–141. https://doi.org/10.1111/j.1574-6941.2009.00663.x
Stegenta-Dabrowska S, Drabczynski G, Sobieraj K, Koziel JA, Bialowiec A (2019) The biotic and abiotic carbon monoxide formation during aerobic co-digestion of dairy cattle manure with green waste and sawdust. Front Bioeng Biotechnol 7:283. https://doi.org/10.3389/fbioe.2019.00283
Thauer RK, Jungermann K, Decker K (1977) Energy conservation in chemotrophic anaerobic bacteria. Bact Rev 41:100–180
Weghoff MC, Müller V (2016) CO metabolism in the thermophilic acetogen Thermoanaerobacter kivui. Appl Environ Microbiol 82:2312–2319. https://doi.org/10.1128/AEM.00122-16
Wilcoxen J, Zhang B, Hille R (2011) Reaction of the molybdenum- and copper-containing carbon monoxide dehydrogenase from Oligotropha carboxydovorans with quinones. Biochemistry 50:1910–1916. https://doi.org/10.1021/bi1017182
Wilkins MR, Atiyeh HK (2011) Microbial production of ethanol from carbon monoxide. Curr Opin Biotechnol 22:326–330. https://doi.org/10.1016/j.copbio.2011.03.005
Acknowledgements
We are obliged to the BMBF (ThermoSynCon, grant no.: 031B0857B) for the continuous support. S.J. was funded by fellowship from the DAAD (German Academic Exchange Service).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
VM, SJ, AK and MB designed the experiments. SJ and AK performed the experiments. SJ, AK, MB and VM wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Communicated by M. Moracci.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jain, S., Katsyv, A., Basen, M. et al. The monofunctional CO dehydrogenase CooS is essential for growth of Thermoanaerobacter kivui on carbon monoxide. Extremophiles 26, 4 (2022). https://doi.org/10.1007/s00792-021-01251-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00792-021-01251-y