New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story

 , , and
, , and 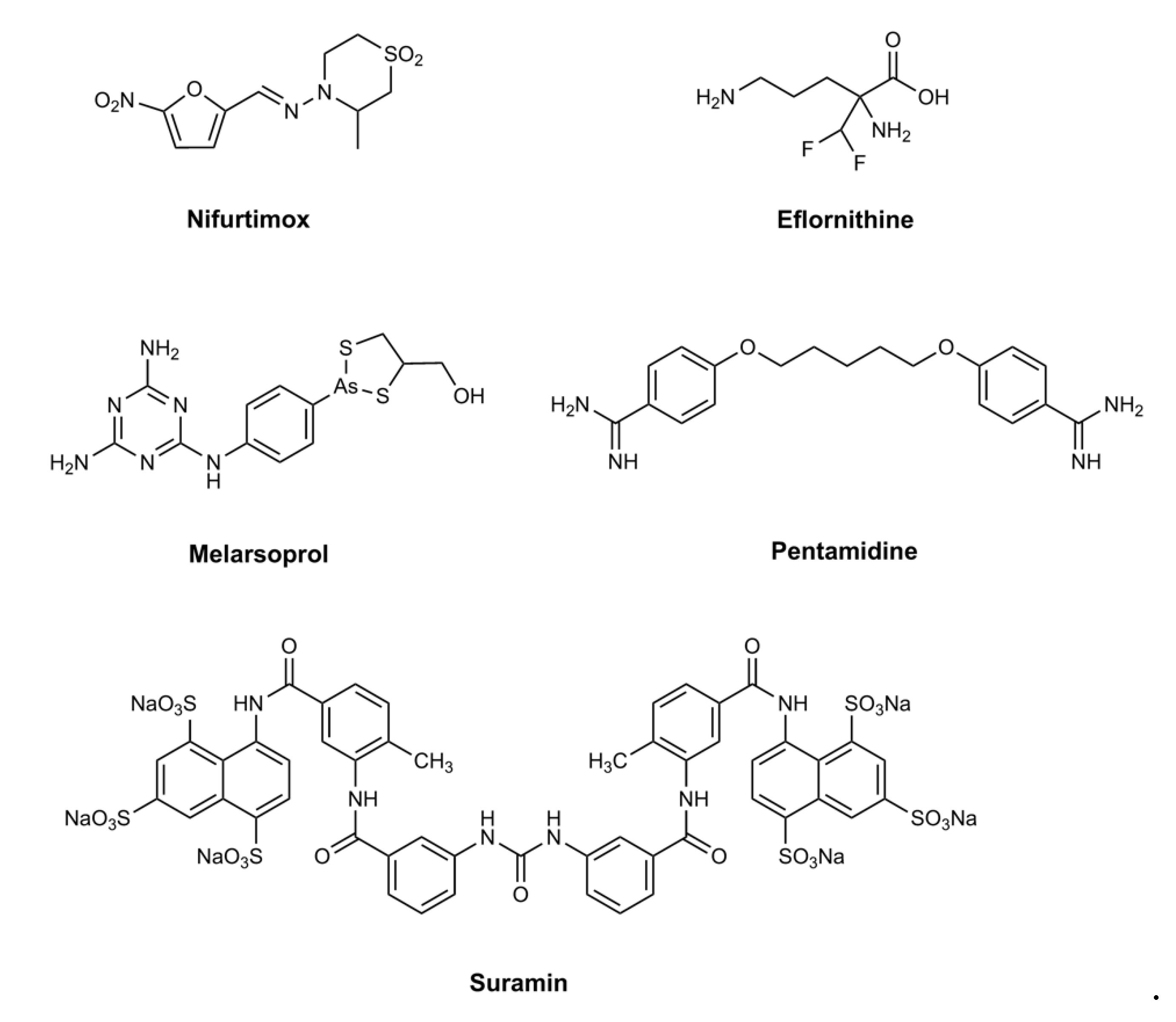{kind=link}
{kind=link}
Abstract
:1. Introduction
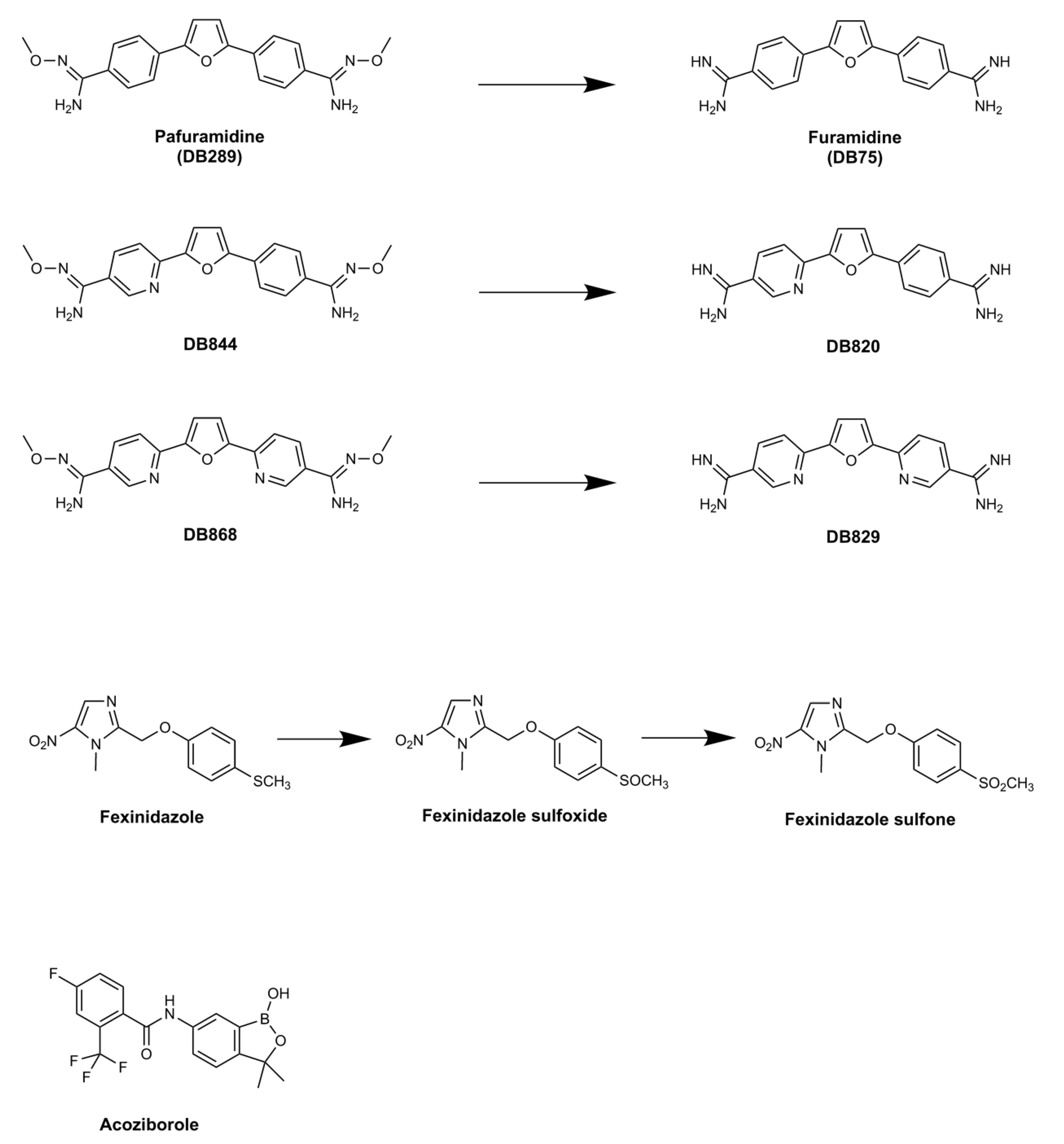
2. Pafuramidine—A New Paradigm in Anti-Trypanosomal Drug Development
2.1. Clinical Trials of Pafuramidine
2.2. Pafuramidine: Mode of Action and Resistance Risk
3. Fexinidazole: the First Oral Treatment for HAT
3.1. Pre-Clinical Development of Fexinidazole
3.2. Fexinidazole: Mode of Action and Resistance Risk
4. Acoziborole: A Single Dose Oral Cure for Stage 2 HAT
Acoziborole: Mode of Action and Resistance Risk
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Available online: https://www.who.int/trypanosomiasis_african/en/ (accessed on 12 January 2020).
- Barrett, M.P.; Boykin, D.W.; Brun, R.; Tidwell, R.R. Human African trypanosomiasis: Pharmacological re-engagement with a neglected disease. Br. J. Pharmacol. 2007, 152, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.; Nkunku, S.; Burri, C. Clinical description of encephalopathic syndromes and risk factors for their occurrence and outcome during melarsoprol treatment of human African trypanosomiasis. Trop. Med. Int. Health 2001, 6, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S.; et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicenter, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Fairlamb, A.H.; Horn, D. Melarsoprol resistance in African trypanosomiasis. Trends Parasitol. 2018, 34, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, I.M.; Creek, D.; Watson, D.G.; Kamleh, M.A.; Woods, D.J.; Wong, P.E.; Burchmore, R.J.; Barrett, M.P. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 2010, 6, e1001204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolova, A.Y.; Wyllie, S.; Patterson, S.; Oza, S.L.; Read, K.D.; Fairlamb, A.H. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54, 2893–2900. [Google Scholar] [CrossRef] [Green Version]
- Brun, R.; Don, R.; Jacobs, R.T.; Wang, M.Z.; Barrett, M.P. Development of novel drugs for human African trypanosomiasis. Future Microbiol. 2011, 6, 677–691. [Google Scholar] [CrossRef]
- Barrett, M.P. The fall and rise of sleeping sickness. Lancet 1999, 353, 1113–1114. [Google Scholar] [CrossRef]
- Kindermans, J.M.; Matthys, F. Introductory note: The access to essential medicines campaign. Trop. Med. Int. Health 2001, 6, 955–956. [Google Scholar] [CrossRef] [Green Version]
- Burri, C.; Baltz, T.; Giroud, C.; Doua, F.; Welker, H.A.; Brun, R. Pharmacokinetic properties of the trypanocidal drug melarsoprol. Chemotherapy 1993, 39, 225–234. [Google Scholar] [CrossRef]
- Burri, C.; Nkunku, S.; Merolle, A.; Smith, T.; Blum, J.; Brun, R. Efficacy of new, concise schedule for melarsoprol in treatment of sleeping sickness caused by Trypanosoma brucei gambiense: A randomised trial. Lancet 2000, 55, 1419–1425. [Google Scholar] [CrossRef]
- Schmid, C.; Nkunku, S.; Merolle, A.; Vounatsou, P.; Burri, C. Efficacy of 10-day melarsoprol schedule 2 years after treatment for late-stage gambiense sleeping sickness. Lancet 2004, 364, 789–790. [Google Scholar] [CrossRef]
- Burri, C.; Brun, R. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 2003, 90 (Suppl. 1), S49–S52. [Google Scholar] [CrossRef] [PubMed]
- Balfour, J.A.; McClellan, K. Topical eflornithine. Am. J. Clin. Dermatol. 2001, 2, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.theguardian.com/world/2001/may/07/medicalscience.businessofresearch (accessed on 12 January 2020).
- McKerrow, J.H. Designing drugs for parasitic diseases of the developing world. PLoS Med. 2005, 2, e210. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.dndi.org/ (accessed on 12 January 2020).
- Available online: https://www.finddx.org/ (accessed on 12 January 2020).
- Tirados, I.; Esterhuizen, J.; Kovacic, V.; Mangwiro, T.N.; Vale, G.A.; Hastings, I.; Solano, P.; Lehane, M.J.; Torr, S.J. Tsetse control and gambian sleeping sickness; implications for control strategy. PLoS Negl. Trop. Dis. 2015, 9, e0003822. [Google Scholar] [CrossRef] [Green Version]
- Ballell, L.; Strange, M.; Cammack, N.; Fairlamb, A.H.; Borysiewicz, L. Open Lab as a source of hits and leads against tuberculosis, malaria and kinetoplastid diseases. Nat. Rev. Drug Discov. 2016, 15, 292. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.P.S.; Barrett, M.P.; Dranoff, G.; Faraday, C.J.; Gimpelewicz, C.R.; Hailu, A.; Jones, C.L.; Kelly, J.M.; Lazdins-Helds, J.K.; Mäser, P.; et al. Drug discovery for kinetoplastid diseases: Future directions. ACS Infect. Dis. 2019, 5, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Pohlig, G.; Bernhard, S.C.; Blum, J.; Burri, C.; Mpanya, A.; Lubaki, J.P.; Mpoto, A.M.; Munungu, B.F.; N’tombe, P.M.; Deo, G.K.; et al. Efficacy and safety of pafuramidine versus pentamidine maleate for treatment of first stage sleeping sickness in a randomized, comparator-controlled, international phase 3 clinical trial. PLoS Negl. Trop. Dis. 2016, 10, e0004363. [Google Scholar] [CrossRef] [Green Version]
- Mesu, V.K.B.K.; Kalonji, W.M.; Bardonneau, C.; Mordt, O.V.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Kuziena, W.; Lubaki, J.F.; et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicenter, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Plattner, J.J.; Nare, B.; Wring, S.A.; Chen, D.; Freund, Y.; Gaukel, E.G.; Orr, M.D.; Perales, J.B.; Jenks, M.; et al. Benzoxaboroles: A new class of potential drugs for human African trypanosomiasis. Future Med. Chem. 2011, 3, 1259–1278. [Google Scholar] [CrossRef] [PubMed]
- Bray, P.G.; Barrett, M.P.; Ward, S.A.; de Koning, H.P. Pentamidine uptake and resistance in pathogenic protozoa: Past, present and future. Trends Parasitol. 2003, 19, 232–239. [Google Scholar] [CrossRef]
- Peregrine, A.S.; Mamman, M. Pharmacology of diminazene: A review. Acta Trop. 1993, 54, 185–203. [Google Scholar] [CrossRef]
- Paine, M.F.; Wang, M.Z.; Generaux, C.N.; Boykin, D.W.; Wilson, W.D.; De Koning, H.P.; Olson, C.A.; Pohlig, G.; Burri, C.; Brun., R. Diamidines for human African trypanosomiasis. Curr. Opin. Investig. Drugs 2010, 11, 876–883. [Google Scholar]
- Das, B.P.; Boykin, D.W. Synthesis and antiprotozoal activity of 2,5-bis(4-guanylphenyl)furans. J. Med. Chem. 1977, 20, 531–536. [Google Scholar] [CrossRef]
- Zhou, L.; Lee, K.; Thakker, D.R.; Boykin, D.W.; Tidwell, R.R.; Hall, J.E. Enhanced permeability of the antimicrobial agent 2,5-bis(4-amidinophenyl)furan across Caco-2 cell monolayers via its methylamidoidme prodrug. Pharm. Res. 2002, 19, 1689–1695. [Google Scholar] [CrossRef]
- Midgley, I.; Fitzpatrick, K.; Taylor, L.M.; Houchen, T.L.; Henderson, S.J.; Wright, S.J.; Cybulski, Z.R.; John, B.A.; McBurney, A.; Boykin, D.W. Pharmacokinetics and metabolism of the prodrug DB289 (2,5-bis[4-(N-methoxyamidino)phenyl]furan monomaleate) in rat and monkey and its conversion to the antiprotozoal/antifungal drug DB75 (2,5-bis(4-guanylphenyl)furan dihydrochloride). Drug. Metab. Dispos. 2007, 35, 955–967. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Z.; Saulter, J.Y.; Usuki, E.; Cheung, Y.L.; Hall, M.; Bridges, A.S.; Loewen, G.; Parkinson, O.T.; Stephens, C.E.; Allen, J.L.; et al. CYP4F enzymes are the major enzymes in human liver microsomes that catalyze the O-demethylation of the antiparasitic prodrug DB289 [2,5-bis(4-amidinophenyl)furan-bis-O-methylamidoxime]. Drug Metab. Dispos. 2006, 34, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Z.; Wu, J.Q.; Bridges, A.S.; Zeldin, D.C.; Kornbluth, S.; Tidwell, R.R.; Hall, J.E.; Paine, M.F. Human enteric microsomal CYP4F enzymes O-demethylate the antiparasitic prodrug pafuramidine. Drug Metab. Dispos. 2007, 35, 2067–2075. [Google Scholar] [CrossRef] [Green Version]
- Ansede, J.H.; Voyksner, R.D.; Ismail, M.A.; Boykin, D.W.; Tidwell, R.R.; Hall, J.E. In vitro metabolism of an orally active O-methyl amidoxime prodrug for the treatment of CNS trypanosomiasis. Xenobiotica 2005, 35, 211–226. [Google Scholar] [CrossRef]
- Saulter, J.Y.; Kurian, J.R.; Trepanier, L.A.; Tidwell, R.R.; Bridges, A.S.; Boykin, D.W.; Stephens, C.E.; Anbazhagan, M.; Hall, J.E. Unusual dehydroxylation of antimicrobial amidoxime prodrugs by cytochrome b5 and NADH cytochrome b5 reductase. Drug Metab. Dispos. 2005, 33, 1886–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansede, J.H.; Anbazhagan, M.; Brun, R.; Easterbrook, J.D.; Hall, J.E.; Boykin, D.W. O-alkoxyamidine prodrugs of furamidine: In vitro transport and microsomal metabolism as indicators of in vivo efficacy in a mouse model of Trypanosoma brucei rhodesiense infection. J. Med. Chem. 2004, 47, 4335–4338. [Google Scholar] [CrossRef] [PubMed]
- Mdachi, R.E.; Thuita, J.K.; Kagira, J.M.; Ngotho, J.M.; Murilla, G.A.; Ndung’u, J.M.; Tidwell, R.R.; Hall, J.E.; Brun, R. Efficacy of the novel diamidine compound 2,5-Bis(4-amidinophenyl)- furan-bis-O-Methlylamidoxime (Pafuramidine, DB289) against Trypanosoma brucei rhodesiense infection in vervet monkeys after oral administration. Antimicrob. Agents Chemother. 2009, 53, 953–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuita, J.K.; Karanja, S.M.; Wenzler, T.; Mdachi, R.E.; Ngotho, J.M.; Kagira, J.M.; Tidwell, R.; Brun, R. Efficacy of the diamidine DB75 and its prodrug DB289, against murine models of human African trypanosomiasis. Acta Trop. 2008, 108, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Burri, C.; Yeramian, P.D.; Allen, J.L.; Merolle, A.; Serge, K.K.; Mpanya, A.; Lutumba, P.; Mesu, V.K.; Bilenge, C.M.; Lubaki, J.P.; et al. Efficacy, Safety, and Dose of Pafuramidine, a New Oral Drug for Treatment of First Stage Sleeping Sickness, in a Phase 2a Clinical Study and Phase 2b Randomized Clinical Studies. PLoS Negl. Trop. Dis. 2016, 10, e0004362. [Google Scholar] [CrossRef] [Green Version]
- Harrill, A.H.; Desmet, K.D.; Wolf, K.K.; Bridges, A.S.; Eaddy, J.S.; Kurtz, C.L.; Hall, J.E.; Paine, M.F.; Tidwell, R.R.; Watkins, P.B. A mouse diversity panel approach reveals the potential for clinical kidney injury due to DB289 not predicted by classical rodent models. Toxicol. Sci. 2012, 130, 416–426. [Google Scholar] [CrossRef]
- Wenzler, T.; Boykin, D.W.; Ismail, M.A.; Hall, J.E.; Tidwell, R.R.; Brun, R. New treatment option for second-stage African sleeping sickness: In vitro and in vivo efficacy of aza analogs of DB289. Antimicrob. Agents Chemother. 2009, 53, 4185–4192. [Google Scholar] [CrossRef] [Green Version]
- Thuita, J.K.; Wang, M.Z.; Kagira, J.M.; Denton, C.L.; Paine, M.F.; Mdachi, R.E.; Murilla, G.A.; Ching, S.; Boykin, D.W.; Tidwell, R.R.; et al. Pharmacology of DB844, an orally active aza analog of pafuramidine, in a monkey model of second stage human African trypanosomiasis. PLoS Negl. Trop. Dis. 2012, 6, e1734. [Google Scholar] [CrossRef] [Green Version]
- Goldsmith, R.B.; Tidwell, R.R. Organ specific accumulation and distribution of structurally related anti-trypanosomal compounds: A possible role in renal toxicity. Am. J. Trop. Med. Hyg. 2009, 81, 1–50. [Google Scholar] [CrossRef]
- Depauw, S.; Lambert, M.; Jambon, S.; Paul, A.; Peixoto, P.; Nhili, R.; Marongiu, L.; Figeac, M.; Dassi, C.; Paul-Constant, C. Heterocyclic diamidine DNA ligands as HOXA9 transcription factor inhibitors: Design, molecular evaluation, and cellular consequences in a HOXA9-dependant leukemia cell model. J. Med. Chem. 2019, 62, 1306–1329. [Google Scholar] [CrossRef]
- Soeiro, M.N.; Werbovetz, K.; Boykin, D.W.; Wilson, W.D.; Wang, M.Z.; Hemphill, A. Novel amidines and analogs as promising agents against intracellular parasites: A systematic review. Parasitology 2013, 140, 929–951. [Google Scholar] [CrossRef] [Green Version]
- Mathis, A.M.; Holman, J.L.; Sturk, L.M.; Ismail, M.A.; Boykin, D.W.; Tidwell, R.R.; Hall, J.E. Accumulation and intracellular distribution of antitrypanosomal diamidine compounds DB75 and DB820 in African trypanosomes. Antimicrob. Agents Chemother. 2006, 50, 2185–2191. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.P.; Wong, P.E.; Burchmore, R.J.; de Koning, H.P.; Barrett, M.P. Trypanocidal furamidine analogs: Influence of pyridine nitrogens on trypanocidal activity, transport kinetics, and resistance patterns. Antimicrob. Agents Chemother. 2011, 55, 2352–2361. [Google Scholar] [CrossRef] [Green Version]
- Lanteri, C.A.; Tidwell, R.R.; Meshnick, S.R. The mitochondrion is a site of trypanocidal action of the aromatic diamidine DB75 in bloodstream forms of Trypanosoma brucei. Antimicrob. Agents Chemother. 2008, 52, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Lanteri, C.A.; Trumpower, B.L.; Tidwell, R.R.; Meshnick, S.R. DB75, a novel trypanocidal agent, disrupts mitochondrial function in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 2004, 48, 3968–3974. [Google Scholar] [CrossRef] [Green Version]
- Dean, S.; Gould, M.K.; Dewar, C.E.; Schnaufer, A.C. Single point mutations in ATP synthase compensate for mitochondrial genome loss in trypanosomes. Proc. Natl. Acad. Sci. USA 2013, 110, 14741–14746. [Google Scholar] [CrossRef] [Green Version]
- Gould, M.K.; Schnaufer, A. Independence from Kinetoplast DNA maintenance and expression is associated with multidrug resistance in Trypanosoma brucei in vitro. Antimicrob. Agents Chemother. 2014, 58, 2925–2928. [Google Scholar] [CrossRef] [Green Version]
- Ludewig, G.; Williams, J.M.; Li, Y.; Staben, C. Effects of pentamidine isethionate on Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 1994, 38, 1123–1128. [Google Scholar] [CrossRef] [Green Version]
- Carter, N.S.; Berger, B.J.; Fairlamb, A.H. Uptake of diamidine drugs by the P2 nucleoside transporter in melarsen-sensitive and -resistant Trypanosoma brucei brucei. J. Biol. Chem. 1995, 270, 28153–28157. [Google Scholar]
- De Koning, H.P. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by three distinct transporters: Implications for cross-resistance with arsenicals. Mol. Pharmacol. 2001, 59, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 2012, 482, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.; Glover, L.; Munday, J.C.; Aguinaga Andrés, D.; Barrett, M.P.; de Koning, H.P.; Horn, D. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc. Natl. Acad. Sci. USA 2012, 109, 10996–11001. [Google Scholar] [CrossRef] [Green Version]
- Lanteri, C.A.; Stewart, M.L.; Brock, J.M.; Alibu, V.P.; Meshnick, S.R.; Tidwell, R.R.; Barrett, M.P. Roles for the Trypanosoma brucei P2 transporter in DB75 uptake and resistance. Mol. Pharmacol. 2006, 70, 1585–1592. [Google Scholar] [CrossRef]
- Barrett, M.P.; Fairlamb, A.H. The biochemical basis of arsenical-diamidine crossresistance in African trypanosomes. Parasitol. Today 1999, 15, 136–140. [Google Scholar] [CrossRef]
- Barrett, M.P.; Gilbert, I.H. Targeting of toxic compounds to the trypanosome’s interior. Adv. Parasitol. 2006, 63, 125–183. [Google Scholar]
- Stewart, M.L.; Bueno, G.J.; Baliani, A.; Klenke, B.; Brun, R.; Brock, J.M.; Gilbert, I.H.; Barrett, M.P. Trypanocidal activity of melamine-based nitroheterocycles. Antimicrob. Agents Chemother. 2004, 48, 1733–1738. [Google Scholar] [CrossRef] [Green Version]
- Tye, C.K.; Kasinathan, G.; Barrett, M.P.; Brun, R.; Doyle, V.E.; Fairlamb, A.H.; Weaver, R.; Gilbert, I.H. An approach to use an unusual adenosine transporter to selectively deliver polyamine analogs to trypanosomes. Bioorg. Med. Chem. Lett. 1998, 8, 811–816. [Google Scholar] [CrossRef]
- Sanderson, L.; Dogruel, M.; Rodgers, J.; De Koning, H.P.; Thomas, S.A. Pentamidine movement across the murine blood-brain and blood-cerebrospinal fluid barriers: Effect of trypanosome infection, combination therapy, P-glycoprotein, and multidrug resistance-associated protein. J. Pharmacol. Exp. Ther. 2009, 329, 967–977. [Google Scholar] [CrossRef]
- Ming, X.; Ju, W.; Wu, H.; Tidwell, R.R.; Hall, J.E.; Thakker, D.R. Transport of dicationic drugs pentamidine and furamidine by human organic cation transporters. Drug Metab. Dispos. 2009, 37, 424–430. [Google Scholar] [CrossRef]
- Sekhar, G.N.; Georgian, A.R.; Sanderson, L.; Vizcay-Barrena, G.; Brown, R.C.; Muresan, P.; Fleck, R.A.; Thomas, S.A. Organic cation transporter 1 (OCT1) is involved in pentamidine transport at the human and mouse blood-brain barrier (BBB). PLoS ONE 2017, 12, e0173474. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wenzler, T.; Miller, P.N.; Wu, H.; Boykin, D.W.; Brun, R.; Wang, M.Z. Pharmacokinetic comparison to determine the mechanisms underlying the differential efficacies of cationic diamidines against first- and second-stage human African trypanosomiasis. Antimicrob. Agents Chemother. 2014, 58, 4064–4074. [Google Scholar] [CrossRef] [Green Version]
- Lindner, A.K.; Lejon, V.; Chappuis, F.; Seixas, J.; Kazumba, L.; Barrett, M.P.; Mwamba, E.; Erphas, O.; Akl, E.A.; Villanueva, G.; et al. New WHO guidelines for treatment of gambiense human African trypanosomiasis including fexinidazole: Substantial changes for clinical practice. Lancet Infect. Dis. 2020, 20, e38–e46. [Google Scholar] [CrossRef]
- Neau, P.; Hänel, H.; Lameyre, V.; Strub-Wourgaft, N.; Kuykens, L. Innovative partnerships for the elimination of Human African Trypanosomiasis and the development of fexinidazole. Trop. Med. Infect. Dis. 2020, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Raether, W.; Hanel, H. Nitroheterocyclic drugs with broad spectrum activity. Parasitol. Res. 2003, 90 (Suppl. 1), S19–S39. [Google Scholar] [CrossRef]
- Jennings, F.W.; Urquhart, G.M. The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice. Z. Parasitenkd. 1983, 69, 577–581. [Google Scholar] [CrossRef]
- Kaiser, M.; Bray, M.A.; Cal, M.; Bourdin Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef] [Green Version]
- Barrett, M.P.; Fairlamb, A.H.; Rousseau, B.; Chauvière, G.; Perié, J. Uptake of the nitroimidazole drug megazol by African trypanosomes. Biochem. Pharmacol. 2000, 59, 615–620. [Google Scholar] [CrossRef]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef]
- Torreele, E.; Bourdin Trunz, B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazué, G.; Bray, M.A.; Pécoul, B. Fexinidazole—A new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e923. [Google Scholar] [CrossRef] [Green Version]
- Tweats, D.; Bourdin Trunz, B.; Torreele, E. Genotoxicity profile of fexinidazole—A drug candidate in clinical development for human African trypanomiasis (sleeping sickness). Mutagenesis 2012, 27, 523–532. [Google Scholar] [CrossRef]
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyllie, S.; Foth, B.J.; Kelner, A.; Sokolova, A.Y.; Berriman, M.; Fairlamb, A.H. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 2016, 71, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.S.; Bot, C.; Wilkinson, S.R. Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J. Biol. Chem. 2011, 286, 13088–13095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahia, M.T.; de Andrade, I.M.; Martins, T.A.; do Nascimento, Á.F.; Diniz Lde, F.; Caldas, I.S.; Talvani, A.; Trunz, B.B.; Torreele, E.; Ribeiro, I. Fexinidazole: A potential new drug candidate for Chagas disease. PLoS Negl. Trop. Dis. 2012, 6, e1870. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.S.; Wilkinson, S.R. Activation of benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob. Agents Chemother. 2012, 56, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Trochine, A.; Creek, D.J.; Faral-Tello, P.; Barrett, M.P.; Robello, C. Benznidazole biotransformation and multiple targets in Trypanosoma cruzi revealed by metabolomics. PLoS Negl. Trop. Dis. 2014, 8, e2844. [Google Scholar] [CrossRef]
- Hall, B.S.; Meredith, E.L.; Wilkinson, S.R. Targeting the substrate preference of a type I nitroreductase to develop antitrypanosomal quinone-based prodrugs. Antimicrob. Agents Chemother. 2012, 56, 5821–5830. [Google Scholar] [CrossRef] [Green Version]
- Menzies, S.K.; Tulloch, L.B.; Florence, G.J.; Smith, T.K. The trypanosome alternative oxidase: A potential drug target? Parasitology 2018, 145, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.A.; Baker, N.; Hutchinson, S.; Dominicus, C.; Trenaman, A.; Glover, L.; Alsford, S.; Horn, D. Insights into antitrypanosomal drug mode-of-action from cytology-based profiling. PLoS Negl. Trop. Dis. 2018, 12, e0006980. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T.; Winum, J.Y. Benzoxaborole compounds for therapeutic uses: A patent review (2010–2018). Expert. Opin. Ther. Pat. 2018, 28, 493–504. [Google Scholar] [CrossRef]
- Ding, D.; Zhao, Y.; Meng, Q.; Xie, D.; Nare, B.; Chen, D.; Bacchi, C.J.; Yarlett, N.; Zhang, Y.K.; Hernandez, V.; et al. Discovery of novel benzoxaborole-based potent antitrypanosomal agents. ACS Med. Chem. Lett. 2010, 1, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Nare, B.; Wring, S.; Bacchi, C.; Beaudet, B.; Bowling, T.; Brun, R.; Chen, D.; Ding, C.; Freund, Y.; Gaukel, E.; et al. Discovery of novel orally bioavailable oxaborole 6-carboxamides that demonstrate cure in a murine model of late-stage central nervous system African trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54, 4379–4388. [Google Scholar] [CrossRef] [Green Version]
- Bacchi, C.J.; Nathan, H.C.; Hutner, S.H.; McCann, P.P.; Sjoerdsma, A. Polyamine metabolism: A potential therapeutic target in trypanosomes. Science 1980, 210, 332–334. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [Green Version]
- Wring, S.; Gaukel, E.; Nare, B.; Jacobs, R.; Beaudet, B.; Bowling, T.; Mercer, L.; Bacchi, C.; Yarlett, N.; Randolph, R. Pharmacokinetics and pharmacodynamics utilizing unbound target tissue exposure as part of a disposition-based rationale for lead optimization of benzoxaboroles in the treatment of Stage 2 Human African Trypanosomiasis. Parasitology 2014, 141, 104–118. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.dndi.org/2012/media-center/press-releases/oxa-phasei/ (accessed on 12 January 2020).
- Available online: https://www.dndi.org/diseases-projects/portfolio/acoziborole/ (accessed on 12 January 2020).
- Jones, D.C.; Foth, B.J.; Urbaniak, M.D.; Patterson, S.; Ong, H.B.; Berriman, M.; Fairlamb, A.H. Genomic and Proteomic Studies on the Mode of Action of Oxaboroles against the African Trypanosome. PLoS Negl. Trop. Dis. 2015, 9, e0004299. [Google Scholar] [CrossRef] [Green Version]
- Sonoiki, E.; Ng, C.L.; Lee, M.C.; Guo, D.; Zhang, Y.K.; Zhou, Y.; Alley, M.R.; Ahyong, V.; Sanz, L.M.; Lafuente-Monasterio, M.J.; et al. A potent antimalarial benzoxaborole targets a Plasmodium falciparum cleavage and polyadenylation specificity factor homologue. Nat. Commun. 2017, 8, 14574. [Google Scholar] [CrossRef]
- Palencia, A.; Bougdour, A.; Brenier-Pinchart, M.P.; Touquet, B.; Bertini, R.L.; Sensi, C.; Gay, G.; Vollaire, J.; Josserand, V.; Easom, E.; et al. Targeting Toxoplasma gondii CPSF3 as a new approach to control toxoplasmosis. EMBO Mol. Med. 2017, 9, 385–394. [Google Scholar] [CrossRef]
- Steketee, P.C.; Vincent, I.M.; Achcar, F.; Giordani, F.; Kim, D.H.; Creek, D.J.; Freund, Y.; Jacobs, R.; Rattigan, K.; Horn, D.; et al. Benzoxaborole treatment perturbs S-adenosyl-L-methionine metabolism in Trypanosoma brucei. PLoS Negl. Trop. Dis. 2018, 12, e0006450. [Google Scholar] [CrossRef]
- Begolo, D.; Vincent, I.M.; Giordani, F.; Pöhner, I.; Witty, M.J.; Rowan, T.G.; Bengaly, Z.; Gillingwater, K.; Freund, Y.; Wade, R.C.; et al. The trypanocidal benzoxaborole AN7973 inhibits trypanosome mRNA processing. PLoS Pathog. 2018, 14, e1007315. [Google Scholar] [CrossRef] [Green Version]
- Wall, R.J.; Rico, E.; Lukac, I.; Zuccotto, F.; Elg, S.; Gilbert, I.H.; Freund, Y.; Alley, M.R.K.; Field, M.C.; Wyllie, S.; et al. Clinical and veterinary trypanocidal benzoxaboroles target CPSF3. Proc. Natl. Acad. Sci. USA 2018, 15, 9616–9621. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zoltner, M.; Leung, K.F.; Scullion, P.; Hutchinson, S.; Del Pino, R.C.; Vincent, I.M.; Zhang, Y.K.; Freund, Y.R.; Alley, M.R.K.; et al. Host-parasite co-metabolic activation of antitrypanosomal aminomethyl-benzoxaboroles. PLoS Pathog. 2018, 14, e1006850. [Google Scholar] [CrossRef] [Green Version]
- Akama, T.; Zhang, Y.K.; Freund, Y.R.; Berry, P.; Lee, J.; Easom, E.E.; Jacobs, R.T.; Plattner, J.J.; Witty, M.J.; Peter, R.; et al. Identification of a 4-fluorobenzyl l-valinate amide benzoxaborole (AN11736) as a potential development candidate for the treatment of Animal African Trypanosomiasis (AAT). Bioorg. Med. Chem. Lett. 2018, 28, 6–10. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dickie, E.A.; Giordani, F.; Gould, M.K.; Mäser, P.; Burri, C.; Mottram, J.C.; Rao, S.P.S.; Barrett, M.P. New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story. Trop. Med. Infect. Dis. 2020, 5, 29. https://doi.org/10.3390/tropicalmed5010029
Dickie EA, Giordani F, Gould MK, Mäser P, Burri C, Mottram JC, Rao SPS, Barrett MP. New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story. Tropical Medicine and Infectious Disease. 2020; 5(1):29. https://doi.org/10.3390/tropicalmed5010029
Chicago/Turabian StyleDickie, Emily A., Federica Giordani, Matthew K. Gould, Pascal Mäser, Christian Burri, Jeremy C. Mottram, Srinivasa P. S. Rao, and Michael P. Barrett. 2020. "New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story" Tropical Medicine and Infectious Disease 5, no. 1: 29. https://doi.org/10.3390/tropicalmed5010029