
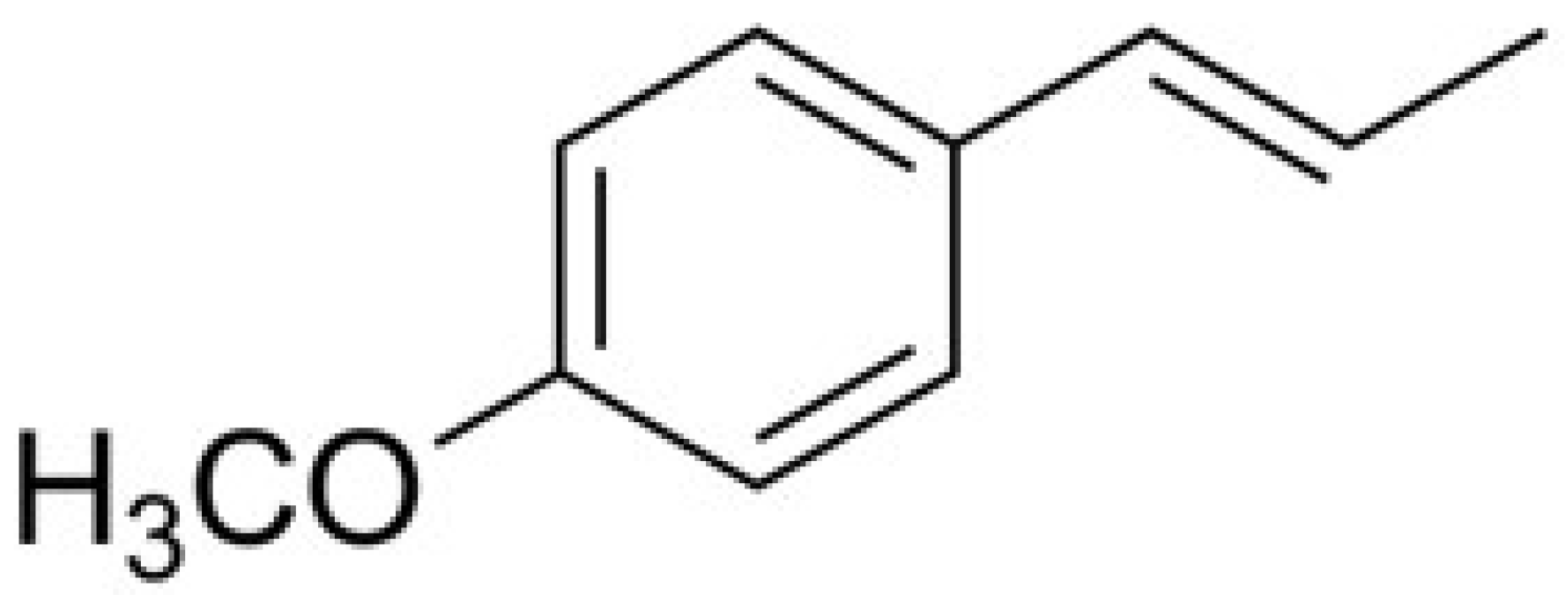
State-Dependent Blockade of Dorsal Root Ganglion Voltage-Gated Na+ Channels by Anethole
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
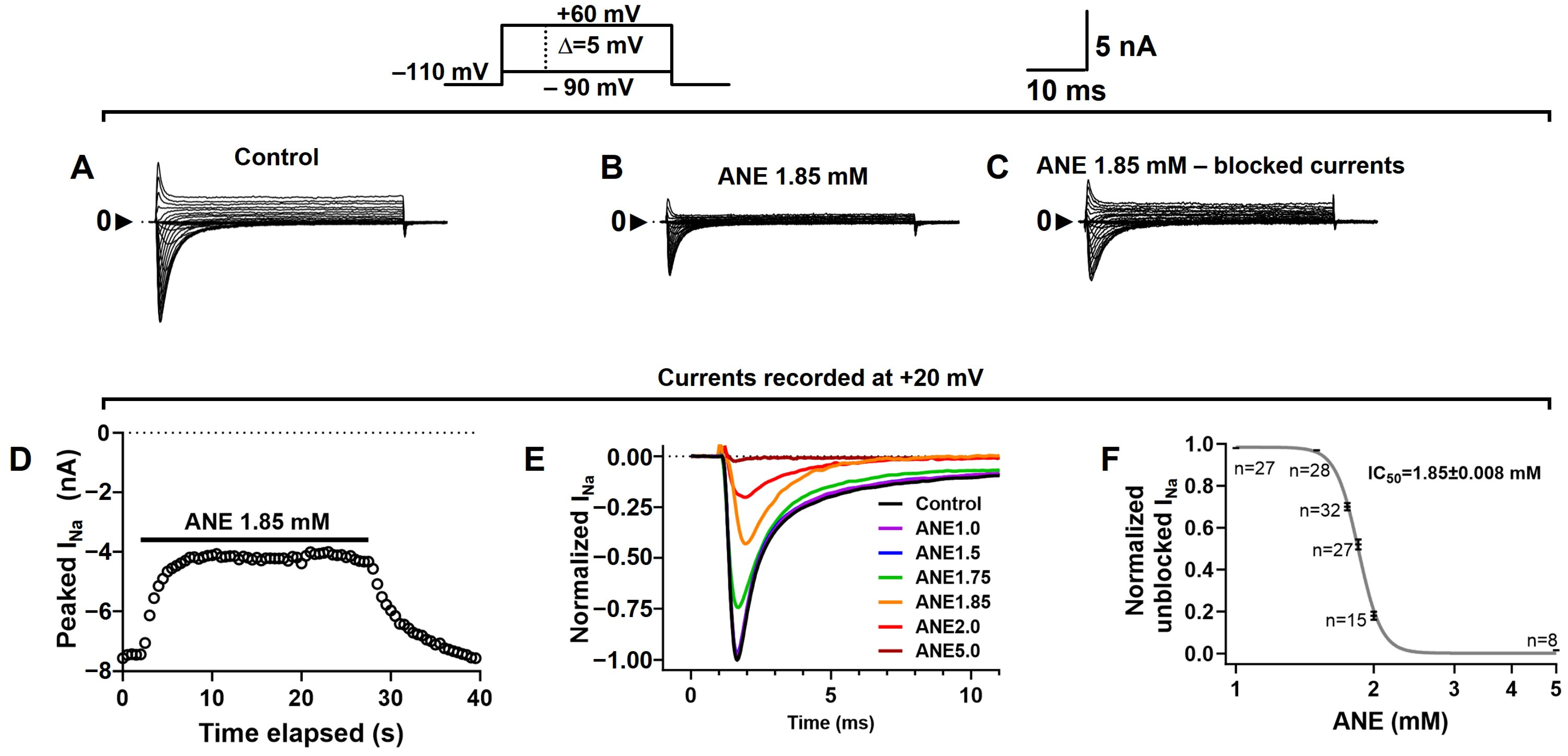
2.1. Anethole Reversibly Inhibited Total Sodium Currents (INa+) in DRG Neurons
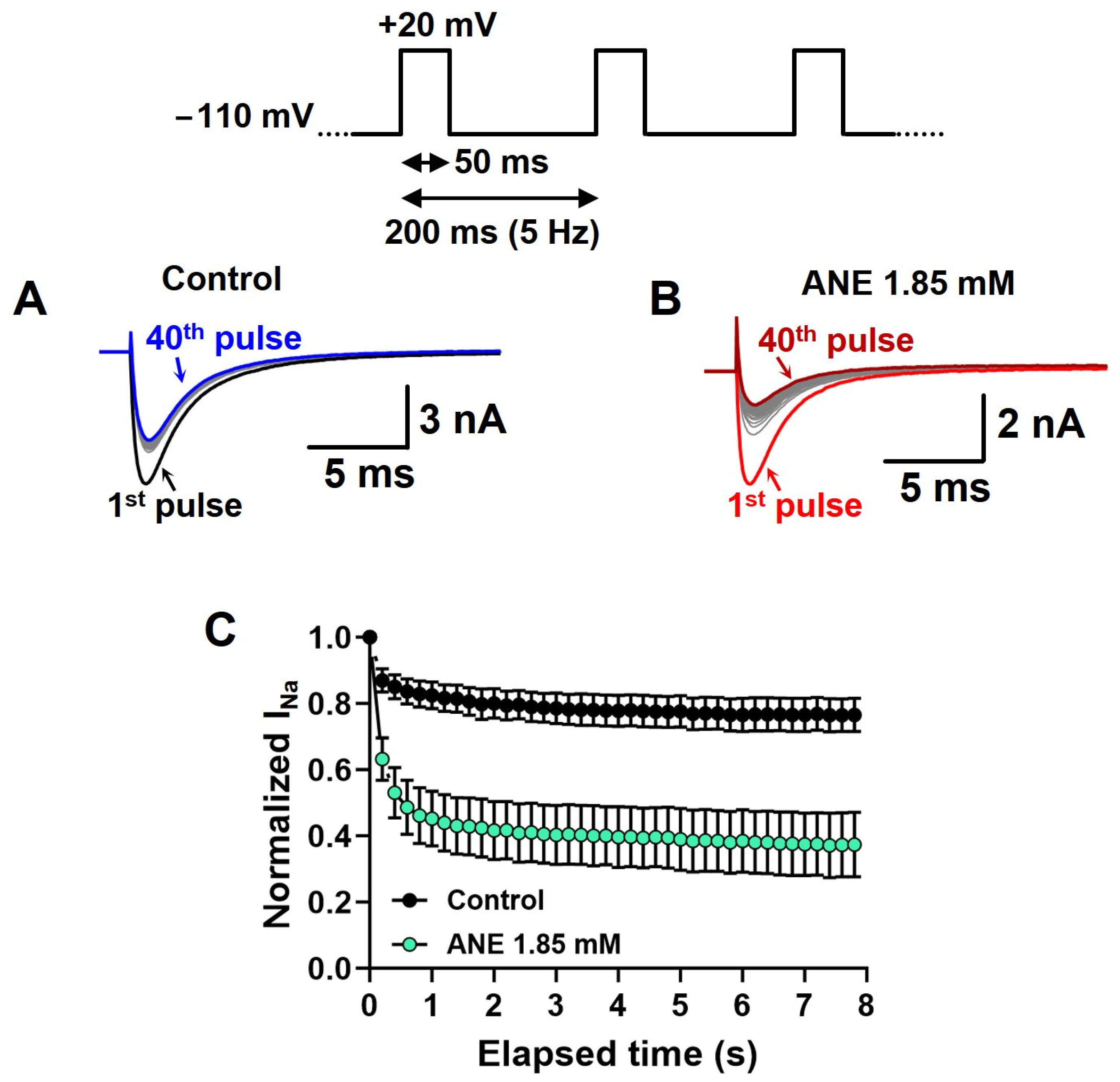
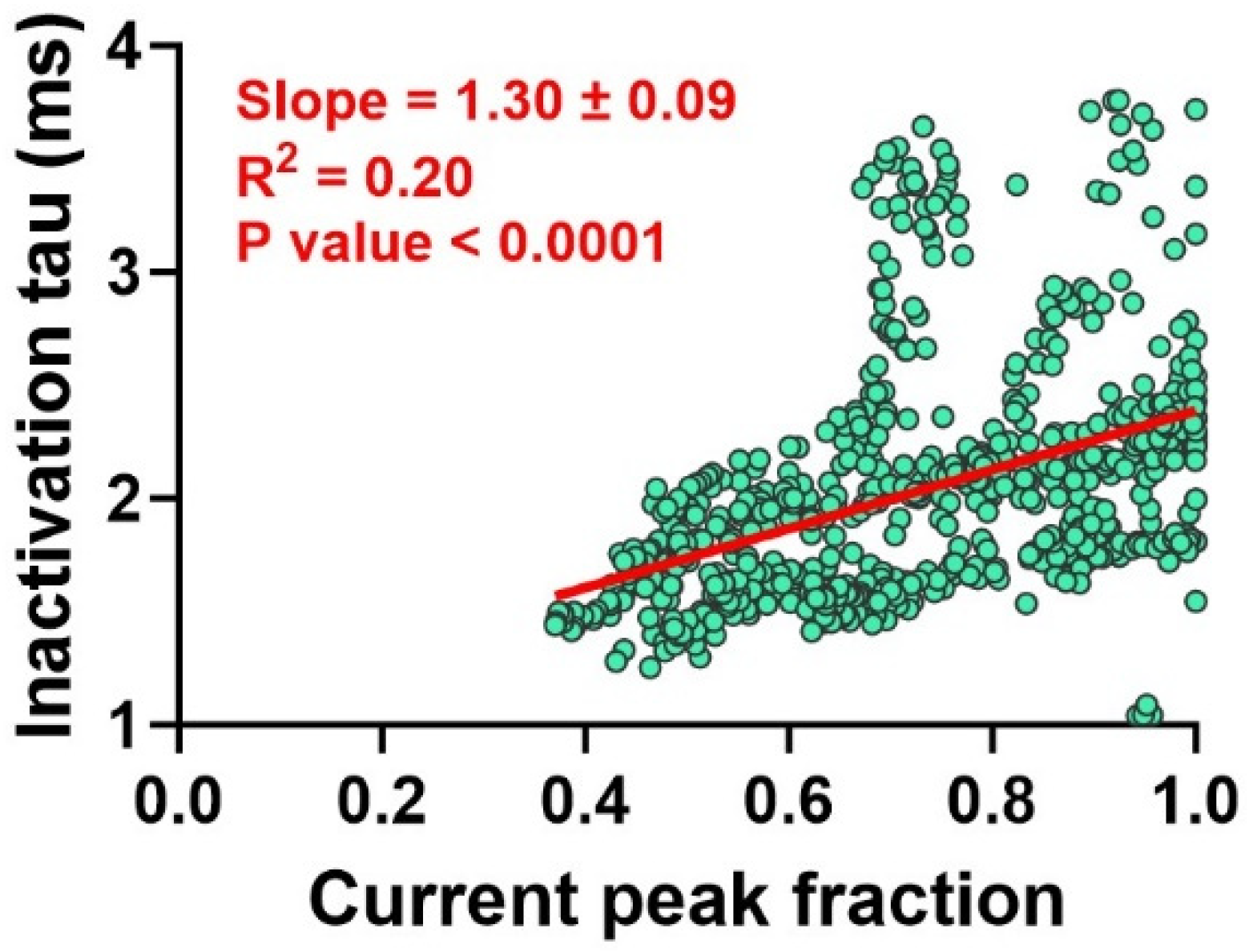
2.2. Anethole Has a Frequency-Dependent Inhibition Effect on INa+
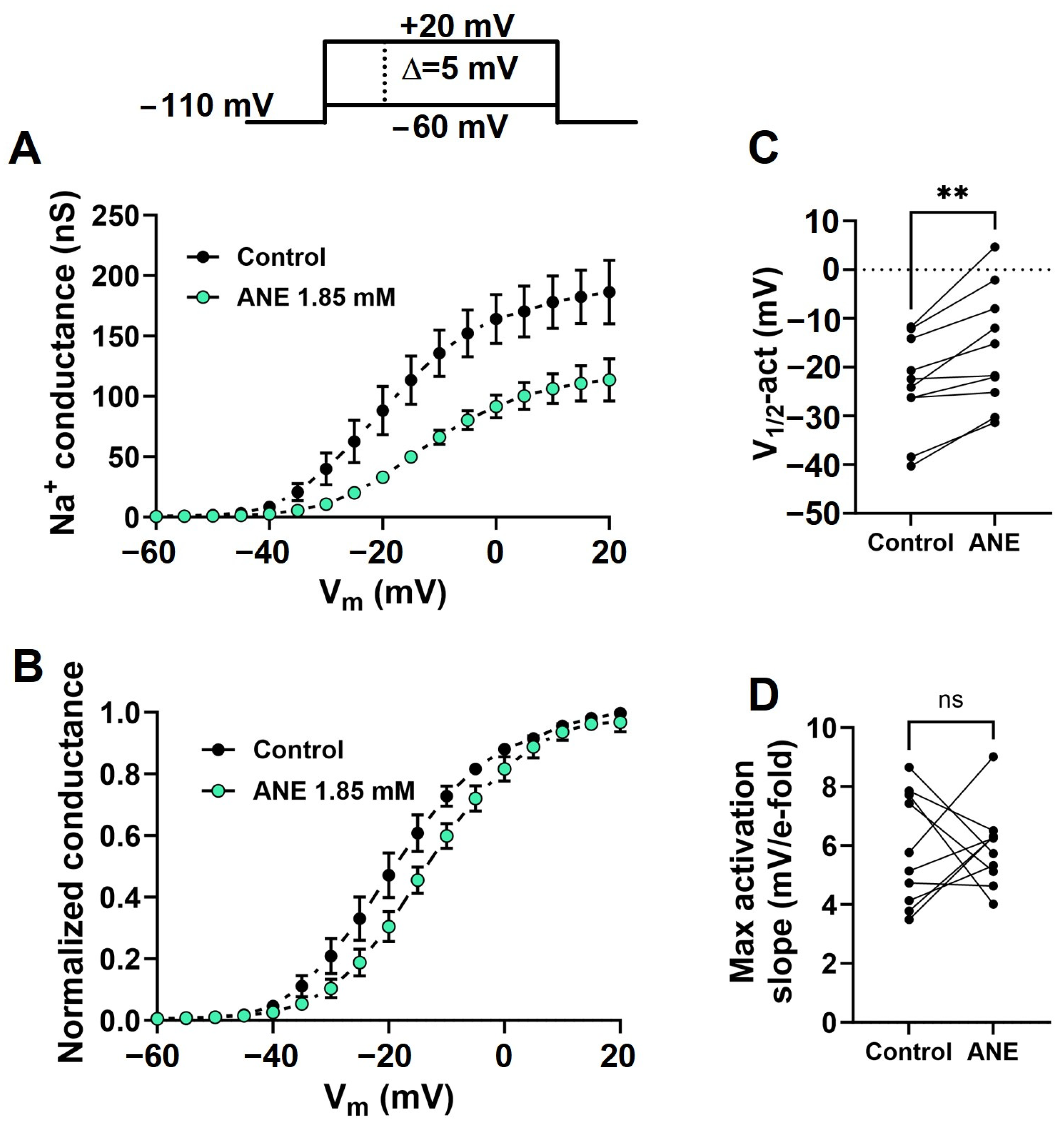
2.3. Anethole Shifts the Voltage Dependence of Na+ Conductance Activation
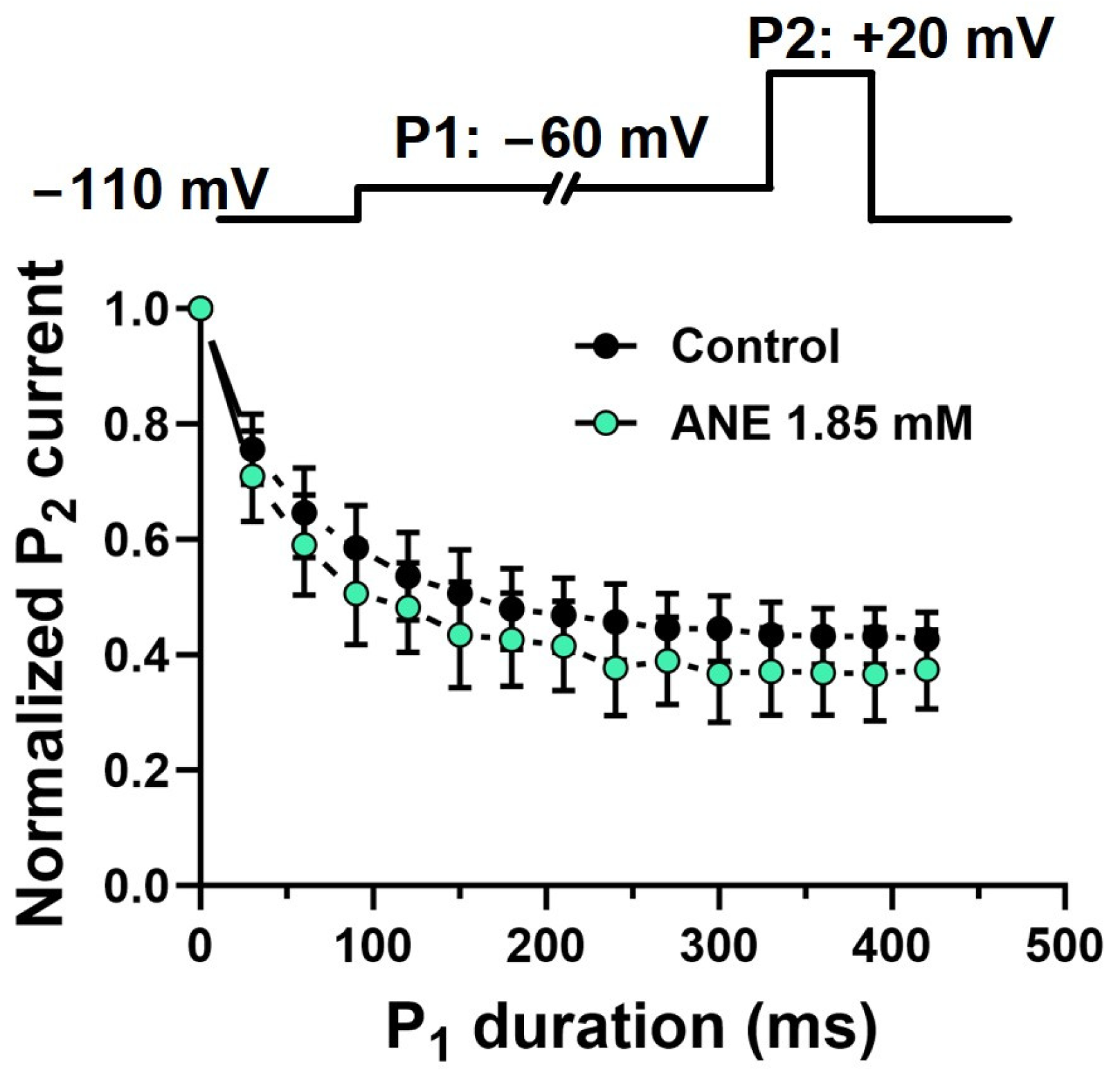
2.4. Anethole Does Not Interact Specifically with Voltage-Gated Na+ Channels in Pre-Open States
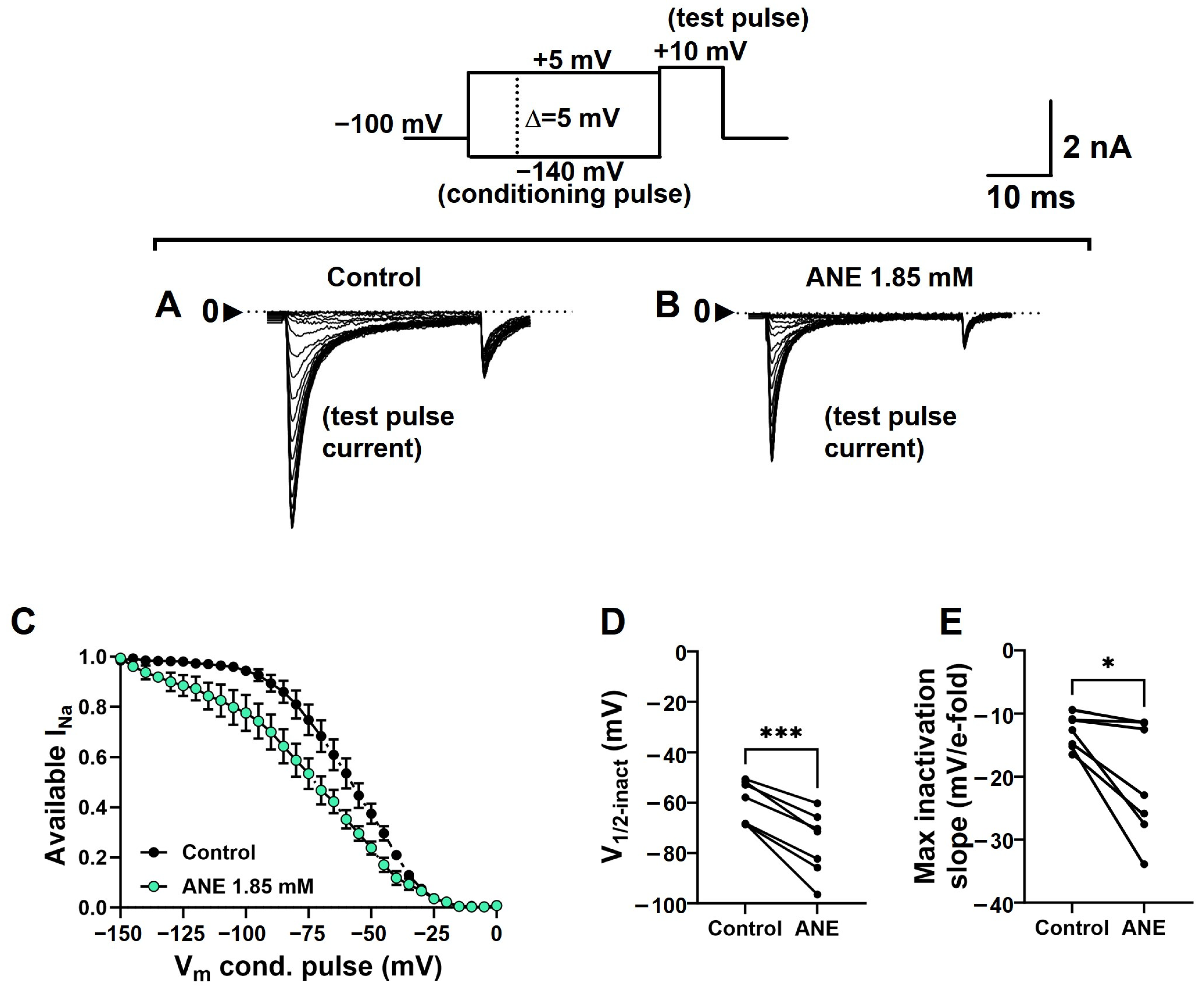
2.5. Anethole Shifts the Steady-State Voltage Dependence of Na+ Conductance Inactivation
3. Discussion
4. Materials and Methods
4.1. Cell Preparation
4.2. Anethole Solutions
4.3. Electrophysiology
4.4. Data Analysis and Graphs
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Acimovic, M.; Tešević, V.; Marina, T.; Jovana, D.; Snezana, O. Compositional characteristics of the essential oil of Pimpinella anisum and Foeniculum vulgare grown in Serbia. Bot. Serbica 2015, 39, 9–14. [Google Scholar]
- Ritter, A.M.V.; Domiciano, T.P.; Verri, W.A.; Zarpelon, A.C.; da Silva, L.G.; Barbosa, C.P.; Natali, M.R.M.; Cuman, R.K.N.; Bersani-Amado, C.A. Antihypernociceptive activity of anethole in experimental inflammatory pain. Inflammopharmacology 2013, 21, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Jongedijk, E.; Bouwmeester, H.; Van Der Krol, A. Monoterpene biosynthesis potential of plant subcellular compartments. New Phytol. 2016, 209, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Aprotosoaie, A.C.; Costache, I.-I.; Miron, A. Anethole and Its Role in Chronic Diseases. In Drug Discovery from Mother Nature; Gupta, S.C., Prasad, S., Aggarwal, B.B., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; pp. 247–267. ISBN 978-3-319-41342-6. [Google Scholar]
- Ponte, E.L.; Sousa, P.L.; Rocha, M.V.A.P.; Soares, P.M.G.; Coelho-de-Souza, A.N.; Leal-Cardoso, J.H.; Assreuy, A.M.S. Comparative study of the anti-edematogenic effects of anethole and estragole. Pharmacol. Rep. PR 2012, 64, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Lima, C.C.; de Holanda-Angelin-Alves, C.M.; Pereira-Gonçalves, Á.; Kennedy-Feitosa, E.; Evangelista-Costa, E.; Bezerra, M.A.C.; Coelho-de-Souza, A.N.; Leal-Cardoso, J.H. Antispasmodic effects of the essential oil of Croton zehnteneri, anethole, and estragole, on tracheal smooth muscle. Heliyon 2020, 6, e05445. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, A.A.; Sorenson, A.L.; Leal-Cardoso, J.H. Effects of essential oil of Croton zehntneri, and of anethole and estragole on skeletal muscles. J. Ethnopharmacol. 1995, 49, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Cabral, P.H.B.; de Morais Campos, R.; Fonteles, M.C.; Santos, C.F.; Leal Cardoso, J.H.; do Nascimento, N.R.F. Effects of the essential oil of Croton zehntneri and its major components, anethole and estragole, on the rat corpora cavernosa. Life Sci. 2014, 112, 74–81. [Google Scholar] [CrossRef]
- da Silva-Alves, K.S.; Ferreira-da-Silva, F.W.; Coelho-de-Souza, A.N.; Albuquerque, A.A.C.; do Vale, O.C.; Leal-Cardoso, J.H. Essential oil of Croton zehntneri and its main constituent anethole block excitability of rat peripheral nerve. Planta Med. 2015, 81, 292–297. [Google Scholar] [CrossRef]
- Coelho-de-Souza, A.N.; Lahlou, S.; Barreto, J.E.F.; Yum, M.E.M.; Oliveira, A.C.; Oliveira, H.D.; Celedônio, N.R.; Feitosa, R.G.F.; Duarte, G.P.; Santos, C.F.; et al. Essential oil of Croton zehntneri and its major constituent anethole display gastroprotective effect by increasing the surface mucous layer. Fundam. Clin. Pharmacol. 2013, 27, 288–298. [Google Scholar] [CrossRef]
- de Siqueira, R.J.B.; Magalhães, P.J.C.; Leal-Cardoso, J.H.; Duarte, G.P.; Lahlou, S. Cardiovascular effects of the essential oil of Croton zehntneri leaves and its main constituents, anethole and estragole, in normotensive conscious rats. Life Sci. 2006, 78, 2365–2372. [Google Scholar] [CrossRef]
- Tognolini, M.; Ballabeni, V.; Bertoni, S.; Bruni, R.; Impicciatore, M.; Barocelli, E. Protective effect of Foeniculum vulgare essential oil and anethole in an experimental model of thrombosis. Pharmacol. Res. 2007, 56, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Domiciano, T.P.; Dalalio, M.M.d.O.; Silva, E.L.; Ritter, A.M.V.; Estevão-Silva, C.F.; Ramos, F.S.; Caparroz-Assef, S.M.; Cuman, R.K.N.; Bersani-Amado, C.A. Inhibitory effect of anethole in nonimmune acute inflammation. Naunyn. Schmiedebergs Arch. Pharmacol. 2013, 386, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Mukherjee, P.K.; Kumar, N.S.; Bandyopadhyay, A. Anticholinesterase activity of standardized extract of Illicium verum Hook. f. fruits. Fitoterapia 2011, 82, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Memon, T.; Yarishkin, O.; Reilly, C.A.; Križaj, D.; Olivera, B.M.; Teichert, R.W. trans-Anethole of Fennel Oil is a Selective and Nonelectrophilic Agonist of the TRPA1 Ion Channel. Mol. Pharmacol. 2019, 95, 433–441. [Google Scholar] [CrossRef]
- Merrill, A.W.; Cuellar, J.M.; Judd, J.H.; Carstens, M.I.; Carstens, E. Effects of TRPA1 agonists mustard oil and cinnamaldehyde on lumbar spinal wide-dynamic range neuronal responses to innocuous and noxious cutaneous stimuli in rats. J. Neurophysiol. 2008, 99, 415–425. [Google Scholar] [CrossRef]
- Ghasemi, Z.; Hassanpour-Ezatti, M.; Kamalinejad, M.; Janahmadi, M. Functional involvement of Ca2+ and Ca2+-activated K+ channels in anethol-induced changes in Ca2+ dependent excitability of F1 neurons in Helix aspersa. Fitoterapia 2011, 82, 750–756. [Google Scholar] [CrossRef]
- Faizi, M.; Janahmadi, M.; Mahmoudian, M. The effect of mebudipine and dibudipine, two new Ca2+ channel blockers, in comparison with nifedipine on Ca2+ spikes of F1 neuronal soma membrane in Helix aspersa. Acta Physiol. Hung. 2003, 90, 243–254. [Google Scholar] [CrossRef]
- Gerschenfeld, H.M.; Paupardin-Tritsch, D.; Yakel, J.L. Muscarinic enhancement of the voltage-dependent calcium current in an identified snail neuron. J. Physiol. 1991, 434, 85–105. [Google Scholar] [CrossRef]
- Luo, T.; Wang, F.; Weng, S.; Chen, H.; Kang, H.; Wang, J.; Luo, S. Anethole compromises human sperm function by affecting the sperm intracellular calcium concentration and tyrosine phosphorylation. Reprod. Toxicol. 2020, 93, 99–105. [Google Scholar] [CrossRef]
- Soares, P.M.G.; Lima, R.F.; de Freitas Pires, A.; Souza, E.P.; Assreuy, A.M.S.; Criddle, D.N. Effects of anethole and structural analogues on the contractility of rat isolated aorta: Involvement of voltage-dependent Ca2+-channels. Life Sci. 2007, 81, 1085–1093. [Google Scholar] [CrossRef]
- Horishita, T.; Harris, R.A. n-Alcohols inhibit voltage-gated Na+ channels expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 2008, 326, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.V.; Kendig, J.J. Differential sensitivities of TTX-resistant and TTX-sensitive sodium channels to anesthetic concentrations of ethanol in rat sensory neurons. J. Neurosci. Res. 1998, 54, 433–443. [Google Scholar] [CrossRef]
- Prinz, H. Hill coefficients, dose-response curves and allosteric mechanisms. J. Chem. Biol. 2010, 3, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Gamal El-Din, T.M.; Lenaeus, M.J.; Zheng, N.; Catterall, W.A. Fenestrations control resting-state block of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 2018, 115, 13111–13116. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, R.; Fedida, D. Closed- and open-state binding of 4-aminopyridine to the cloned human potassium channel Kv1.5. J. Pharmacol. Exp. Ther. 1995, 275, 864–876. [Google Scholar] [PubMed]
- Bassetto Junior, C.A.Z.; Passianoto, L.V.G.; González, E.R.P.; Varanda, W.A. Benzenesulfonamides act as open-channel blockers on KV3.1 potassium channel. Amino Acids 2019, 51, 355–364. [Google Scholar] [CrossRef]
- Wang, G.K.; Wang, S.-Y. Block of human cardiac sodium channels by lacosamide: Evidence for slow drug binding along the activation pathway. Mol. Pharmacol. 2014, 85, 692–702. [Google Scholar] [CrossRef]
- Chevrier, P.; Vijayaragavan, K.; Chahine, M. Differential modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by the local anesthetic lidocaine. Br. J. Pharmacol. 2004, 142, 576–584. [Google Scholar] [CrossRef]
- Shao, J.; Liu, Y.; Gao, D.; Tu, J.; Yang, F. Neural Burst Firing and Its Roles in Mental and Neurological Disorders. Front. Cell. Neurosci. 2021, 15, 741292. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317–322. [Google Scholar] [CrossRef]
- Yuan, Y.; Wu, W.; Chen, M.; Cai, F.; Fan, C.; Shen, W.; Sun, W.; Hu, J. Reward Inhibits Paraventricular CRH Neurons to Relieve Stress. Curr. Biol. CB 2019, 29, 1243–1251.e4. [Google Scholar] [CrossRef] [PubMed]
- Sanabria, E.R.; Su, H.; Yaari, Y. Initiation of network bursts by Ca2+-dependent intrinsic bursting in the rat pilocarpine model of temporal lobe epilepsy. J. Physiol. 2001, 532, 205–216. [Google Scholar] [CrossRef]
- Fremont, R.; Calderon, D.P.; Maleki, S.; Khodakhah, K. Abnormal high-frequency burst firing of cerebellar neurons in rapid-onset dystonia-parkinsonism. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 11723–11732. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.M.; Tyson, J.R.; Choi, H.-B.; Ko, R.; Lin, P.J.C.; LeDue, J.M.; Powell, K.L.; Bernier, L.-P.; Rungta, R.L.; Yang, Y.; et al. CaV 3.2 drives sustained burst-firing, which is critical for absence seizure propagation in reticular thalamic neurons. Epilepsia 2018, 59, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 35–51. [Google Scholar] [CrossRef]
- Marban, E.; Yamagishi, T.; Tomaselli, G.F. Structure and function of voltage-gated sodium channels. J. Physiol. 1998, 508 Pt 3, 647–657. [Google Scholar] [CrossRef]
- Falqueto, E.B.; Massensini, A.R.; Moraes-Santos, T.; Gomez, M.V.; Romano-Silva, M.A. Modulation of Na+-channels by neurotoxins produces different effects on [3H]ACh release with mobilization of distinct Ca2+-channels. Cell. Mol. Neurobiol. 2002, 22, 819–826. [Google Scholar] [CrossRef]
- Ho, C.; O’Leary, M.E. Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol. Cell. Neurosci. 2011, 46, 159–166. [Google Scholar] [CrossRef]
- Berta, T.; Poirot, O.; Pertin, M.; Ji, R.-R.; Kellenberger, S.; Decosterd, I. Transcriptional and functional profiles of voltage-gated Na+ channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol. Cell. Neurosci. 2008, 37, 196–208. [Google Scholar] [CrossRef]
- Shiers, S.; Klein, R.M.; Price, T.J. Quantitative differences in neuronal subpopulations between mouse and human dorsal root ganglia demonstrated with RNAscope in situ hybridization. Pain 2020, 161, 2410–2424. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Yamamoto, M.; Matsutomi, T.; Zheng, T.; Nakata, Y.; Wood, J.N.; Ogata, N. Electrophysiological characterization of the tetrodotoxin-resistant Na+ channel, Na(v)1.9, in mouse dorsal root ganglion neurons. Pflugers Arch. 2004, 449, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.L.; Clark, A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wood, J.N. The roles of sodium channels in nociception: Implications for mechanisms of neuropathic pain. Pain Med. Malden Mass 2011, 12 (Suppl. S3), S93–S99. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Dobrev, D. Ion channels as part of macromolecular multiprotein complexes: Clinical significance. Herzschrittmacherther. Elektrophysiol. 2018, 29, 30–35. [Google Scholar] [CrossRef]
- Abriel, H.; Rougier, J.-S.; Jalife, J. Ion channel macromolecular complexes in cardiomyocytes: Roles in sudden cardiac death. Circ. Res. 2015, 116, 1971–1988. [Google Scholar] [CrossRef]
- Vacher, H.; Mohapatra, D.P.; Trimmer, J.S. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 2008, 88, 1407–1447. [Google Scholar] [CrossRef]
- Subramanyam, P.; Colecraft, H.M. Ion channel engineering: Perspectives and strategies. J. Mol. Biol. 2015, 427, 190–204. [Google Scholar] [CrossRef]
- Baronas, V.A.; McGuinness, B.R.; Brigidi, G.S.; Gomm Kolisko, R.N.; Vilin, Y.Y.; Kim, R.Y.; Lynn, F.C.; Bamji, S.X.; Yang, R.; Kurata, H.T. Use-dependent activation of neuronal Kv1.2 channel complexes. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 3515–3524. [Google Scholar] [CrossRef]
- Newberne, P.; Smith, R.L.; Doull, J.; Goodman, J.I.; Munro, I.C.; Portoghese, P.S.; Wagner, B.M.; Weil, C.S.; Woods, L.A.; Adams, T.B.; et al. The FEMA GRAS assessment of trans-anethole used as a flavouring substance. Flavour and Extract Manufacturer’s Association. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 1999, 37, 789–811. [Google Scholar] [CrossRef]
- Han, C.; Estacion, M.; Huang, J.; Vasylyev, D.; Zhao, P.; Dib-Hajj, S.D.; Waxman, S.G. Human Na(v)1.8: Enhanced persistent and ramp currents contribute to distinct firing properties of human DRG neurons. J. Neurophysiol. 2015, 113, 3172–3185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Priest, B.T.; Belfer, I.; Gold, M.S. Voltage-gated Na+ currents in human dorsal root ganglion neurons. eLife 2017, 6, e23235. [Google Scholar] [CrossRef] [PubMed]
- Estacion, M.; Waxman, S.G. The response of Na(V)1.3 sodium channels to ramp stimuli: Multiple components and mechanisms. J. Neurophysiol. 2013, 109, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Mok, W.M.; Wang, G.K. Use-dependent inhibition of Na+ currents by benzocaine homologs. Biophys. J. 1996, 70, 194–201. [Google Scholar] [CrossRef]
- Lee, H.-S. Recent advances in topical anesthesia. J. Dent. Anesth. Pain Med. 2016, 16, 237–244. [Google Scholar] [CrossRef]
- Heavner, J.E. Local anesthetics. Curr. Opin. Anaesthesiol. 2007, 20, 336–342. [Google Scholar] [CrossRef]
- Bezanilla, F.; Armstrong, C. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 1977, 70, 549–566. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira-Junior, L.; Leal-Cardoso, J.H.; Cassola, A.C.; Carvalho-de-Souza, J.L. State-Dependent Blockade of Dorsal Root Ganglion Voltage-Gated Na+ Channels by Anethole. Int. J. Mol. Sci. 2024, 25, 1034. https://doi.org/10.3390/ijms25021034
Moreira-Junior L, Leal-Cardoso JH, Cassola AC, Carvalho-de-Souza JL. State-Dependent Blockade of Dorsal Root Ganglion Voltage-Gated Na+ Channels by Anethole. International Journal of Molecular Sciences. 2024; 25(2):1034. https://doi.org/10.3390/ijms25021034
Chicago/Turabian StyleMoreira-Junior, Luiz, Jose Henrique Leal-Cardoso, Antonio Carlos Cassola, and Joao Luis Carvalho-de-Souza. 2024. "State-Dependent Blockade of Dorsal Root Ganglion Voltage-Gated Na+ Channels by Anethole" International Journal of Molecular Sciences 25, no. 2: 1034. https://doi.org/10.3390/ijms25021034