
Synthetic Study of Natural Metabolites Containing a Benzo[c]oxepine Skeleton: Heterocornol C and D
 , and
, and
Abstract
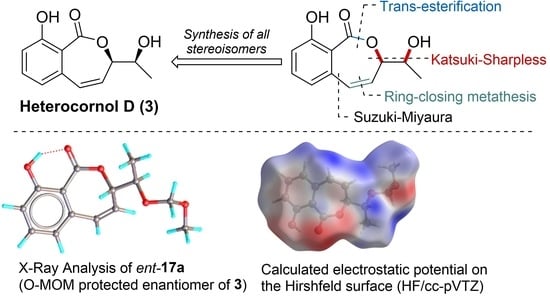
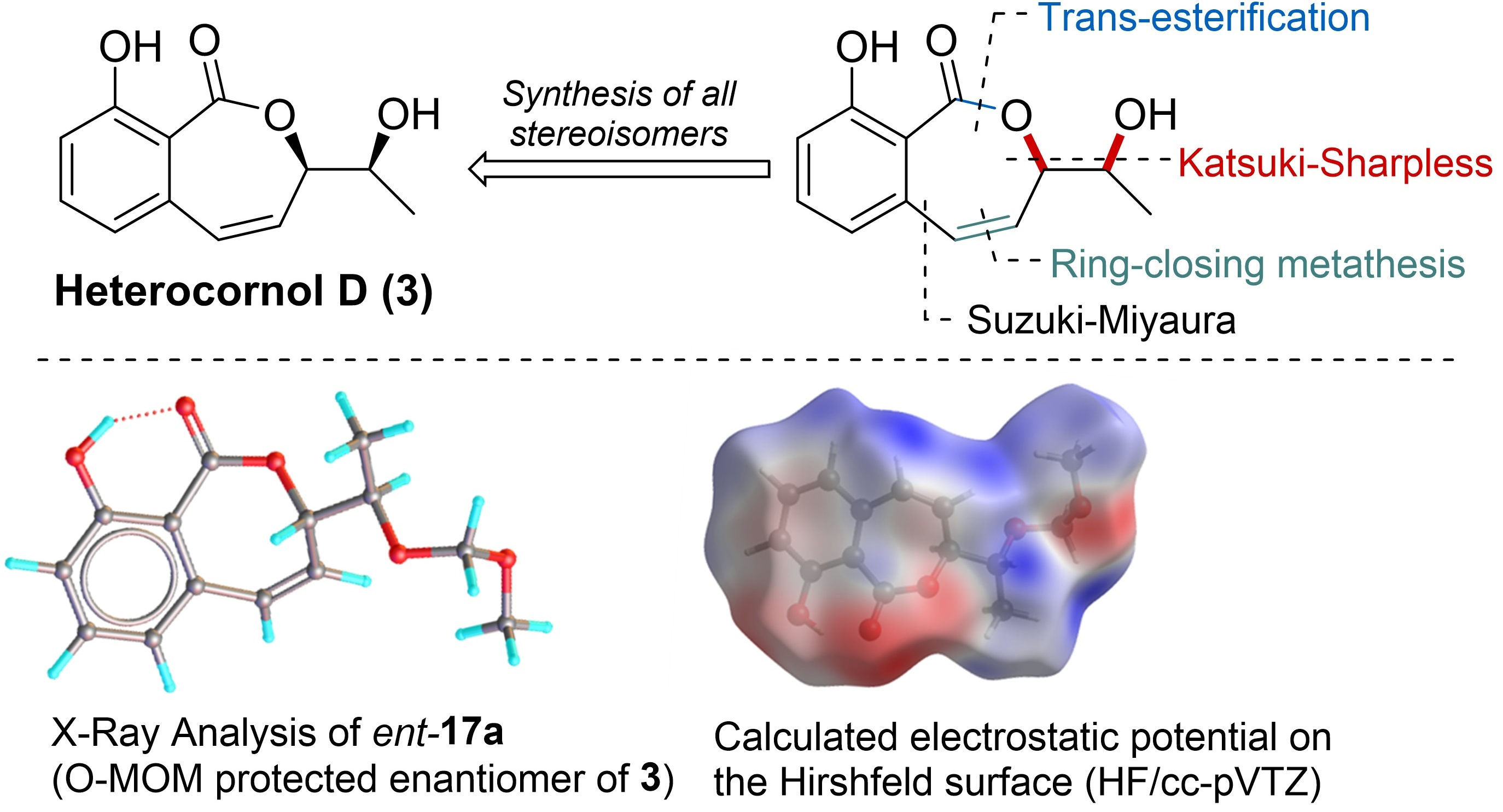
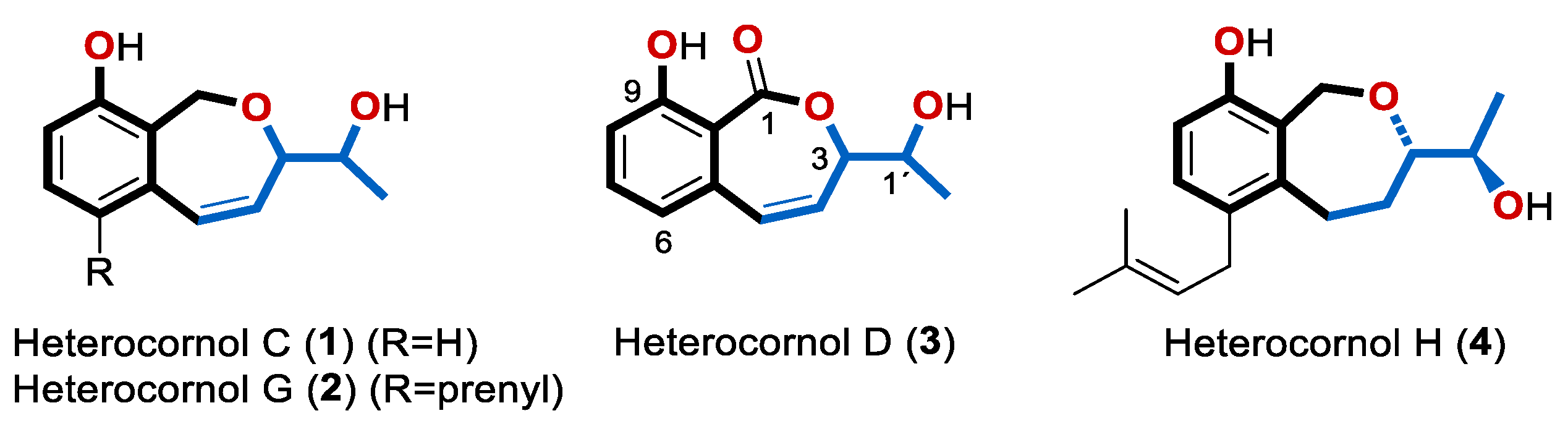
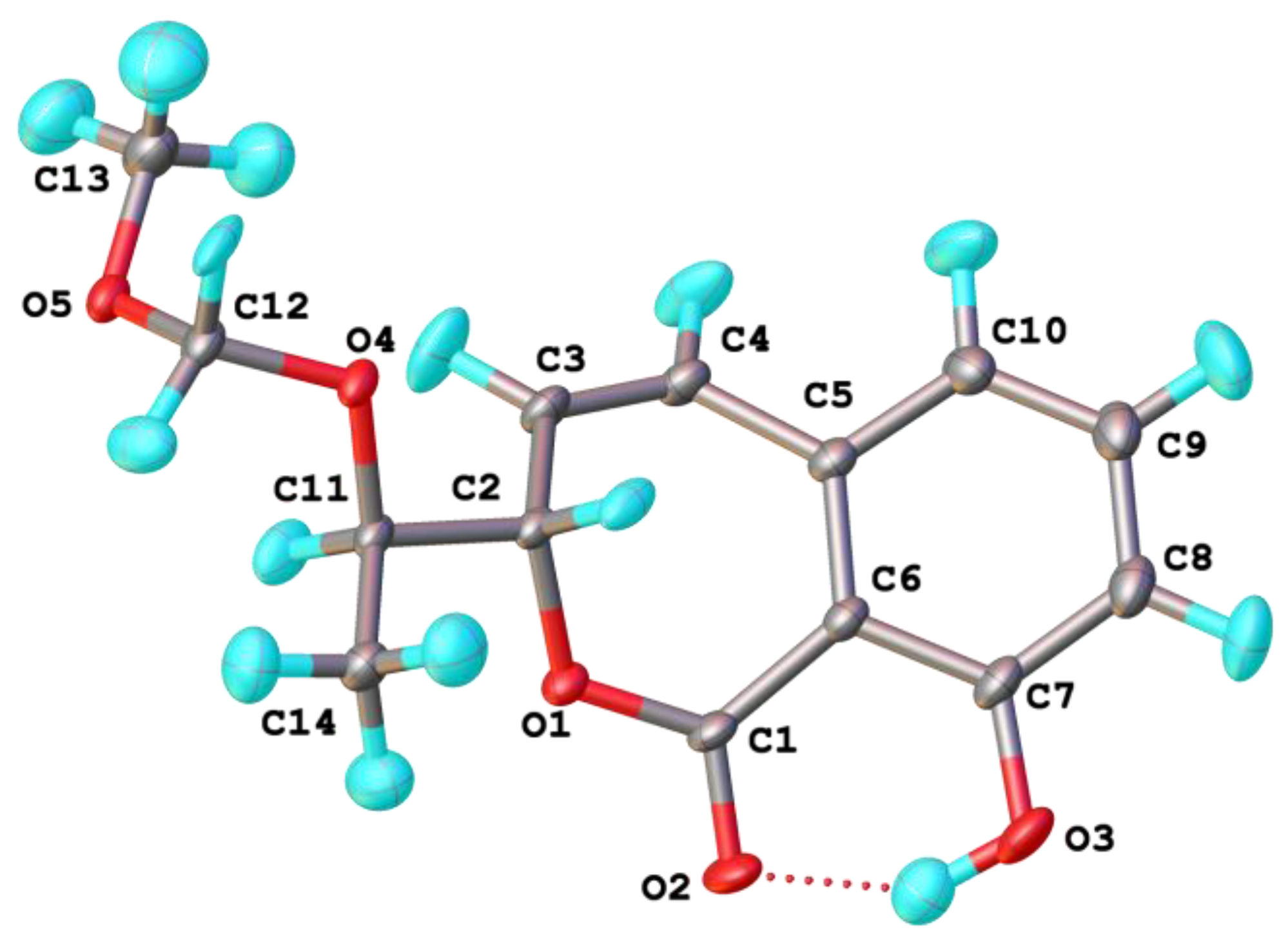
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
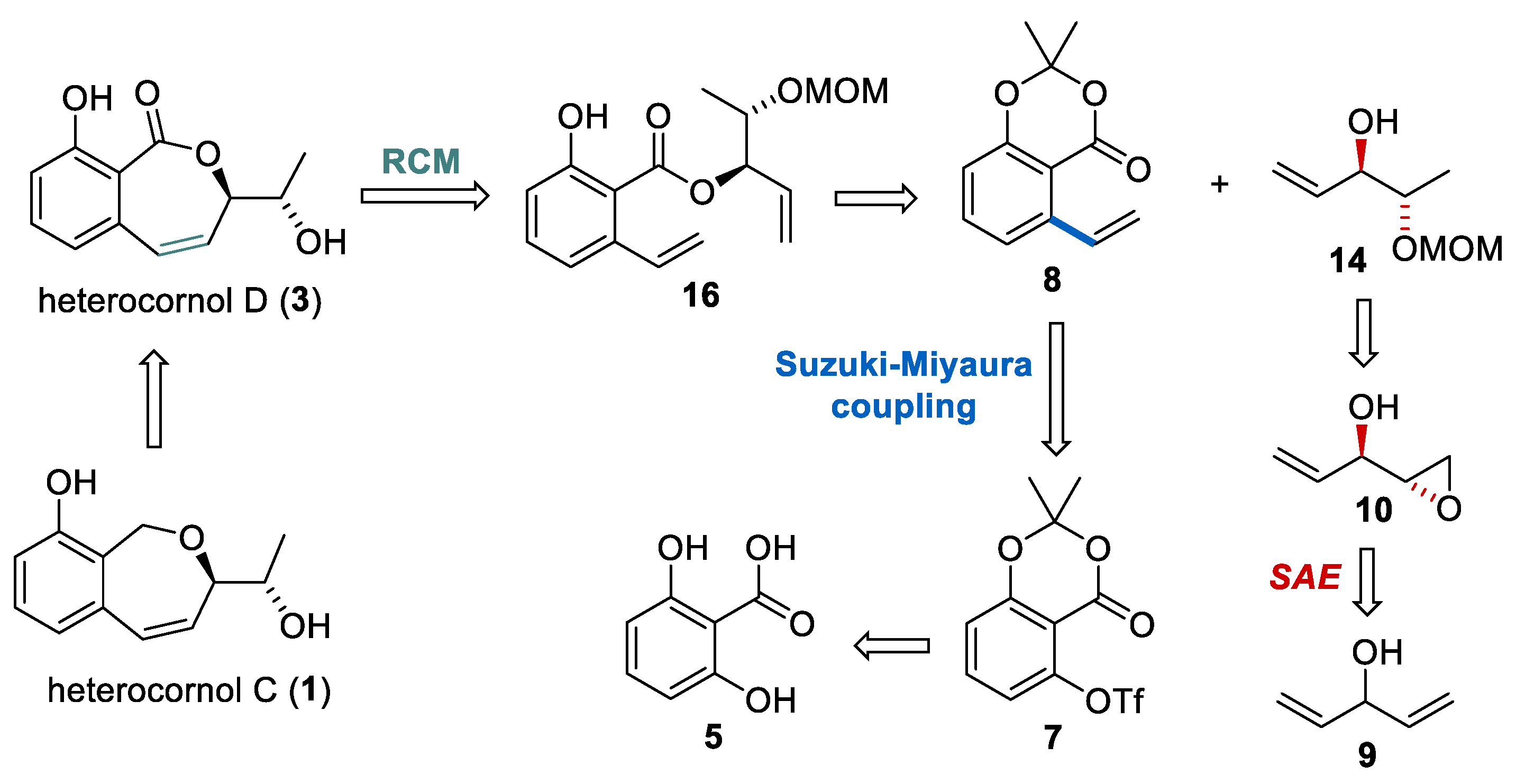
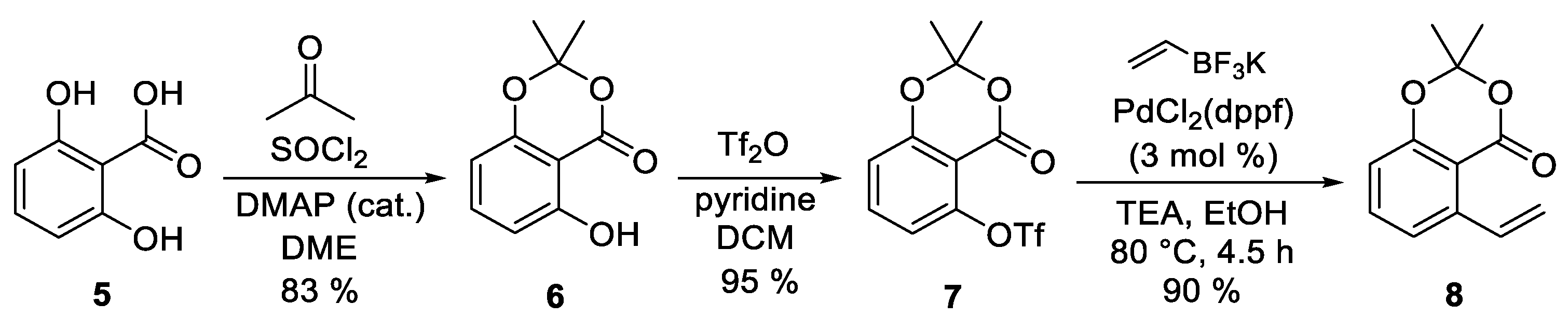
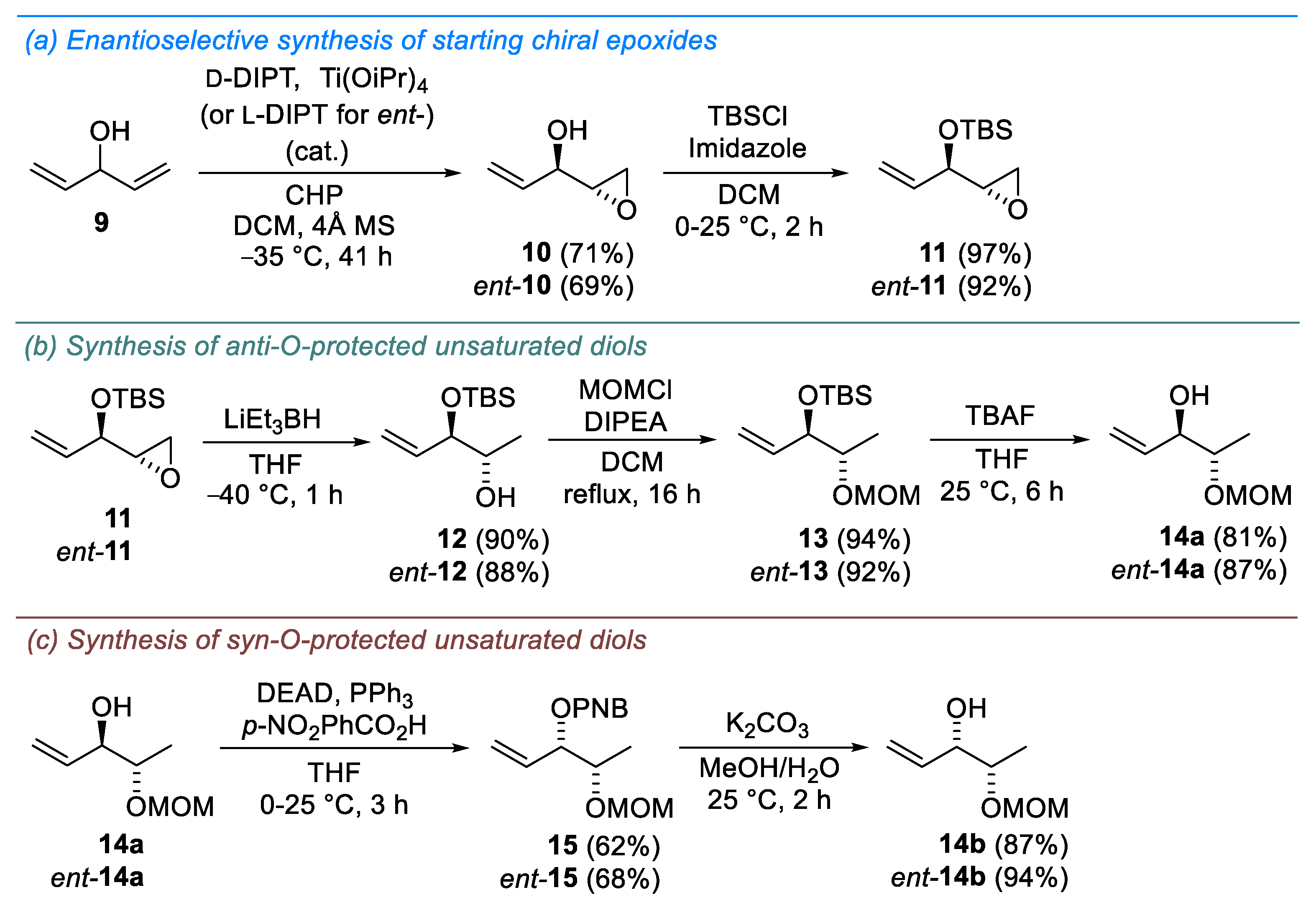
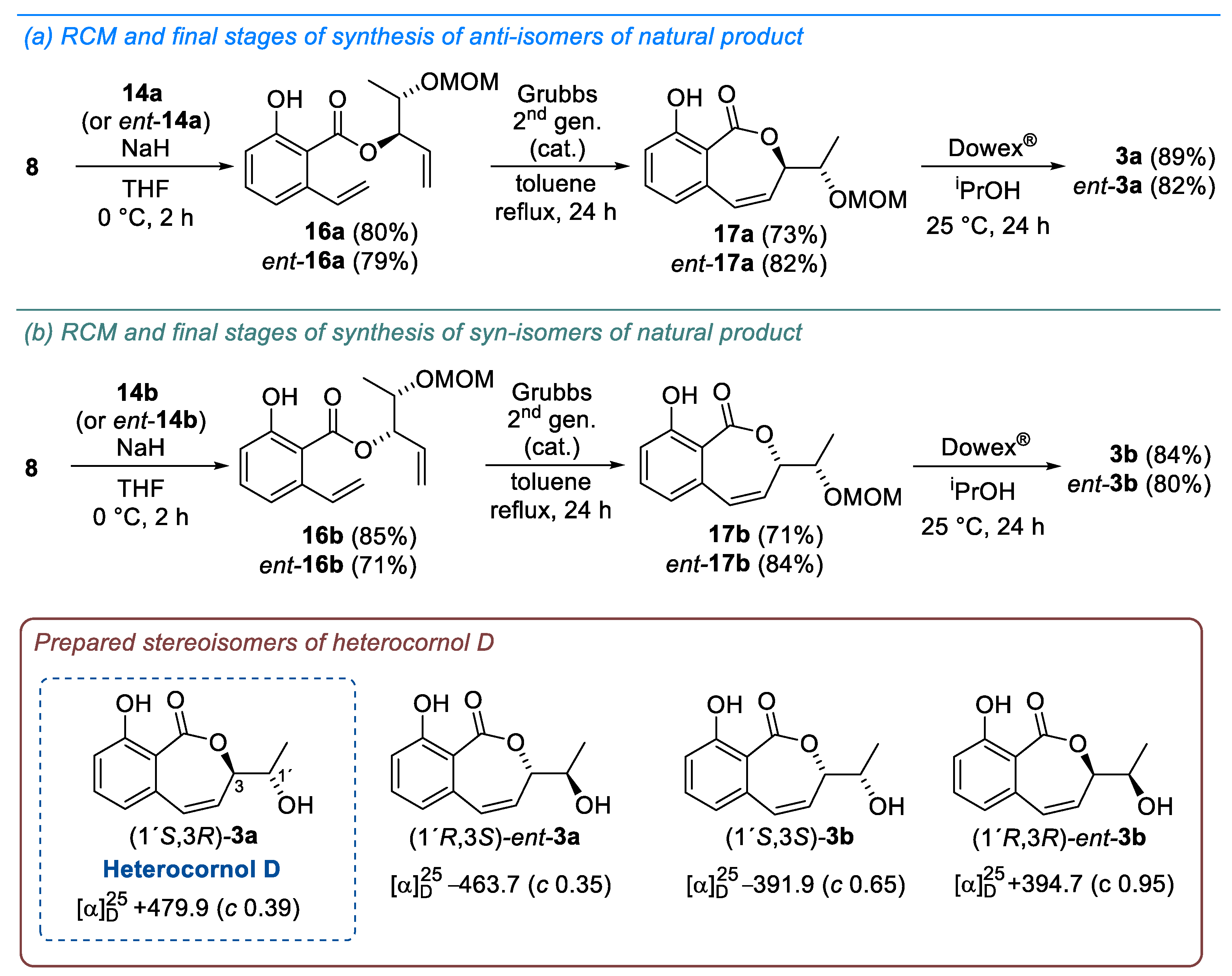
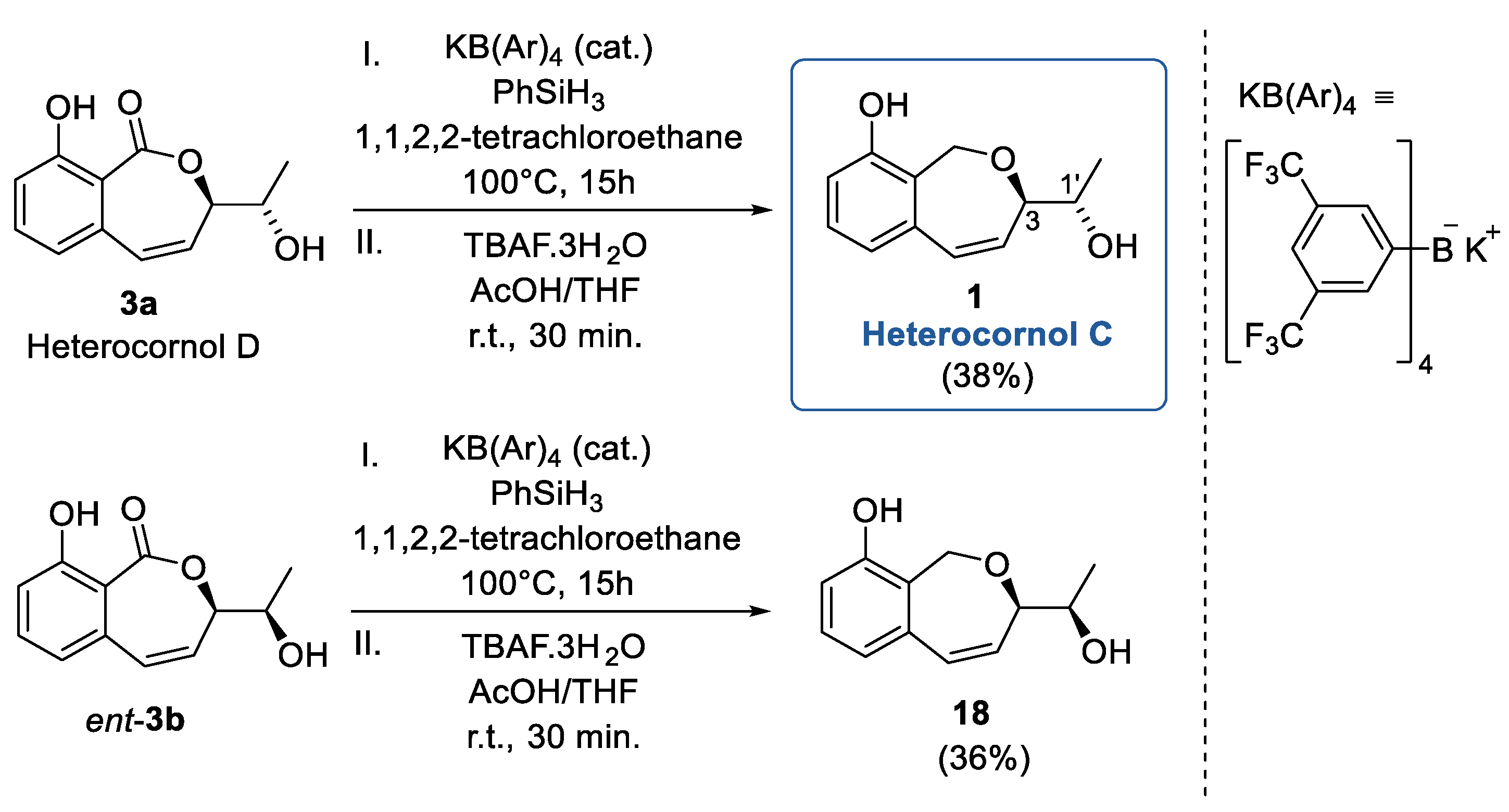
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References and Notes
- Schueffler, A.; Anke, T. Fungal natural products in research and development. Nat. Prod. Rep. 2014, 31, 1425–1448. [Google Scholar] [CrossRef] [PubMed]
- Baerson, S.R.; Rimando, A.M. A Plethora of Polyketides: Structures, Biological Activities, and Enzymes. In Polyketides; ACS Symposium Series; Baerson, S.R., Rimando, A.M., Eds.; American Chemical Society: Washington, DC, USA, 2007; Volume 955, pp. 2–14. [Google Scholar] [CrossRef] [Green Version]
- Štibláriková, M.; Lásiková, A.; Gracza, T. Benzyl Alcohol/Salicylaldehyde-Type Polyketide Metabolites of Fungi: Sources, Biosynthesis, Biological Activities, and Synthesis. Mar. Drugs 2023, 21, 19. [Google Scholar] [CrossRef] [PubMed]
- Dechert-Schmitt, A.M.R.; Schmitt, D.C.; Gao, X.; Itoh, T.; Krische, M. Polyketide construction via hydrohydroxyalkylation and related alcohol C-H functionalizations: Reinventing the chemistry of carbonyl addition. J. Nat. Prod. Rep. 2014, 31, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wei, X.; Lu, X.; Xu, F.; Wan, J.; Lin, X.; Zhou, X.; Liao, S.; Yang, B.; Tu, Z.; et al. Eight new polyketide metabolites from the fungus Pestalotiopsis vaccinii endogenous with mangrove plant Kandelia candel (L.) Druce. Tetrahedron 2014, 70, 9695–9701. [Google Scholar] [CrossRef]
- Wang, J.; Wei, X.; Qin, X.; Chen, H.; Lin, X.; Zhang, T.; Yang, X.; Liao, S.; Yang, B.; Tu, Z.; et al. Two new prenylated phenols from endogenous fungus Pestalotiopsis vaccinii of mangrove plant Kandelia candel (L.) Druce. Phytochem. Lett. 2015, 12, 59–62. [Google Scholar] [CrossRef]
- Zhao, Z.; Ying, Y.; Hung, Y.-S.; Tang, Y. Genome Mining Reveals Neurospora crassa Can Produce the Salicylaldehyde Sordarial. J. Nat. Prod. 2018, 82, 1029–1033. [Google Scholar] [CrossRef]
- Lei, H.; Lin, X.; Han, L.; Ma, J.; Dong, K.; Wang, X.; Zhong, J.; Mu, Y.; Liu, Y.; Huang, X. Polyketide derivatives from a marine-sponge-associated fungus Pestalotiopsis heterocornins. Phytochemistry 2017, 142, 51–59. [Google Scholar] [CrossRef]
- More, A.A.; Ramana, C.V. Total synthesis of the putative structure of xylarinol B. Chem. Asian J. 2014, 9, 1557–1562. [Google Scholar] [CrossRef]
- Schreiber, S.L.; Schreiber, T.S.; Smith, D.B. Reactions that proceed with a combination of enantiotopic group and diastereotopic face selectivity can deliver products with very high enantiomeric excess: Experimental support of a mathematical model. J. Am. Chem. Soc. 1987, 109, 1525–1529. [Google Scholar] [CrossRef]
- Jäger, V.; Schröter, D.; Koppenhoefer, B. Asymmetric Sharpless epoxidation of divinylcarbinol. Erythro-D- and -L-4-pentenitols by hydrolysis of regioisomeric epoxy-4-pentenols. Tetrahedron 1991, 47, 2195–2210. [Google Scholar] [CrossRef]
- Clemens, R.T.; Jennings, M.P. An efficient total synthesis and absolute configuration determination of varitriol. Chem. Commun. 2006, 25, 2720–2721. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Wong, C.-H. Chemo-Enzymatic Total Synthesis of 3-Epiaustraline, Australine, and 7-Epialexine. J. Org. Chem. 2000, 65, 8264–8268. [Google Scholar] [CrossRef] [PubMed]
- Markovič, M.; Koóš, P.; Sokoliová, S.; Boháčiková, N.; Vyskočil, T.; Moncoľ, J.; Gracza, T. A universal strategy for polyketide synthesis. Total synthesis of agropyrenol, sordarial and heterocornol A and B. J. Org. Chem. 2022, 87, 15947–15962. [Google Scholar] [CrossRef] [PubMed]
- For comparison of optical rotation with known literature values, see refs [10,11,12,13].
- CCDC-2222838 and 2222839 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (accessed on 28 November 2022).
- Rysak, V.; Dixit, R.; Trivelli, X.; Merle, M.; Agbossou-Niedercorn, F.; Vanka, K.; Michon, C. Catalytic reductive deoxygenation of esters to ethers driven by hydrosilane activation through non-covalent interactions with a fluorinated borate salt. Catal. Sci. Technol. 2020, 10, 4586–4592. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.; Gildea, R.I.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Crystallogr. 2015, 71, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Kleemiss, F.; Dolomanov, O.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genomi, A.; Malaspina, L.A.; Jayalitaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X—H bond lengths from neutron diffraction data. Acta Crystallogr. 2010, 66, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.; Bourhis, L.J.; Gildea, R.I.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. 2013, 69, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Crystallogr. 2008, 41, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
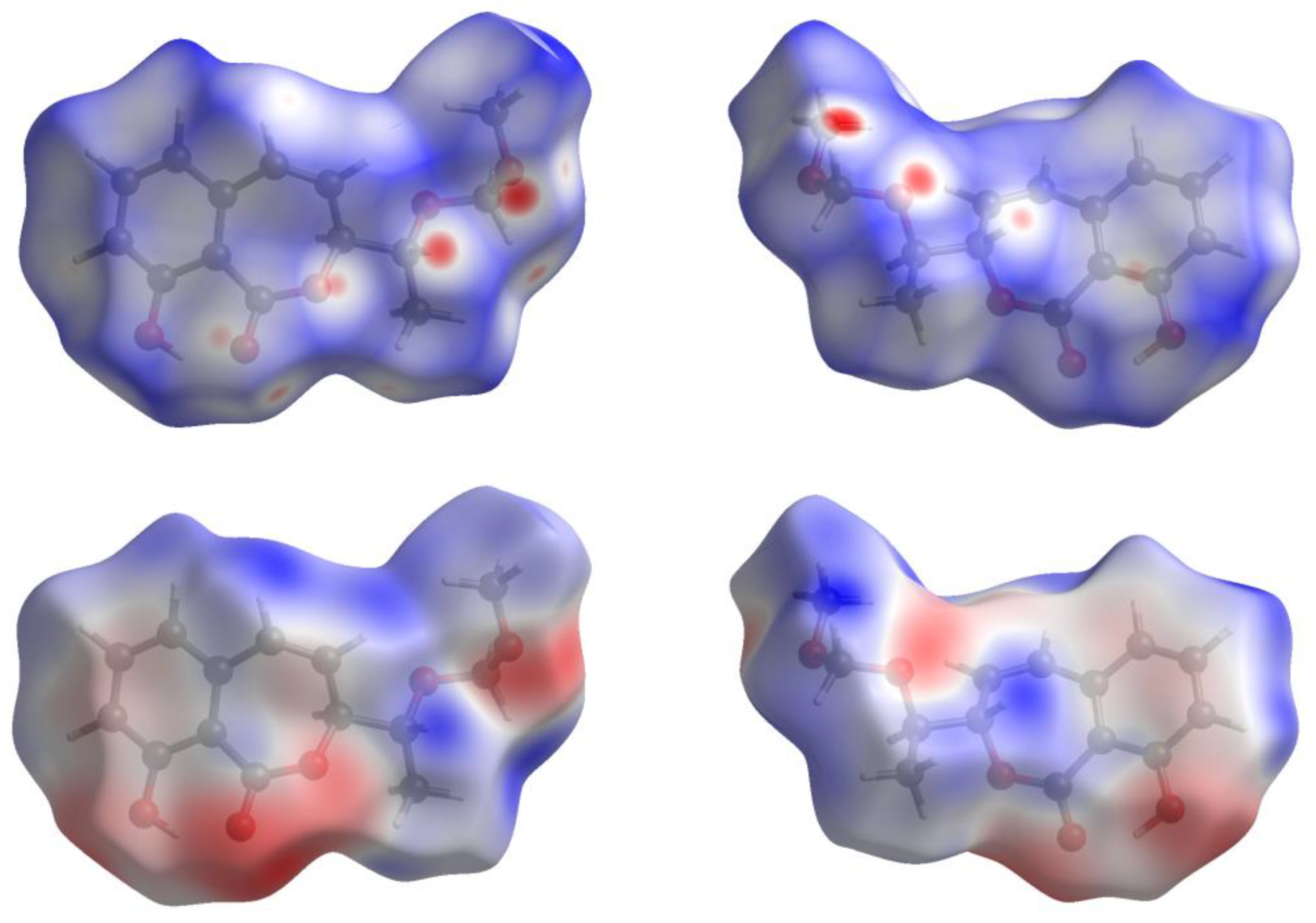
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayalitaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayalitaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.A.; Mckinnon, J.J.; Jayalitaka, D. Analysis of the compression of molecular crystal structures using Hirshfeld surfaces. CrystEngComm 2008, 10, 368–376. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gettler, J.; Čarný, T.; Markovič, M.; Koóš, P.; Samoľová, E.; Moncoľ, J.; Gracza, T. Synthetic Study of Natural Metabolites Containing a Benzo[c]oxepine Skeleton: Heterocornol C and D. Int. J. Mol. Sci. 2023, 24, 10331. https://doi.org/10.3390/ijms241210331
Gettler J, Čarný T, Markovič M, Koóš P, Samoľová E, Moncoľ J, Gracza T. Synthetic Study of Natural Metabolites Containing a Benzo[c]oxepine Skeleton: Heterocornol C and D. International Journal of Molecular Sciences. 2023; 24(12):10331. https://doi.org/10.3390/ijms241210331
Chicago/Turabian StyleGettler, Ján, Tomáš Čarný, Martin Markovič, Peter Koóš, Erika Samoľová, Ján Moncoľ, and Tibor Gracza. 2023. "Synthetic Study of Natural Metabolites Containing a Benzo[c]oxepine Skeleton: Heterocornol C and D" International Journal of Molecular Sciences 24, no. 12: 10331. https://doi.org/10.3390/ijms241210331