
VirtualFlow Ants—Ultra-Large Virtual Screenings with Artificial Intelligence Driven Docking Algorithm Based on Ant Colony Optimization

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Ant Intelligence in Molecular Docking with PLANTS
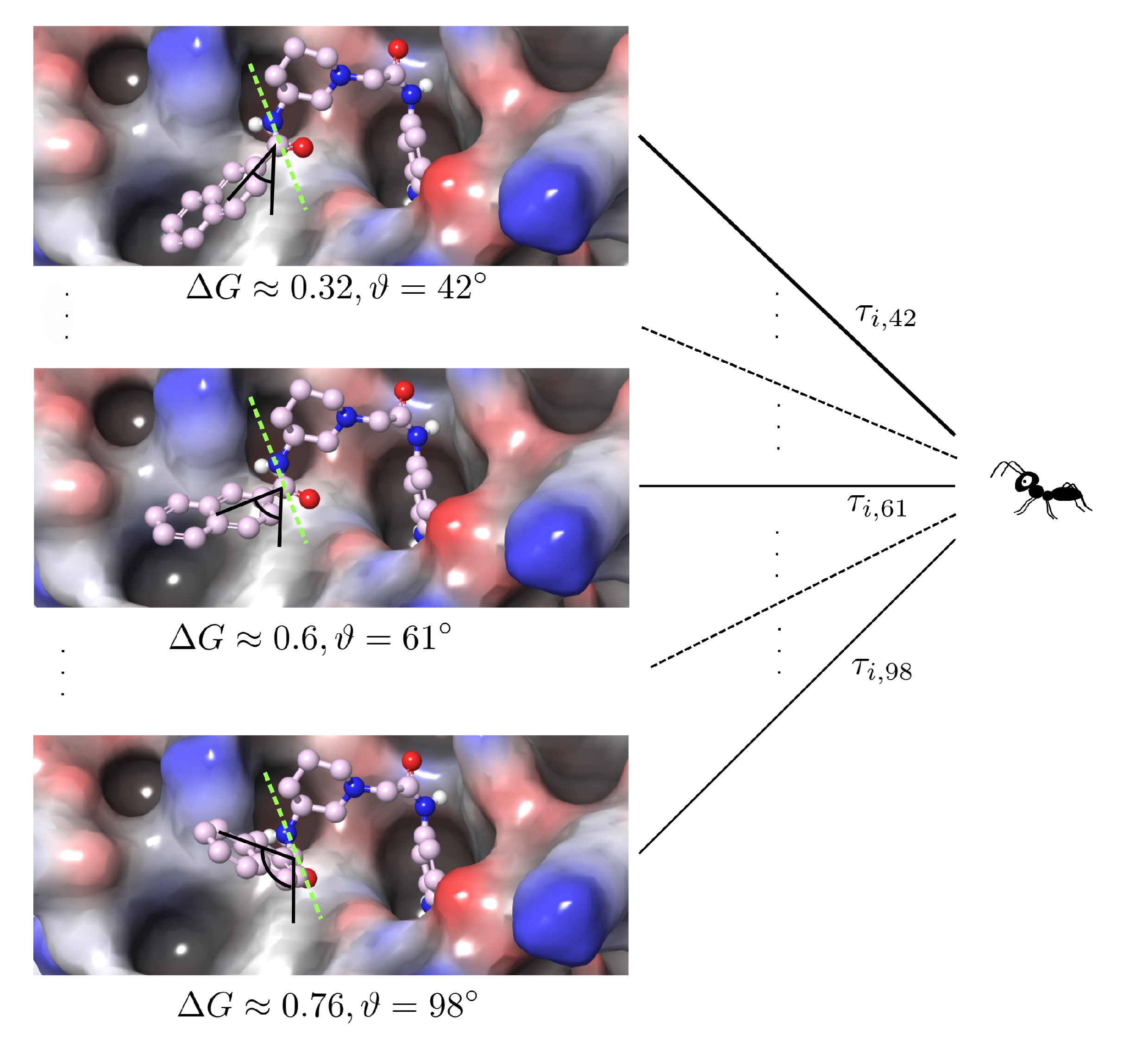
2.1. Discretization

2.2. Iteration Rule of the Ants
2.3. PLANTS Features for Molecular Docking
3. VirtualFlow Ants—Virtual Screenings Using Ant Intelligence via PLANTS
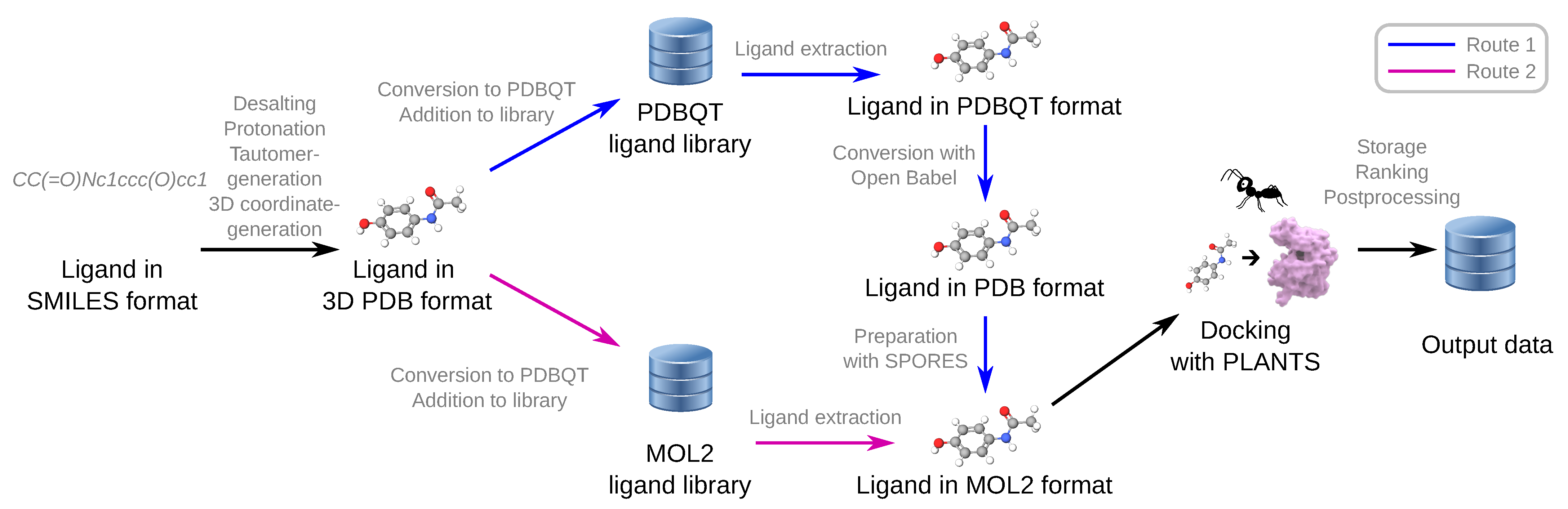
3.1. Ligand Preparation and Chemical File Formats
3.2. I/O and File Management
3.3. Configuration and Set Up of VirtualFlow Ants
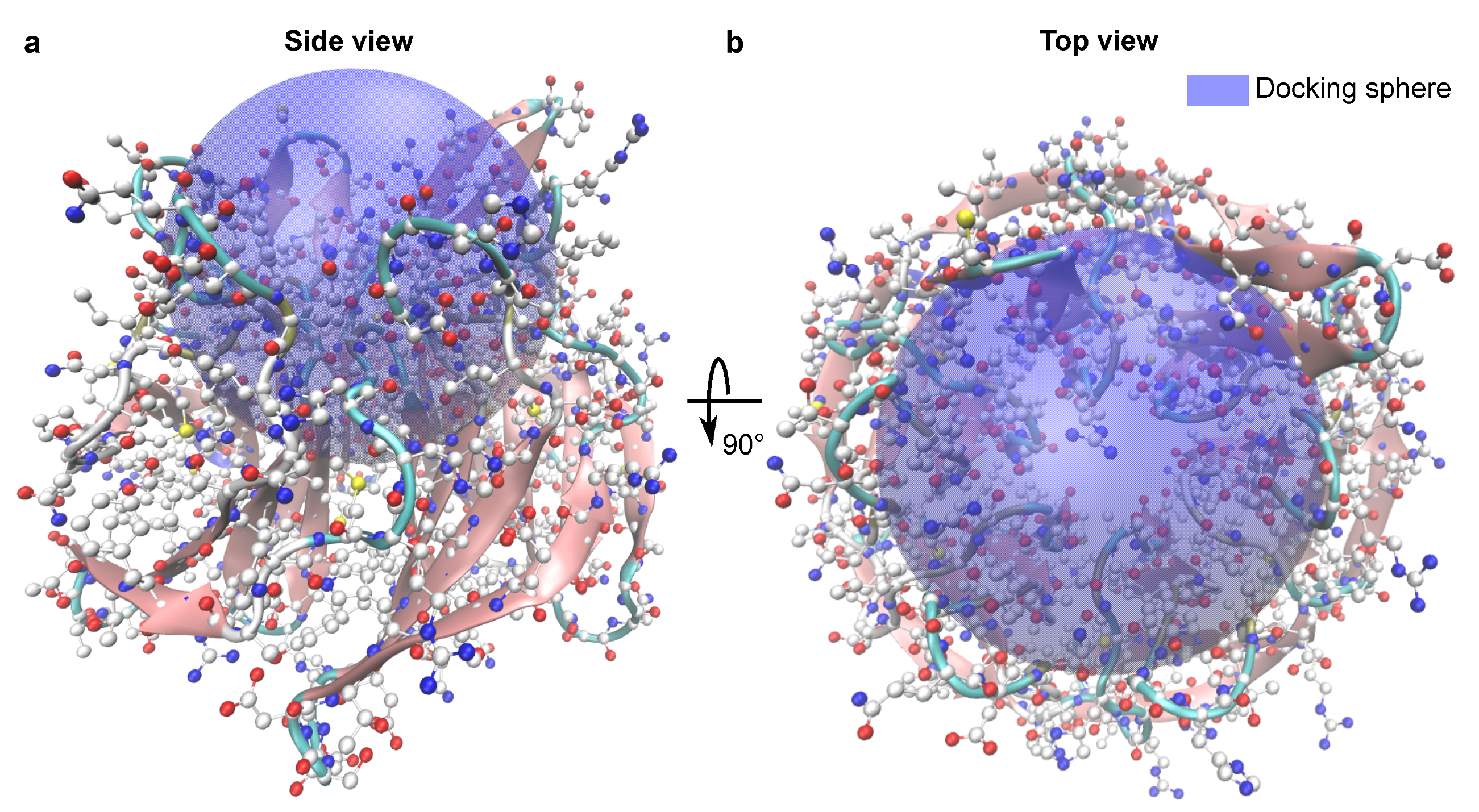
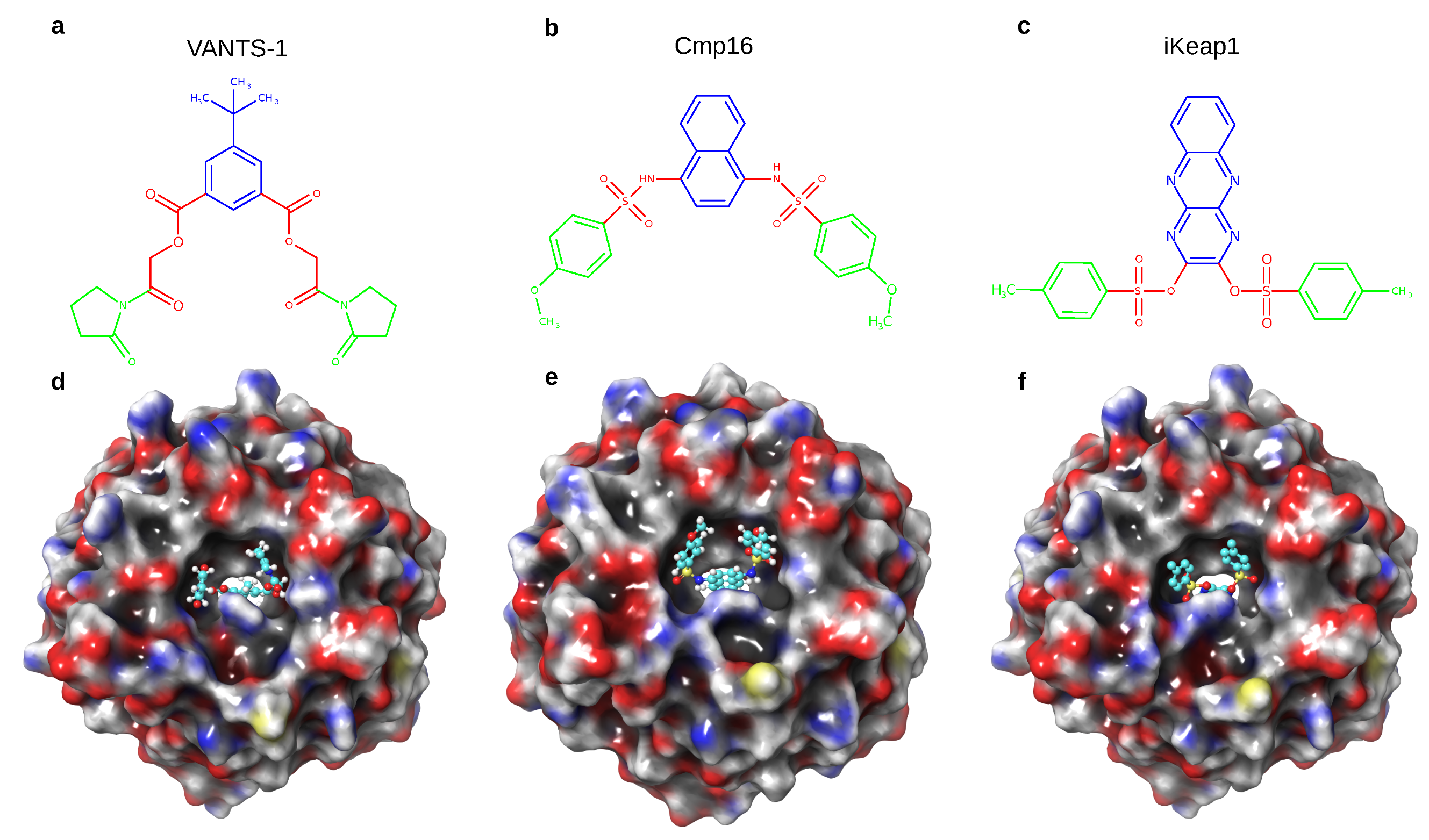
3.4. Test System
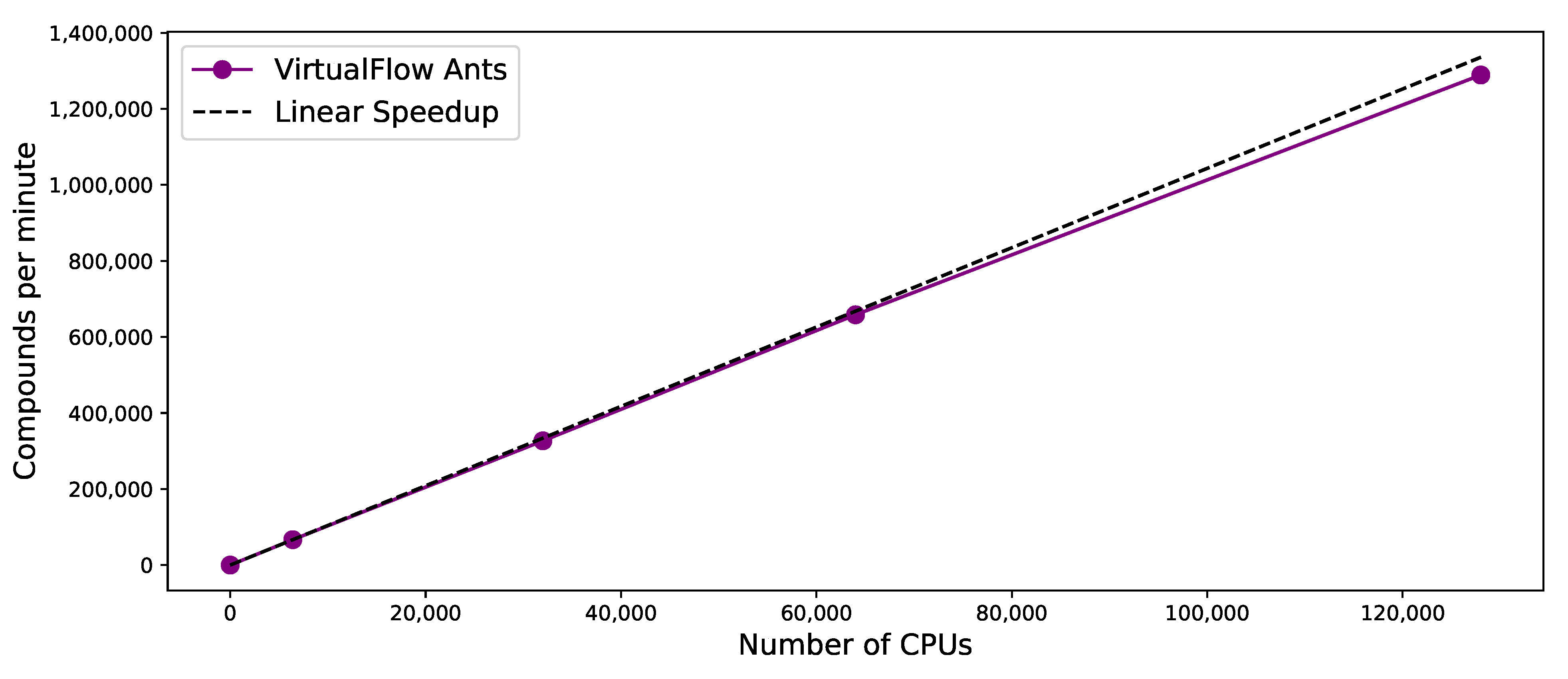
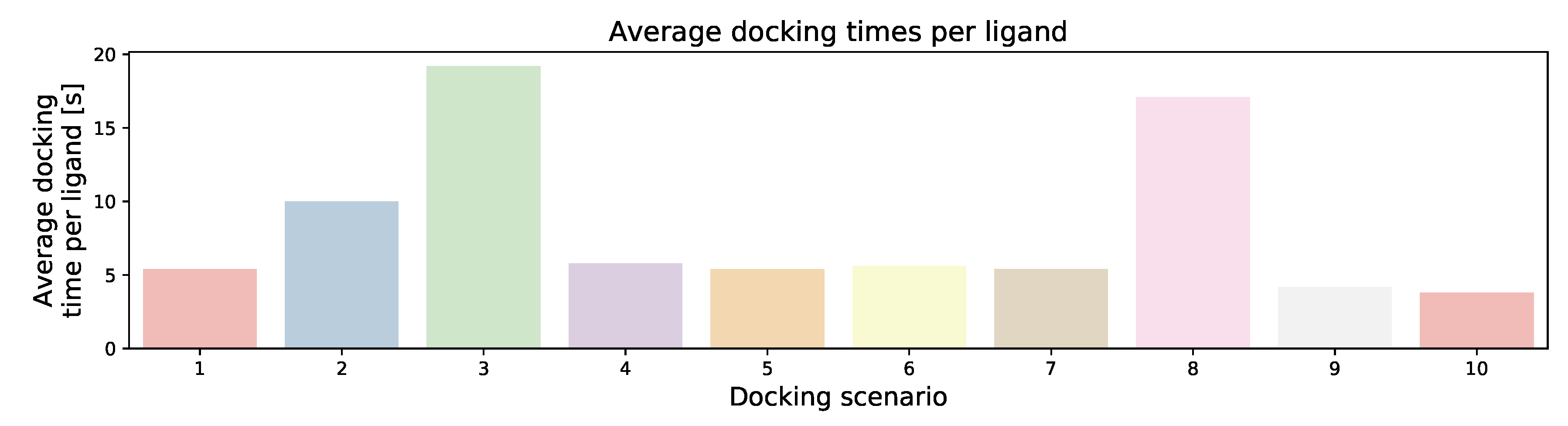
3.5. Scaling Behavior
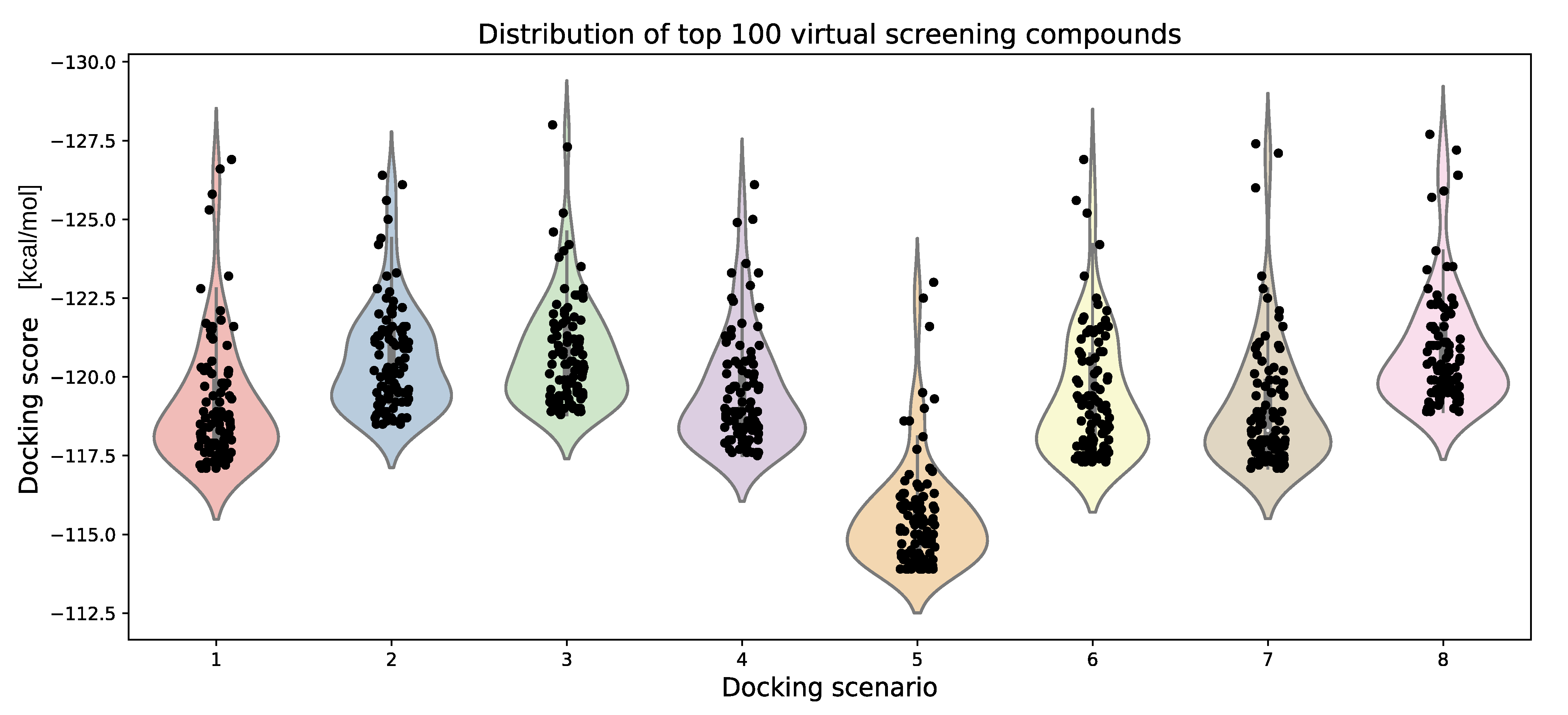
3.6. Parameter Variation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | Linear dichroism |
| AVE100 | Average docking scores of the top 100 ranking compounds |
| IFP | Interaction fingerprints |
| STD | Saturation transfer difference |
| ACO | Ant colony optimization |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
References
- Ewing, T.J.A.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Lauble, H.; Stout, C.D.; Olson, A.J. Automated docking in crystallography: Analysis of the substrates of aconitase. Proteins Struct. Funct. Bioinform. 1993, 17, 1–10. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Nguyen, D.T.; Case, D.A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible Docking Method using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. In Ant Colony Optimization and Swarm Intelligence; Dorigo, M., Gambardella, L.M., Birattari, M., Martinoli, A., Poli, R., Stützle, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 247–258. [Google Scholar]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. An Ant Colony Optimization Approach to Flexible Protein-Ligand Docking. Swarm Intell. 2007, 1, 115–134. [Google Scholar] [CrossRef]
- Dorigo, M.; Birattari, M.; Stützle, T. Ant Colony Optimization. Comput. Intell. Mag. IEEE 2006, 1, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Dorigo, M.; Stützle, T. Ant Colony Optimization; MIT Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Verkhivker, G.M. Computational analysis of ligand binding dynamics at the intermolecular hot spots with the aid of simulated tempering and binding free energy calculations. J. Mol. Graph. Model. 2004, 22, 335–348. [Google Scholar] [CrossRef]
- Clark, M.; Cramer, R.D., III; Van Opdenbosch, N. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Auton, T.R.; Eldridge, M.D. Empirical scoring functions. II. The testing of an empirical scoring function for the prediction of ligand-receptor binding affinities and the use of Bayesian regression to improve the quality of the model. J. Comput. Aided Mol. Des. 1998, 12, 503–519. [Google Scholar] [CrossRef]
- Boittier, E.D.; Tang, Y.Y.; Buckley, M.E.; Schuurs, Z.P.; Richard, D.J.; Gandhi, N.S. Assessing Molecular Docking Tools to Guide Targeted Drug Discovery of CD38 Inhibitors. Int. J. Mol. Sci. 2020, 21, 5183. [Google Scholar] [CrossRef]
- Ballante, F.; Marshall, G.R. An Automated Strategy for Binding-Pose Selection and Docking Assessment in Structure-Based Drug Design. J. Chem. Inf. Model. 2016, 56, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Çınaroğlu, S.S.; Timuçin, E. Comparative Assessment of Seven Docking Programs on a Nonredundant Metalloprotein Subset of the PDBbind Refined. J. Chem. Inf. Model. 2019, 59, 3846–3859. [Google Scholar] [CrossRef]
- Ren, X.; Shi, Y.S.; Zhang, Y.; Liu, B.; Zhang, L.H.; Peng, Y.B.; Zeng, R. Novel Consensus Docking Strategy to Improve Ligand Pose Prediction. J. Chem. Inf. Model. 2018, 58, 1662–1668. [Google Scholar] [CrossRef]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzym. Inhib. Med. Chem. 2016, 31, 167–173. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Freire, E. Finding a better path to drug selectivity. Drug Discov. Today 2011, 16, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today 2008, 13, 869–874. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K. Exploiting ordered waters in molecular docking. J. Med. Chem. 2008, 51, 4862–4865. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Recent advances in scaffold hopping: Miniperspective. J. Med. Chem. 2017, 60, 1238–1246. [Google Scholar] [CrossRef]
- Fejzo, J.; Lepre, C.A.; Peng, J.W.; Bemis, G.W.; Murcko, M.A.; Moore, J.M. The SHAPES strategy: An NMR-based approach for lead generation in drug discovery. Chem. Biol. 1999, 6, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Mizukoshi, Y.; Abe, A.; Takizawa, T.; Hanzawa, H.; Fukunishi, Y.; Shimada, I.; Takahashi, H. An Accurate Pharmacophore Mapping Method by NMR Spectroscopy. Angew. Chem. Int. Ed. 2012, 51, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Pedregal, V.M.; Reese, M.; Meiler, J.; Blommers, M.J.J.; Griesinger, C.; Carlomagno, T. The INPHARMA Method: Protein-Mediated Interligand NOEs for Pharmacophore Mapping. Angew. Chem. Int. Ed. 2005, 44, 4172–4175. [Google Scholar] [CrossRef]
- Korb, O.; Möller, H.M.; Exner, T.E. NMR-Guided Molecular Docking of a Protein–Peptide Complex Based on Ant Colony Optimization. ChemMedChem 2010, 5, 1001–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onila, I.; ten Brink, T.; Fredriksson, K.; Codutti, L.; Mazur, A.; Griesinger, C.; Carlomagno, T.; Exner, T.E. On-the-Fly Integration of Data from a Spin-Diffusion-Based NMR Experiment into Protein–Ligand Docking. J. Chem. Inf. Model. 2015, 55, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Exner, T. Protein-Ligand ANT System User Manual for Version 1.2; Universität Konstanz.
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.F.; Fischer, P.D.; Coote, P.W.; Padmanabha Das, K.M.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Gorgulla, C.; Fackeldey, K.; Wagner, G.; Arthanari, H. Accounting of Receptor Flexibility in Ultra-Large Virtual Screens with VirtualFlow Using a Grey Wolf Optimization Method. Supercomput. Front. Innov. 2020, 7, 4–12. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.K. Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Open Babel Package. 2020. Available online: http://openbabel.org (accessed on 15 March 2021).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- ten Brink, T.; Exner, T.E. Influence of Protonation, Tautomeric, and Stereoisomeric States on Protein-Ligand Docking Results. J. Chem. Inf. Model. 2009, 49, 1535–1546. [Google Scholar] [CrossRef]
- ten Brink, T.; Exner, T.E. pKa based protonation states and microspecies for protein–ligand docking. J. Comput. Aided Mol. Des. 2010, 24, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [Green Version]
- Hassan, N.M.; Alhossary, A.A.; Mu, Y.; Kwoh, C.K. Protein-Ligand Blind Docking Using QuickVina-W With Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017, 7, 15451. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [Green Version]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J. Cheminform. 2016, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Nivedha, A.K.; Thieker, D.F.; Makeneni, S.; Hu, H.; Woods, R.J. Vina-Carb: Improving Glycosidic Angles during Carbohydrate Docking. J. Chem. Theory Comput. 2016, 12, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Yonchuk, J.G.; Foley, J.P.; Bolognese, B.J.; Logan, G.; Wixted, W.E.; Kou, J.P.; Chalupowicz, D.G.; Feldser, H.G.; Sanchez, Y.; Nie, H.; et al. Characterization of the Potent, Selective Nrf2 Activator, 3-(Pyridin-3-Ylsulfonyl)-5-(Trifluoromethyl)-2 H -Chromen-2-One, in Cellular and In Vivo Models of Pulmonary Oxidative Stress. J. Pharmacol. Exp. Ther. 2017, 363, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Pallesen, J.S.; Tran, K.T.; Bach, A. Non-covalent Small-Molecule Kelch-like ECH-Associated Protein 1–Nuclear Factor Erythroid 2-Related Factor 2 (Keap1–Nrf2) Inhibitors and Their Potential for Targeting Central Nervous System Diseases. J. Med. Chem. 2018, 61, 8088–8103. [Google Scholar] [CrossRef]
- Davies, T.G.; Wixted, W.E.; Coyle, J.E.; Griffiths-Jones, C.; Hearn, K.; McMenamin, R.; Norton, D.; Rich, S.J.; Richardson, C.; Saxty, G.; et al. Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein–Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J. Med. Chem. 2016, 59, 3991–4006. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcotte, D.; Zeng, W.; Hus, J.C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Chessari, G.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Modeling Water Molecules in Protein-Ligand Docking Using GOLD. J. Med. Chem. 2005, 48, 6504–6515. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Zhao, H. Enriching screening libraries with bioactive fragment space. Bioorg. Med. Chem. Lett. 2016, 26, 3594–3597. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docking Scenario | scoring_Function | Search_Speed | aco_Ants | aco_Eevap | aco_Sigma |
|---|---|---|---|---|---|
| 1 | chemplp | 4 | default (20) | default (0.15) | default (0.25) |
| 2 | chemplp | 2 | default (20) | default (0.20) | default (0.5) |
| 3 | chemplp | 1 | default (20) | default (0.20) | default (1.25) |
| 4 | chemplp | 4 | 10 | default (0.15) | default (0.25) |
| 5 | chemplp | 4 | 50 | default (0.15) | default (0.25) |
| 6 | chemplp | 4 | default (20) | 0.10 | default (0.25) |
| 7 | chemplp | 4 | default (20) | 0.25 | default (0.25) |
| 8 | chemplp | 4 | default (20) | default (0.15) | 1 |
| 9 | plp | 4 | default (20) | default (0.2) | default (0.5) |
| 10 | plp95 | 4 | default (20) | default (0.2) | default (1.25) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorgulla, C.; Çınaroğlu, S.S.; Fischer, P.D.; Fackeldey, K.; Wagner, G.; Arthanari, H. VirtualFlow Ants—Ultra-Large Virtual Screenings with Artificial Intelligence Driven Docking Algorithm Based on Ant Colony Optimization. Int. J. Mol. Sci. 2021, 22, 5807. https://doi.org/10.3390/ijms22115807
Gorgulla C, Çınaroğlu SS, Fischer PD, Fackeldey K, Wagner G, Arthanari H. VirtualFlow Ants—Ultra-Large Virtual Screenings with Artificial Intelligence Driven Docking Algorithm Based on Ant Colony Optimization. International Journal of Molecular Sciences. 2021; 22(11):5807. https://doi.org/10.3390/ijms22115807
Chicago/Turabian StyleGorgulla, Christoph, Süleyman Selim Çınaroğlu, Patrick D. Fischer, Konstantin Fackeldey, Gerhard Wagner, and Haribabu Arthanari. 2021. "VirtualFlow Ants—Ultra-Large Virtual Screenings with Artificial Intelligence Driven Docking Algorithm Based on Ant Colony Optimization" International Journal of Molecular Sciences 22, no. 11: 5807. https://doi.org/10.3390/ijms22115807