
Autosomal Dominant Gyrate Atrophy-Like Choroidal Dystrophy Revisited: 45 Years Follow-Up and Association with a Novel C1QTNF5 Missense Variant

 , , and
, , and
Abstract
:1. Introduction
2. Results
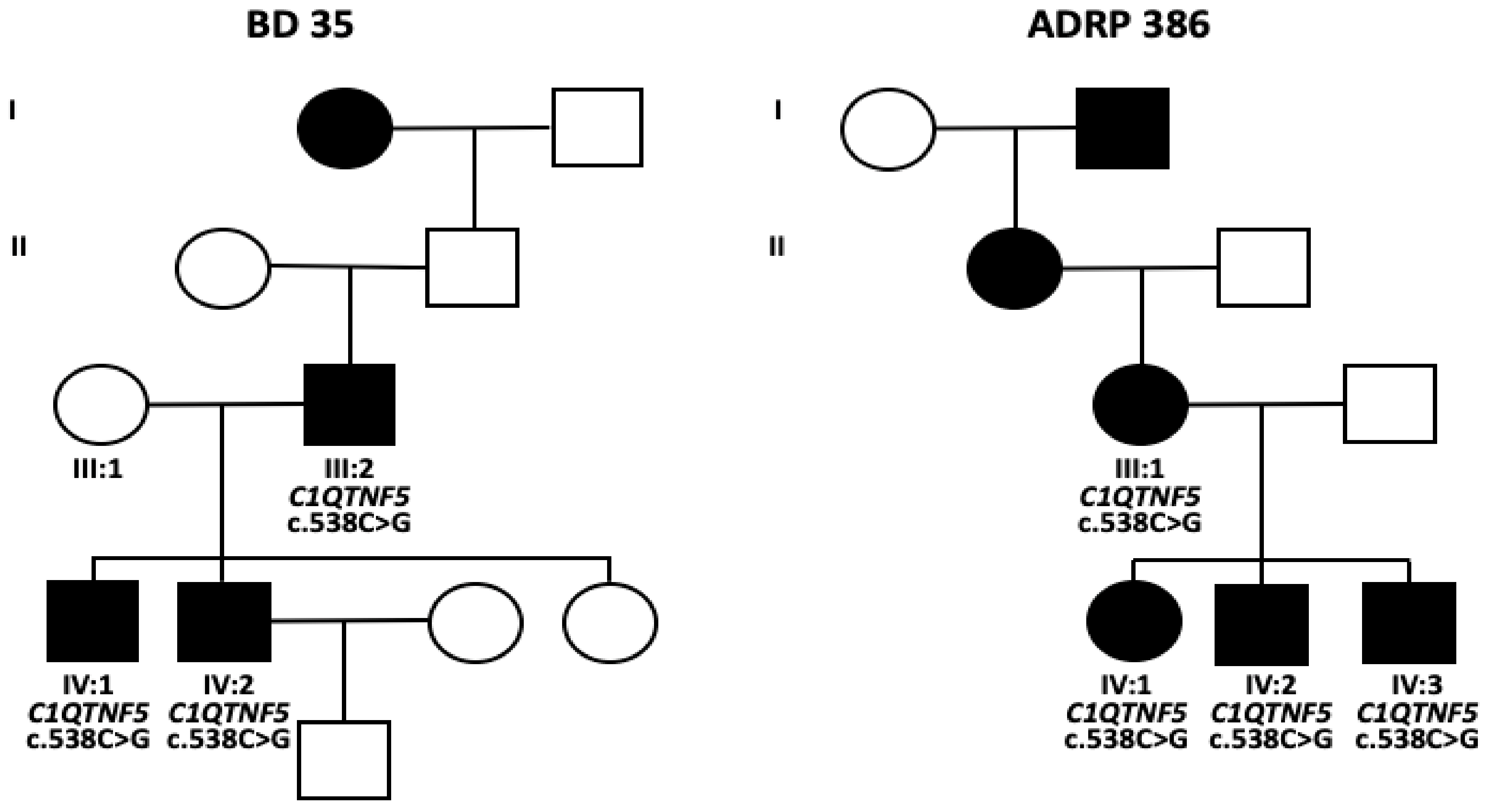
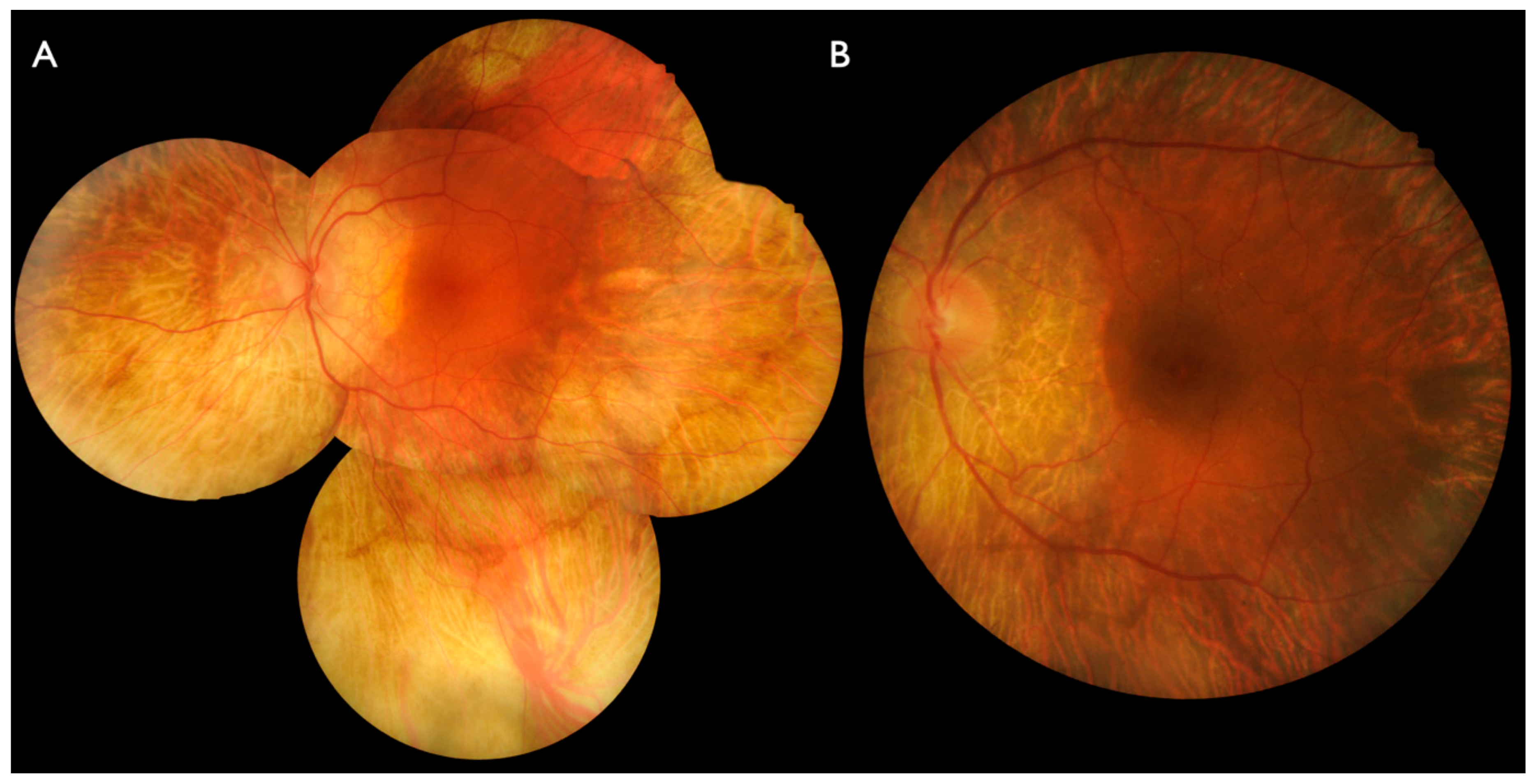
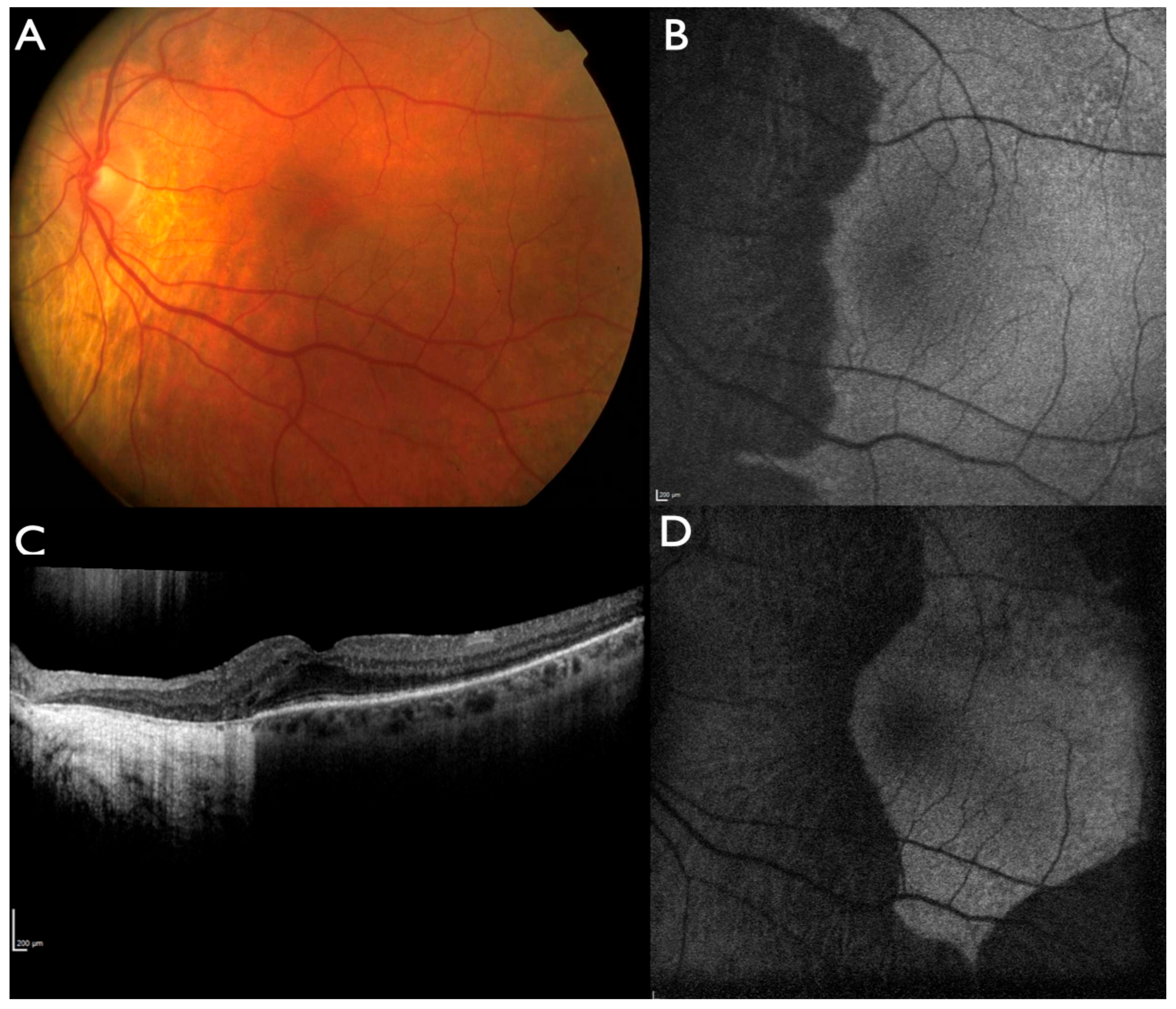
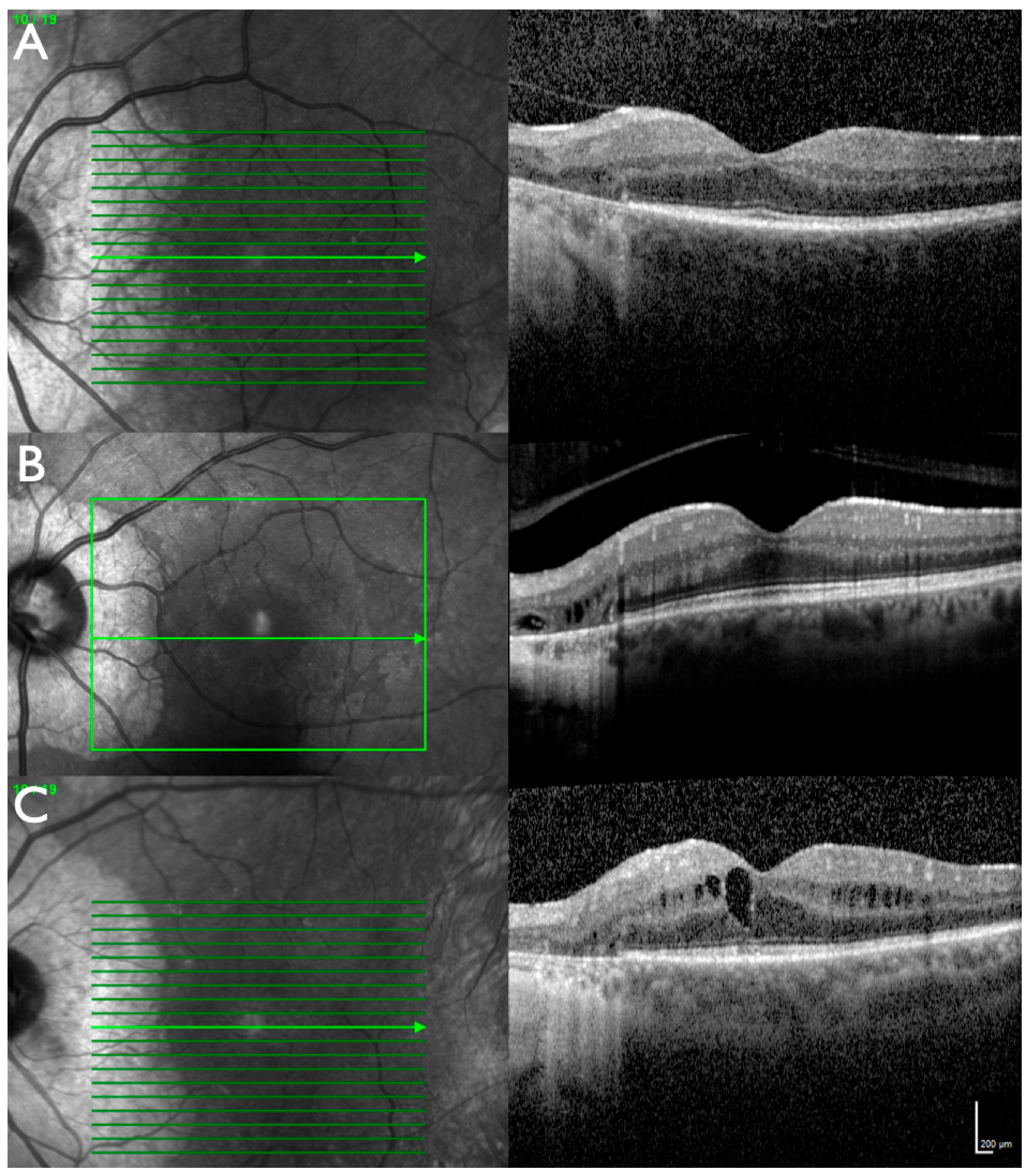
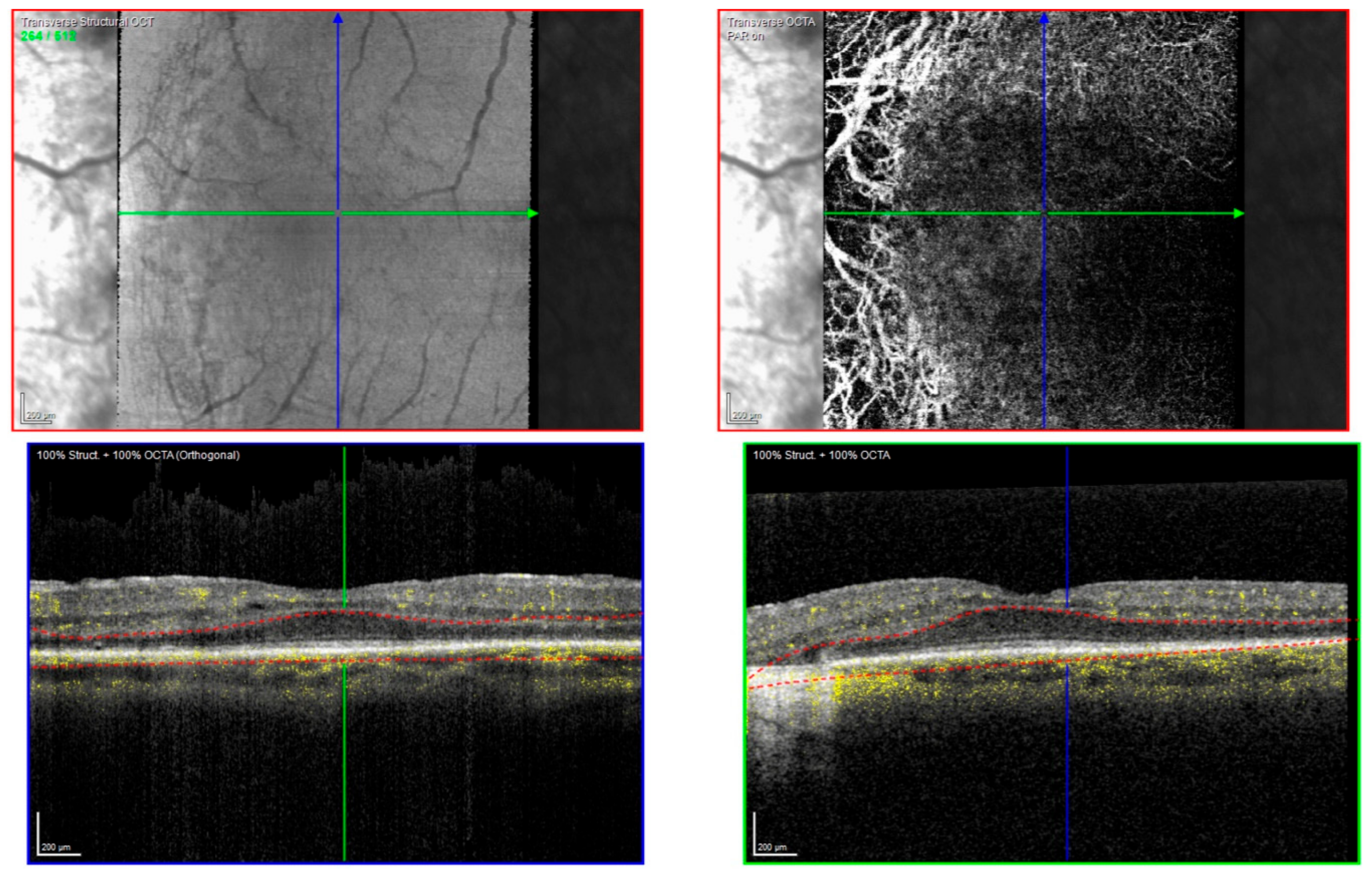
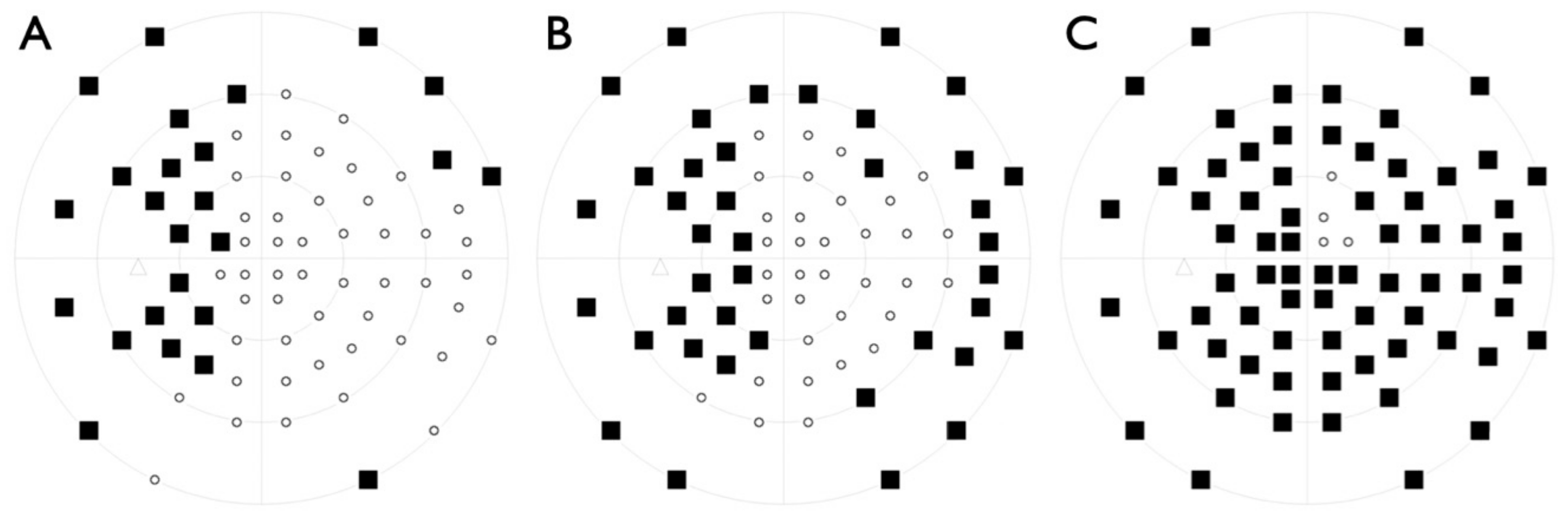
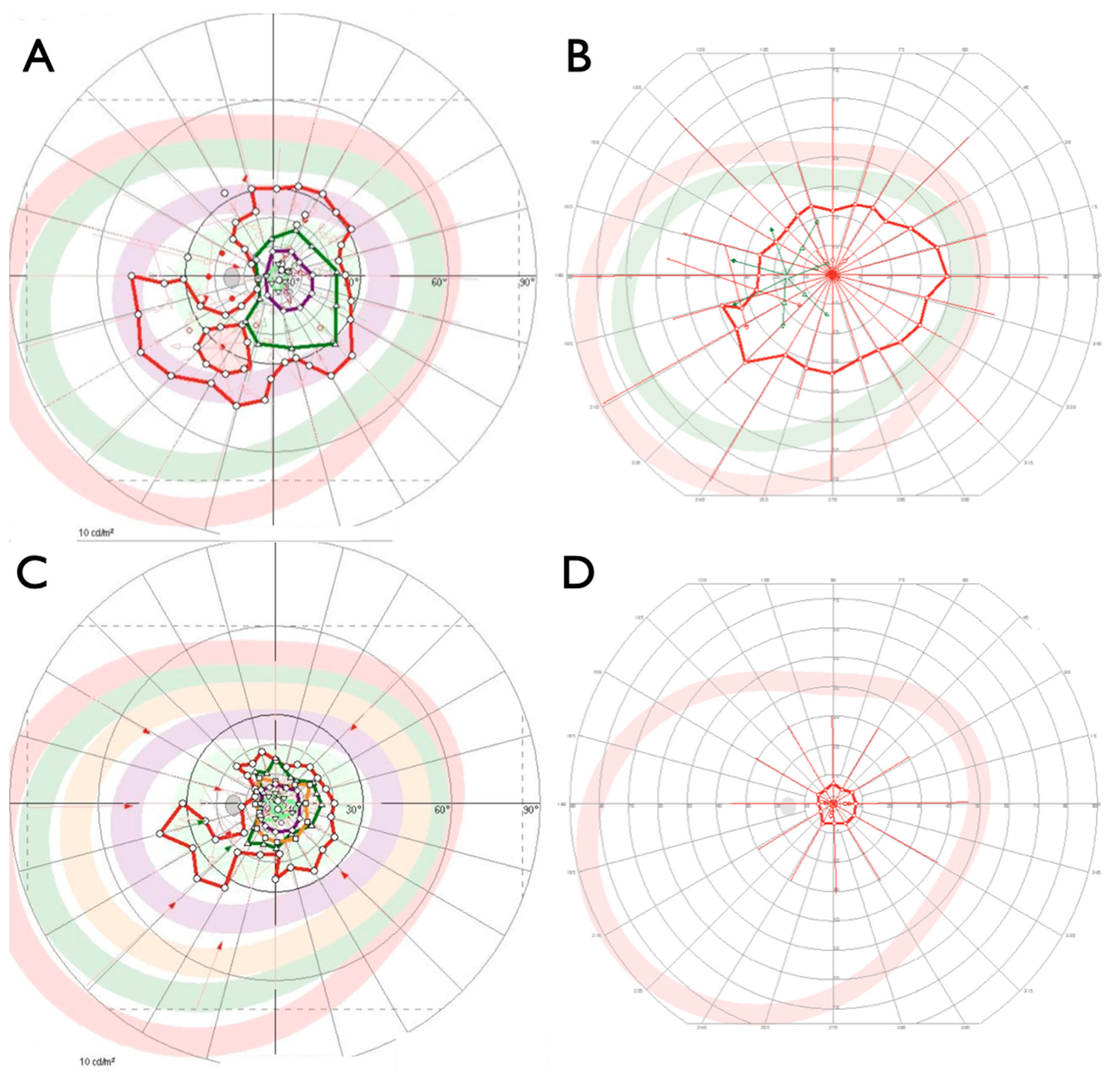
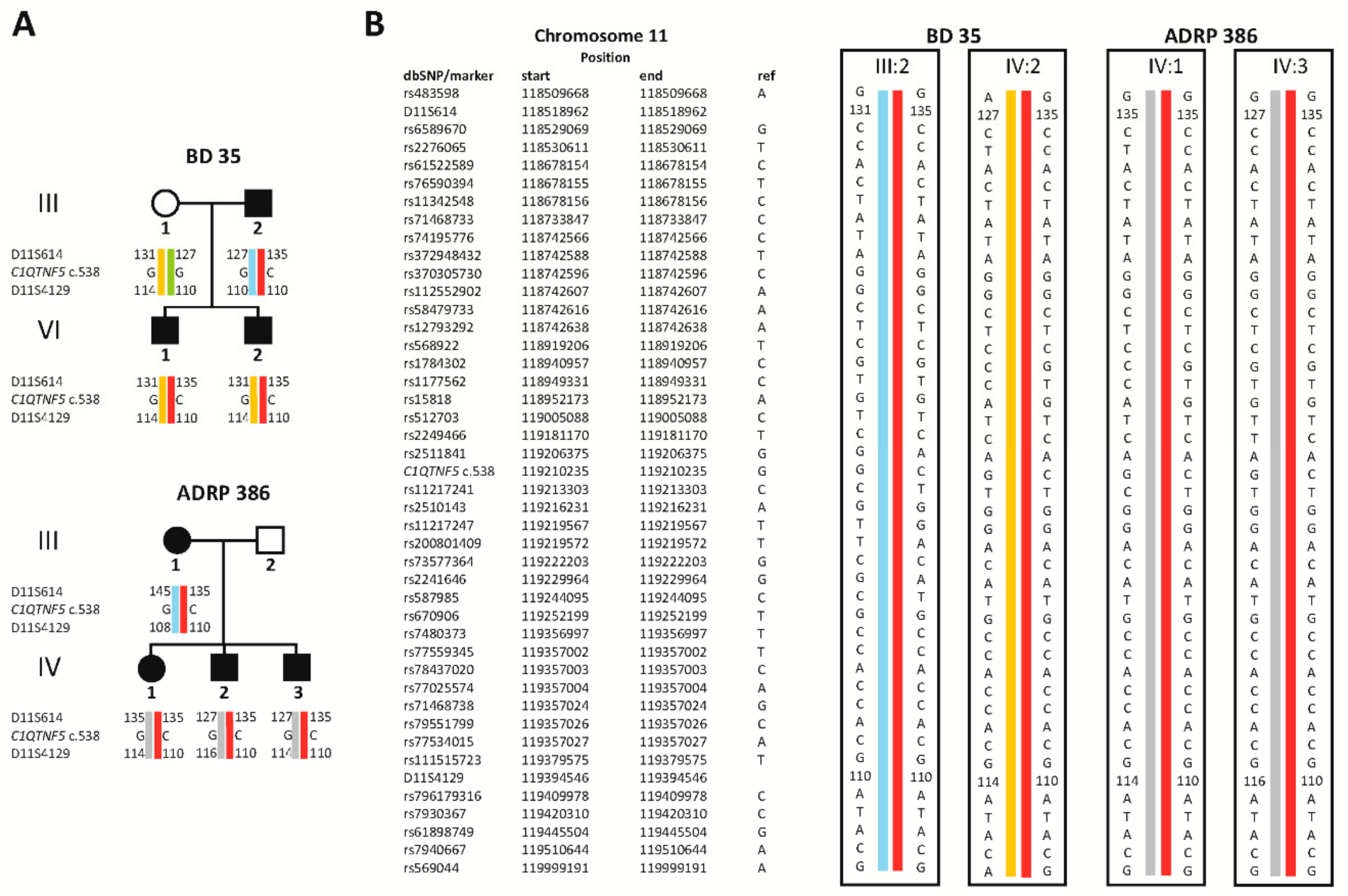
2.1. Clinical Evaluation of Family Members
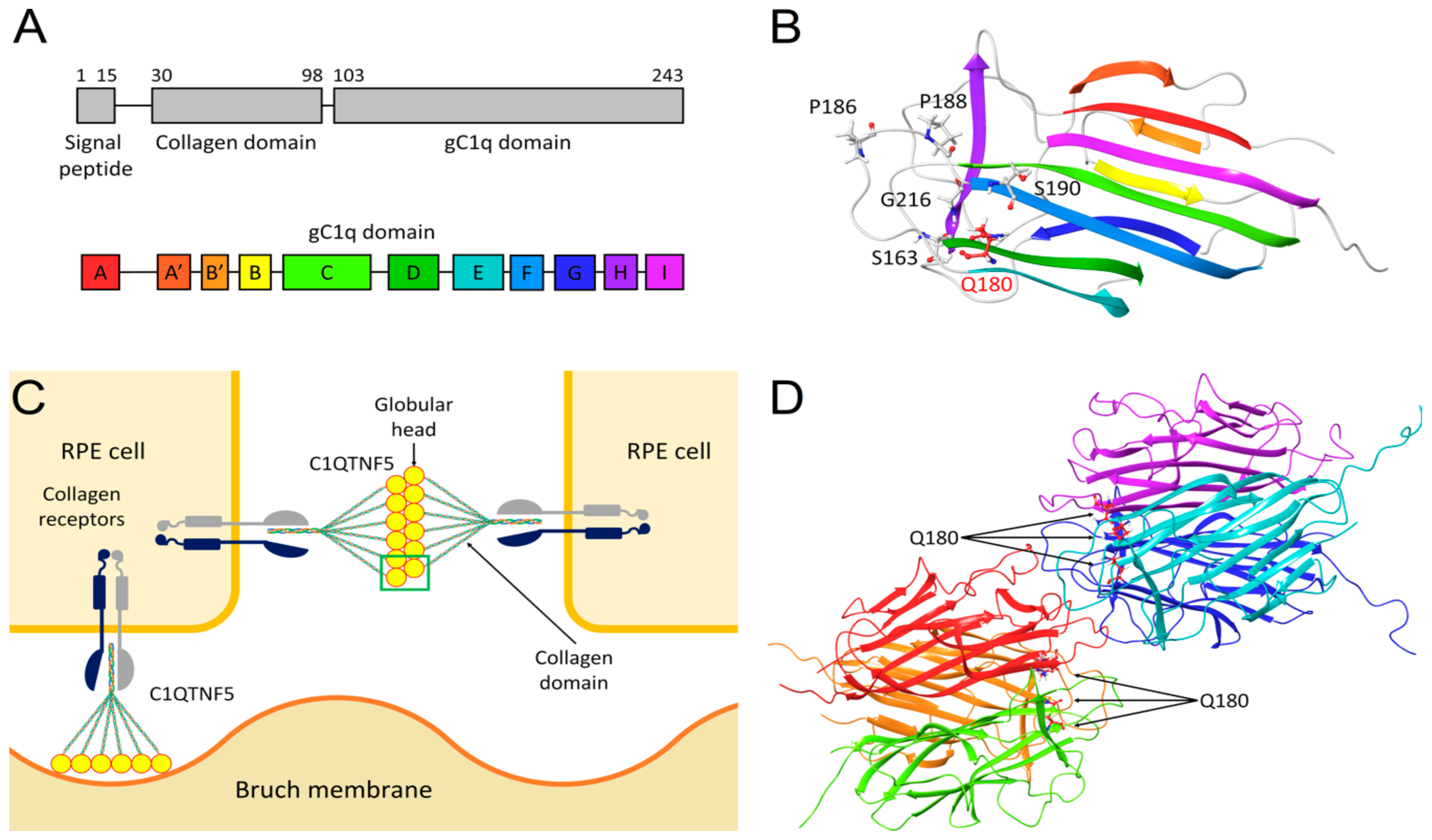
2.2. Identification of a Novel Missense Variant in C1QTNF5
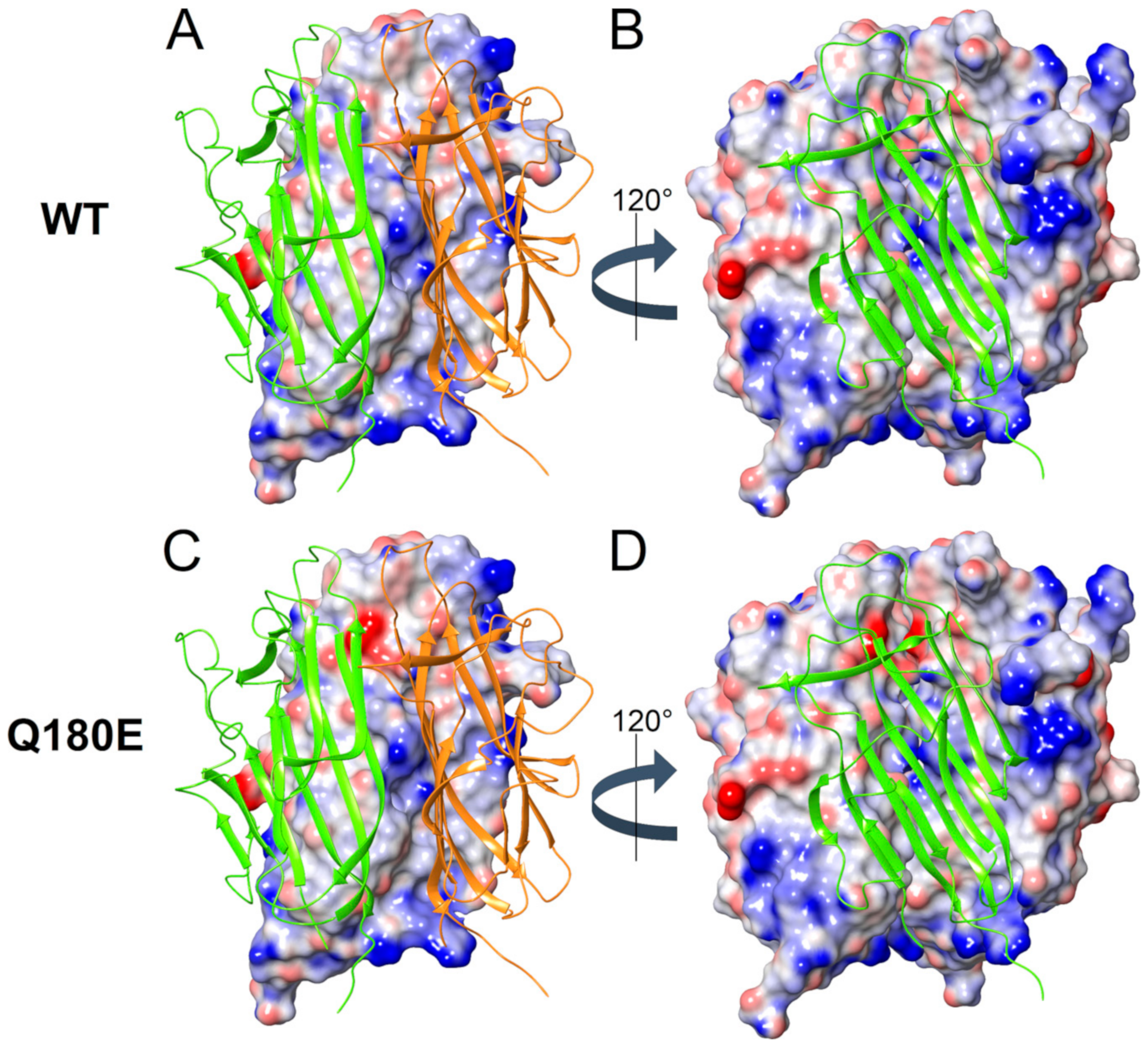
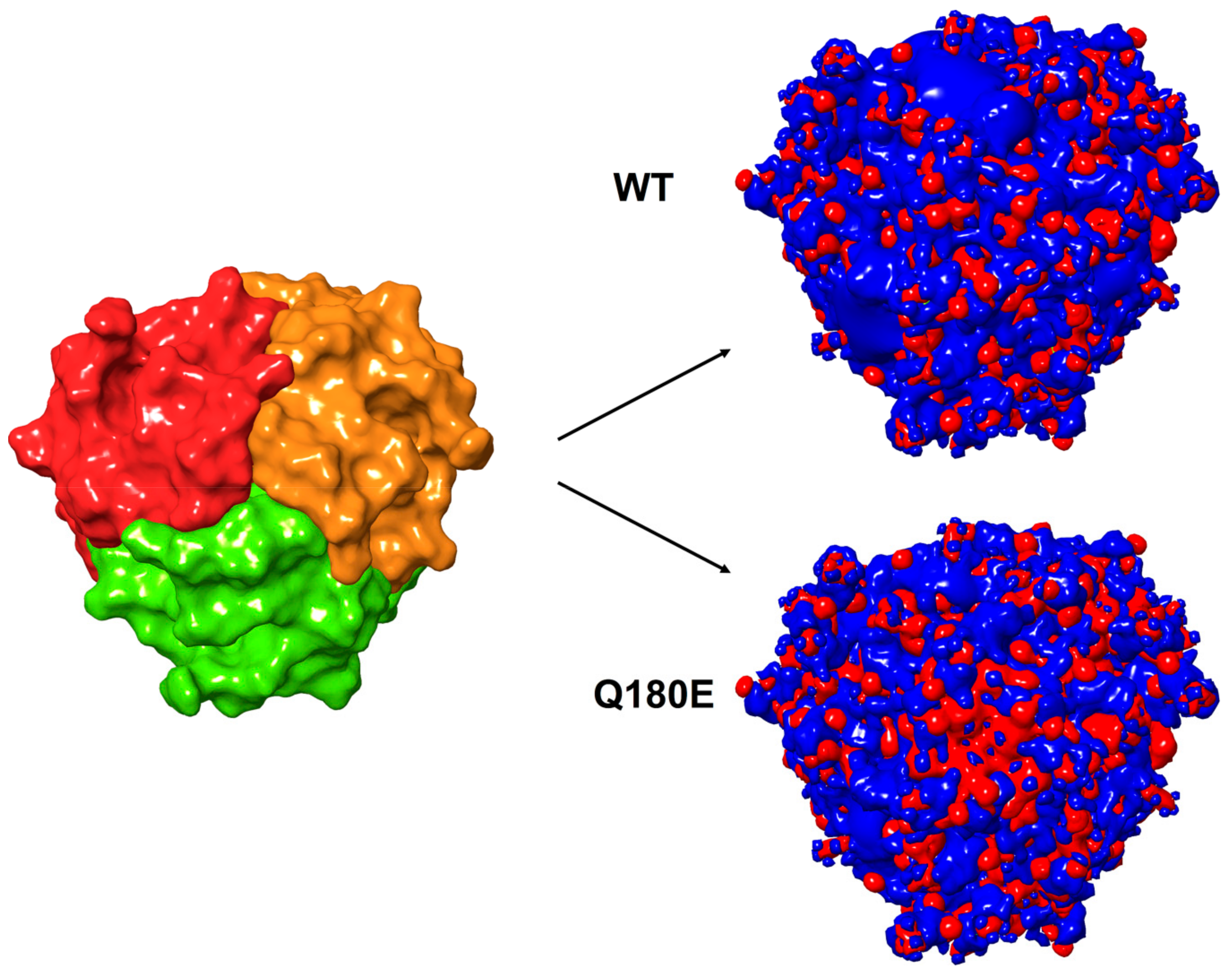
2.3. Molecular Modeling of C1QTNF5 and Analysis of the Structural and Functional Effects of the Missense Mutation p.(Q180E)
3. Discussion
4. Materials and Methods
4.1. Patient Enrollment and Retrieval of Blood Samples
4.2. Clinical Examination
4.3. Diagnostic Genetic Testing
4.4. Haplotype Analysis
4.5. In Silico Predictions of Pathogenicity
4.6. Molecular Modeling of C1QTNF5 Protein Structure and Analysis of the p.(Q180E) Effects on Stability and Affinity
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| adGALCD | Autosomal dominant gyrate atrophy-like choroidal dystrophy |
| ERG | Electroretinography |
| FAF | Fundus autofluorescence |
| LORD | Late onset retinal dystrophy |
| mfERG | Multifocal electroretinography |
| OCT | Optical coherence tomography |
| RPE | Retinal pigment epithelium |
| WT | Wild-type |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PolyPhen2 | MutationTaster | PROVEAN | SIFT | phyloP | CADD | FATHMM-MKL |
|---|---|---|---|---|---|---|
| Possibly damaging (0.692) | Disease causing (0.999) | Neutral (0.38) | Tolerated (0.254) | 9.071 | 25.00 | 0.98 |
| Species | Amino Acid Snippet |
|---|---|
| Homo sapiens | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Acanthisitta chloris | 178 FFQYYGNWPKPTSLSGGTLVR 198 |
| Acinonyx jubatus | 92 FFQFFGGWPKPASLSGGAMVR 112 |
| Ailuropoda melanoleuca | 88 FFQFFGGWPKPASLSGGAMVR 108 |
| Alligator mississippiensis | 176 FFQFYGNWPKPTSLSGGSLVR 196 |
| Alligator sinensis | 176 FFQFYGNWPKPTSLSGGSLVR 196 |
| Anas platyrhynchos | 353 FFQYYGNWPKPTSLSGGALVR 373 |
| Anolis carolinensis | 180 FFQFYGNWPKPTSLSGGALVR 200 |
| Antrostomus carolinensis | 64 FFQYYGNWPKPTSLSGGALVR 84 |
| Aotus nancymaae | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Aptenodytes forsteri | 178 FFQYYGNWPKPTSLSGGSLVR 198 |
| Astyanax mexicanus | 183 YFQFFGNWSKPASLSGGTLMH 203 |
| Balaenoptera acutorostrata scammoni | 151 FFQFFGGWPKPASLSGGAMVR 171 |
| Boleophthalmus pectinirostris | 184 YFQFYGNWPKPVSLSGGSLLH 204 |
| Bos indicus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Bos mutus | 134 FFQFFGGWPKPASLSGGAMVR 154 |
| Bos taurus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Bubalus bubalis | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Calidris pugnax | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Callithrix jacchus | 289 FFQFFGGWPKPASLSGGAMVR 309 |
| Callorhinchus milii | 180 FFQFYGNWTKPVSLSGGSLVH 200 |
| Calypte anna | 180 FFQYYGNWPKPTSLSGGALVR 200 |
| Camelus bactrianus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Camelus ferus | 90 FFQFFGGWPKPASLSGGAMVR 110 |
| Canis lupus | 189 FFQFFGGWPKPASLSGGAMVR 209 |
| Capra hircus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Castor canadensis | 238 FFQFFGGWPKPASLSGGAMVR 258 |
| Cavia porcellus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Ceratotherium simum simum | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Cercocebus atys | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Chaetura pelagica | 150 FFQYYGNWPKPTSLSGGALVR 170 |
| Charadrius vociferus | 177 FFQYYGNWPKPTSLSGGALVR 197 |
| Chelonia mydas | 176 FFQFYGNWPKPTSLSGGALVR 196 |
| Chinchilla lanigera | 178 FFQFFGGWPKPASLSGGTMVR 198 |
| Chlorocebus sabaeus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Chrysemys picta | 181 FFQFYGNWPKPTSLSGGALVR 201 |
| Chrysochloris asiatica | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Clupea harengus | 183 YFQFFGNWSKPASLSGGTLAH 203 |
| Columba livia | 178 FFQYYGNWPKPTSLSGGSLVR 198 |
| Condylura cristata | 276 FFQFFGGWPKPTSLSGGAMVR 296 |
| Corvus brachyrhynchos | 178 FFQYYGNWPKPTSLSGGTLVR 198 |
| Coturnix japonica | 182 FFQYYGNWPKPTSLSGGALVR 202 |
| Cricetulus griseus | 273 FFQFFGGWPKPASLSGGAMVR 293 |
| Crocodylus porosus | 176 FFQFYGNWPKPASLSGGSLVR 196 |
| Cuculus canorus | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Cynoglossus semilaevis | 187 YFQFYGNWPKPASLSGGSLLH 207 |
| Cyprinodon variegatus | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Cyprinus carpio | 183 YFQIFGNWSKPASLSGGTLVH 203 |
| Danio rerio | 183 YFQIFGNWSKPASLSGGTLVH 203 |
| Dasypus novemcinctus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Dipodomys ordii | 130 FFQFFGGWPKPASLSGGAMVR 150 |
| Egretta garzetta | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Elephantulus edwardii | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Eptesicus fuscus | 80 FFQVFGGWPKPASLSGGAMVR 100 |
| Equus asinus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Equus caballus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Equus przewalskii | 60 FFQFFGGWPKPASLSGGAMVR 80 |
| Erinaceus europaeus | 322 FFQFFGGWPKPTSLSGGAMVR 342 |
| Esox lucius | 183 YFQFYGNWPKPVSLTGGSLLH 203 |
| Falco cherrug | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Falco peregrinus | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Felis catus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Ficedula albicollis | 178 FFQYYGNWPKPTSLSGGTLVR 198 |
| Fukomys damarensis | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Fundulus heteroclitus | 185 YFQYYGNWSKPASLSGGTMLH 205 |
| Gadus morhua | 245 YFQFYGNWPKPASLSGGTMLH 265 |
| Galeopterus variegatus | 92 FFQFFGGWPKPASLSGGAMVR 112 |
| Gallus gallus | 182 FFQYYGNWPKPTSLSGGALVR 202 |
| Gavialis gangeticus | 176 FFQFYGNWPKPASLSGGSLVR 196 |
| Geospiza fortis | 178 FFQYYGNWPKPTSLSGGTLVR 198 |
| Gorilla gorilla | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Haliaeetus leucocephalus | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Haplochromis burtoni | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Heterocephalus glaber | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Hippocampus comes | 185 YFQFFGNWPKPVSLSGGSLLH 205 |
| Hipposideros armiger | 236 FFQFFGGWPKPASLSGGTMVR 256 |
| Ictalurus punctatus | 183 YFQMFGNWSKPASLSGGTLLH 203 |
| Ictidomys tridecemlineatus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Jaculus jaculus | 179 FFQFFGGWPKPASLSGGAMVR 199 |
| Kryptolebias marmoratus | 224 YFQIYGNWSKPASLSGGSLLH 244 |
| Labrus bergylta | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Larimichthys crocea | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Lates calcarifer | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Latimeria chalumnae | 179 FFQFYGNWPKPSSLSGGTLLH 199 |
| Lepidothrix coronata | 178 FFQYYGNWPKPTSLSGGTLVR 198 |
| Lepisosteus oculatus | 183 YFQYYGNWPKPASLSGGSLLH 203 |
| Leptonychotes weddellii | 153 FFQFFGGWPKPASLSGGAMVR 173 |
| Leptosomus discolor | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Lipotes vexillifer | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Loxodonta africana | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Macaca fascicularis | 327 FFQFFGGWPKPASLSGGAMVR 347 |
| Macaca mulatta | 327 FFQFFGGWPKPASLSGGAMVR 347 |
| Macaca nemestrina | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Manacus vitellinus | 178 FFQYYGNWPKPTSLSGGTLVR 198 |
| Mandrillus leucophaeus | 166 FFQFFGGWPKPASLSGGAMVR 186 |
| Manis javanica | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Marmota marmota marmota | 88 FFQFFGGWPKPASLSGGAMVR 108 |
| Maylandia zebra | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Meleagris gallopavo | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Melopsittacus undulatus | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Mesitornis unicolor | 182 FFQYYGNWPKPTSLSGGALVR 202 |
| Mesocricetus auratus | 203 FFQFFGGWPKPASLSGGAMVR 223 |
| Microcebus murinus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Microtus ochrogaster | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Monodelphis domestica | 261 FFQFFGGWPKPASLSGGALVR 281 |
| Monopterus albus | 185 YFQFYGNWSKPASLSGGSLLH 205 |
| Mus musculus | 178 FFQYFGGWPKPASLSGGAMVR 198 |
| Mustela putorius furo | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Myotis brandtii | 51 FFQFFGGWPKPASLSGGAMVR 71 |
| Myotis lucifugus | 120 FFQFFGGWPKPASLSGGAMVR 140 |
| Nannospalax galili | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Nipponia nippon | 178 FFQYYGNWPKPTSLSGGALVR 198 |
| Nomascus leucogenys | 201 FFQFFGGWPKPASLSGGAMVR 221 |
| Nothobranchius furzeri | 198 YFQFYGNWPKPASLSGGSLLH 218 |
| Notothenia coriiceps | 185 YFQYYGNWPKPASLSGGSLLH 205 |
| Ochotona princeps | 179 FFQFFGGWPKPASLSGGAMVR 199 |
| Octodon degus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Odobenus rosmarus divergens | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Oncorhynchus kisutch | 186 YFQFYGNWPKPASLSGGSLLH 206 |
| Oncorhynchus mykiss | 184 YFQFYGNWPKPASLSGGSLLH 204 |
| Orcinus orca | 178 FFQFFGGWPKPASLSGGTMVR 198 |
| Oreochromis niloticus | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Ornithorhynchus anatinus | 181 FFQFFGGWPKPASLSGGALVR 201 |
| Orycteropus afer afer | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Oryctolagus cuniculus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Oryzias latipes | 183 YFQFYGSWPKPASLSGGSLLH 203 |
| Otolemur garnettii | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Ovis aries | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Pan paniscus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Pan troglodytes | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Panthera pardus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Panthera tigris altaica | 79 FFQFFGGWPKPASLSGGAMVR 99 |
| Pantholops hodgsonii | 91 FFQFFGGWPKPASLSGGAMVR 111 |
| Papio anubis | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Paralichthys olivaceus | 185 YFQFYGNWPKPGSLSGGSLLH 205 |
| Parus major | 178 FFQYYGNWPKPTSLSGGSLVR 198 |
| Pelodiscus sinensis | 176 FFQFYGNWPKPTSLSGGALVR 196 |
| Peromyscus maniculatus bairdii | 273 FFQFFGGWPKPASLSGGAMVR 293 |
| Phascolarctos cinereus | 178 FFQFFGGWPKPASLSGGALVR 198 |
| Physeter catodon | 234 FFQFFGGWPKPASLSGGAMVR 254 |
| Picoides pubescens | 182 FFQYYGNWPKPTSLSGGALVR 202 |
| Poecilia formosa | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Poecilia latipinna | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Poecilia reticulata | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Pogona vitticeps | 182 FFQFYGNWPKPTSLSGGVLVR 202 |
| Pongo abelii | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Propithecus coquereli | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Pseudopodoces humilis | 178 FFQYYGNWPKPTSLSGGSLVR 198 |
| Pteropus alecto | 225 FFQFFGGWPKPASLSGGAMVR 245 |
| Pteropus vampyrus | 225 FFQFFGGWPKPASLSGGAMVR 245 |
| Pundamilia nyererei | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Pygocentrus nattereri | 193 YFQIFGNWSKPASLSGGTLLH 213 |
| Python bivittatus | 189 FFQFYGNWPKPTSLSGGSLVR 209 |
| Rattus norvegicus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Rhinolophus sinicus | 178 FFQFFGGWPKPASLSGGTMVR 198 |
| Rhinopithecus bieti | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Rhinopithecus roxellana | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Rousettus aegyptiacus | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Saimiri boliviensis | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Salmo salar | 186 YFQFYGNWPKPASLSGGSLLH 206 |
| Sarcophilus harrisii | 154 FFQFFGGWPKPASLSGGALVR 174 |
| Scleropages formosus | 182 YFQFYANWPKPASLSGGSLLH 202 |
| Sorex araneus | 178 FFQFFGGWPKPASLSGGTMVR 198 |
| Stegastes partitus | 185 YFQFYGNWPKPASLSGGSLLH 205 |
| Sturnus vulgaris | 178 FFQYYGNWPKPTSLSGGTLVR 198 |
| Sus scrofa | 195 FFQFFGGWPKPASLSGGAMVR 215 |
| Taeniopygia guttata | 251 FFQYYGNWPKPTSLSGGTLVR 271 |
| Takifugu rubripes | 231 YFQFYGNWPKPVSLSGSSLLH 251 |
| Tauraco erythrolophus | 168 FFQYYGNWPKPTSLSGGALVR 188 |
| Tinamus guttatus | 182 FFQYYGNWPKPTSLSGGALVR 202 |
| Trichechus manatus latirostris | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Tupaia chinensis | 178 FFQFFGGWPKPASLSGGAMVR 198 |
| Ursus maritimus | 55 FFQFFGGWPKPASLSGGAMVR 75 |
| Vicugna pacos | 131 FFQFFGGWPKPASLSGGAMVR 151 |
| Xenopus tropicalis | 179 FFQL-GDAKKPVGLCGGAALR 198 |
| Xiphophorus maculatus | 185 YFQFYGNWPKPASLSGGSLLH 205 |

References
- Montioli, R.; Bellezza, I.; Desbats, M.A.; Voltattorni, C.B.; Salviati, L.; Cellini, B. Deficit of human ornithine aminotransferase in gyrate atrophy: Molecular, cellular, and clinical aspects. Biochim. Biophys. Acta BBA Proteins Proteom. 2021, 1869, 140555. [Google Scholar] [CrossRef]
- Shen, L.L.; Ahluwalia, A.; Sun, M.; Young, B.K.; Nardini, H.K.G.; Del Priore, L.V. Long-term natural history of visual acuity in eyes with choroideremia: A systematic review and meta-analysis of data from 1004 individual eyes. Br. J. Ophthalmol. 2021, 105, 271–278. [Google Scholar] [CrossRef]
- Bowne, S.J.; Humphries, M.M.; Sullivan, L.S.; Kenna, P.F.; Tam, L.C.S.; Kiang, A.S.; Campbell, M.; Weinstock, G.M.; Koboldt, D.C.; Ding, L.; et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur. J. Hum. Genet. 2011, 19, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, K.; Fujinami, K.; Yoshitake, K.; Iwata, T. Late-onset night blindness with peripheral flecks accompanied by progressive trickle-like macular degeneration. Doc. Ophthalmol. 2019, 139, 171–184. [Google Scholar] [CrossRef]
- Hayasaka, S.; Shoji, K.; Kanno, C.-I.; Oura, F.; Mizuno, K. Differential diagnosis of diffuse choroidal atrophies. Retina 1985, 5, 30–37. [Google Scholar] [CrossRef]
- Marchesani, O.; Sautter, H. Atlas des Augenhintergrundes; Urban & Schwarzenberg: München, Germany, 1957; Volume 2. [Google Scholar]
- Kellner, U.; Weleber, R.G.; Kennaway, N.G.; Fishman, G.A.; Foerster, M.H. Gyrate atrophy-like phenotype with normal plasma ornithine. Retina 1997, 17, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, A.C.; Jacobson, S.G.; Cideciyan, A.V.; Li, Z.Y.; Stone, E.M.; Possin, D.; Milam, A.H. Sub-retinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1772–1782. [Google Scholar]
- Duvall, J.; McKechnie, N.M.; Lee, W.R.; Rothery, S.; Marshall, J. Extensive subretinal pigment epithelial deposit in two brothers suffering from dominant retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. 1986, 224, 299–309. [Google Scholar] [CrossRef]
- Brosnahan, D.M.; Kennedy, S.M.; Converse, C.A.; Lee, W.R.; Hammer, H.M. Pathology of hereditary retinal degeneration associated with hypobetalipoproteinemia. Ophthalmology 1994, 101, 38–45. [Google Scholar] [CrossRef]
- Hayward, C.; Shu, X.; Cideciyan, A.V.; Lennon, A.; Barran, P.; Zareparsi, S.; Sawyer, L.; Hendry, G.; Dhillon, B.; Milam, A.H.; et al. Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: A genetic model for age-related macular degeneration. Hum. Mol. Genet. 2003, 12, 2657–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanton, C.M.; Borooah, S.; Drake, C.; Marsh, J.A.; Campbell, S.; Lennon, A.; Soares, D.C.; Vallabh, N.A.; Sahni, J.; Cideciyan, A.V.; et al. Novel pathogenic mutations in C1QTNF5 support a dominant negative disease mechanism in late-onset retinal degeneration. Sci. Rep. 2017, 7, 12147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borooah, S.; Stanton, C.M.; Marsh, J.; Carss, K.J.; Waseem, N.; Biswas, P.; Agorogiannis, G.; Raymond, L.; Arno, G.; Webster, A.R. Whole genome sequencing reveals novel mutations causing autosomal dominant inherited macular degeneration. Ophthalmic Genet. 2018, 39, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.W.; Krawczyk, S.A.; Kitidis-Mitrokostas, C.; Revett, T.; Gimeno, R.; Lodish, H.F. Molecular, biochemical and functional characterizations of C1q/TNF family members: Adipose-tissue-selective expression patterns, regulation by PPAR-γ agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem. J. 2008, 416, 161–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, M.N.A.; Vasireddy, V.; Jablonski, M.M.; Ayyagari, R.; Reddy, G.B.; Wang, X.; Moroi, S.E.; Pattnaik, B.R.; Hughes, B.A.; Heckenlively, J.R.; et al. CTRP5 is a membrane-associated and secretory protein in the rpe and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Investig. Opthalmol. Vis. Sci. 2006, 47, 5505–5513. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Palczewski, K. Crystal structure of the globular domain of C1QTNF5: Implications for late-onset retinal macular degeneration. J. Struct. Biol. 2012, 180, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Shu, X.; Tulloch, B.; Lennon, A.; Vlachantoni, D.; Zhou, X.; Hayward, C.; Wright, A.F. Disease mechanisms in late-onset retinal macular degeneration associated with mutation in C1QTNF5. Hum. Mol. Genet. 2006, 15, 1680–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, X.; Palczewski, K. The macular degeneration-linked C1QTNF5 (S163) mutation causes higher-order structural rearrangements. J. Struct. Biol. 2014, 186, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, L.; Scherer, P.E. The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Curr. Biol. 1998, 8, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Dell’Orco, D.; Xue, W.-F.; Thulin, E.; Linse, S. Electrostatic contributions to the kinetics and thermodynamics of protein assembly. Biophys. J. 2005, 88, 1991–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milam, A.H.; Curcio, A.C.; Cideciyan, A.V.; Saxena, S.; John, S.K.; Kruth, H.S.; Malek, G.; Heckenlively, J.R.; Weleber, R.G.; Jacobson, S.G. Dominant late-onset retinal degeneration with regional variation of sub-retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology 2000, 107, 2256–2266. [Google Scholar] [CrossRef]
- Borooah, S.; Collins, C.; Wright, A.; Dhillon, B. Late-onset retinal macular degeneration: Clinical insights into an inherited retinal degeneration. Postgrad. Med. J. 2009, 85, 495–500. [Google Scholar] [CrossRef]
- Vincent, A.; Munier, F.L.; VandenHoven, C.C.; Wright, T.; Westall, C.A.; Héon, E. The characterization of retinal phenotype in a family with c1qtnf5-related late-onset retinal degeneration. Retina 2012, 32, 1643–1651. [Google Scholar] [CrossRef]
- Mandal, N.; Lotery, A.J. Multimodal imaging of late-onset retinal degeneration complicated by bilateral choroidal neovascularization. Eye 2019, 33, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Papastavrou, V.T.; O’Brien, J.M.; Regan, A.J.; Aftab, A.M.; Browning, A.C. The progression of macular structural and func-tional changes in late-onset retinal degeneration. Retin. Cases Brief. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Borooah, S.; Papastavrou, V.T.; Browning, A.C.; Lando, L.; Moghimi, S.; Lin, T.; Dans, K.; Motevasseli, T.; Cameron, J.R.; Freeman, W.R.; et al. Characterizing the natural history of foveal-sparing atrophic late-onset retinal degeneration. Retina 2020. [Google Scholar] [CrossRef]
- Borooah, S.; Collins, C.; Wright, A.; Dhillon, B. Late-onset retinal macular degeneration: Clinical insights into an inherited retinal degeneration. Br. J. Ophthalmol. 2009, 93, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Ayyagari, R.; Mandal, N.A.; Maumenee, I.H.; Sieving, P.A.; Karoukis, A.J.; Chen, L.; McLaren, N.C.; Lichter, M.; Wong, D.T.; Hitchcock, P.F.; et al. Late-onset macular degeneration and long anterior lens zonules result from ACTRP5gene mutation. Investig. Opthalmol. Vis. Sci. 2005, 46, 3363–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.N.; Borooah, S.; Webster, A.R.; Moore, A.T.; McKibbin, M.; Michaelides, M.; Lando, L.; Dans, K.; Mahroo, O.A.; Meshi, A.; et al. Quantifying the separation between the retinal pigment epithelium and bruch’s membrane using optical coherence tomography in patients with inherited macular degeneration. Transl. Vis. Sci. Technol. 2020, 9, 26. [Google Scholar] [CrossRef]
- Ramtohul, P.; Gascon, P.; Matonti, F. Choroidal neovascularization in late-onset retinal macular degeneration. Ophthalmol. Retin. 2019, 3, 153. [Google Scholar] [CrossRef]
- Cukras, C.; Flamendorf, J.; Wong, W.T.; Ayyagari, R.; Cunningham, D.; Sieving, P.A. Longitudinal structural changes in late-onset retinal degeneration. Retina 2016, 36, 2348–2356. [Google Scholar] [CrossRef] [Green Version]
- Soumplis, V.; Sergouniotis, P.I.; Robson, A.G.; Michaelides, M.; Moore, A.T.; Holder, G.E.; Webster, A.R. Phenotypic findings inC1QTNF5retinopathy (late-onset retinal degeneration). Acta Ophthalmol. 2013, 91, e191–e195. [Google Scholar] [CrossRef]
- Papastavrou, V.T.; Bradshaw, K.R.; Aye, K.H.; Turney, C.; Browning, A.C. Improvement of retinal function in L-ORD after prolonged dark adaptation. Can. J. Ophthalmol. 2015, 50, 112–118. [Google Scholar] [CrossRef]
- Troumani, Y.; Beral, L.; David, T. Traitement d’une membrane néovasculaire choroïdienne chez un patient présentant une dystrophie rétinienne autosomique dominante rare par aflibercept: À propos d’un cas. J. Français d’Ophtalmol. 2015, 38, e67–e69. [Google Scholar] [CrossRef] [PubMed]
- Aye, K.H.; Gupta, R.; Talks, S.J.; Browning, A.C. Treatment of a choroidal neovascular membrane in a patient with late-onset retinal degeneration (L-ORD) with intravitreal Ranibizumab. Eye 2010, 24, 1528–1530. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Sumaroka, A.; Roman, A.J.; Wright, A.F. Late-onset retinal degeneration caused byC1QTNF5mutation. JAMA Ophthalmol. 2014, 132, 1252. [Google Scholar] [CrossRef] [Green Version]
- Almoguera, B.; Li, J.; Garcia-Sandoval, B.; Guo, Y.; Tian, L.; Liu, X.; Guan, L.; Zhang, J.; Keating, B.; Xu, X.; et al. Application of whole exome sequencing in six families with an initial diagnosis of autosomal dominant retinitis pigmentosa: Lessons learned. PLoS ONE 2015, 10, e0133624. [Google Scholar] [CrossRef] [PubMed]
- Ayyagari, R.; Griesinger, I.B.; Bingham, E.; Lark, K.K.; Moroi, S.E.; Sieving, P.A. Autosomal dominant hemorrhagic macular dystrophy not associated with the TIMP3 gene. Arch. Ophthalmol. 2000, 118, 85–92. [Google Scholar] [CrossRef]
- Subrayan, V.; Morris, B.; Armbrecht, A.M.; Wright, A.F.; Dhillon, B. Long anterior lens zonules in late-onset retinal degeneration (L-ORD). Am. J. Ophthalmol. 2005, 140, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.K.; Ayyagari, R.; Moroi, S.E. Possible association between long anterior lens zonules and plateau iris configuration. J. Glaucoma 2008, 17, 393–396. [Google Scholar] [CrossRef]
- Papastavrou, V.T.; Borooah, S.; O’Brien, J.M.; Ray-Chaudhuri, N.; Dhillon, B.; Vieira, R.V.; Browning, A.C. Cataract surgery in patients with late-onset retinal degeneration. J. Cataract. Refract. Surg. 2017, 43, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- Mandal, N.A.; Vasireddy, V.; Jablonski, M.M.; Wang, X.; Heckenlively, J.R.; Hughes, B.A.; Reddy, G.B.; Ayyagari, R. Spatial and temporal expression of MFRP and its interaction with CTRP5. Investig. Opthalmol. Vis. Sci. 2006, 47, 5514–5521. [Google Scholar] [CrossRef]
- Chekuri, A.; Zientara-Rytter, K.; Soto-Hermida, A.; Borooah, S.; Voronchikhina, M.; Biswas, P.; Kumar, V.; Goodsell, D.; Hayward, C.; Shaw, P.; et al. Late-onset retinal degeneration pathology due to mutations in CTRP5 is mediated through HTRA1. Aging Cell 2019, 18, e13011. [Google Scholar] [CrossRef] [Green Version]
- Dinculescu, A.; Dyka, F.M.; Min, S.-H.; Stupay, R.M.; Hooper, M.J.; Smith, W.C.; Hauswirth, W.W. Co-expression of wild-type and mutant S163R C1QTNF5 in retinal pigment epithelium. Results Probl. Cell Differ. 2018, 1074, 61–66. [Google Scholar] [CrossRef]
- Dinculescu, A.; Min, S.-H.; Dyka, F.M.; Deng, W.-T.; Stupay, R.M.; Chiodo, V.; Smith, W.C.; Hauswirth, W.W. Pathological effects of mutant C1QTNF5 (S163R) expression in murine retinal pigment epithelium. Investig. Opthalmol.Vis. Sci. 2015, 56, 6971–6980. [Google Scholar] [CrossRef] [Green Version]
- Lewis, T.R.; Makia, M.S.; Kakakhel, M.; Al-Ubaidi, M.R.; Arshavsky, V.Y.; Naash, M.I. Photoreceptor disc enclosure occurs in the absence of normal peripherin-2/RDS oligomerization. Front. Cell. Neurosci. 2020, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Okuda, A.; Naganuma, T.; Ohno, Y.; Abe, K.; Yamagata, M.; Igarashi, Y.; Kihara, A. Hetero-oligomeric interactions of an ELOVL4 mutant protein: Implications in the molecular mechanism of Stargardt-3 macular dystrophy. Mol. Vis. 2010, 16, 2438–2445. [Google Scholar] [PubMed]
- Kuehlewein, L.; Zobor, D.; Andreasson, S.O.; Ayuso, C.; Banfi, S.; Bocquet, B.; Bernd, A.S.; Biskup, S.; Boon, C.J.F.; Downes, S.M.; et al. Clinical phenotype and course of pde6a-associated retinitis pigmentosa disease, characterized in preparation for a gene supplementation trial. JAMA Ophthalmol. 2020, 138, 1241–1250. [Google Scholar] [CrossRef]
- Kellner, S.; Stöhr, H.; Fiebig, B.; Weinitz, S.; Farmand, G.; Kellner, U.; Weber, B.H.F. Fundus autofluorescence and SD-oct document rapid progression in autosomal dominant vitreoretinochoroidopathy (ADVIRC) Associated with a C.256G> A mutation in BEST1. Ophthalmic Genet. 2016, 37, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Marmor, M.F.; McCulloch, D.L.; Palmowski-Wolfe, A.M.; International Society for Clinical Electrophysiology of Vision. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. 2011, 124, 1–13. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef]
- Weisschuh, N.; Mazzola, P.; Heinrich, T.; Haack, T.; Wissinger, B.; Tonagel, F.; Kelbsch, C. First submicroscopic inversion of the OPA1 gene identified in dominant optic atrophy–a case report. BMC Med. Genet. 2020, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.K.; Van Cauwenbergh, C.; Rother, C.; Baumann, B.; Reuter, P.; De Baere, E.; Wissinger, B.; Kohl, S.; ACHM Study Group. CNGB3 mutation spectrum including copy number variations in 552 achromatopsia patients. Hum. Mutat. 2017, 38, 1579–1591. [Google Scholar] [CrossRef] [PubMed]
- Sankar, K.; Krystek, S.R.; Carl, S.M.; Day, T.; Maier, J.K.X. AggScore: Prediction of aggregation-prone regions in proteins based on the distribution of surface patches. Proteins Struct. Funct. Bioinform. 2018, 86, 1147–1156. [Google Scholar] [CrossRef] [PubMed]










| ID | Age First Exam | Age Last Exam | Follow-Up in Years | Age of Onset | Eye | Refraction First Exam | VA First Exam | Refraction Last Exam | VA Last Exam |
|---|---|---|---|---|---|---|---|---|---|
| BD 35 | |||||||||
| III:2 | 40 | 70 | 30 | 40 NB | OD | NA | 1.0 | +1.0 | 0.16 |
| OS | NA | 1.0 | +0.50 −1.25/64° | 0.16 | |||||
| IV:1 | 41 | - | 0 | 40 NB | OD | NA | 1.0 | - | - |
| OS | NA | 1.0 | - | - | |||||
| IV:2 | 18 | 63 | 45 | 18 RA, VFD | OD | +2.25 −2.25/11° | 1.0 | −0.50 −0.75/75° | 0.63 |
| OS | +2.25 −1.75/172° | 1.0 | +3.25 −1.25/166° | 0.63 | |||||
| ADRP 386 | |||||||||
| IV:1 | 58 | - | 0 | OD | −0.25 −2.25/168° | 0.5 | - | - | |
| OS | ±0.00 −0.50/25° | 0.63 | - | - | |||||
| IV:2 | 53 | - | 0 | 24 RA 39 NB 53 VFD, P | OD | +1.75 −1.50/94° | 1.0 | - | - |
| OS | +1.50 −1.50/95° | 1.0 | - | - | |||||
| IV:3 | 50 | 56 | 6 | 40 NB 50 VFD, AP, P | OD | −0.75 −1.75/90° | 0.63 | −0.75 −2.50/87° | 0.4 |
| OS | −0.25 −1.50/85° | 0.63 | −1.75 −1.50/95° | 0.25 | |||||
| ID | Anterior Segment | Retina | Optic Disc | ERG | Dark Adaptation | mfERG | Color Vision |
|---|---|---|---|---|---|---|---|
| BD 35 | |||||||
| III:2 | IOL OU (63) | Residual foveal island, no pigmentation | pale | No residual response (52/62) | ND | ND | ND |
| IV:1 | normal | Peripheral and peripapillary atrophic lesions, no pigmentation | vital | ND | ND | ND | ND |
| IV:2 | normal, IOL OD (63) | Progressive peripheral and peripapillary atrophic lesions, no pigmentation | vital | slightly abnormal (18); Severely reduced (42); No residual responses (54) | Mildly increased threshold (38) | Centrally preserved but reduced responses, normal implicit time (54) | Nagel anomaloscope: normal; PD15 desaturated: minor errors |
| ADRP 386 | |||||||
| IV:1 | IOL OU (54) | Peripheral and peripapillary atrophy, no pigmentation | vital | Minimal photopic residual response | Increased threshold for white, red, blue | ND | ND |
| IV:2 | normal | Peripheral and peripapillary atrophy, Minimal pigmentation OCT: OD mild ERM | vital | Residual responses | ND | Centrally preserved but reduced, normal implicit time | PD15 desaturated and saturated: minor errors |
| IV:3 | normal | Progressive peripheral and peripapillary atrophic lesions, limited pigmentation, cystoid macular edema | vital | No residual responses (50) | Markedly increased threshold for white, red, blue (56) | Centrally preserved but reduced responses, normal implicit time (50) | PD15 desaturated and saturated: minor errors (56) |
| Number of Mutations [n] | ∆∆Gbapp (kcal/mol) | ∆∆Gbapp C+ (kcal/mol) | ∆∆Gbapp S * (kcal/mol) | ∆∆Gfapp (kcal/mol) | ∆∆Gfapp C+ (kcal/mol) | ∆∆Gfapp S * (kcal/mol) |
|---|---|---|---|---|---|---|
| Protomer–protomer | ||||||
| 1 mut [9] | 12.2 ± 5.5 | −0.43 ± 0.06 | 12.9 ± 4.7 | 30.5 ± 1.0 | 44.7 ± 2.6 | −9.0 ± 4.8 |
| 2 mut [9] | 27.7 ± 6.6 | 11.4 ± 9.8 | 16.5 ± 5.2 | 64.8 ± 1.6 | 95.3 ± 9.8 | −24.1 ± 5.2 |
| 3 mut [3] | 44.7 ± 1.4 | 35.3 ± 1.9 | 9.4 ± 1.2 | 101.12 ± 0.04 | 151.8 ± 1.9 | −45.5 ± 1.2 |
| Trimer–trimer | ||||||
| 1 mut [6] | 9.0 ± 9.8 | −42.9 ± 5.5 | 52.4 ± 12.3 | 26.7 ± 2.8 | −90.7 ± 13.6 | 127.4 ± 25.8 |
| 2 mut [15] | 18.6 ± 12.4 | −83.4 ± 6.7 | 103.2 ± 15.4 | 55.6 ± 4.4 | −173.7 ± 18.7 | 248.5 ± 32.7 |
| 3 mut [20] | 28.6 ± 13.6 | −121.1 ± 7.1 | 151.6 ± 16.9 | 86.3 ± 4.9 | −249.1 ± 20.3 | 362.9 ± 35.6 |
| 4 mut [15] | 38.9 ± 13.2 | −156.3 ± 7.1 | 197.8 ± 16.2 | 118.8 ± 4.7 | −316.7 ± 18.1 | 470.8 ± 34.3 |
| 5 mut [6] | 49.5 ± 11.2 | −190.1 ± 5.6 | 242.9 ± 13.0 | 152.7 ± 4.2 | −376.7 ± 12.3 | 572.0 ± 28.4 |
| 6 mut [1] | 60.0 | −221.7 | 285.9 | 187.8 | −429.0 | 666.3 |
| adGALCD | LORD | |
|---|---|---|
| C1QTNF5 | c.538C>G, p.(Q180E) | c.489C>G, p.(S163R) c.489C>A, p.(S163R) c.556C>T, p.(P186S) c.562C>A, p.(P188W) c.569C>G, p.(S190W) c.646G>T, p.(G216C) |
| Age at onset symptoms | 40 years | 40–50 years |
| Initial functional deficits | Problems with adaption in the dark, night blindness | Problems with adaption in the dark, night blindness |
| Age at onset of retinal alterations | 18–24 years | 44–50 years |
| Area of onset | Peripheral and peripapillary atrophy | Midperipheral, temporal to the macula |
| Pseudodrusen-like changes | No | Yes |
| Sub-RPE deposits | No | Yes |
| Choroidal atrophy | Large and confluent, sharply demarcated | Scalloped beginning temporal of the macula in areas with previous pseudodrusen, irregular borders |
| Pigmentation | No or minimal | Moderate to marked |
| Choroidal neovascularization | Not observed | Frequent during progression |
| Macular edema | May occur | Secondary to choroidal neovascularization |
| FAF | Large, sharply demarcated areas of absent/severely reduced FAF | Fleck-like irregular or scalloped midperipheral loss bordered by increased FAF, irregular in macular lesions |
| OCT | No deposits, absence of RPE and photoreceptors in affected areas | Subretinal deposits, irregular photoreceptor loss |
| ERG | Markedly reduced at age 38, residual or not measurable responses at age >50 years | Normal or well-preserved ERG up to 60–67 years of age |
| mfERG | Reduced amplitude, normal implicit time in preserved areas, no response in affected areas | Not reported |
| Anterior segment | Normal | Long anterior lens zonules in most patients |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kellner, U.; Weisschuh, N.; Weinitz, S.; Farmand, G.; Deutsch, S.; Kortüm, F.; Mazzola, P.; Schäferhoff, K.; Marino, V.; Dell’Orco, D. Autosomal Dominant Gyrate Atrophy-Like Choroidal Dystrophy Revisited: 45 Years Follow-Up and Association with a Novel C1QTNF5 Missense Variant. Int. J. Mol. Sci. 2021, 22, 2089. https://doi.org/10.3390/ijms22042089
Kellner U, Weisschuh N, Weinitz S, Farmand G, Deutsch S, Kortüm F, Mazzola P, Schäferhoff K, Marino V, Dell’Orco D. Autosomal Dominant Gyrate Atrophy-Like Choroidal Dystrophy Revisited: 45 Years Follow-Up and Association with a Novel C1QTNF5 Missense Variant. International Journal of Molecular Sciences. 2021; 22(4):2089. https://doi.org/10.3390/ijms22042089
Chicago/Turabian StyleKellner, Ulrich, Nicole Weisschuh, Silke Weinitz, Ghazaleh Farmand, Sebastian Deutsch, Friederike Kortüm, Pascale Mazzola, Karin Schäferhoff, Valerio Marino, and Daniele Dell’Orco. 2021. "Autosomal Dominant Gyrate Atrophy-Like Choroidal Dystrophy Revisited: 45 Years Follow-Up and Association with a Novel C1QTNF5 Missense Variant" International Journal of Molecular Sciences 22, no. 4: 2089. https://doi.org/10.3390/ijms22042089