
The Burden of Post-Translational Modification (PTM)—Disrupting Mutations in the Tumor Matrisome

 , , ,
, , ,  and
and 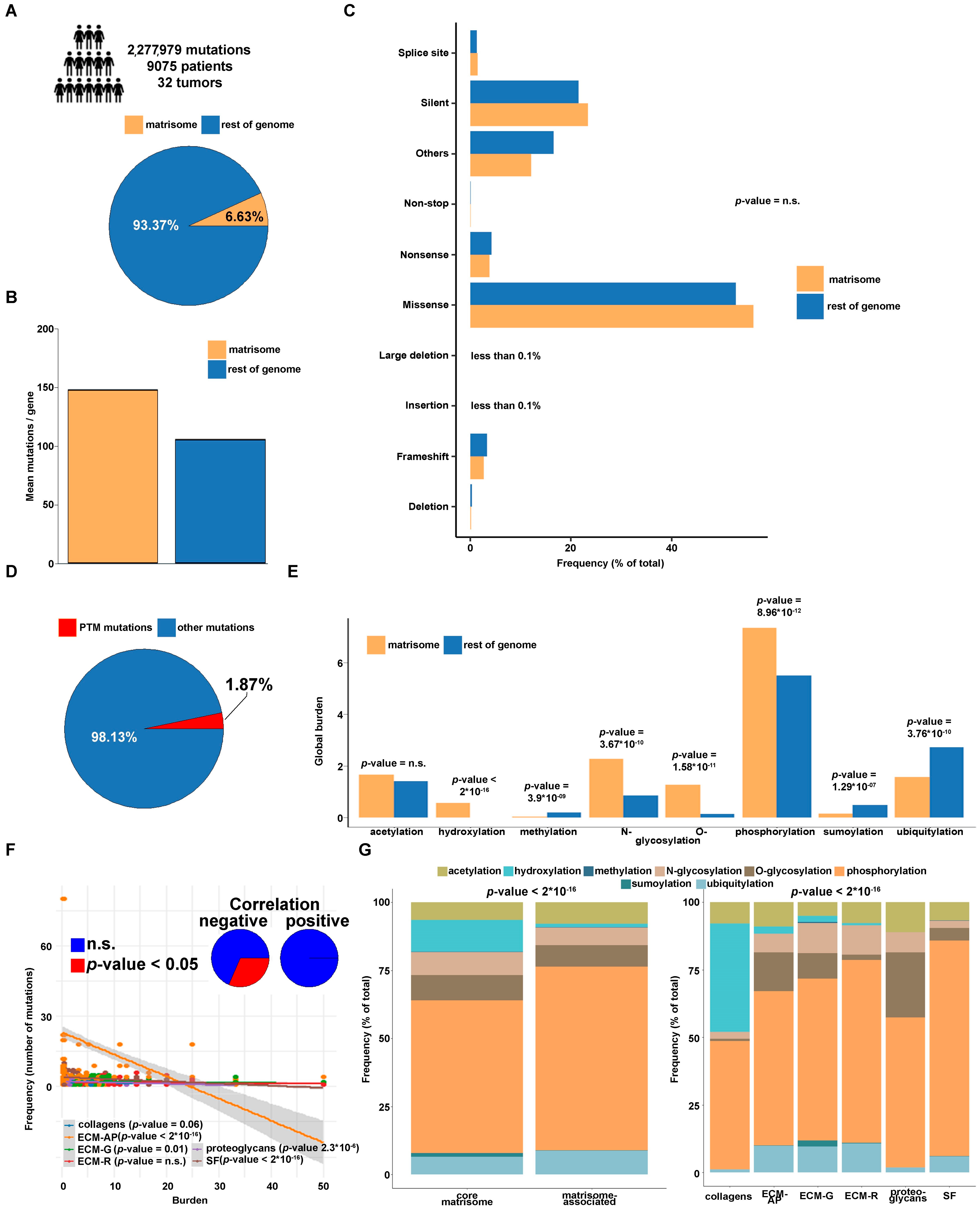{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Source Data
2.2. Statistical Analysis
3. Results
3.1. Genomic Features of Overall Mutations and PTM-Affecting Mutations (PTMmut) in the Tumor Matrisome and in the Rest of the Genome
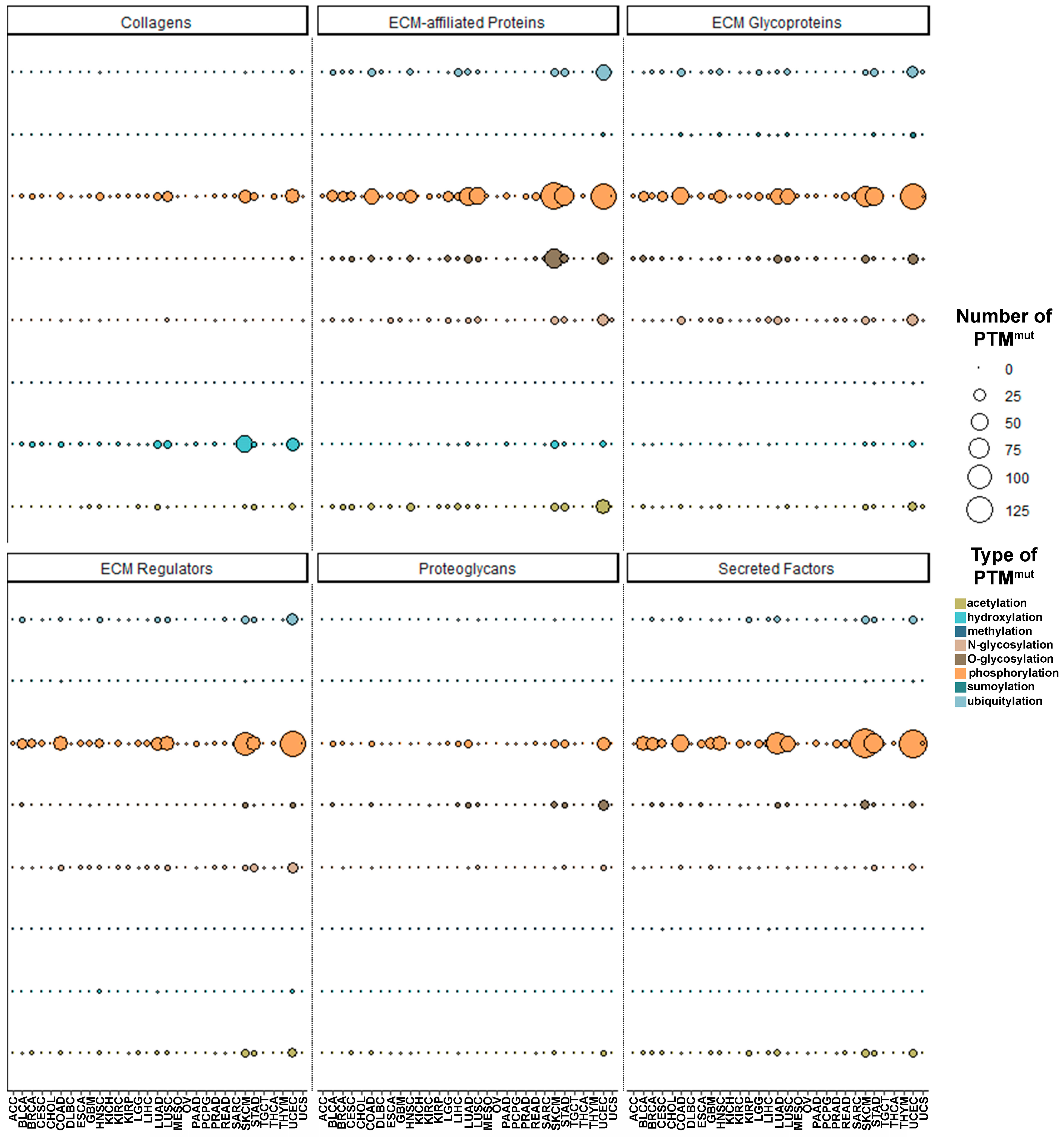
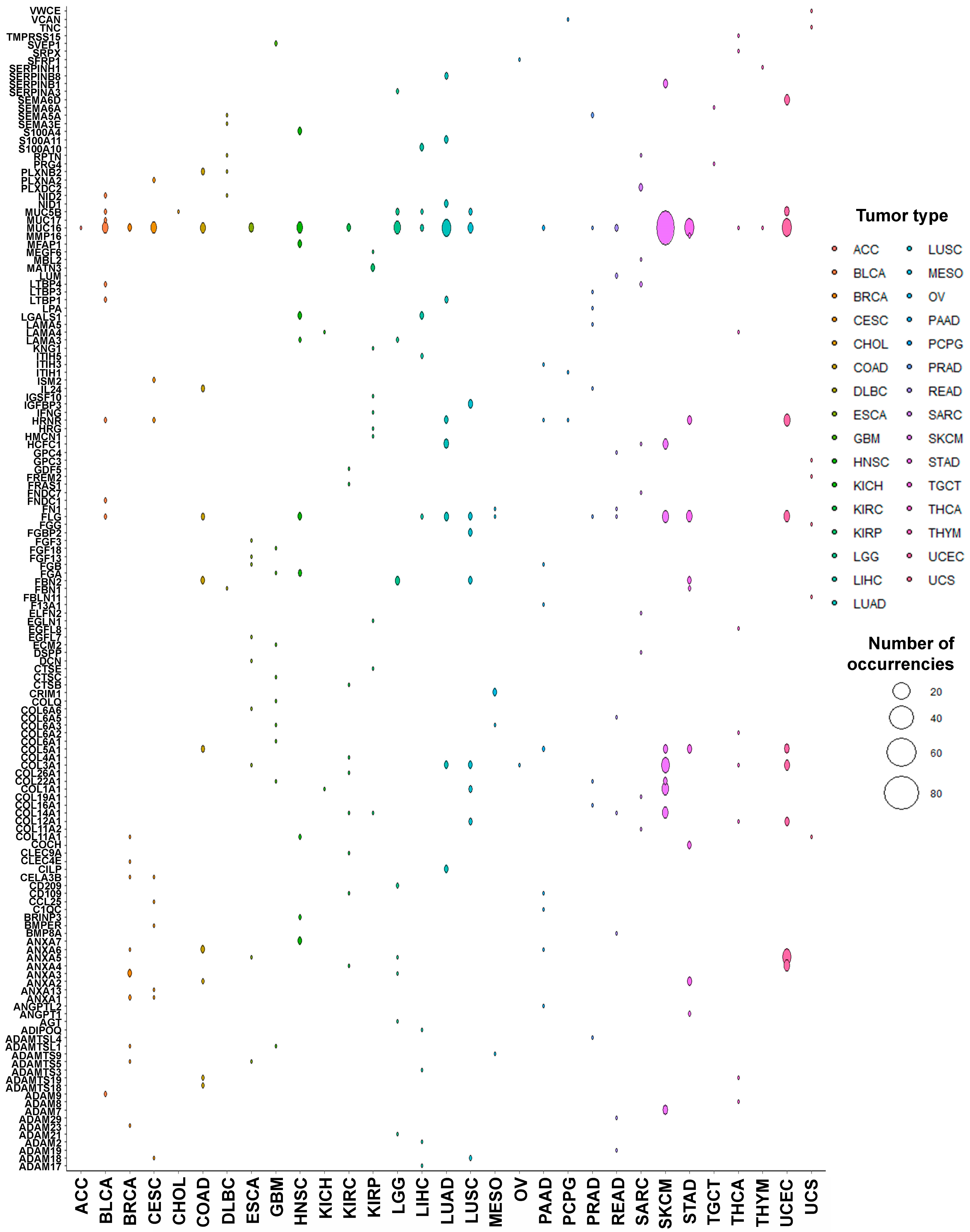
3.2. PTMmut in the Tumor Matrisome
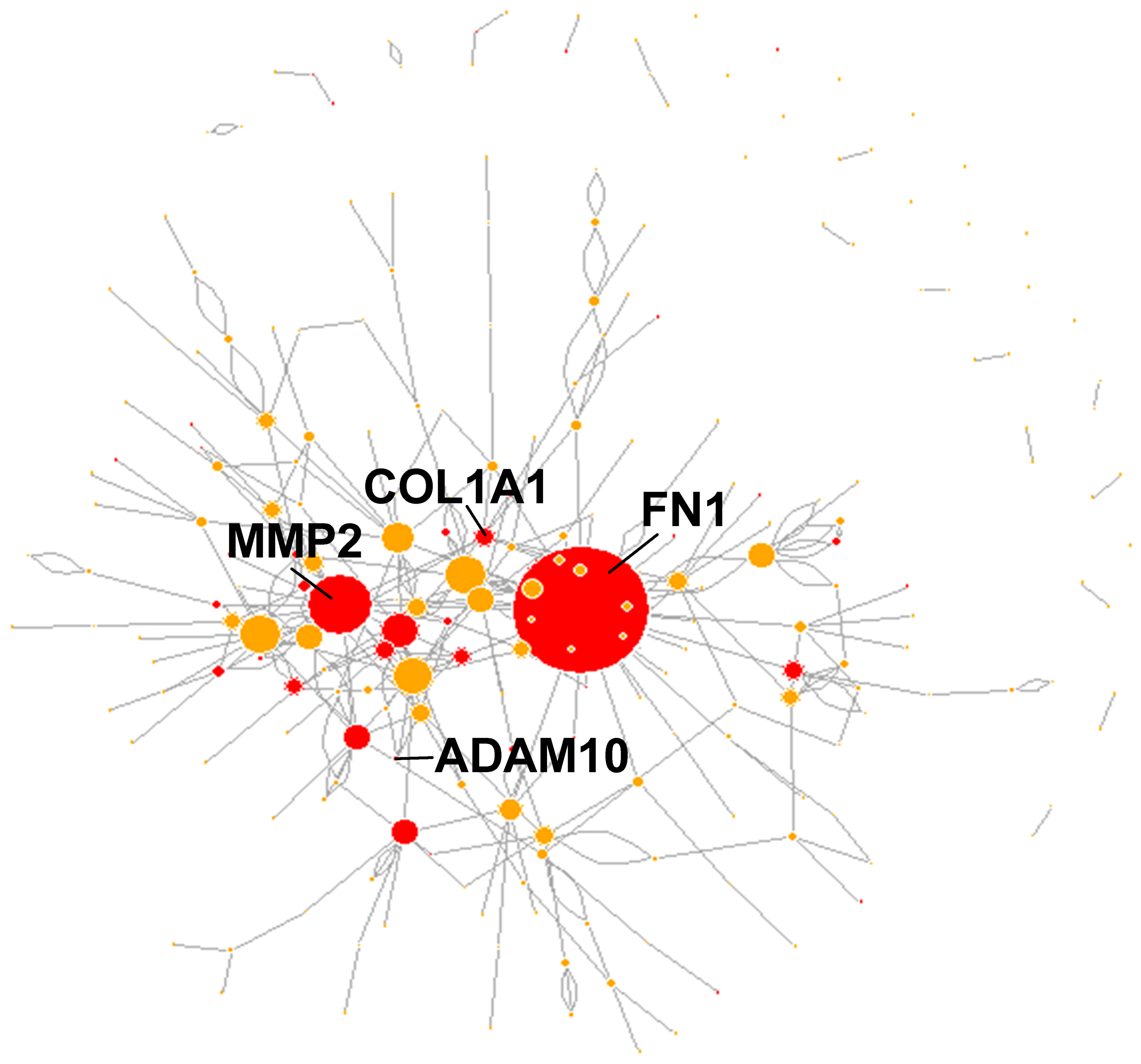
3.3. Functional Characterization of Matrisome PTMmut
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol. 2015, 19, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The Tumor Microenvironment Underlies Acquired Resistance to CSF-Inhibition in Gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell. Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Rianna, C.; Kumar, P.; Radmacher, M. The Role of the Microenvironment in the Biophysics of Cancer. Semin. Cell Dev. Biol. 2018, 73, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. The Hypoxic Tumor Microenvironment: A Driving Force for Breast Cancer Progression. Biochim. Biophys. Acta 2016, 1863, 382–391. [Google Scholar] [CrossRef] [PubMed]
- DeClerck, Y.A.; Pienta, K.J.; Woodhouse, E.C.; Singer, D.S.; Mohla, S. The Tumor Microenvironment at a Turning Point Knowledge Gained over the Last Decade, and Challenges and Opportunities Ahead: A White Paper from the NCI TME Network. Cancer Res. 2017, 77, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef] [Green Version]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The Extracellular Matrix: Tools and Insights for the “Omics” Era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The Matrisome: In Silico Definition and in Vivo Characterization by Proteomics of Normal and Tumor Extracellular Matrices. Mol. Cell Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzhalin, A.E.; Urbonas, T.; Silva, M.A.; Muschel, R.J.; Gordon-Weeks, A.N. A Core Matrisome Gene Signature Predicts Cancer Outcome. Br. J. Cancer 2018, 118, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Liu, Z.; Deng, N.; Tan, T.Z.; Huang, R.Y.; Taylor-Harding, B.; Cheon, D.J.; Lawrenson, K.; Wiedemeyer, W.R.; Walts, A.E.; et al. A COL11A1-Correlated Pan-Cancer Gene Signature of Activated Fibroblasts for the Prioritization of Therapeutic Targets. Cancer Lett. 2016, 382, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzi, V.; Lakkala, J.; Devarajan, R.; Kääriäinen, A.; Koivunen, J.; Heljasvaara, R.; Pihlajaniemi, T. Pan-Cancer Analysis of the Expression and Regulation of Matrisome Genes across 32 Tumor Types. Matrix Biol. Plus 2019, 1, 100004. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socovich, A.M.; Naba, A. The Cancer Matrisome: From Comprehensive Characterization to Biomarker Discovery. Semin. Cell Dev. Biol. 2019, 89, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, J.; Collignon, E.; Fuks, F. DNA Methylome Profiling Beyond Promoters—Taking an Epigenetic Snapshot of the Breast Tumor Microenvironment. FEBS J. 2015, 282, 1801–1814. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, H.; Ushijima, T. Accumulation of Genetic and Epigenetic Alterations in Normal Cells and Cancer Risk. NPJ Precis. Oncol. 2019, 3, 1–8. [Google Scholar]
- Jin, M.; Jin, W. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.M. Post-Translational Modifications of Protein Backbones: Unique Functions, Mechanisms, and Challenges. Biochemistry 2018, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Karve, T.M.; Cheema, A.K. Small Changes Huge Impact: The Role of Protein Posttranslational Modifications in Cellular homeostasis and disease. J. Amino. Acids 2011, 2011, 207691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virág, D.; Dalmadi-Kiss, B.; Vékey, K.; Drahos, L.; Klebovich, I.; Antal, I.; Ludányi, K. Current Trends in the Analysis of Post-Translational Modifications. Chromatographia 2020, 83, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Barber, K.W.; Rinehart, J. The ABCs of PTMs. Nat. Chem. Biol. 2018, 14, 188–192. [Google Scholar] [CrossRef]
- Kam, R.K.T.; Poon, T.C.W. The Potentials of Glycomics in Biomarker Discovery. Clin. Proteom. 2008, 4, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Shriver, Z.; Raguram, S.; Sasisekharan, R. Glycomics: A Pathway to a Class of New and Improved Therapeutics. Nat. Rev. Drug Discov. 2004, 3, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological Roles of Glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.L.; Lindner, A.B. Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxid. Med. Cell Longev. 2017, 2017, 5716409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimand, J.; Wagih, O.; Bader, G.D. Evolutionary Constraint and Disease Associations of Post-Translational Modification Sites in Human Genomes. PLoS Genet. 2015, 11, e1004919. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Peng, X.; Ying, P.; Tian, J.; Li, J.; Ke, J.; Zhu, Y.; Gong, Y.; Zou, D.; Yang, N.; et al. AWESOME: A Database of SNPs that Affect Protein Post-Translational Modifications. Nucleic Acids Res. 2019, 47, D874–D880. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kang, C.; Min, B.; Yi, G. Detection and Analysis of Disease-Associated Single Nucleotide Polymorphism Influencing Post-Translational Modification. BMC Med. Genom. 2015, 8 (Suppl. 2), S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeming, D.J.; Bay-Jensen, A.C.; Vassiliadis, E.; Larsen, M.R.; Henriksen, K.; Karsdal, M.A. Post-Translational Modifications of the Extracellular Matrix are Key Events in Cancer Progression: Opportunities for Biochemical Marker Development. Biomarkers 2011, 16, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Nielsen, M.J.; Sand, J.M.; Henriksen, K.; Genovese, F.; Bay-Jensen, A.; Smith, V.; Adamkewicz, J.I.; Christiansen, C.; Leeming, D.J. Extracellular Matrix Remodeling: The Common Denominator in Connective Tissue Diseases. Possibilities for Evaluation and Current Understanding of the Matrix as More than a Passive Architecture, but a Key Player in Tissue Failure. Assay Drug Dev. Technol. 2013, 11, 70–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappu, P.; Salo, A.M.; Myllyharju, J.; Heino, J. Role of Prolyl Hydroxylation in the Molecular Interactions of Collagens. Essays Biochem. 2019, 63, 325–335. [Google Scholar] [PubMed] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nature reviews. Nephrology 2019, 15, 346–366. [Google Scholar]
- Hsiao, C.; Cheng, H.; Huang, C.; Li, H.; Ou, M.; Huang, J.; Khoo, K.; Yu, H.W.; Chen, Y.; Wang, Y.; et al. Fibronectin in Cell Adhesion and Migration Via N-Glycosylation. Oncotarget 2017, 8, 70653–70668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Miao, Y.; Liu, M.; Zeng, Y.; Gao, Z.; Peng, D.; Hu, B.; Li, X.; Zheng, Y.; Xue, Y.; et al. Pan-Cancer Analysis Reveals the Functional Importance of Protein Lysine Modification in Cancer Development. Front. Genet. 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, S.; Tao, Y. Regulating Tumor Suppressor Genes: Post-Translational Modifications. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef]
- Jin, H.; Zangar, R.C. Protein Modifications as Potential Biomarkers in Breast Cancer. Biomark Insights 2009, 4, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Izzi, V.; Davis, M.N.; Naba, A. Pan-Cancer Analysis of the Genomic Alterations and Mutations of the Matrisome. Cancers 2020, 12, 2046. [Google Scholar] [CrossRef]
- Trinh, A.; Del Alcazar, C.R.G.; Shukla, S.A.; Chin, K.; Chang, Y.H.; Thibault, G.; Eng, J.; Jovanović, B.; Aldaz, C.M.; Park, S.Y.; et al. Genomic Alterations during the in Situ to Invasive Ductal Breast Carcinoma Transition Shaped by the Immune System. Mol. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value Beyond its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef]
- Zapata, L.; Pich, O.; Serrano, L.; Kondrashov, F.A.; Ossowski, S.; Schaefer, M.H. Negative Selection in Tumor Genome Evolution Acts on Essential Cellular Functions and the Immunopeptidome. Genome Biol. 2018, 19, 67. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Engel, J. Domain structure and organisation in extracellular matrix proteins. Matrix Biol. 2002, 21, 115–128. [Google Scholar] [CrossRef]
- Xu, J.; Liao, K.; Wang, X.; He, J.; Wang, X. Combining Bioinformatics Techniques to Explore the Molecular Mechanisms Involved in Pancreatic Cancer Metastasis and Prognosis. J. Cell Mol. Med. 2020, 24, 14128–14138. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Liu, X.; Zheng, H.; Wang, H.; Hong, D. Integrated Bioinformatics Analysis Identified COL11A1 as an Immune Infiltrates Correlated Prognosticator in Pancreatic Adenocarcinoma. Int. Immunopharmacol. 2020, 90, 106982. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.; Bitter, C.; Winkler, S.; Weichenhan, D.; Thavamani, A.; Hengstler, J.G.; Borkham-Kamphorst, E.; Kohlbacher, O.; Plass, C.; Geffers, R.; et al. Identification of Pparγ-Modulated miRNA Hubs that Target the Fibrotic Tumor Microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Zhang, X.; Kang, X.; Jin, L.; Wang, P.; Wang, Z. Screening of Core Genes and Pathways in Breast Cancer Development via Comprehensive Analysis of Multi Gene Expression Datasets. Oncol. Lett. 2019, 18, 5821–5830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Vila, M.; Takahashi, R.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix Metalloproteinases (MMPs), the Main Extracellular Matrix (ECM) Enzymes in Collagen Degradation, as a Target for Anticancer Drugs. J. Enzyme. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berisio, R.; Vitagliano, L.; Mazzarella, L.; Zagari, A. Crystal Structure of the Collagen Triple Helix Model [(Pro-Pro-Gly)10]3. Protein Sci. 2002, 11, 262–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caswell, C.C.; Barczyk, M.; Keene, D.R.; Lukomska, E.; Gullberg, D.E.; Lukomski, S. Identification of the First Prokaryotic Collagen Sequence Motif that Mediates Binding to Human Collagen Receptors, Integrins alpha2beta1 and alpha11beta1. J. Biol. Chem. 2008, 283, 36168–36175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manwar Hussain, M.R.; Iqbal, Z.; Qazi, W.M.; Hoessli, D.C. Charge and Polarity Preferences for N -Glycosylation: A Genome-Wide in Silico Study and its Implications regarding Constitutive Proliferation and Adhesion of Carcinoma Cells. Front. Oncol. 2018, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhajosyula, R.R.; Chakravarti, D.; Lutfeali, S.; Ray, A.; Raval, A. Identifying Hubs in Protein Interaction Networks. PLoS ONE 2009, 4, e5344. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomko, L.A.; Hill, R.C.; Barrett, A.; Szulczewski, J.M.; Conklin, M.W.; Eliceiri, K.W.; Keely, P.J.; Hansen, K.C.; Ponik, S.M. Targeted Matrisome Analysis Identifies Thrombospondin-2 and Tenascin-C in Aligned Collagen Stroma from Invasive Breast Carcinoma. Sci. Rep. 2018, 8, 12941. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.R.; Ryall, K.A.; Vyse, S.; Wong, J.P.; Natrajan, R.C.; Yuan, Y.; Tan, A.C.; Huang, P.H. Systematic Analysis of Tumour Cell-Extracellular Matrix Adhesion Identifies Independent Prognostic Factors in Breast Cancer. Oncotarget 2016, 7, 62939–62953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoye, A.M.; Erler, J.T. Structural ECM Components in the Premetastatic and Metastatic Niche. Am. J. Physiol. Cell Physiol. 2016, 310, 955–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzi, V.; Lakkala, J.; Devarajan, R.; Ruotsalainen, H.; Savolainen, E.R.; Koistinen, P.; Heljasvaara, R.; Pihlajaniemi, T. An Extracellular Matrix Signature in Leukemia Precursor Cells and Acute Myeloid Leukemia. Haematologica 2017, 102, e245–e248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Ricard-Blum, S. The Extracellular Matrix Goes-Omics: Resources and Tools, 7th ed.; Springer: Cham, Switzerland, 2020; pp. 1–16. [Google Scholar]
- King, R.J.; Yu, F.; Singh, P.K. Genomic Alterations in Mucins Across Cancers. Oncotarget 2017, 8, 67152–67168. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Jiang, C.; De, S. Dissecting the Sources of Gene Expression Variation in a Pan-Cancer Analysis Identifies Novel Regulatory Mutations. Nucleic Acids Res. 2018, 46, 4370–4381. [Google Scholar] [CrossRef]
- Kanwal, M.; Ding, X.J.; Song, X.; Zhou, G.B.; Cao, Y. MUC16 Overexpression Induced by Gene Mutations Promotes Lung Cancer Cell Growth and Invasion. Oncotarget 2018, 9, 12226–12239. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic Pan-Cancer Analysis of Tumour Purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicens, A.; Posada, D. Selective Pressures on Human Cancer Genes Along the Evolution of Mammals. Genes 2018, 9, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, K.A.; Barber, L.J.; Davies, M.N.; Ashenden, M.; Sottoriva, A.; Gerlinger, M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer 2016, 2, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The Evolutionary History of 2,658 Cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Auslander, N.; Wolf, Y.I.; Koonin, E.V. In Silico Learning of Tumor Evolution through Mutational Time Series. Proc. Natl. Acad Sci. USA 2019, 116, 9501–9510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kääriäinen, A.; Pesola, V.; Dittmann, A.; Kontio, J.; Koivunen, J.; Pihlajaniemi, T.; Izzi, V. Machine Learning Identifies Robust Matrisome Markers and Regulatory Mechanisms in Cancer. Int. J. Mol. Sci. 2020, 21, 8837. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Salvadores, M.; Mas-Ponte, D.; Supek, F. Passenger Mutations Accurately Classify Human Tumors. PLoS Comput. Biol. 2019, 15, e1006953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salo, A.M.; Myllyharju, J. Prolyl and Lysyl Hydroxylases in Collagen Synthesis. Exp. Dermatol. 2021, 30, 38–49. [Google Scholar] [CrossRef]
- Myllyharju, J.; Kivirikko, K.I. Collagens, Modifying Enzymes and their Mutations in Humans, Flies and Worms. Trends Genet. 2004, 20, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Gjaltema, R.A.F.; Bank, R.A. Molecular Insights into Prolyl and Lysyl Hydroxylation of Fibrillar Collagens in Health and Disease. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Vitkup, D.; Sander, C.; Church, G.M. The Amino-Acid Mutational Spectrum of Human Genetic Disease. Genome Biol. 2003, 4, R72. [Google Scholar] [CrossRef] [Green Version]
- Creixell, P.; Schoof, E.M.; Tan, C.H.S.; Linding, R. Mutational Properties of Amino Acid Residues: Implications for Evolvability of Phosphorylatable Residues. Philosophical transactions. Biol. Sci. 2012, 367, 2584–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, C.; Rost, B. Predict Impact of Single Amino Acid Change upon Protein Structure. BMC Genom. 2012, 13 (Suppl. 4), S4. [Google Scholar] [CrossRef] [Green Version]
- Holehouse, A.S.; Naegle, K.M. Reproducible Analysis of Post-Translational Modifications in Proteomes—Application to Human Mutations. PLoS ONE 2015, 10, e0144692. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Sricholpech, M. Lysine Post-Translational Modifications of Collagen. Essays Biochem. 2012, 52, 113–133. [Google Scholar] [PubMed] [Green Version]
- Wu, Y.J.; La Pierre, D.P.; Jin, W.U.; Albert, J.Y.; Burton, B.Y. The Interaction of Versican with its Binding Partners. Cell Res. 2005, 15, 483–494. [Google Scholar] [CrossRef]
- Li, Y.; Sun, C.; Yates, E.A.; Jiang, C.; Wilkinson, M.C.; Fernig, D.G. Heparin Binding Preference and Structures in the Fibroblast Growth Factor Family Parallel their Evolutionary Diversification. Open Biol. 2016, 6, 150275. [Google Scholar] [CrossRef] [Green Version]
- Dalton, B.A.; McFarland, C.D.; Underwood, P.A.; Steele, J.G. Role of the Heparin Binding Domain of Fibronectin in Attachment and Spreading of Human Bone-Derived Cells. J. Cell Sci. 1995, 108, 2083–2092. [Google Scholar] [PubMed]
- Madzharova, E.; Kastl, P.; Sabino, F.; auf dem Keller, U. Post-Translational Modification-Dependent Activity of Matrix Metalloproteinases. Int. J. Mol. Sci. 2019, 20, 3077. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holstein, E.; Dittmann, A.; Kääriäinen, A.; Pesola, V.; Koivunen, J.; Pihlajaniemi, T.; Naba, A.; Izzi, V. The Burden of Post-Translational Modification (PTM)—Disrupting Mutations in the Tumor Matrisome. Cancers 2021, 13, 1081. https://doi.org/10.3390/cancers13051081
Holstein E, Dittmann A, Kääriäinen A, Pesola V, Koivunen J, Pihlajaniemi T, Naba A, Izzi V. The Burden of Post-Translational Modification (PTM)—Disrupting Mutations in the Tumor Matrisome. Cancers. 2021; 13(5):1081. https://doi.org/10.3390/cancers13051081
Chicago/Turabian StyleHolstein, Elisa, Annalena Dittmann, Anni Kääriäinen, Vilma Pesola, Jarkko Koivunen, Taina Pihlajaniemi, Alexandra Naba, and Valerio Izzi. 2021. "The Burden of Post-Translational Modification (PTM)—Disrupting Mutations in the Tumor Matrisome" Cancers 13, no. 5: 1081. https://doi.org/10.3390/cancers13051081