
Abstract
Stenotopic specialization to a fragmented habitat promotes the evolution of genetic structure. It is not yet clear whether small-scale population structure generally translates into large-scale intraspecific divergence. In the present survey of mitochondrial genetic structure in the Lake Tanganyika endemic Altolamprologus (Teleostei, Cichlidae), a rock-dwelling cichlid genus comprising A. compressiceps and A. calvus, habitat-induced population fragmentation contrasts with weak phylogeographic structure and recent divergence among genetic clades. Low rates of dispersal, perhaps along gastropod shell beds that connect patches of rocky habitat, and periodic secondary contact during lake level fluctuations are apparently sufficient to maintain genetic connectivity within each of the two Altolamprologus species. The picture of genetic cohesion was interrupted by a single highly divergent haplotype clade in A. compressiceps restricted to the northern part of the lake. Comparisons between mitochondrial and nuclear phylogenetic reconstructions suggested that the divergent mitochondrial clade originated from ancient interspecific introgression. Finally, ‘isolation-with-migration’ models indicated that divergence between the two Altolamprologus species was recent (67–142 KYA) and proceeded with little if any gene flow. As in other rock-dwelling cichlids, recent population expansions were inferred in both Altolamprologus species, which may be connected with drastic lake level fluctuations.
Similar content being viewed by others


Introduction
The genetic structure within a species is often predicted by components of the species’ ecology and life history traits (Wiens & Donoghue, 2004). For instance, generalist species and species with good dispersal abilities tend to show little phylogeographic structure (e.g. Cadena et al., 2011; Koblmüller et al., 2012; Diedericks & Daniels, 2014), whereas species that are specialized to particular, fragmented habitats or food sources are often divided into distinct populations and develop marked phylogeographic patterns (e.g. Young et al., 1996; Row et al., 2010; Kajtoch et al., 2014). This contrast is particularly evident when the different species occur in the same region. In the two East African lakes Malawi and Tanganyika, stenotopic cichlid species inhabiting the discontinuous rocky littoral consistently show remarkable genetic population differentiation, even across very short geographic distances (e.g. van Oppen et al., 1997; Markert et al., 1999; Taylor et al., 2001; Rico & Turner, 2002; Pereyra et al., 2004; Duftner et al., 2006; Koblmüller et al., 2011; Sefc et al., 2007; Wagner & McCune, 2009; Nevado et al., 2013; Van Steenberge et al., 2015; Sefc et al., 2016). Differentiation is lower and only occurs on a larger geographic scale in less specialized species and species that inhabit the intermediate habitat, i.e. the transition between rocky and sandy habitat surrounding the rocky habitat patches (Koblmüller et al., 2007a, 2009; Sefc et al., 2007; Kotrschal et al., 2012). Finally, the sandy and the open-water habitat contain virtually no barriers to dispersal, and cichlid species specialized to these habitats show little if any population genetic structure at all (Shaw et al., 2000; Taylor & Verheyen, 2001; Pereyra et al., 2004; Genner et al., 2008, 2010a; Anseeuw et al., 2011; Koblmüller et al., 2015a).
The majority of studies addressing the genetic structure of cichlid species in the two lakes did so by sampling populations within a small portion of the species’ entire distribution range (see references above). It is not yet clear whether findings regarding small-scale population structure can be extrapolated to large-scale phylogeographic patterns. Significant genetic differentiation can evolve among fragmented populations within relatively short periods of time. However, large-scale phylogeographic patterns have been shown to be influenced by the long-term history of the species, including range shifts and introgression among differentiated lineages during secondary contact in the course of lake level fluctuations (Verheyen et al., 1996; Sturmbauer et al., 2001; Egger et al., 2007; Nevado et al., 2009). Therefore, distinct population genetic structure may, but need not, go along with a clear-cut phylogeographic pattern; and vice versa, species with high levels of gene flow across the geographical scale of previous population genetic studies may still be genetically structured on a larger scale.
Due to logistic and sometimes political reasons, lake-wide sampling in Lakes Malawi and Tanganyika can be difficult, and only few species have been investigated throughout large parts of their distribution ranges. The stenotopic rock-dwelling genus Tropheus from Lake Tanganyika is one example in which both population genetic and lake-wide phylogeographic studies have been conducted. In this fish, strong small-scale population differentiation contrasts with evidence for ancient introgression among phylogenetically old lineages (Sturmbauer et al., 2005; Sefc et al., 2007; Egger et al., 2007; Koblmüller et al., 2011; Sefc et al., 2016). A similar picture—deep intraspecific divergences and past introgression among geographically distant populations—emerged in a phylogeographic study of another rock-dwelling cichlid of Lake Tanganyika, the Neolamprologus brichardi/pulcher complex (Duftner et al., 2007). In contrast, two species that are less strictly restricted to rocky habitat (Lamprologus callipterus and Neolamprologus fasciatus) exhibit clear phylogeographic structure but shallower intraspecific divergence (Nevado et al., 2009), and a highly mobile benthopelagic species (Boulengerochromis microlepis) shows no phylogeographic structure on a lake-wide scale and very shallow intraspecific divergence (Koblmüller et al., 2015a). Similarly, in Lake Malawi, the pelagic species Diplotaxodon limnothrissa shows no phylogeographic structure on a lake-wide scale (Shaw et al., 2000).
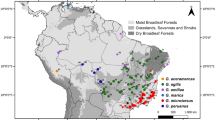
Here, we investigate the phylogeographic structure in two closely related species of the Lake Tanganyika cichlid genus Altolamprologus, a member of the ‘ossified group’ within the tribe Lamprologini (Stiassny, 1997). The genus diverged from its sister group >1 MYA (Sturmbauer et al., 2010) and includes only two described species, Altolamprologus compressiceps and A. calvus. A. compressiceps has a lake-wide distribution, whereas A. calvus is restricted to the southwestern part of Lake Tanganyika, where it occurs in sympatry with A. compressiceps (Fig. 1; Konings, 1998; Van Steenberge et al., 2011). Both species typically inhabit the shallow rocky habitat, but can also be found down to a depth of 30–40 m (Konings, 1998; Muschick et al., 2012). In suitable habitat, Altolamprologus spp. are common, but less abundant than many other rock-dwelling cichlid species (Sturmbauer et al., 2008; Takeuchi et al., 2010). Both species are characterized by deep and laterally compressed bodies enabling them to enter narrow cracks and shallow caves (Fig. 1). They predominately prey on atyid shrimps, but also on other arthropods and fish fry (Hori, 1991; Yuma & Kondo, 1997; Konings, 1998; Muschick et al., 2012). In addition to the two described species, a dwarf ecotype, Altolamprologus sp. “shell”, lives and breeds in mollusc shell beds at a few locations in Burundi, Tanzania and Zambia. The taxonomic status of this ecotype is uncertain. A detailed morphological study of shell-breeding dwarf Altolamprologus from Burundi concluded that this fish is not a distinct species but merely a dwarf morph of A. compressiceps (Gashagaza et al., 1995). While our sampling of A. compressiceps and A. calvus covers their entire respective distributions, only one individual of the shell-breeding ecotype could be analysed in the present study, which certainly is not enough to fully uncover its evolutionary history but gives a first hint as to where it stands within the genus.

A Typical representatives of A. calvus, A. compressiceps and A. sp. “shell”. Photographs courtesy of Wolfgang Gessl (www.pisces.at). B Map of Lake Tanganyika with sampling localities. Numbers in parentheses refer to sample sizes; different colours indicate different species (red, A. calvus; blue, A. compressiceps; green, A. sp. “shell”). The three deepwater basins are indicated by grey shading
Like many other rock-dwelling cichlids, Altolamprologus spp. exhibit considerable geographic variation in colour pattern (Kohda et al., 1996; Konings, 1998), and comparisons among three populations in the very south of Lake Tanganyika detected significant population genetic and morphometric differentiation in A. compressiceps (Spreitzer et al., 2012), a pattern potentially indicating pronounced levels of lake-wide phylogeographic structuring. Furthermore, if habitat preferences correlate with phylogeographic structure, we expect to see a similar phylogeographic pattern in Altolamprologus as in the rock-dwellers Tropheus spp. and N. pulcher/brichardi, with ancient introgression among deeply divergent genetic lineages (Sturmbauer et al., 2005; Duftner et al., 2007; Egger et al., 2007). In this study, we use mitochondrial and nuclear sequence data to uncover the phylogeographic structure and demographic history of A. calvus and A. compressiceps, and their phylogenetic relationships to other ‘ossified group’ lamprologines. In doing so and by comparing the inferred pattern with those of other taxa, we test whether the similar ecological and life history traits in cichlid species from different tribes can predict the degree of genetic structure attained over large geographic scales.
Materials and methods
Sample collection and DNA extraction
A total of 150 Altolamprologus samples (30 A. calvus, 119 A. compressiceps, 1 A. sp. “shell”) from 41 localities around Lake Tanganyika were included in this study (Fig. 1; Supplementary Table 1). The majority of samples were collected during several expeditions between 1992 and 2010 by snorkelling and chasing individual fish into gill nets. Three samples were obtained via the ornamental fish trade. Whole genomic DNA was extracted from finclips or muscle tissue preserved in 96% ethanol by means of a Chelex protocol (Richlen & Barber, 2005) or by applying a proteinase K digestion followed by a standard ammonium acetate extraction protocol (Sambrook & Russell, 2001).
PCR amplification and sequencing
Phylogeographic inference was based on sequences of the most variable part of the mitochondrial control region (358 bp in the aligned dataset) obtained according to the protocols in Koblmüller et al. (2011; PCR) and Duftner et al. (2005; Sanger sequencing) and using primers L-Pro-F_Tropheus (Koblmüller et al., 2011) and TDK-D (Lee et al., 1995).
The phylogenetic position of Altolamprologus spp. in relation to the other species of the ‘ossified group’ of Lamprologini was investigated using comprehensive mitochondrial and nuclear sequence datasets. The Altolamprologus spp. samples included in these mtDNA and nucDNA datasets were selected based on their assignment to distinct lineages in the control region data. The complete mitochondrial NADH dehydrogenase subunit 2 gene (ND2; 1,047 bp) was amplified and sequenced using the primers Met, ND2.2A, Trp (Kocher et al., 1995) and ND2.T-R (Duftner et al., 2005) in four A. calvus and nine A. compressiceps samples. Furthermore, four nuclear genes (ITS, 1,306 bp; LSU, 676 bp; RAG1 intron 2, 928 bp; RAG1 exon 3, 828 bp) were amplified and sequenced for three A. calvus and eight A. compressiceps according to Nevado et al. (2009), using the primers ITS_18D, ITS_28C, ITS_5.8C (ITS; Nevado et al., 2009), LSU_F1, LSU_R1 (LSU; Sonnenberg et al., 2007), KALIF1, CR1, CF1 and CR5 (RAG1 intron 2 and RAG1 exon 3; Clabaut et al., 2005). DNA fragments were purified with Sephadex™ G-50 (Amersham Biosciences) and visualized on an ABI 3130xl capillary sequencer (Applied Biosystems). Sequences were aligned by eye (protein coding sequences) or using MUSCLE (Edgar, 2004) (for non-coding sequences and rDNA) as implemented in Mega 6.05 (Tamura et al., 2013). Our newly generated sequence data were complemented with previously published sequences of ‘ossified group’ lamprologines and outgroup taxa (Duftner et al., 2005; Schelly et al., 2006; Koblmüller et al., 2007b; Nevado et al., 2009), downloaded from Genbank (Supplementary Table 2). New sequences are deposited in GenBank (for GenBank accession numbers, see Supplementary Tables 1 and 2).
Phylogeographic and phylogenetic analysis within Altolamprologus
Phylogenetic relationships among mitochondrial control region haplotypes were inferred by means of Bayesian phylogenetic inference (BI), implemented in MrBayes 3.2.1 (Ronquist et al., 2012). Posterior probabilities were obtained from a Metropolis-coupled Markov chain Monte Carlo simulation (2 independent runs; 10 chains with 5 million generations each; trees sampled every 1,000 generations; the first 25% of trees were discarded as burn-in) employing the best substitution model suggested by the Bayesian Information Criterion (BIC) in jModelTest 0.1 (Posada, 2008). Chain stationarity and run parameter convergence were assessed in Tracer 1.6 (Rambaut et al., 2013; all Effective Sample Size (ESS) >200). The post-burn-in trees were summarized in a 50% majority rule consensus tree. Furthermore, a haplotype network was constructed based on statistical parsimony using TCS 1.2 (Clement et al., 2000), with gaps treated as 5th character. Haplotype (H d) and nucleotide diversities (π) were calculated in DnaSP 5.10 (Librado & Rozas, 2009).
Spatial patterns of genetic differentiation were inferred and visualized by genetic landscape shape interpolation analysis as implemented in Alleles in Space 1.0 (AIS: Miller, 2005). The genetic landscape analysis was based on a Delauny triangulation connectivity network and residual genetic distances derived from the linear regression of genetic versus geographic distances (as recommended for data sets with substantial variation in geographic distances between sampling sites; Manni et al., 2004). Grid size was set to 0.04 × 0.04 latitude and longitude degrees, respectively, and a distance weighting parameter α = 0.5 was used (for more details on the method see Miller et al., 2006). Qualitatively similar results were obtained with different grid sizes and a range of distance weighting parameters (α = 0.1–2; not shown).
Phylogenetic relationships of Altolamprologus within the ‘ossified group’ of lamprologines
Phylogenetic analyses of the ND2 and concatenated nuclear data employed BI as implemented in MrBayes using non-ossified group lamprologines as outgroup taxa (Julidochromis ornatus, Telmatochromis vittatus and Variabilichromis moorii for the ND2 data; Neolamprologus cunningtoni, N. modestus, N. savoryi, N. tetracanthus and V. moorii for the nuclear data). The most appropriate data partitioning schemes and the best fitting molecular evolution models for BI tree search were selected based on the BIC criterion in PartitionFinder (Lanfear et al., 2012). Posterior probabilities were obtained from a Metropolis-coupled Markov chain Monte Carlo simulation (2 independent runs; 10 chains with 5 million generations each for both the ND2 and the concatenated nuclear dataset; trees sampled every 1,000 generations; the first 25% of trees were discarded as burn-in). Chain stationarity and run parameter convergence were assessed in Tracer 1.6 (Rambaut et al., 2013; all ESS >200). A 50% majority rule consensus tree was calculated from all post-burn-in trees.
Species divergence and demographic history
The program IMa2 (Hey, 2010) was used to infer divergence time and the level of gene flow between A. compressiceps and A. calvus based on an isolation-with-migration (IM) coalescent model (Hey & Nielsen, 2004, 2007). Isolation-with-migration models and pure isolation models were each fit to datasets including lake-wide samples or a subset of samples from the area of sympatry. Each of these four analyses was replicated twice with different random number seeds and >10,000,000 steps (until ESS for each estimated parameter were >200; Hey & Nielsen, 2004) and a burn-in of 100,000 steps under a finite-sites model (HKY; Hasegawa et al., 1985). Analyses were considered to have converged upon the stationary distribution if independent runs yielded virtually identical posterior distributions. The values with the highest posterior probability were scored as best estimates. Credibility intervals for each parameter are represented by the 95% highest posterior density (HPD) interval which is the shortest span that includes 95% of the probability density of a parameter. Parameter estimates for divergence time, rate of gene flow, as well as current and ancestral effective population sizes were translated to absolute values by employing a minimum and maximum substitution rate of 0.0324 (Genner et al., 2007, 2010b) and 0.057 (Koblmüller et al., 2009) per site per million years, respectively.
Past population size trajectories and time to the most recent common ancestory (TMRCA) for A. compressiceps and A. calvus were inferred by means of a Bayesian coalescent approach (Bayesian skyline tree prior; Drummond et al., 2005) as implemented in BEAST 1.8.0 (Drummond & Rambaut, 2007). We employed the model of molecular evolution selected by the Bayesian information criterion (BIC) in jModelTest 0.1 (Posada, 2008), assuming a strict molecular clock and the same two substitution rates as above. Two independent MCMC runs of ten million generations each were conducted, sampling every 1000th step with a burn-in of the first 10% of sampled generations. Verification of effective sample sizes (ESS >200 for all parameters), trace of MCMC runs and visualization of past demographic changes by means of Bayesian skyline plots (BSPs) were done in Tracer 1.6 (Rambaut et al., 2013).
Results
Phylogenetic structure of Altolamprologus
In total, 77 hayplotypes were detected in 150 samples; 12 were exclusive to A. calvus, 62 to A. compressiceps, 1 to A. sp. “shell” and 2 haplotypes were shared between A. calvus and A. compressiceps. The statistical parsimony network and the BI tree revealed two peculiar features of the phylogenetic relationships among mitochondrial control region haplotypes of Altolamprologus, which had to be explored before enquiring into the phylogeography of Altolamprologus (Fig. 2). First, A. compressiceps from northern Lake Tanganyika (including Congolese and Tanzanian localities) grouped in a distinct clade that is highly divergent from the remaining Altolamprologus samples (7.1% net divergence based on uncorrected p-distances). Second, A. calvus haplotypes did not form a distinct cluster, but were shared with or closely related to A. compressiceps haplotypes.

Phylogenetic relationships among Altolamprologus haplotypes based on a 348 bp long segment of the mitochondrial control region. A Statistical parsimony network. Each haplotype has a number (see Supplementary Table 1) and is represented by a circle, the diameter of which correlates with the number of individuals sharing the same haplotype. Small bars indicate the number of mutations between haplotypes. B Bayesian inference (BI) tree. Numbers in bold denote haplotype ID, numbers in parentheses refer to sample sizes and numbers above branches indicate posterior probabilities (PP) of the respective nodes (only PP > 0.7 are shown). Geographic clusters PP > 0.7 are indicated by different colours (both in the network and in the BI tree)
ND2 and nuclear DNA sequence data were used to investigate the phylogenetic relationship between the two Altolampologus species and between the two clades of A. compressiceps in relation to other species in the ossified group of Lamprologini. Consistent with the mitochondrial control region data, there was no statistical support for monophyly of either A. compressiceps or A. calvus with respect to each other (Fig. 3). These results suggest that the two species diverged only recently. Indeed, divergence between the two species was dated to 67–142 KYA in ‘isolation-with-migration’ and pure isolation models (Table 1). Comparable divergence time estimates were obtained from models with and without gene flow between species, regardless of whether all A. compressiceps samples except for the divergent northern clade were included in the analyses or only samples from the southwestern region in which both species occur. When gene flow was included in the model, it was estimated to occur at very low rates (Table 2).

Phylogenetic placement of aberrant northern A. compressiceps within the “ossified group” of lamprologines. A Bayesian inference tree based on 1,047 bp of the mitochondrial ND2 gene. B Bayesian inference tree based on a concatenated data set of four nuclear genes (ITS, 1,306 bp; LSU, 676 bp; RAG1 intron 2, 928 bp; RAG1 exon 3, 828 bp). Nodal support in form of posterior probabilities (PP) is shown above the branches (only PP > 0.7 are shown). Aberrant northern A. compressiceps are indicated by bold lettering
The position of the northern A. compressiceps clade differed between mitochondrial and nuclear trees (Fig. 3). In the ND2 tree, Altolamprologus excepting the northern samples formed a well-supported (PP 1.00) clade that clustered with L. callipterus, Lamprologus ocellatus and N. fasciatus (PP 0.97). The northern A. compressiceps branched off basal to this group, but this sister group relationship was only weakly supported. In contrast, the nucDNA tree included the northern A. compressiceps into a well-supported monophyletic Altolamprologus clade. The discrepancy between mtDNA and nucDNA sequence relationships can be explained by ancient interspecific introgression into the northern A. compressiceps (see “Discussion” section).
Phylogeographic structure
Except for the divergent clade of northern samples, there was little structure in the control region haplotype network, and the BI tree identified only few well-supported clades (Fig. 2). A fair number of haplotypes detected in samples from southern Lake Tanganyika, including those shared between A. compressiceps and A. calvus, belonged to an unresolved central cluster from which several geographically restricted clades derived, including two separate southeastern, two separate central Tanzanian and a well-supported central Congolese clade. A genetic landscape shape interpolation analysis (Fig. 4) corroborated the impression of only weak phylogeographic structure given by the phylogenetic reconstructions. In A. compressiceps (excepting the northern clade), elevated genetic differentiation was inferred only between the central Congolese coast (Kalemie, Magogoro, Murega) and the remaining locations. In A. calvus, whose restricted distribution range offers less opportunity for geographic structuring, elevated differentiation was indicated across the Lufubu river estuary (between Kasaba and Chaitika), which imposes a major dispersal barrier to rock-dwelling cichlids (Sefc et al., unpublished). Our single sample of an Altolamprologus ecotype inhabiting gastropod shell beds, A. sp. “shell” from Sumbu, had a private haplotype that clustered with Altolamprologus spp. from the same geographical region.

Results of the genetic landscape shape interpolation analysis (the degree of shading reflects genetic distance: dark, large pairwise distance; light, low pairwise distance). Grid size was set to 0.04 × 0.04 latitude and longitude degrees, respectively, and a distance weighting parameter α = 0.5 was used. A Genetic landscape for A. compressiceps excluding the aberrant northern haplogroup. B Genetic landscape for A. calvus. Numbers in parentheses refer to sample size
Demographic history
With many different closely related low-frequency haplotypes detected in the two species, high haplotype diversity (A. compressiceps excepting the northern clade, H d = 0.976; A. calvus, H d = 0.847) contrasted with only moderate nucleotide diversity (A. compressiceps excepting the northern clade, π = 0.01679; A. calvus, π = 0.01033). BSP analysis estimated the TMRCA of Altolamprologus (northern A. compressiceps haplotypes excluded) between 292 and 512 KYA (95% HPDs: 160–448 and 282–784 with substitution rates of 0.0324 and 0.057 per site per million years, respectively; Genner et al., 2007, 2010b; Koblmüller et al., 2009). The TMRCA of the northern A. compressiceps sequences was dated to 210–370 KYA (respective 95% HPDs: 94–343, 165–603). Despite the small sample size of the northern clade (N = 11), ESS >20,000 for the TMRCA parameter suggests that the parameter estimates have converged.
Reconstructions of population size changes through time in A. compressiceps (northern clade excluded) and A. calvus indicated recent population growth in both species during the last 20–35 KY predated by a period of stable population sizes (Fig. 5). A more ancient period of population expansion in A compressiceps ended about 70–125 KYA.

Bayesian skyline plot (BSP) reconstruction of past population size trajectories assuming substitution rates of 3.24–5.7% per site per MY (Koblmüller et al., 2009; Genner et al., 2010b). Blue and red refer to the A. compressiceps (but excluding the aberrant northern haplogroup) and A. calvus groups, respectively. Thick lines denote median estimates, thin lines indicate 95% HPD intervals. The y-axis represents the population size parameter (female effective population size, fN e, times the mutation rate, µ)
Discussion
Shallow divergence and weak phylogeographic structure
A previous population genetic study in A. compressiceps detected low but significant nuclear genetic differentiation on a small geographic scale at the southern end of Lake Tanganyika (F ST = 0.01–0.02 at microsatellite loci with mean H e = 0.80; Spreitzer et al., 2012). In rock-dwelling cichlids, dispersal is often restricted by habitat barriers such as sandy beaches (Rico & Turner, 2002; Sefc et al., 2007, 2016; Koblmüller et al., 2011) and fish remain confined within their native rock patch. Because populations are isolated from each other, different genetic lineages can persist and diverge within species. However, periods of population isolation were interrupted by secondary contact due to displacements during lake level fluctuations (McGlue et al., 2008), which left signatures of introgression among distinct lineages (Verheyen et al., 1996; Rüber et al., 1999, 2001; Duftner et al., 2007; Egger et al., 2007). Extrapolating from other stenotopic rock-dwellers, we predicted that the phylogeographic structure of Altolamprologus spp. would be characterized by deep intraspecific divergence, geographic clusters and introgression between distinct lineages. Opposite to our expectations, recent ancestry and only weak phylogeographic structure in the two Altolamprologus species rather corresponded with observations in mobile cichlid species less dependent on rocky substrate (Nevado et al., 2009) and revealed a contradiction between large-scale genetic cohesion and small-scale genetic differentiation. Altolamprologus spp. occur at lower densities than many other rock-dwelling cichlids (Sturmbauer et al., 2008; Takeuchi et al., 2010; personal observations). Genetic drift in the small populations can therefore lead to significant differentiation within relatively short periods of time despite occasional gene flow. Observations of juvenile A. compressiceps (of the common rock-dwelling type) in the large shell beds that aggregate on the sandy lake bottom (SK, pers. obs.) suggest that Altolamprologus may bridge the gaps between rocky patches with the help of these structures, and are therefore not as curtailed in their dispersal as are many other rock specialists. In the long run, the exploitation of such dispersal opportunities as well as periodic secondary contact during lake level fluctuations will transport haplotypes across long geographic distances. The small overall population size of Altolamprologus may then contribute to accelerated lineage sorting and frequent replacements of original haplotypes.
The picture of genetic connectivity was interrupted by a highly divergent haplotype lineage restricted to the northern part of the lake. Apparently, the Malagarazi estuary (between Masaka and Kibwe) impedes gene flow between this distinct northern clade and more southern populations along the eastern shore. The comparatively sparse sampling along the west coast does not allow for pinpointing a similar phylogeographic break on the Congolese side. The high divergence of this lineage from other Altolamprologus spp. haplotypes can be explained by its heterospecific origin (see below) and does not necessarily reflect longstanding isolation. However, the northern A. compressiceps clearly represent another geographically defined clade within the species.
Ancient interspecific introgression into northern A. compressiceps
Considering the comprehensive taxon coverage of ‘ossified group’—lamprologines for phylogenetic analyses based on mitochondrial and nuclear DNA sequences, the disparate nuclear and mitochondrial phylogenetic placements of the northern A. compressiceps in relation to other Lamprologini (Fig. 3) suggest ancient mitochondrial introgression from a now extinct species and subsequent replacement of the original mitogenome throughout the very northern part of Lake Tanganyika. Alternatively, incomplete lineage sorting could underlie the mitochondrial non-monophyly of Altolamprologus, but nuclear monophyly of the genus Altolamprologus and the substantial length of the branches separating the involved species and clades make this unlikely (McCracken & Sorenson, 2005). Mitochondrial replacement in isolated populations and even in entire species is being inferred in an increasing number of taxa (e.g. Nevado et al., 2011; Tang et al., 2012; Melo-Ferreira et al., 2014; Good et al., 2015), including lamprologines (Schelly et al., 2006; Nevado et al., 2009). Lamprologine species can produce interspecific offspring for long periods of time following their divergence (Koblmüller et al., 2007b; Sturmbauer et al., 2010), which may explain the frequency of interspecific introgressions detected in this group. When small population size contributes to rapid sorting of lineages, as in A. compressiceps, mitochondrial replacement by the introgressed lineage is not an altogether unlikely outcome (Leonard et al., 2007; Ray et al., 2007).
Recently diverged species with similar demographic histories
The two nominal species of Altolamprologus differ in morphometric characters such as relative body depth, head length and head shape (Fig. 1), and—when sympatric—also in colour pattern (Fig. 1). Despite these pronounced differences, their divergence was dated to only 67–142 KYA. Such recent speciation is unusual for Lake Tanganyika cichlids, in which species typically represent reciprocally monophyletic lineages separated by substantial genetic distances (Koblmüller et al., 2004, 2005, 2007c, 2010; Duftner et al., 2005), except when subject to interspecific introgression (e.g. Rüber et al., 2001; Koblmüller et al., 2007b; Sturmbauer et al., 2010; Nevado et al., 2011). Divergence between the two Altolamprologus species was inferred to have proceeded with little if any gene flow (Table 2), which attributes their polyphyletic relationship mainly to incomplete lineage sorting. The structure of the haplotype network suggests that A. calvus evolved from A. compressiceps in southern Lake Tanganyika, where both species are found today. However, a sympatric speciation scenario is challenged by the ecological similarity of the two species (Konings, 1998; Muschick et al., 2012), which rather suggests that divergence was initiated in geographic separation, and the two species came into secondary contact only later due to distribution range shifts in the wake of drastic lake level fluctuations (McGlue et al., 2008).
Reconstructions of past population size changes reveal clear signatures of congruent recent population growth in both species, which can be expected given their similar ecology and habitat preferences. Simultaneous expansions have also been inferred in other rock-dwelling cichlids from Lake Tanganyika (Koblmüller et al., 2011; Nevado et al., 2013; Winkelmann et al., 2016; Sefc et al., 2016) and Lake Malawi (Genner & Turner, 2015; Husemann et al., 2015), suggesting synchronization by an external trigger, such as habitat expansion during a lake level rise after a major drought period. Throughout their history, both Lake Tanganyika and Lake Malawi experienced numerous severe lake level fluctuations with drops of several hundred metres below the current level, which drastically altered the distribution and extent of littoral habitat types (e.g. McGlue et al., 2008; Lyons et al., 2015). In Lake Tanganyika, severe lake level drops down to 435 below the present level associated with the megadrought events at 75–135 KYA affected the distributions of littoral cichlid communities (Cohen et al., 2007; McGlue et al., 2008). Smaller fluctuations in the more recent past, especially the lake level drop of ~260 m during the last glacial maximum (~20 KYA), also had a profound impact on the current genetic structure of rock-dwelling cichlid species (Cohen et al., 2007; Scholz et al., 2007; McGlue et al., 2008). We have previously noted difficulties in aligning molecular datings of population divergence and growth with the geological datings of lake level fluctuations (Koblmüller et al., 2011; Sefc et al., 2016); in particular, the age of recent demographic events may be overestimated due to the potential time dependency of the molecular clock (Ho et al., 2007) in combination with comparatively ancient calibration points (Koblmüller et al., 2009) and large confidence intervals around substitution rates inferred from recent events (Genner et al., 2010b). In this light, although the reconstructed dates do not match exactly, divergence between A. compressiceps and A. calvus and the first wave of population expansion in A. compressiceps may be connected with lake level fluctuations during the megadroughts, and the more recent population size expansions in A. compressiceps and A. calvus may be associated with fluctuations during the last glacial maximum. To tackle the issue of time dependency of the molecular clock for inferring recent events, estimating substitution rates using alternative methods, such as an expansion dating approach (e.g. Crandall et al., 2012; Koblmüller et al., 2015b; Hoareau, 2016), rather than the typically employed divergence dating might yield more reliable estimates. Because of their often high degrees of stenotopy and hence sensitivity towards increase and decrease of available habitats in the wake of lake fluctuations and the increasingly precise estimates of time and extent of these fluctuations in both Lakes Tanganyika and Malawi (McGlue et al., 2008; Lyons et al., 2015), the rock-dwelling cichlid fishes of the East African Great Lakes Tanganyika and Malawi should represent ideal model systems to establish a recent clock for cichlid fishes based on an expansion dating approach.
Conclusions
In numerous studies of lacustrine cichlids, the species’ ecological specialization predicts population genetic differentiation on small geographic scales. The present study shows that population fragmentation and small-scale differentiation do not necessarily translate into distinct lake-wide structure. The discrepancy between population genetic and phylogeographic structures may arise from the different time scales relevant for the evolution of either. In particular, population differentiation—i.e. significant differences in allele and haplotype frequencies between populations—can evolve despite low gene flow within relatively short periods of time. Over longer periods of time, however, even low levels of gene flow can mediate genetic connectivity across substantial geographic distances.
References
Anseeuw, D., J. A. M. Raeymaekers, P. Busslen, E. Verheyen & J. Snoeks, 2011. Low genetic and morphometric intraspecific divergence in peripheral Copadichromis populations (Perciformes: Cichlidae) in the Lake Malawi basin. International Journal of Evolutionary Biology 2011: 835946.
Cadena, C. D., N. Gutiérrez-Pinto, N. Dávila & R. T. Chesser, 2011. No population genetic structure in a widespread aquatic songbird from the Neotropics. Molecular Phylogenetics and Evolution 58: 540–545.
Clabaut, C., W. Salzburger & A. Meyer, 2005. Comparative phylogenetic analyses of the adaptive radiation of Lake Tanganyika cichlid fish: nuclear sequences are less homoplasious but also less informative than mitochondrial DNA. Journal of Molecular Evolution 61: 666–681.
Clement, M., D. Posada & K. A. Crandall, 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1659.
Cohen, A. S., J. R. Stone, K. R. M. Beuning, L. E. Park, P. N. Reinthal, D. Dettman, C. A. Scholz, T. C. Johnson, J. W. King, M. R. Talbot, E. T. Brown & S. J. Ivory, 2007. Ecological consequences of early Late Pleistocene megadroughts in tropical Africa. Proceedings of the National Academy of Sciences USA 104: 16422–16427.
Crandall, E. D., E. J. Sbrocco, T. S. Deboer, P. H. Barber & K. E. Carpenter, 2012. Expansion dating: calibrating molecular clocks in marine species from expansions onto the Sunda Shelf following the Last Glacial Maximum. Molecular Biology and Evolution 29: 707–719.
Diedericks, G. & S. R. Daniels, 2014. Ain’t no mountain high enough, ain’t no valley low enough? Phylogeography of the rupicolous Cape girdled lizard (Cordylus cordylus) reveals a generalist pattern. Molecular Phylogenetics and Evolution 71: 234–248.
Drummond, A. J., A. Ramabut, B. Shapiro & O. G. Pybus, 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular Biology and Evolution 22: 1185–1192.
Drummond, A. J. & A. Rambaut, 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology 7: 214.
Duftner, N., S. Koblmüller & C. Sturmbauer, 2005. Evolutionary relationships of the Limnochromini, a tribe of benthic deepwater cichlid fish endemic to Lake Tanganyika, East Africa. Journal of Molecular Evolution 60: 277–289.
Duftner, N., K. M. Sefc, S. Koblmüller, B. Nevado, E. Verheyen, H. Phiri & C. Sturmbauer, 2006. Distinct population structure in a phenotypically homogeneous rock-dwelling cichlid fish from Lake Tanganyika. Molecular Ecology 15: 2381–2396.
Duftner, N., K. M. Sefc, S. Koblmüller, W. Salzburger, M. Taborsky & C. Sturmbauer, 2007. Parallel evolution of facial stripe patterns in the Neolamprologus brichardi/pulcher species complex endemic to Lake Tanganyika. Molecular Phylogenetics and Evolution 45: 706–715.
Edgar, R. C., 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nuleic Acids Research 32: 1792–1797.
Egger, B., S. Koblmüller, C. Sturmbauer & K. M. Sefc, 2007. Nuclear and mitochondrial data reveal different evolutionary process in the Lake Tanganyika cichlid genus Tropheus. BMC Evolutionary Biology 7: 137.
Gashagaza, M., K. Nakaya & T. Sato, 1995. Taxonomy of small-sized cichlid fishes in the shell-bed area of Lake Tanganyika. Japanese Journal of Ichthyology 42: 291–302.
Genner, M. J. & G. F. Turner, 2015. Timing of population expansions within the Lake Malawi haplochromini cichlid fish radiation. Hydrobiologia 748: 121–132.
Genner, M. J., O. Seehausen, D. H. Lunt, D. A. Joyce, P. W. Shaw, G. R. Carvalho & G. F. Turner, 2007. Age of cichlids: new dates for ancient lake fish radiations. Molecular Biology and Evolution 24: 1268–1282.
Genner, M. J., P. Nichols, P. W. Shaw, G. R. Carvalho, R. L. Robinson & G. F. Turner, 2008. Genetic homogeneity among breeding grounds and nursery areas of an exploited Lake Malawi cichlid fish. Freshwater Biology 53: 1823–1831.
Genner, M. J., P. Nichols, P. W. Shaw, G. R. Carvalho, R. L. Robinson & G. F. Turner, 2010a. Population structure on breeding grounds of Lake Malawi’s ‘twilight zone’ cichlid fishes. Journal of Biogeography 37: 258–269.
Genner, M. J., M. E. Knight, M. P. Haeseler & G. F. Turner, 2010b. Establishment and expansion of Lake Malawi rock fish populations after a dramatic Late Pleistocene lake level rise. Molecular Ecology 19: 170–182.
Good, J. M., D. Vanderpool, S. Keeble & K. Bi, 2015. Neglible nuclear introgression despite complete mitochondrial capture between two species of chipmunks. Evolution 69: 1961–1972.
Hasegawa, M., H. Kishino & T. Yano, 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22: 160–174.
Hey, J., 2010. Isolation with migration models for more than two populations. Molecular Biology and Evolution 27: 905–920.
Hey, J. & R. Nielsen, 2004. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167: 747–760.
Hey, J. & R. Nielsen, 2007. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences USA 104: 2785–2790.
Ho, S. Y. W., B. Shapiro, M. J. Phillips, A. Cooper & A. J. Drummond, 2007. Evidence for time dependency of molecular rate estimates. Systematic Biology 56: 515–522.
Hoareau, T. B., 2016. Late-glacial demographic expansion motivates a clock overhaul for population genetics. Systematic Biology 65(3): 449–464.
Hori, M., 1991. Feeding relationships among cichlid fishes in Lake Tanganyika: effects of intra- and interspecific variations o feeding behaviour on their coexistence. Ecology International Bulletin 19: 89–101.
Husemann, M., R. Nguyen, B. Ding & P. D. Danley, 2015. A genetic demographic analysis of Lake Malawi rock-dwelling cichlids using spatio-temporal sampling. Molecular Ecology 24: 2686–2701.
Kajtoch, Ł., M. Mazur, D. Kubisz, M. A. Mazur & W. Babik, 2014. Low effective population sizes and limited connectivity in xerothermic beetles: implications for the conservation of an endangered habitat. Animal Conservation 17: 454–466.
Koblmüller, S., W. Salzburger & C. Sturmbauer, 2004. Evolutionary relationships in the sand-dwelling cichlid lineage of Lake Tanganyika suggest multiple colonization of rocky habitats and convergent evolution of biparental mouthbrooding. Journal of Molecular Evolution 58: 79–96.
Koblmüller, S., N. Duftner, C. Katongo, H. Phiri & C. Sturmbauer, 2005. Ancient divergence in bathypelagic Lake Tanganyika deepwater cichlids: mitochondrial phylogeny of the tribe Bathybatini. Journal of Molecular Evolution 60: 297–314.
Koblmüller, S., K. M. Sefc, N. Duftner, M. Warum & C. Sturmbauer, 2007a. Genetic population structure as indirect measure of dispersal ability in a Lake Tanganyika cichlid. Genetica 130: 121–131.
Koblmüller, S., N. Duftner, K. M. Sefc, M. Aibara, M. Stipacek, M. Blanc, B. Egger & C. Sturmbauer, 2007b. Reticulate phylogeny of gastropod-shell-breeding cichlids from Lake Tanganyika – the result of repeated introgressive hybridization. BMC Evolutionary Biology 7: 7.
Koblmüller, S., B. Egger, C. Sturmbauer & K. M. Sefc, 2007c. Evolutionary history of Lake Tanganyika’s scale eating cichlid fishes. Molecular Phylogenetics and Evolution 44: 1295–1305.
Koblmüller, S., N. Duftner, K. M. Sefc, U. Aigner, M. Rogetzer & C. Sturmbauer, 2009. Phylogeographic structure and gene flow in the scale-eating cichlid Perissodus microlepis (Teleostei, Perciformes, Cichlidae) in southern Lake Tanganyika. Zoologica Scripta 38: 257–268.
Koblmüller, S., B. Egger, C. Sturmbauer & K. M. Sefc, 2010. Rapid radiation, ancient incomplete lineage sorting and ancient hybridization in the endemic Lake Tanganyika cichlid tribe Tropheini. Molecular Phylogenetics and Evolution 55: 318–334.
Koblmüller, S., W. Salzburger, B. Obermüller, E. Eigner, C. Sturmbauer & K. M. Sefc, 2011. Separated by sand, fused by dropping water: habitat barriers and fluctuating water levels steer the evolution of rock-dwelling cichlid populations. Molecular Ecology 20: 2272–2290.
Koblmüller, S., R. K. Wayne & J. A. Leonard, 2012. Impact of Quaternary climatic changes and interspecific competition on the demographic history of a highly mobile generalist carnivore, the coyote. Biology Letters 8: 644–647.
Koblmüller, S., E. A. Odhiambo, D. Sinyinza, C. Sturmbauer & K. M. Sefc, 2015a. Big fish, little divergence: phylogeography of Lake Tanganyika’s giant cichlid, Boulengerochromis microlepis. Hydrobiologia 748: 29–38.
Koblmüller, S., B. Steinwender, S. Weiß & K. M. Sefc, 2015b. Gene flow, population growth and a novel substitution rate estimate in a subtidal rock specialist, the black-faced blenny Tripterygion delaisi (Perciformes, Blennioidei, Tripterygiidae) from the Adriatic Sea. Journal of Zoological Systematics and Evolutionary Research 53: 291–299.
Kocher, T. D., J. A. Conroy, K. R. McKay, J. R. Stauffer & S. F. Lockwood, 1995. Evolution of NADH dehydrogenase subunit 2 in East African cichlid fish. Molecular Phylogenetics and Evolution 4: 420–432.
Kohda, M., Y. Yanagisawa, T. Sato, K. Nakaya, Y. Nimura, K. Matsumoto & H. Ochi, 1996. Geographical color variation in cichlid fishes at the southern end of Lake Tanganyika. Environmental Biology of Fishes 45: 237–248.
Konings, A., 1998. Tanganyika Cichlids in their Natural Habitat. Cichlid Press, El Paso.
Kotrschal, A., G. Heckel, D. Bonfils & B. Taborsky, 2012. Life-stage specific environments in a cichlid fish: implications for inducible maternal effects. Evolutionary Ecology 26: 123–137.
Lanfear, R., B. Calcott, S. Y. W. Ho & S. Guindon, 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution 29: 1695–1701.
Lee, W., J. Conroy, W. H. Howell & T. D. Kocher, 1995. Structure and evolution of teleost mitochondrial control regions. Journal of Molecular Evolution 41: 54–66.
Leonard, J. A., C. Vilà, K. Fox-Dobbs, P. L. Koch, R. K. Wayne & B. Van Valkenburgh, 2007. Megafaunal extinctions and the disappearance of a specialized wolf ecomorph. Current Biology 17: 1146–1150.
Librado, P. & J. Rozas, 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Lyons, R. P., C. A. Scholz, A. S. Cohen, J. W. King, E. T. Brown, S. J. Ivory, T. C. Johnson, A. L. Deino, P. N. Reinthal, M. M. McGlue & M. W. Blome, 2015. Continuous 1.3-million-year record of East African hydroclimate, and implications for patterns of evolution and biodiversity. Proceedings of the National Academy of Sciences USA 112: 15568–15573.
Manni, F., E. Guerard & E. Heyer, 2004. Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Human Biology 76: 173–190.
Markert, J. A., M. E. Arnegard, P. D. Danley & T. D. Kocher, 1999. Biogeography and population genetics of the Lake Malawi cichlid Melanochromis auratus: habitat transience, philopatry and speciation. Molecular Ecology 8: 1013–1026.
McCracken, K. G. & M. D. Sorenson, 2005. Is homoplasy or lineage sorting the source of incongruent mtDNA and nuclear gene trees in the stiff-tailed ducks (Nomonyx-Oxyura). Systematic Biology 54: 35–55.
McGlue, M. M., K. E. Lezzar, A. S. Cohen, J. M. Russell, J.-J. Tiercelin, A. A. Felton, E. Mbede & H. H. Nkotagu, 2008. Seismic records of late Pleistocene aridity in Lake Tanganyika, tropical Africa. Journal of Paleolimnology 40: 635–653.
Melo-Ferreira, J. F., A. Seixas, E. Cheng, L. S. Mills & P. C. Alves, 2014. The hidden history of the snowshoe hare, Lepus americanus: extensive mitochondrial DNA introgression inferred from multilocus genetic variation. Molecular Ecology 18: 4617–4630.
Miller, M. P., 2005. Alleles in Space (AIS): computer software for the joint analysis of individual spatial and genetic information. Journal of Heredity 96: 722–724.
Miller, M. P., M. R. Bellinger, E. D. Forsman & S. M. Haig, 2006. Effects of historical climate change, habitat connectivity, and vicariance on genetic structure and diversity across the range of the red tree vole (Phenacomys longicaudus) in the Pacific Northwestern United States. Molecular Ecology 15: 134–159.
Muschick, M., A. Indermaur & W. Salzburger, 2012. Convergent evolution within an adaptive radiation of cichlid fishes. Current Biology 22: 2352–2368.
Nevado, B., S. Koblmüller, C. Sturmbauer, J. Snoeks, J. Usano-Alemany & E. Verheyen, 2009. Complete mitochondrial DNA replacement in a Lake Tanganyika cichlid fish. Molecular Ecology 18: 4240–4255.
Nevado, B., V. Fazalova, T. Backeljau, M. Hanssens & E. Verheyen, 2011. Repeated unidirectional introgression of nuclear and mitochondrial DNA between four congeneric Tanganyikan cichlids. Molecular Biology and Evolution 28: 2253–2267.
Nevado, B., S. Mautner, C. Sturmbauer & E. Verheyen, 2013. Water-level fluctuations and metapopulation dynamics as drivers of genetic diversity in populations of three Tanganyikan cichlid fish species. Molecular Ecology 22: 3933–3948.
Pereyra, R., M. I. Taylor, G. F. Turner & C. Rico, 2004. Variation in habitat preferences and population structure among three species of the Lake Malawi cichlid genus Protomelas. Molecular Ecology 13: 2691–2697.
Posada, D., 2008. jModelTest: phylogenetic model averaging. Molecular Biology and Evolution 25: 1253–1256.
Rambaut, A., M. Suchard, A. Drummond, 2013. Tracer v1.6. http://beast.bio.ed.ac.uk/Tracer.
Ray, J. M., N. J. Lang, R. M. Wood & R. L. Mayden, 2007. History repeated: recent and historical mitochondrial introgression between the current darter Etheostoma suniporum and rainbow darter Etheostoma caeruleum (Teleostei: Percidae). Journal of Fish Biology 72: 418–434.
Richlen, M. L. & P. H. Barber, 2005. A technique for the rapid extraction of microalgal DNA from single live and preserved cells. Molecular Ecology Notes 5: 688–691.
Rico, C. & G. F. Turner, 2002. Extreme microallopatric divergence in a cichlid species from Lake Malawi. Molecular Ecology 11: 1585–1590.
Ronquist, F., M. Teslenko, P. van der Mark, D. L. Ayres, A. Darling, S. Hohna, B. Larget, L. Liu, M. A. Suchard & J. P. Huelsenbeck, 2012. MrBayes3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542.
Row, J. R., G. Blouin-Demers & S. C. Lougheed, 2010. Habitat distribution influences dispersal and fine-scale genetic population structure of eastern foxsnakes (Mintonius gloydi) across a fragmented landscape. Molecular Ecology 19: 5157–5171.
Rüber, L., E. Verheyen & A. Meyer, 1999. Replicated evolution of trophic specializations in an endemic cichlid lineage from Lake Tanganyika. Proceedings of the National Academy of Sciences USA 96: 10230–10235.
Rüber, L., A. Meyer, C. Sturmbauer & E. Verheyen, 2001. Population structure in two sympatric species of the Lake Tanganyika cichlid tribe Eretmodini: evidence for introgression. Molecular Ecology 11: 619–625.
Sambrook, J. & D. W. Russell, 2001. Molecular Cloning: A Laboratory Manual, 3rd ed. Cold Springs Harbor Laboratory Press, Cold Springs Harbor, NY.
Schelly, R., W. Salzburger, S. Koblmüller, N. Duftner & C. Sturmbauer, 2006. Phylogenetic relationships of the lamprologine cichlid genus Lepidiolamprologus (Teleostei: Perciformes) based on mitochondrial and nuclear sequences, suggesting introgressive hybridization. Molecular Phylogenetics and Evolution 38: 426–438.
Scholz, C. A., T. C. Johnson, A. S. Cohen, J. W. King, J. A. Peck, J. T. Overpeck, M. R. Talbot, E. T. Brown, L. Kalindekafe, P. Y. A. Amoako, R. P. Lyons, T. M. Shanahan, I. S. Castaneda, C. W. Heil, S. L. Forman, L. R. McHargue, K. R. Beuning, J. Gomez & J. Pierson, 2007. East African megadroughts between 135 and 75 thousand years ago and bearing on early-modern human origins. Proceedings of the National Academy of Sciences USA 104: 16416–16421.
Sefc, K. M., S. Baric, W. Salzburger & C. Sturmbauer, 2007. Species-specific population structure in rock-specialized sympatric cichlid species in Lake Tanganyika, East Africa. Journal of Molecular Evolution 64: 33–49.
Sefc, K. M., K. Mattersdorfer, C. M. Hermann & S. Koblmüller, 2016. Past lake shore dynamics explain present pattern of unidirectional introgression across a habitat barrier. Hydrobiologia. doi:10.1007/s10750-016-2791-x.
Shaw, P. W., G. F. Turner, M. R. Idid, R. L. Robinson & G. R. Carvalho, 2000. Genetic population structure indicates sympatric speciation of Lake Malawi pelagic cichlids. Proceedings of the Royal Society London Series B, Biological Sciences 267: 2273–2280.
Sonnenberg, R., A. W. Nolte & D. Tautz, 2007. An evaluation of LSU rDNA D1-D2 sequences for their use in species identification. Frontiers in Zoology 4: 6.
Spreitzer, M. L., S. Mautner, L. Makasa & C. Sturmbauer, 2012. Genetic and morphological population differentiation in the rock-dwelling and specialized shrimp-feeding cichlid fish species Altolamprologus compressiceps from Lake Tanganyika, East Africa. Hydrobiologia 682: 143–154.
Stiassny, M. L. J., 1997. A phylogenetic overview of the lamprologine cichlids of Africa (Teleostei, Cichlidae): a morphological perspective. South African Journal of Science 93: 513–523.
Sturmbauer, C., S. Baric, W. Salzburger, L. Rüber & E. Verheyen, 2001. Lake level fluctuations synchronize genetic divergences of cichlid fishes in African lakes. Molecular Biology and Evolution 18: 144–154.
Sturmbauer, C., S. Koblmüller, K. M. Sefc & N. Duftner, 2005. Phylogeographic history of the genus Tropheus, a lineage of rock-dwelling cichlid fishes endemic to Lake Tanganyika. Hydrobiologia 542: 335–366.
Sturmbauer, C., C. Fuchs, G. Harb, E. Damm, N. Duftner, M. Maderbacher, M. Koch & S. Koblmüller, 2008. Abundance, distribution and territory areas of rock-dwelling Lake Tanganyika cichlid fish species. Hydrobiologia 615: 57–68.
Sturmbauer, C., W. Salzburger, N. Duftner, R. Schelly & S. Koblmüller, 2010. Evolutionary history of the Lake Tanganyika cichlid tribe Lamprologini (Teleostei: Perciformes) derived from mitochondrial and nuclear DNA data. Molecular Phylogenetics and Evolution 57: 266–284.
Takeuchi, Y., H. Ochi, M. Kohda, D. Sinyinza & M. Hori, 2010. A 20-year census of a rocky littoral fish community in Lake Tanganyika. Ecology of Freshwater Fish 19: 239–248.
Tamura, K., G. Stecher, D. Peterson, A. Filipski & S. Kumar, 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30: 2725–2729.
Tang, Q.-Y., S.-Q. Liu, D. Yu, H.-Z. Liu & P. Danley, 2012. Mitochondrial capture and incomplete lineage sorting in the diversification of balitorine loaches (Cypriniformes, Balitoridae) revealed by mitochondrial and nuclear genes. Zoologica Scripta 41: 233–247.
Taylor, M. I. & E. Verheyen, 2001. Microsatellite data reveals weak population substructuring in Copadichromis sp. “virginalis kajose”, a demersal cichlid from Lake Malawi, Africa. Journal of Fish Biology 59: 593–604.
Taylor, M. I., L. Rüber & E. Verheyen, 2001. Microsatellites reveal high levels of population substructuring in the species-poor eretmodine cichlid lineage from Lake Tanganyika. Proceedings of the Royal Society London, B 268: 803–808.
Van Oppen, M. J. H., G. F. Turner, C. Rico, J. C. Deutsch, K. M. Ibrahim, R. L. Robinson & G. M. Hewitt, 1997. Unusually fine-scale genetic structuring found in rapidly speciating Malawi cichlid fishes. Proceedings of the Royal Society London Series B, Biological Sciences 264: 1803–1812.
Van Steenberge, M., M. P. M. Vanhove, D. Muzumani Risasi, T. Mulimbwa N’sibula, F. Muterezi Bukinga, A. Pariselle, C. Gillardin, E. Vreven, J. A. M. Raeymaekers, T. Huyse, F. A. M. Volckaert, V. Nshombo Muderhwa & J. Snokes, 2011. A recent inventory of the fishes of the north-western and central western coast of Lake Tanganyika (Democratic Republic Congo). Acta Ichthyologica et Piscatoria 41: 201–214.
Van Steenberge, M., M. P. M. Vanhove, F. C. Breman & J. Snoeks, 2015. Complex geographical variation patterns in Tropheus duboisi Marlier, 1959 (Perciformes, Cichlidae) from Lake Tanganyika. Hydrobiologia 748: 39–60.
Verheyen, E., L. Rüber, J. Snoeks & A. Meyer, 1996. Mitochondrial phylogeography of rock-dwelling cichlid fishes reveals evolutionary influence of historical lake level fluctuations of Lake Tanganyika, Africa. Philosophical Transactions: Biological Sciences 351: 797–805.
Wagner, C. E. & A. R. McCune, 2009. Contrasting patterns of spatial genetic structure in sympatric rock-dwelling cichlid fishes. Evolution 63: 1312–1326.
Wiens, J. J. & M. J. Donoghue, 2004. Historical biogeography, ecology and species richness. Trends in Ecology and Evolution 19: 639–644.
Winkelmann, K., L. Rüber & M. J. Genner, 2016. Lake level fluctuations and divergence of cichlid fish ecomorphs in Lake Tanganyika. Hydrobiologia. doi:10.1007/s10750-016-2839-y.
Young, A., T. Boyle & T. Brown, 1996. The population genetic consequences of habitat fragmentation for plants. Trends in Ecology and Evolution 11: 413–418.
Yuma, M. & T. Kondo, 1997. Interspecific Relationships and Habitat Utilization Among Benthivorous Cichlids. In Kawanabe, H., M. Hori & M. Nagoshi (eds), Fish Communities in Lake Tanganyika. Kyoto University Press, Kyoto: 87–103.
Acknowledgements
A. Avenant-Oldewage, C. Gillardin, T. Huyse, V. Lumami Kapepula, J. Mbirize Ndalozibwa, V. Muderhwa Nshombo, T. Mulimbwa N’sibula, F. Muterezi Bukinga, D. Muzumani Risasi, A. Pariselle, J.A.M. Raeymaekers, S. Risch, J. Snoeks, D. Sinyinza, T. Veall and F.A.M. Volckaert are acknowledged for their help in organizing sampling. This study was supported by the Austrian Science Fund (FWF): grants P20883-B16 to K.M.S. and P22737-B09 to C.S., and the Belgian Science Policy BRAIN project GENBAS to E.V. Part of the fieldwork was financed by the Research Foundation—Flanders (FWO-Vlaanderen) (research programme G.0553.10) and by travel grants from FWO-Vlaanderen (to M.P.M.V.) and from the King Leopold III Fund for Nature Exploration and Conservation (to M.V.S.). M.P.M.V. and M.V.S. were PhD fellows of FWO-Vlaanderen. M.P.M.V. is partly supported by the Czech Science Foundation, project no. P505/12/G112 (ECIP). Open access funding provided by University of Graz.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest editors: S. Koblmüller, R. C. Albertson, M. J. Genner, K. M. Sefc & T. Takahashi / Advances in Cichlid Research II: Behavior, Ecology and Evolutionary Biology
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Koblmüller, S., Nevado, B., Makasa, L. et al. Phylogeny and phylogeography of Altolamprologus: ancient introgression and recent divergence in a rock-dwelling Lake Tanganyika cichlid genus. Hydrobiologia 791, 35–50 (2017). https://doi.org/10.1007/s10750-016-2896-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-016-2896-2