
Abstract
Background
A breast cancer polygenic risk score (PRS) comprising 313 common variants reliably predicts disease risk. We examined possible relationships between genetic variation, regulation, and expression to clarify the molecular alterations associated with these variants.
Methods
Genome-wide methylomic variation was quantified (MethylationEPIC) in Asian breast cancer patients (1152 buffy coats from peripheral whole blood). DNA methylation (DNAm) quantitative trait loci (mQTL) mapping was performed for 235 of the 313 variants with minor allele frequencies > 5%. Stability of identified mQTLs (p < 5e-8) across lifetime was examined using a public mQTL database. Identified mQTLs were also mapped to expression quantitative trait loci (eQTLs) in the Genotype-Tissue Expression Project and the eQTLGen Consortium.
Results
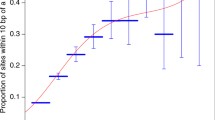
Breast cancer PRS was not associated with DNAm. A higher proportion of significant cis-mQTLs were observed. Of 822 significant cis-mQTLs (179 unique variants) identified in our dataset, 141 (59 unique variants) were significant (p < 5e-8) in a public mQTL database. Eighty-six percent (121/141) of the matched mQTLs were consistent at multiple time points (birth, childhood, adolescence, pregnancy, middle age, post-diagnosis, or treatment). Ninety-three variants associated with DNAm were also cis-eQTLs (35 variants not genome-wide significant). Multiple loci in the breast cancer PRS are associated with DNAm, contributing to the polygenic nature of the disease. These mQTLs are mostly stable over time.
Conclusions
Consistent results from DNAm and expression data may reveal new candidate genes not previously associated with breast cancer.




Similar content being viewed by others
Data availability
The data that support the findings of this study are available on request from Mikael Hartman (ephbamh@nus.edu.sg). The data are not publicly available due to privacy or ethical restrictions.
References
Frayling TM (2014) Genome-wide association studies: the good, the bad and the ugly. Clin Med (Lond) 14(4):428–431
Maurano MT et al (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337(6099):1190–1195
Ernst J et al (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473(7345):43–49
Schaub MA et al (2012) Linking disease associations with regulatory information in the human genome. Genome Res 22(9):1748–1759
Biernacka JM, Cordell HJ (2007) Exploring causality via identification of SNPs or haplotypes responsible for a linkage signal. Genet Epidemiol 31(7):727–740
Husquin LT et al (2018) Exploring the genetic basis of human population differences in DNA methylation and their causal impact on immune gene regulation. Genome Biol 19(1):222
Michailidou K et al (2017) Association analysis identifies 65 new breast cancer risk loci. Nature 551(7678):92–94
Milne RL et al (2017) Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet 49(12):1767–1778
Bahcall O (2013) Functional annotation of susceptibility loci identified by COGS. Nature Genetics
Mavaddat N et al (2019) Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet 104(1):21–34
Ho WK et al (2020) European polygenic risk score for prediction of breast cancer shows similar performance in Asian women. Nat Commun 11(1):3833
Amos CI et al (2017) The oncoarray consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev 26(1):126–135
Delaneau O, Coulonges C, Zagury JF (2008) Shape-IT: new rapid and accurate algorithm for haplotype inference. BMC Bioinformatics 9:540
Delaneau O, Zagury JF, Marchini J (2013) Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods 10(1):5–6
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5(6):e1000529
Delaneau O et al (2014) Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun 5:3934
Muller, F., et al., RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol, 2019. 20(1): p. 55.
Pidsley R et al (2016) Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol 17(1):208
Triche TJ Jr et al (2013) Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res 41(7):e90
Teschendorff AE et al (2013) A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29(2):189–196
Assenov Y et al (2014) Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods 11(11):1138–1140
Aran D, Sirota M, Butte AJ (2015) Systematic pan-cancer analysis of tumour purity. Nat Commun 6:8971
Mansell G et al (2019) Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics 20(1):366
Gaunt TR et al (2016) Systematic identification of genetic influences on methylation across the human life course. Genome Biol 17:61
Watanabe K et al (2017) Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8(1):1826
Võsa U et al (2018) Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv:447367.
Kuleshov MV et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44(W1):W90–W97
Chen EY et al (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14:128
Rivandi M, Martens JWM, Hollestelle A (2018) Elucidating the underlying functional mechanisms of breast cancer susceptibility through post-GWAS analyses. Front Genet 9:280
Fachal L et al (2020) Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat Genet 52(1):56–73
Feng H et al (2020) Transcriptome-wide association study of breast cancer risk by estrogen-receptor status. Genet Epidemiol 44(5):442–468
Liu Y et al (2019) An analysis about heterogeneity among cancers based on the DNA methylation patterns. BMC Cancer 19(1):1259
Lin D et al (2018) Characterization of cross-tissue genetic-epigenetic effects and their patterns in schizophrenia. Genome Med 10(1):13
Smith AK et al (2014) Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics 15:145
Hannon E et al (2018) Leveraging DNA-methylation quantitative-trait loci to characterize the relationship between methylomic variation, gene expression, and complex traits. Am J Hum Genet 103(5):654–665
Zhu Z et al (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48(5):481–487
Wu Y et al (2018) Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun 9(1):918
Parton M, Dowsett M, Smith I (2001) Studies of apoptosis in breast cancer. BMJ 322(7301):1528–1532
Liu H, Ye H (2017) Screening of the prognostic targets for breast cancer based co-expression modules analysis. Mol Med Rep 16(4):4038–4044
Wu D et al (2016) Molecular mechanisms associated with breast cancer based on integrated gene expression profiling by bioinformatics analysis. J Obstet Gynaecol 36(5):615–621
Holbrook J et al (2019) Tumour necrosis factor signalling in health and disease. F1000Res 8.
Acknowledgements
SGBCC thanks the participants and all research coordinators for their excellent help with recruitment, data, and sample collection. The breast cancer genome-wide association analyses were supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the ‘Ministère de l’Économie, de la Science et de l’Innovation du Québec’ through Genome Québec and grant PSR-SIIRI-701, The National Institutes of Health (U19 CA148065, X01HG007492), Cancer Research UK (C1287/A10118, C1287/A16563, C1287/A10710) and The European Union (HEALTH-F2-2009-223175 and H2020 633784 and 634935). All studies and funders are listed in Michailidou et al. (2017).
Funding
This study was supported by the National Research Foundation Singapore (NRF-NRFF2017-02), NUS start-up Grant, National University Cancer Institute Singapore (NCIS) Centre Grant [NMRC/CG/NCIS/2010, NMRC/CG/012/2013, CGAug16M005], Saw Swee Hock School of Public Health Research Programme of Research Seed Funding (Breast Cancer Prevention Program), Asian Breast Cancer Research Fund, and the NMRC Clinician Scientist Award (SI Category) [NMRC/CSA-SI/0015/2017]. The breast cancer genome-wide association analyses were supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the ‘Ministère de l’Économie, de la Science et de l’Innovation du Québec’ through Genome Québec and grant PSR-SIIRI-701, The National Institutes of Health (U19 CA148065, X01HG007492), Cancer Research UK (C1287/A10118, C1287/A16563, C1287/A10710) and The European Union (HEALTH-F2-2009-223175 and H2020 633784 and 634935). All studies and funders are listed in Michailidou et al. (2017).
Author information
Authors and Affiliations
Contributions
Conceptualization: JLi, Data curation: MH, Formal Analysis: PJH, Funding acquisition: JLi, MH, Methodology: RD, JLi, PJH, Project administration: PJH, Resources: MH, Writing – original draft: JLi, PJH, Writing – review & editing: All co-authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Ethical approval
This study is approved by the Agency of Science, Technology and Research, Human Biomedical Research Office (Reference: 2020-005). Recruitment for the Singapore Breast Cancer Cohort is approved by the National Healthcare Group Domain Specific Review Board (Reference: 2009/000501) and the SingHealth Centralised Institutional Review Board (Reference: 2019/2246).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Ho, P.J., Dorajoo, R., Ivanković, I. et al. DNA methylation and breast cancer-associated variants. Breast Cancer Res Treat 188, 713–727 (2021). https://doi.org/10.1007/s10549-021-06185-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-021-06185-9