
Explorative Meta-Analysis of 417 Extant Archaeal Genomes to Predict Their Contribution to the Total Microbiome Functionality

 ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Metadata Collection of the Total Known Archaeal Microbiome Functions
2.2. Accumulation Curves (AC)
2.3. Statistical Analysis
3. Results
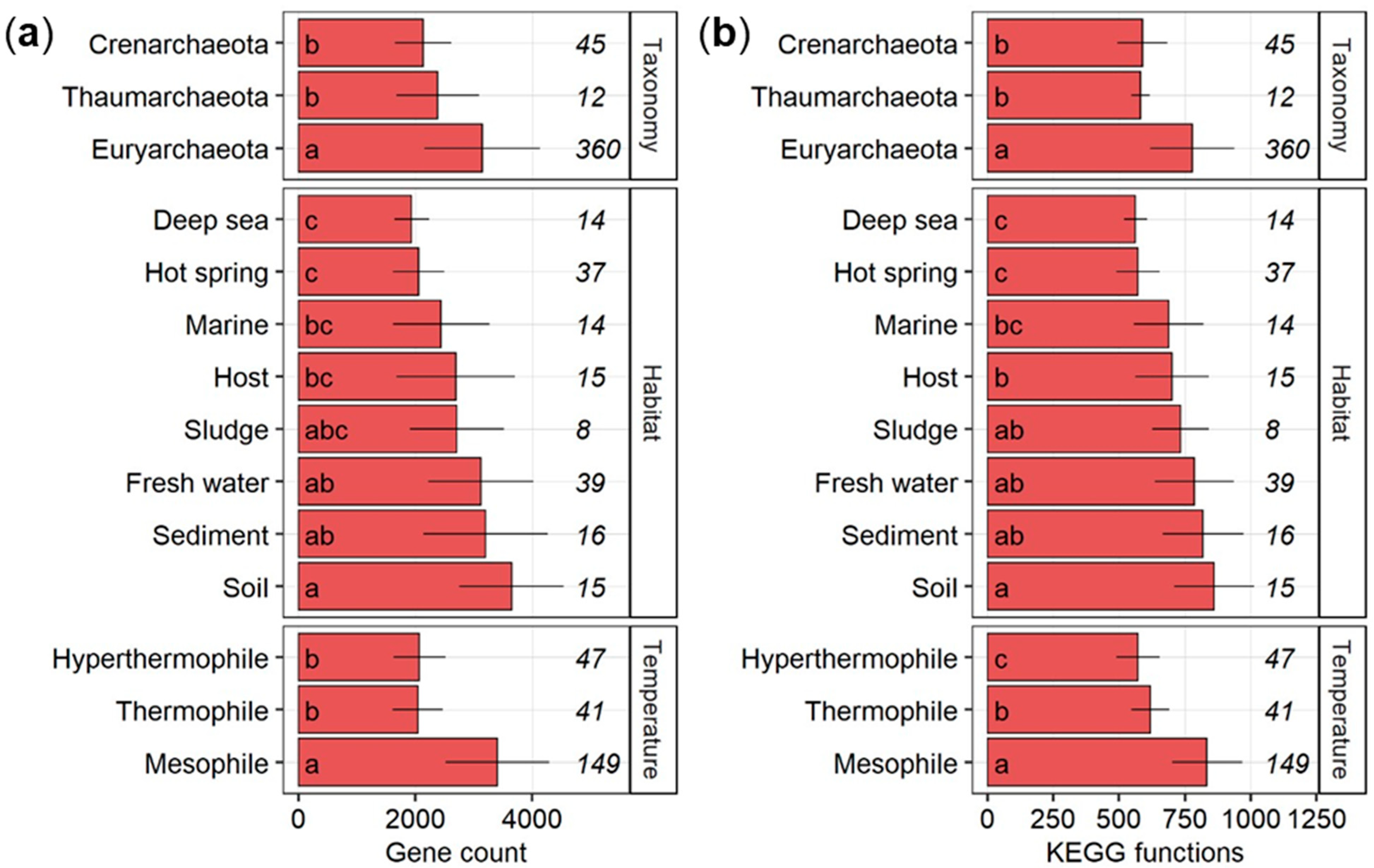
3.1. Gene Counts and Number of KEGG Level 3 Functions
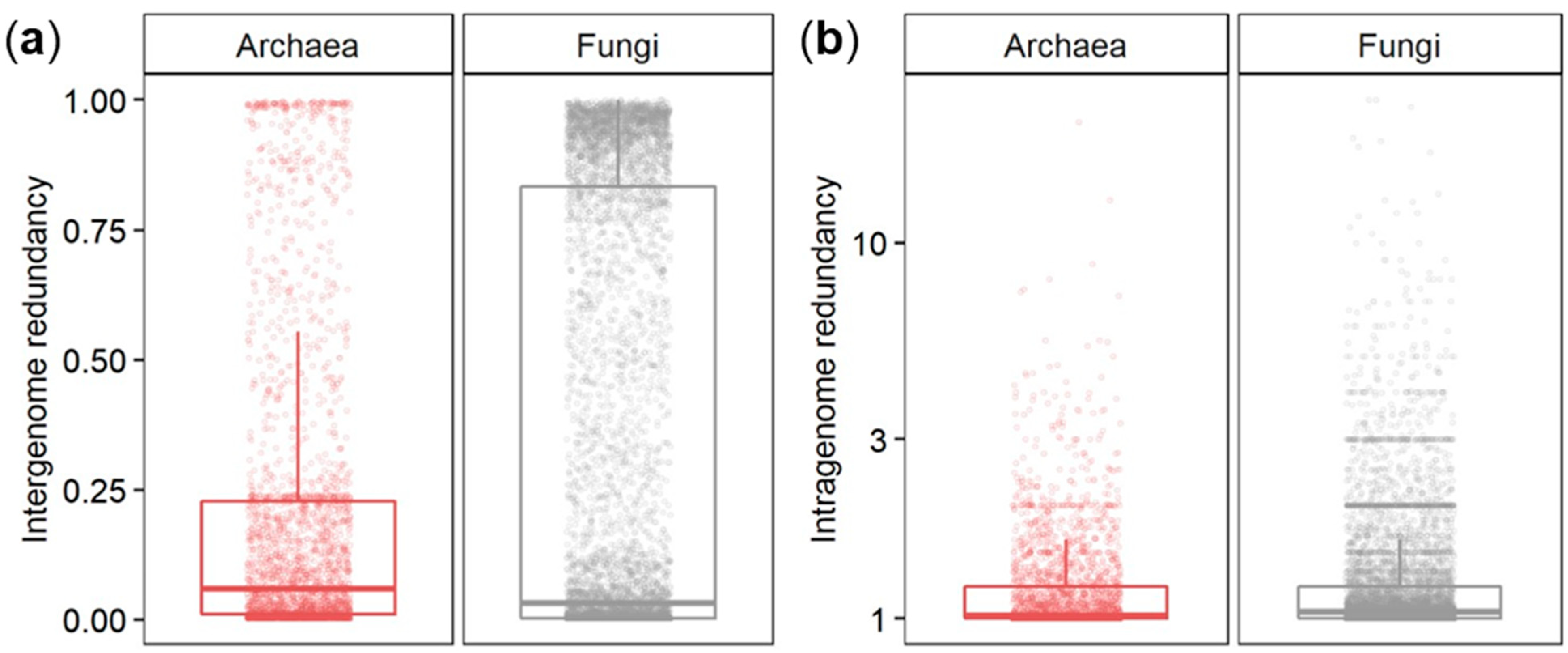
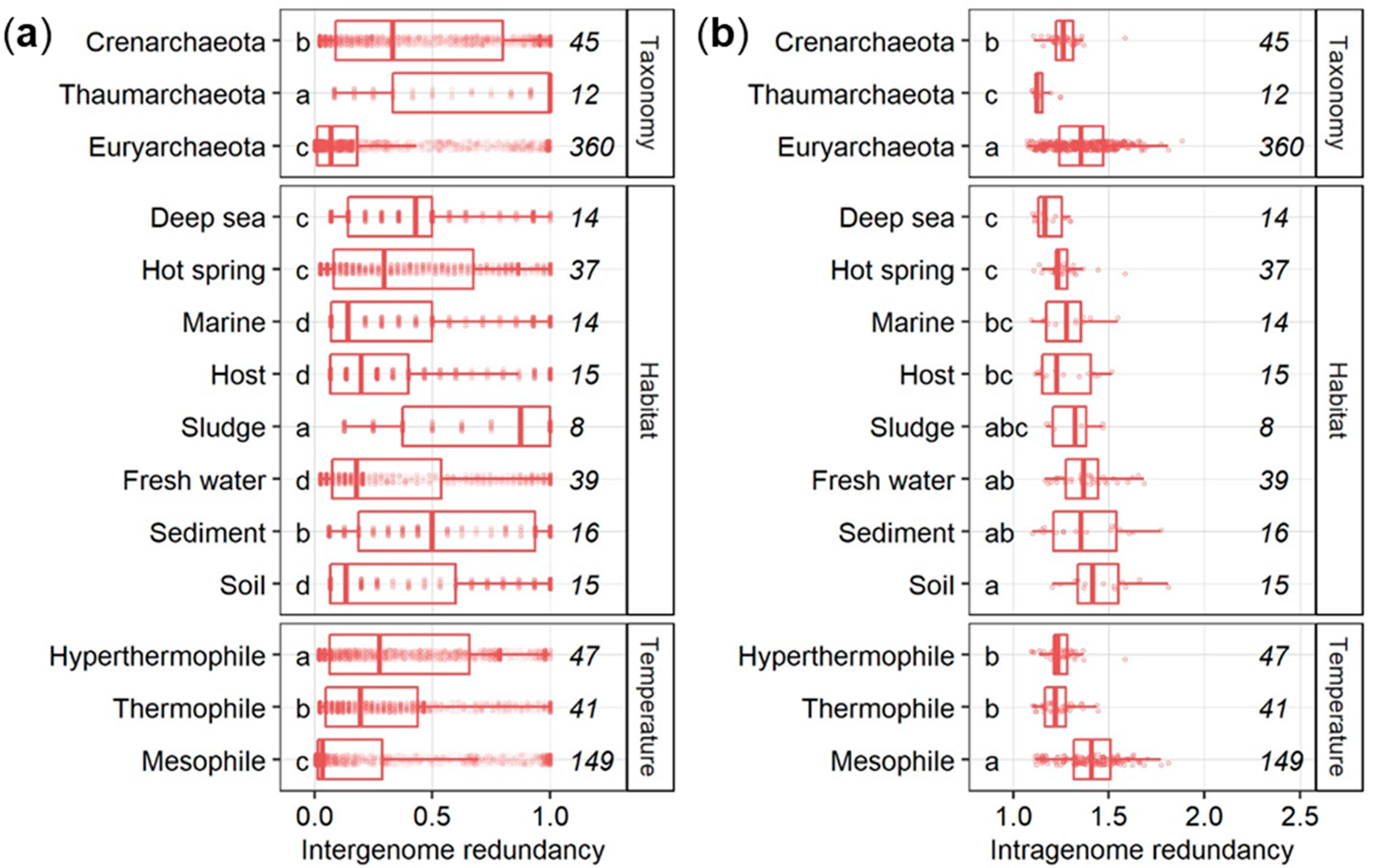
3.2. Inter- and Intragenome Functional Redundancy
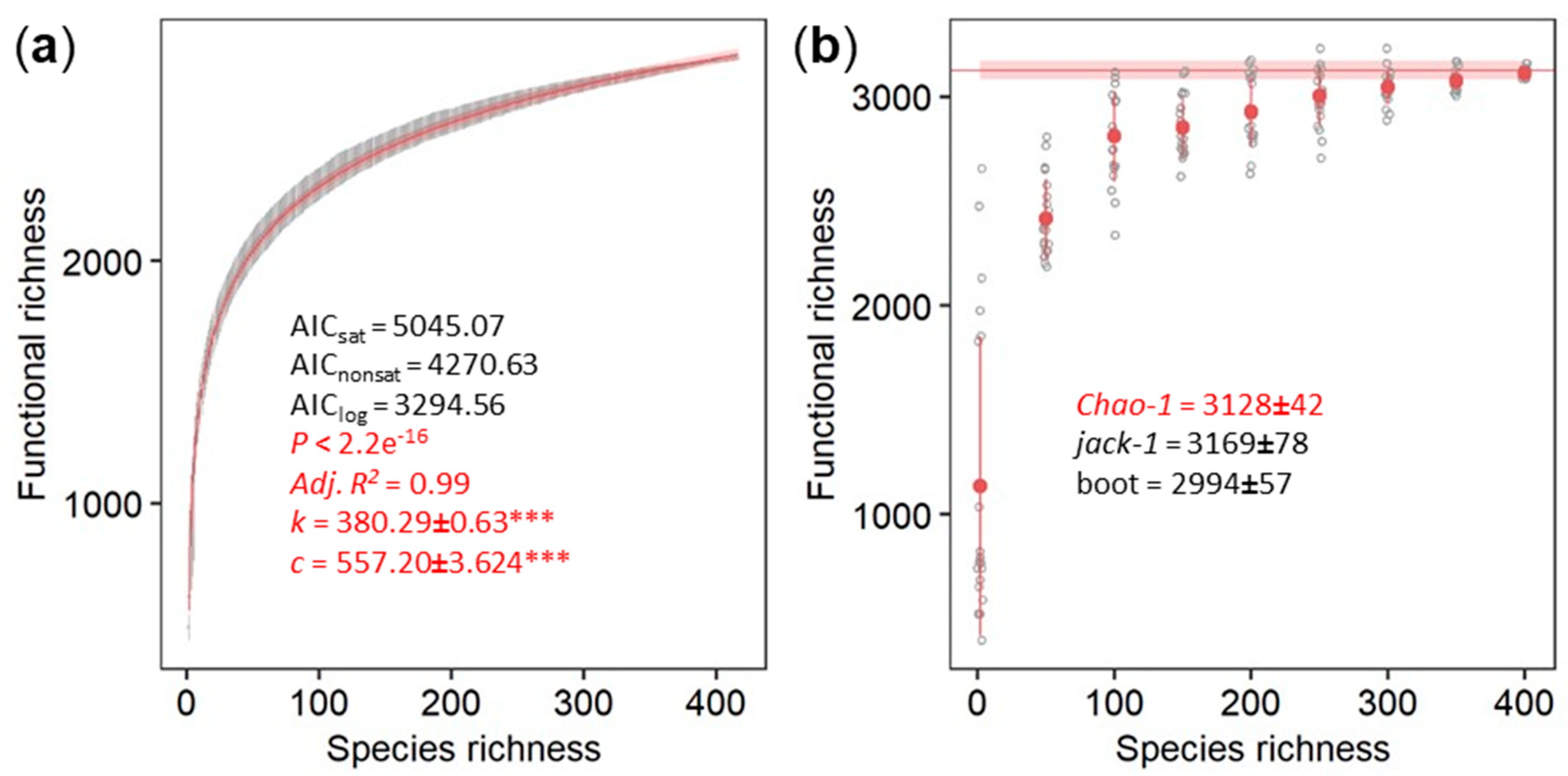
3.3. Parametric and Non-Parametric Estimation of the Archaeal Contribution to the Total Microbiome Functionality
4. Discussion
4.1. Genome Content
4.2. Functional Redundancy
4.3. Archaeal Contribution to the Total Microbiome Functionality
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delong, E.F. Everything in moderation: Archaea as “non-extremophiles.”. Curr. Opin. Genet. Dev. 1998, 8, 649–654. [Google Scholar] [CrossRef]
- Timonen, S.; Bomberg, M. Archaea in dry soil environments. Phytochem. Rev. 2009, 8, 505–518. [Google Scholar] [CrossRef]
- DeLong, E.F.; Pace, N.R. Environmental diversity of bacteria and archaea. Syst. Biol. 2001, 50, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Stoica, E.; Herndl, G.J. Contribution of Crenarchaeota and Euryarchaeota to the prokaryotic plankton in the coastal northwestern Black Sea. J. Plankton Res. 2007, 29, 699–706. [Google Scholar] [CrossRef]
- Bates, S.T.; Berg-Lyons, D.; Caporaso, J.G.; Walters, W.A.; Knight, R.; Fierer, N. Examining the global distribution of dominant archaeal populations in soil. Isme J. 2011, 5, 907–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korzhenkov, A.A.; Toshchakov, S.V.; Bargiela, R.; Gibbard, H.; Ferrer, M.; Teplyuk, A.V.; Jones, D.L.; Kublanov, I.V.; Golyshin, P.N.; Golyshina, O.V. Archaea dominate the microbial community in an ecosystem with low-to-moderate temperature and extreme acidity. Microbiome 2019, 7, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leininger, S.; Urich, T.; Schloter, M.; Schwark, L.; Qi, J.; Nicol, G.W.; Prosser, J.I.; Schuster, S.C.; Schleper, C. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 2006, 442, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Bengtson, P.; Sterngren, A.E.; Rousk, J. Archaeal abundance across a pH gradient in an arable soil and its relationship to bacterial and fungal growth rates. Appl. Env. Microbiol. 2012, 78, 5906–5911. [Google Scholar] [CrossRef] [Green Version]
- Hubbell, S.P. Neutral theory in community ecology and the hypothesis of functional equivalence. Funct. Ecol. 2005, 19, 166–172. [Google Scholar] [CrossRef]
- Žifčáková, L.; Větrovský, T.; Lombard, V.; Henrissat, B.; Howe, A.; Baldrian, P. Feed in summer, rest in winter: Microbial carbon utilization in forest topsoil. Microbiome 2017, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Starke, R.; Capek, P.; Morais, D.; Jehmlich, N.; Baldrian, P. Explorative Meta-Analysis of 377 Extant Fungal Genomes Predicted a Total Mycobiome Functionality of 42.4 Million KEGG Functions. Front. Microbiol. 2020, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.; Capek, P.; Morais, D.; Callister, S.J.; Jehmlich, N. The total microbiome functions in bacteria and fungi. J. Proteom. 2020, 2013, 103623. [Google Scholar] [CrossRef]
- Starke, R.; Capek, P.; Morais, D.K.; Jehmlich, N.; Baldrian, P. The Total Fungal Microbiome Functionality. 2019. Available online: https://www.biorxiv.org/content/biorxiv/early/2020/08/04/2020.08.04.236075.full.pdf (accessed on 17 July 2020).
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Pham, V.H.T.; Kim, J. Cultivation of unculturable soil bacteria. Trends Biotechnol. 2012, 30, 475–484. [Google Scholar] [CrossRef]
- Martiny, A.C. High proportions of bacteria are culturable across major biomes. ISME J. 2019, 13, 2125–2128. [Google Scholar] [CrossRef]
- Starke, R.; Jehmlich, N.; Bastida, F. Using proteins to study how microbes contribute to soil ecosystem services: The current state and future perspectives of soil metaproteomics. J. Proteom. 2018, 198, 50–58. [Google Scholar] [CrossRef]
- Větrovský, T.; Kohout, P.; Kopecký, M.; Macháč, A.; Man, M.; Bahnmann, B.D.; Brabcová, V.; Choi, J.; Meszárošová, L.; Human, Z.R.; et al. A meta-analysis of global fungal distribution reveals climate-driven patterns. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotelli, N.J.; Colwell, R.K. Quantifying biodiversity: Procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 2001, 4, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Thompson, G.G.; Withers, P.C.; Pianka, E.R.; Thompson, S.A. Assessing biodiversity with species accumulation curves; inventories of small reptiles by pit-trapping in Western Australia. Austral. Ecol. 2003, 28, 361–383. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2015. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 17 July 2020).
- Čapek, P.; Kotas, P.; Manzoni, S.; Šantrůčková, H. Drivers of phosphorus limitation across soil microbial communities. Funct. Ecol. 2016, 30, 1705–1713. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, P.V.; Sakamoto, Y.; Ishiguro, M.; Kitagawa, G. Akaike Information Criterion Statistics. J. R. Stat. Soc. Ser. A 2006, 151, 567–568. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Spiess, A.N. Propagate: Propagation of Uncertainty. 2018. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 17 July 2020).
- Chao, A. Non-parametric estimation of the classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar] [CrossRef]
- Chao, A. Estimating Population Size for Sparse Data in Capture-Recapture Experiments. Biometrics 1989, 45, 427–438. [Google Scholar] [CrossRef]
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99–114. [Google Scholar] [CrossRef]
- De Mendiburu, F. Agricolae: Statistical Procedures for Agricultural Research. 2014. Available online: https://cran.r-project.org/web/packages/agricolae/index.html (accessed on 17 July 2020).
- Petrov, D.A. Evolution of genome size: New approaches to an old problem. Trends Genet. 2001, 17, 23–28. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Nuclear volume control by nucleoskeletal DNA, selection for cell volume and cell growth rate, and the solution of the DNA C-value paradox. J. Cell Sci. 1978, 24, 247–278. [Google Scholar]
- Vinogradov, A.E. Buffering: A possible passive-homeostasis role for redundant DNA. J. Biol. 1998, 193, 197–199. [Google Scholar] [CrossRef]
- Wang, H.Y.; Guo, S.Y.; Huang, M.R.; Thorsten, L.H.; Wei, J.C. Ascomycota has a faster evolutionary rate and higher species diversity than Basidiomycota. Sci. China Life Sci. 2010, 53, 1163–1169. [Google Scholar] [CrossRef]
- Větrovský, T.; Baldrian, P. The Variability of the 16S rRNA Gene in Bacterial Genomes and Its Consequences for Bacterial Community Analyses. PLoS ONE 2013, 8, e57923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive earth’s biogeochemical cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [Green Version]
- Rineau, F.; Courty, P.E. Secreted enzymatic activities of ectomycorrhizal fungi as a case study of functional diversity and functional redundancy. Anna. For. Sci. 2011, 68, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Gotelli, N.; Colwell, R. Estimating species richness. Biol. Divers. Front. Meas. Assess. 2011, 12, 39–54. [Google Scholar] [CrossRef]
- Chao, A.; Colwell, R.K.; Lin, C.W.; Gotelli, N.J. Sufficient sampling for asymptotic minimum species richness estimators. Ecology 2009, 90, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.A.; Szöllosi, G.J.; Spang, A.; Foster, P.G.; Heaps, S.E.; Boussau, B.; Ettema, T.J.G.; Martin Embley, T. Integrative modeling of gene and genome evolution roots the archaeal tree of life. Proc. Natl. Acad. Sci. USA 2017, 114, E4602–E4611. [Google Scholar] [CrossRef] [Green Version]
- Castelle, C.J.; Banfield, J.F. Major New Microbial Groups Expand Diversity and Alter our Understanding of the Tree of Life. Cell 2018, 172, 1181–1197. [Google Scholar] [CrossRef] [Green Version]
- Amann, R.; Rosselló-Móra, R. After all, only millions? MBio 2016, 7, e00201-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Mai, U.; Pfeiffer, W.; Janssen, S.; Asnicar, F.; Sanders, J.G.; Belda-Ferre, P.; Al-Ghalith, G.A.; Kopylova, E.; McDonald, D.; et al. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, R.; Fernandes, M.L.P.; Morais, D.K.; Odriozola, I.; Jehmlich, N.; Baldrian, P. Explorative Meta-Analysis of 417 Extant Archaeal Genomes to Predict Their Contribution to the Total Microbiome Functionality. 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.08.04.236075v1 (accessed on 17 July 2020).




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starke, R.; Fernandes, M.L.P.; Morais, D.K.; Odriozola, I.; Baldrian, P.; Jehmlich, N. Explorative Meta-Analysis of 417 Extant Archaeal Genomes to Predict Their Contribution to the Total Microbiome Functionality. Microorganisms 2021, 9, 381. https://doi.org/10.3390/microorganisms9020381
Starke R, Fernandes MLP, Morais DK, Odriozola I, Baldrian P, Jehmlich N. Explorative Meta-Analysis of 417 Extant Archaeal Genomes to Predict Their Contribution to the Total Microbiome Functionality. Microorganisms. 2021; 9(2):381. https://doi.org/10.3390/microorganisms9020381
Chicago/Turabian StyleStarke, Robert, Maysa Lima Parente Fernandes, Daniel Kumazawa Morais, Iñaki Odriozola, Petr Baldrian, and Nico Jehmlich. 2021. "Explorative Meta-Analysis of 417 Extant Archaeal Genomes to Predict Their Contribution to the Total Microbiome Functionality" Microorganisms 9, no. 2: 381. https://doi.org/10.3390/microorganisms9020381