DNA- and RNA- Derived Fungal Communities in Subsurface Aquifers Only Partly Overlap but React Similarly to Environmental Factors

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Groundwater Sample Collection
2.3. Physico-Chemical Analysis of Groundwater Samples
2.4. Nucleic Acid Extraction, Amplicon Library Preparation and Pair-End Illumina Sequencing
2.5. Bioinformatics Processing of Sequence Data
2.6. Statistical Processing
2.7. Functional Analysis
3. Results
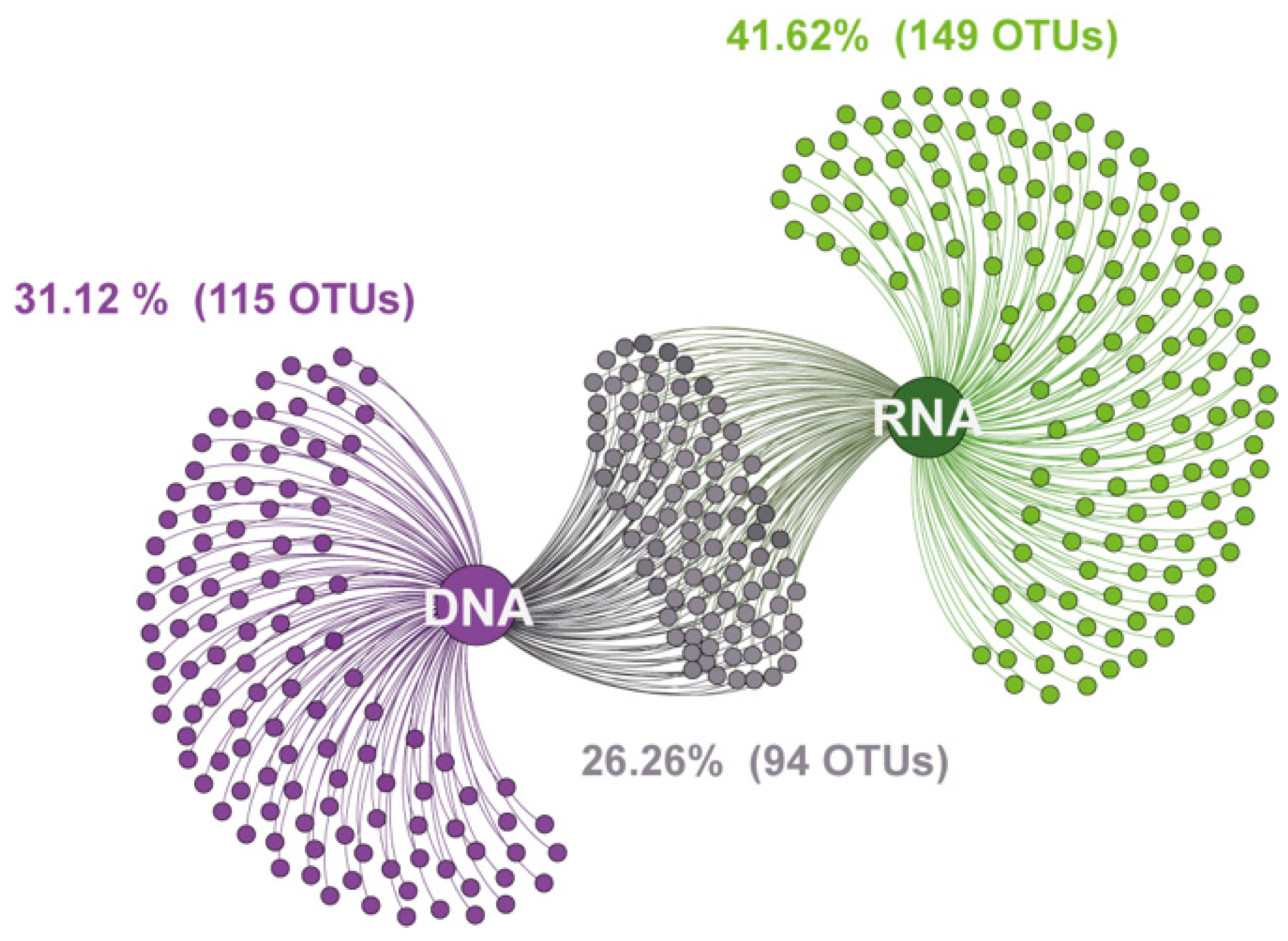
3.1. Illumina Sequencing of DNA- and RNA-Derived Fungal Communities
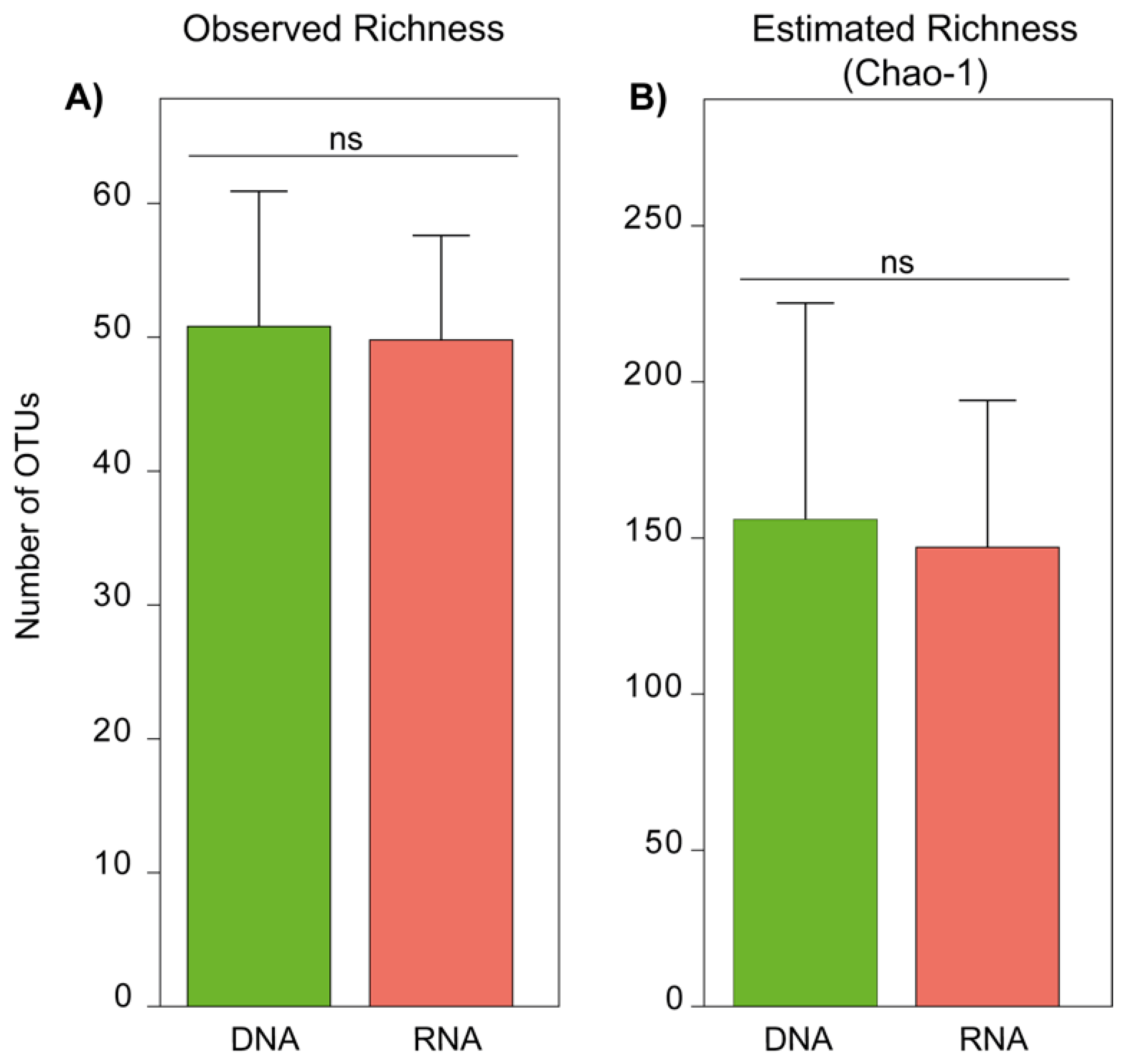
3.2. Fungal Species Richness of DNA- and RNA-Derived Communities
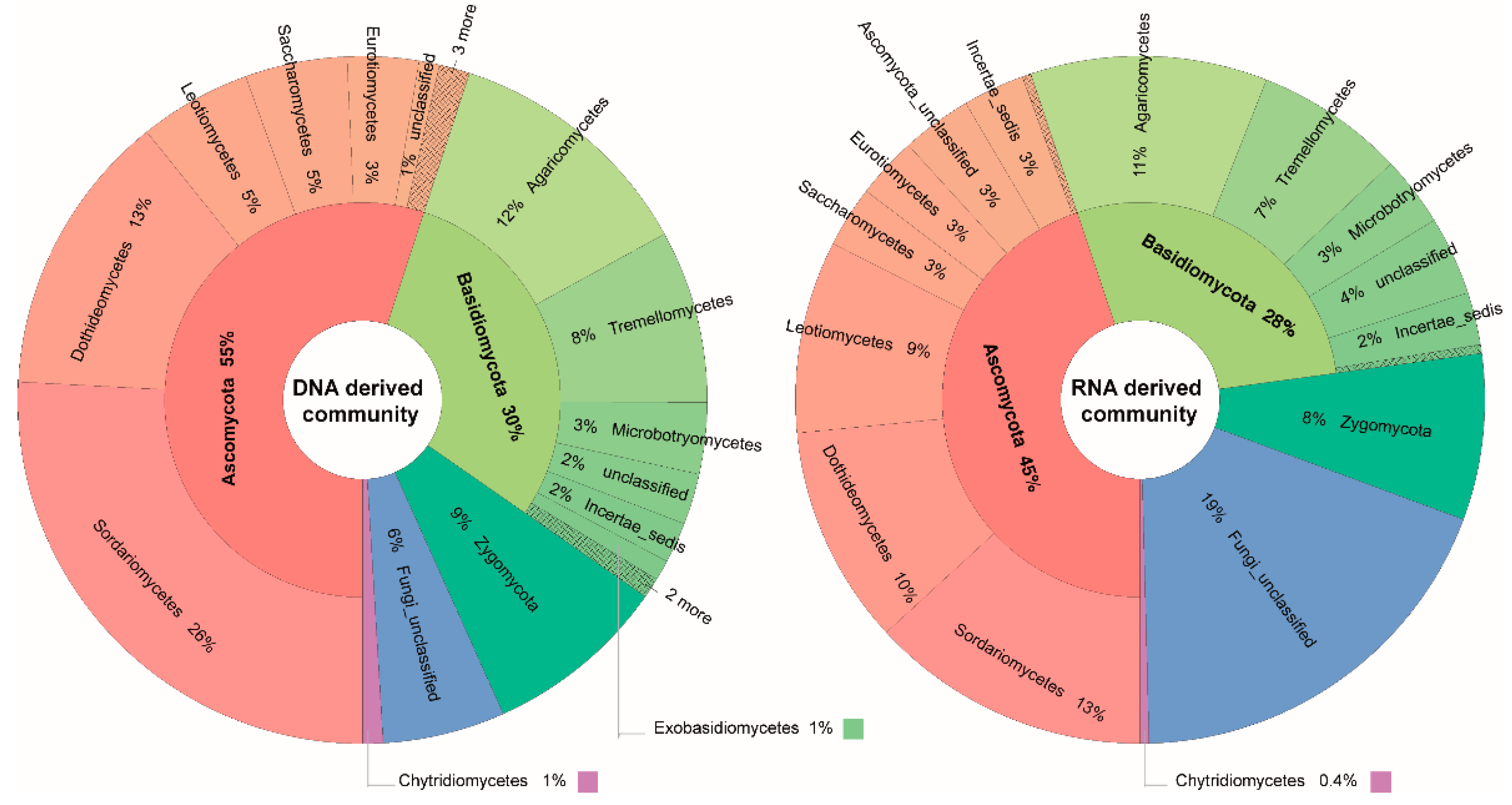
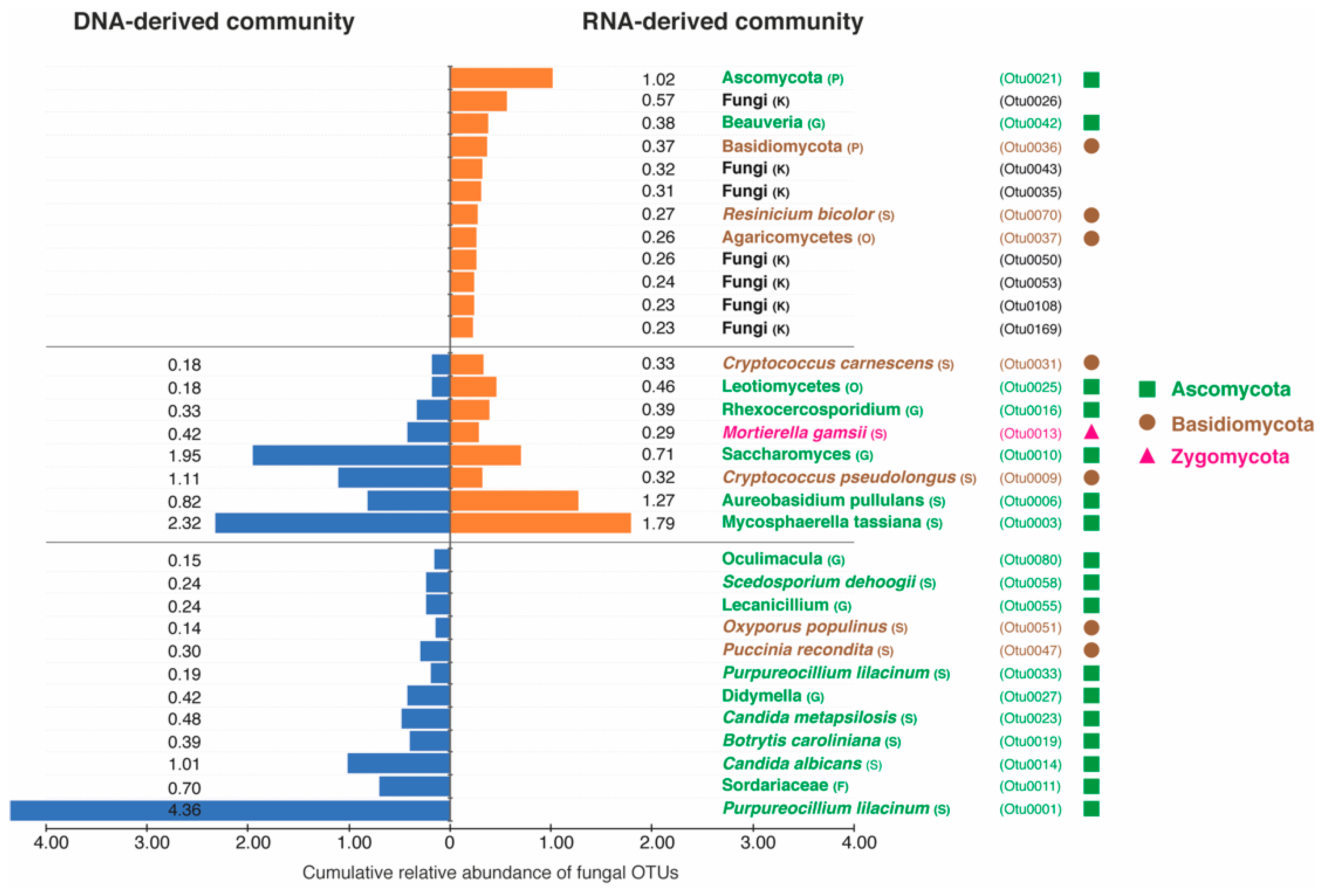
3.3. Taxonomic Differences between DNA- and RNA-Derived Fungal Communities
3.4. DNA- and RNA-Derived Fungal Communities and Significant Environmental Factors
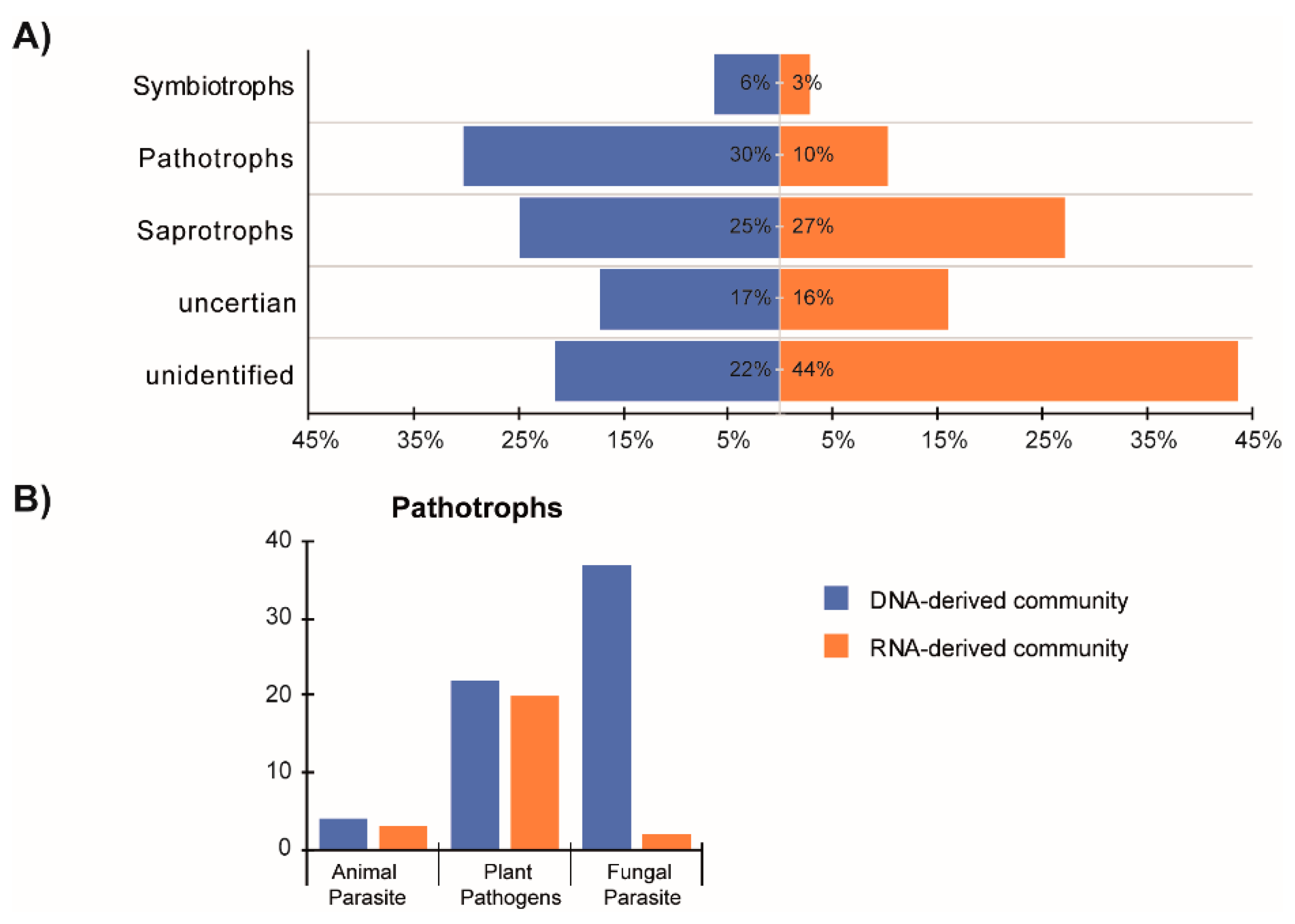
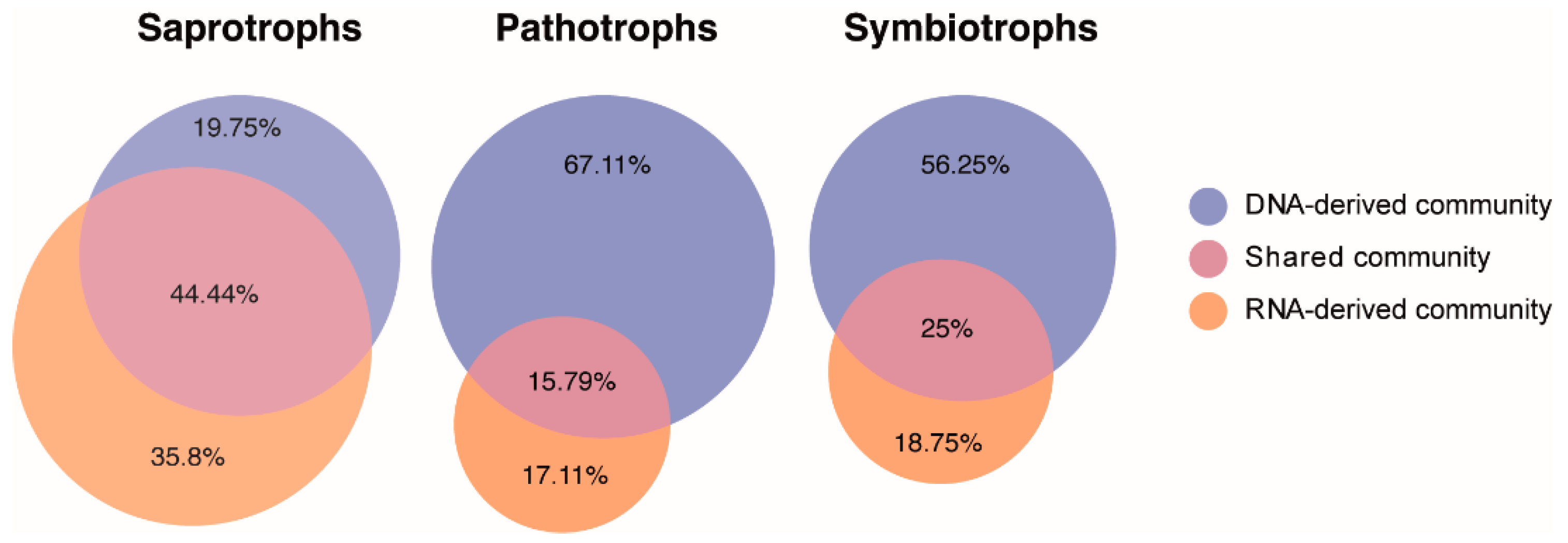
3.5. Distinct Functional Guilds of DNA- and RNA-Derived Fungal Community
4. Discussion
4.1. The Total and Active Fungal Communities Have Similar Structure but Different Community Composition
4.2. Does the DNA-Derived Fungal Community Represent the Total Fungal Community?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hawksworth, D.L.; Lucking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Index Fungorum. 2019. Available online: http://www.speciesfungorum.org/Names/Names.asp (accessed on 27 August 2019).
- Hyde, K.D. Where are the missing fungi? Does Hong Kong have any answers? Mycol. Res. 2001, 105, 1514–1518. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.; Williams, P.D. Karst Hydrogeology and Geomorphology; John Wiley & Sons Ltd.: West Suxxex, UK, 2013. [Google Scholar]
- Martin, J.B.; White, W.B. (Eds.) Frontiers of Karst Research, Special Publication Martin; Karst Water Institute: Lessburg, VA, USA, 2008; p. 118. [Google Scholar]
- Grossart, H.P.; Van den Wyngaert, S.; Kagami, M.; Wurzbacher, C.; Cunliffe, M.; Rojas-Jimenez, K. Fungi in aquatic ecosystems. Nat. Rev. Microbiol. 2019, 17, 339–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, A.; Purahong, W.; Lehmann, R.; Herrmann, M.; Kusel, K.; Totsche, K.U.; Buscot, F.; Wubet, T. Superimposed pristine limestone aquifers with marked hydrochemical differences exhibit distinct fungal communities. Front. Microbiol. 2016, 7, 666. [Google Scholar] [CrossRef] [PubMed]
- Lategan, M.J.; Torpy, F.R.; Newby, S.; Stephenson, S.; Hose, G.C. Fungal diversity of shallow aquifers in Southeastern Australia. Geomicrobiol. J. 2012, 29, 352–361. [Google Scholar] [CrossRef]
- Panzer, K.; Yilmaz, P.; Weiss, M.; Reich, L.; Richter, M.; Wiese, J.; Schmaljohann, R.; Labes, A.; Imhoff, J.F.; Glockner, F.O.; et al. Identification of habitat-specific biomes of aquatic fungal communities using a comprehensive nearly full-length 18s rrna dataset enriched with contextual data. PLoS ONE 2015, 10, e0134377. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Adams, R.I.; Roman, C.M.B.; Brooks, B.; Coil, D.A.; Dahlhausen, K.; Ganz, H.H.; Hartmann, E.M.; Hsu, T.; Justice, N.B.; et al. Schrodinger’s microbes: Tools for distinguishing the living from the dead in microbial ecosystems. Microbiome 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Carini, P.; Marsden, P.J.; Leff, J.W.; Morgan, E.E.; Strickland, M.S.; Fierer, N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol. 2016, 2, 16242. [Google Scholar] [CrossRef]
- Anderson, I.C.; Parkin, P.I. Detection of active soil fungi by RT-PCR amplification of precursor rRNA molecules. J. Microbiol. Methods 2007, 68, 248–253. [Google Scholar] [CrossRef]
- Baldrian, P.; Kolarik, M.; Stursova, M.; Kopecky, J.; Valaskova, V.; Vetrovsky, T.; Zifcakova, L.; Snajdr, J.; Ridl, J.; Vlcek, C.; et al. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012, 6, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Cox, F.; Newsham, K.K.; Robinson, C.H. Endemic and cosmopolitan fungal taxa exhibit differential abundances in total and active communities of Antarctic soils. Environ. Microbiol. 2019, 21, 1586–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanowicz, K.J.; Freedman, Z.B.; Upchurch, R.A.; Argiroff, W.A.; Zak, D.R. Active microorganisms in forest soils differ from the total community yet are shaped by the same environmental factors: the influence of pH and soil moisture. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [PubMed]
- Womack, A.M.; Artaxo, P.E.; Ishida, F.Y.; Mueller, R.C.; Saleska, S.R.; Wiedemann, K.T.; Bohannan, B.J.M.; Green, J.L. Characterization of active and total fungal communities in the atmosphere over the Amazon rainforest. Biogeosci. Discuss. 2015, 12, 7177–7207. [Google Scholar] [CrossRef]
- Kuramae, E.E.; Verbruggen, E.; Hillekens, R.; de Hollander, M.; Roling, W.F.; van der Heijden, M.G.; Kowalchuk, G.A. Tracking fungal community responses to maize plants by DNA- and RNA-based pyrosequencing. PLoS ONE 2013, 8, e69973. [Google Scholar] [CrossRef]
- Wutkowska, M.; Vader, A.; Mundra, S.; Cooper, E.J.; Eidesen, P.B. Dead or alive; or does it really matter? Level of congruency between trophic modes in total and active fungal communities in high arctic soil. Front. Microbiol. 2019, 9, 3243. [Google Scholar] [CrossRef]
- McGee, C.F.; Byrne, H.; Irvine, A.; Wilson, J. Diversity and dynamics of the DNA- and cDNA-derived compost fungal communities throughout the commercial cultivation process for Agaricus bisporus. Mycologia 2017, 109, 475–484. [Google Scholar] [CrossRef]
- Küsel, K.; Totsche, K.U.; Trumbore, S.E.; Lehmann, R.; Steinhäuser, C.; Herrmann, M. How deep can surface signals be traced in the critical zone? Merging biodiversity with biogeochemistry research in a Central German Muschelkalk Landscape. Front. Earth Sci. 2016, 4, 32. [Google Scholar] [CrossRef]
- Kohlhepp, B.; Lehmann, R.; Seeber, P.; Kusel, K.; Trumbore, S.E.; Totsche, K.U. Aquifer configuration and geostructural links control the groundwater quality in thin-bedded carbonate-siliciclastic alternations of the Hainich CZE, central Germany. Hydrol. Earth Syst. Sci. 2017, 21, 6091–6116. [Google Scholar] [CrossRef]
- Nawaz, A.; Purahong, W.; Lehmann, R.; Herrmann, M.; Totsche, K.U.; Kusel, K.; Wubet, T.; Buscot, F. First insights into the living groundwater mycobiome of the terrestrial biogeosphere. Water Res. 2018, 145, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Ihrmark, K.; Bodeker, I.T.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandstrom-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region--evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef] [PubMed]
- White, T.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Boyer, F.; Mercier, C.; Bonin, A.; Le Bras, Y.; Taberlet, P.; Coissac, E. OBITOOLS: A unix-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 2016, 16, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Blaxter, M.; Mann, J.; Chapman, T.; Thomas, F.; Whitton, C.; Floyd, R.; Abebe, E. Defining operational taxonomic units using DNA barcode data. Philosophical Transactions of the Royal Society B-Biological Sciences 2005, 360, 1935–1943. [Google Scholar] [CrossRef]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glockner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.; Sanchez-Garcia, M.; Ebersberger, I.; de Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: http://www.R-project.org (accessed on 11 September 2019).
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Hurlbert, S.H. The nonconcept of species diversity: A critique and alternative parameters. Ecology 1971, 52, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jackson, J.A. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Weblogs and Social Media, Paris, France, 7 July 2019. [Google Scholar]
- Oksanen, J.; Blanchet, F.; Kindt, R.; Legendre, P.; O’Hara, R.; Simpson, G.; Solymos, P.; Stevens, M.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.3–1. 2015. Available online: https://cran.r-project.org/web/packages/vegan/ (accessed on 11 September 2019).
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.W.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- De Vrieze, J.; Pinto, A.J.; Sloan, W.T.; Ijaz, U.Z. The active microbial community more accurately reflects the anaerobic digestion process: 16S rRNA (gene) sequencing as a predictive tool. Microbiome 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Wegner, C.E.; Gaspar, M.; Geesink, P.; Herrmann, M.; Marz, M.; Kusel, K. Biogeochemical regimes in shallow aquifers reflect the metabolic coupling of the elements nitrogen, sulfur, and carbon. Appl. Environ. Microbiol. 2019, 85, e02346-18. [Google Scholar] [CrossRef]
- Li, H.; Su, J.Q.; Yang, X.R.; Zhu, Y.G. Distinct rhizosphere effect on active and total bacterial communities in paddy soils. Sci. Total Environ. 2019, 649, 422–430. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Z.H.; Dai, M.H.; Jiao, N.Z.; Herndl, G.J. Drivers shaping the diversity and biogeography of total and active bacterial communities in the South China Sea. Mol. Ecol. 2014, 23, 2260–2274. [Google Scholar] [CrossRef]
- Franchi, O.; Bovio, P.; Ortega-Martinez, E.; Rosenkranz, F.; Chamy, R. Active and total microbial community dynamics and the role of functional genes bamA and mcrA during anaerobic digestion of phenol and p-cresol. Bioresour. Technol. 2018, 264, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Cassio, F.; Pascoal, C. Environmental drivers are more important for structuring fungal decomposer communities than the geographic distance between streams. Limnetica 2017, 36, 491–506. [Google Scholar]
- Amend, A.; Burgaud, G.; Cunliffe, M.; Edgcomb, V.P.; Ettinger, C.L.; Gutierrez, M.H.; Heitman, J.; Hom, E.F.Y.; Ianiri, G.; Jones, A.C.; et al. Fungi in the marine environment: Open questions and unsolved problems. mBio 2019, 10, e01189-18. [Google Scholar] [CrossRef]
- Tian, J.Q.; Zhu, D.; Wang, J.Z.; Wu, B.; Hussain, M.; Liu, X.Z. Environmental factors driving fungal distribution in freshwater lake sediments across the Headwater Region of the Yellow River. China Sci. Rep. 2018, 8, 3768. [Google Scholar] [CrossRef] [PubMed]
- Krauss, G.J.; Sole, M.; Krauss, G.; Schlosser, D.; Wesenberg, D.; Barlocher, F. Fungi in freshwaters: Ecology, physiology and biochemical potential. Fems Microbiol. Rev. 2011, 35, 620–651. [Google Scholar] [CrossRef] [PubMed]
- Tisthammer, K.H.; Cobian, G.M.; Amend, A.S. Global biogeography of marine fungi is shaped by the environment. Fungal Ecol. 2016, 19, 39–46. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Hillmann, B.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Zhu, Q.Y.; Gohl, D.M.; Beckman, K.B.; Knight, R.; Knights, D. Evaluating the information content of shallow shotgun metagenomics. Msystems 2018, 3, e00069-18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Fuhrman, J.A.; Sun, F.; Zhu, S. Assessment of metagenomic assemblers based on hybrid reads of real and simulated metagenomic sequences. Brief. Bioinform 2019. [Google Scholar] [CrossRef] [PubMed]
- Laudadio, I.; Fulci, V.; Stronati, L.; Carissimi, C. Next-generation metagenomics: Methodological challenges and opportunities. OMICS: J. Integr. Biol. 2019, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | DNA-derived Community | RNA-derived Community | ||
|---|---|---|---|---|
| R2 | P | R2 | P | |
| Extraction °T | 0.16 | 0.10 | 0.15 | 0.11 |
| EC25 | 0.14 | 0.14 | 0.15 | 0.11 |
| pH | 0.02 | 0.86 | 0.03 | 0.82 |
| ORP | 0.11 | 0.24 | 0.10 | 0.28 |
| O2 | 0.09 | 0.33 | 0.10 | 0.27 |
| NH4+ | 0.24 | 0.01 * | 0.26 | 0.01 * |
| PO4 | 0.04 | 0.68 | 0.04 | 0.72 |
| TIC | 0.03 | 0.81 | 0.01 | 0.91 |
| NO3− | 0.06 | 0.49 | 0.06 | 0.50 |
| SO42− | 0.10 | 0.27 | 0.11 | 0.22 |
| Cl− | 0.22 | 0.03 * | 0.24 | 0.02 * |
| Ca2+ | 0.18 | 0.05 | 0.20 | 0.04 * |
| Fet | 0.08 | 0.41 | 0.08 | 0.38 |
| K+ | 0.21 | 0.03 * | 0.23 | 0.03 * |
| Mg2+ | 0.19 | 0.06 | 0.21 | 0.03 * |
| Na+ | 0.16 | 0.10 | 0.18 | 0.07 |
| St | 0.10 | 0.28 | 0.10 | 0.23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nawaz, A.; Purahong, W.; Herrmann, M.; Küsel, K.; Buscot, F.; Wubet, T. DNA- and RNA- Derived Fungal Communities in Subsurface Aquifers Only Partly Overlap but React Similarly to Environmental Factors. Microorganisms 2019, 7, 341. https://doi.org/10.3390/microorganisms7090341
Nawaz A, Purahong W, Herrmann M, Küsel K, Buscot F, Wubet T. DNA- and RNA- Derived Fungal Communities in Subsurface Aquifers Only Partly Overlap but React Similarly to Environmental Factors. Microorganisms. 2019; 7(9):341. https://doi.org/10.3390/microorganisms7090341
Chicago/Turabian StyleNawaz, Ali, Witoon Purahong, Martina Herrmann, Kirsten Küsel, François Buscot, and Tesfaye Wubet. 2019. "DNA- and RNA- Derived Fungal Communities in Subsurface Aquifers Only Partly Overlap but React Similarly to Environmental Factors" Microorganisms 7, no. 9: 341. https://doi.org/10.3390/microorganisms7090341