
ASCL1 Is Involved in the Pathogenesis of Schizophrenia by Regulation of Genes Related to Cell Proliferation, Neuronal Signature Formation, and Neuroplasticity
 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Interaction of the ASCL1 Promoter with SZ-Associated Loci
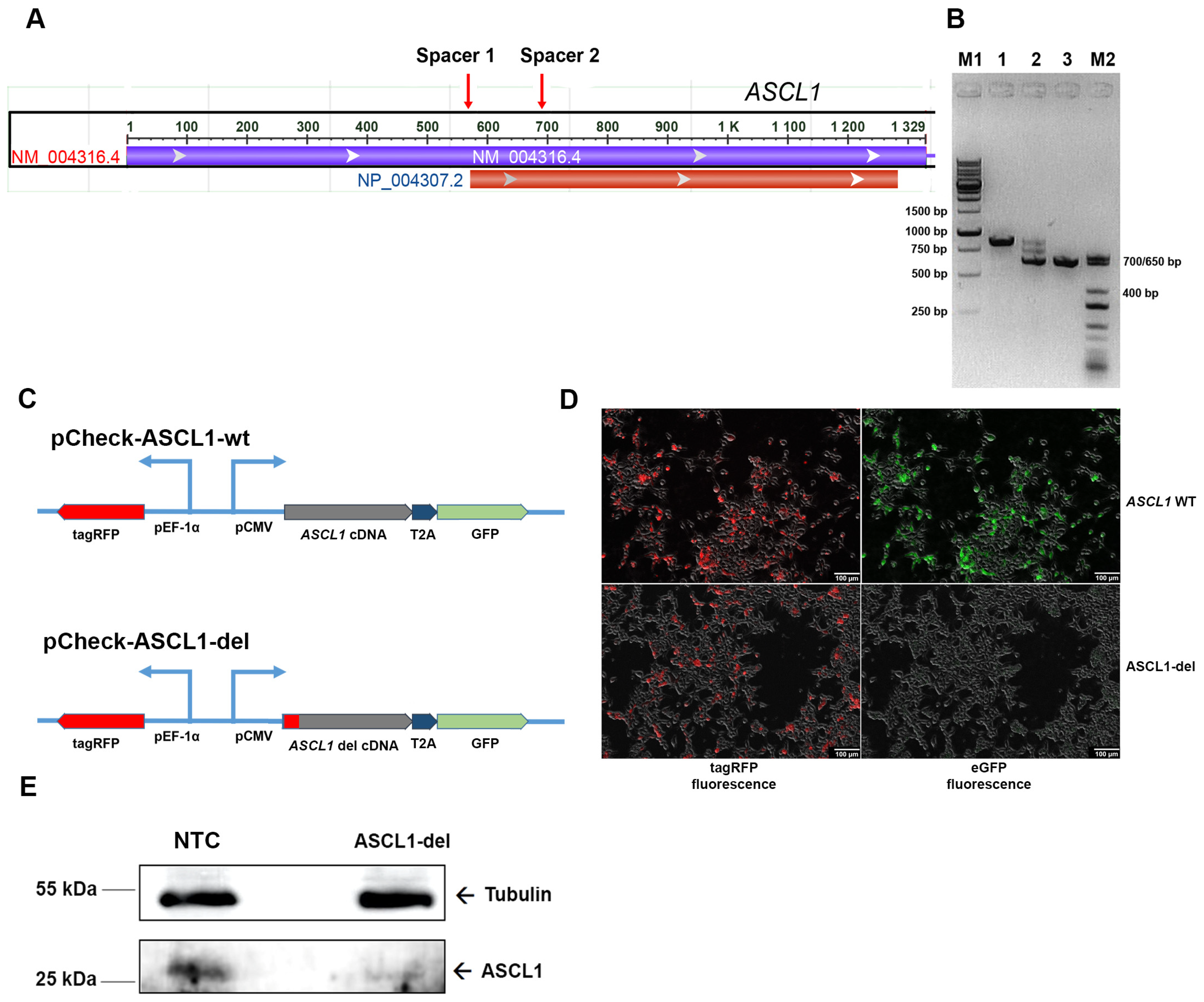
2.2. Generation of the Mutant SH-SY5Y Line with ASCL1 Partial Deletion
2.3. General Characterization of Transcriptomes of SH-SY5Y Cell Lines
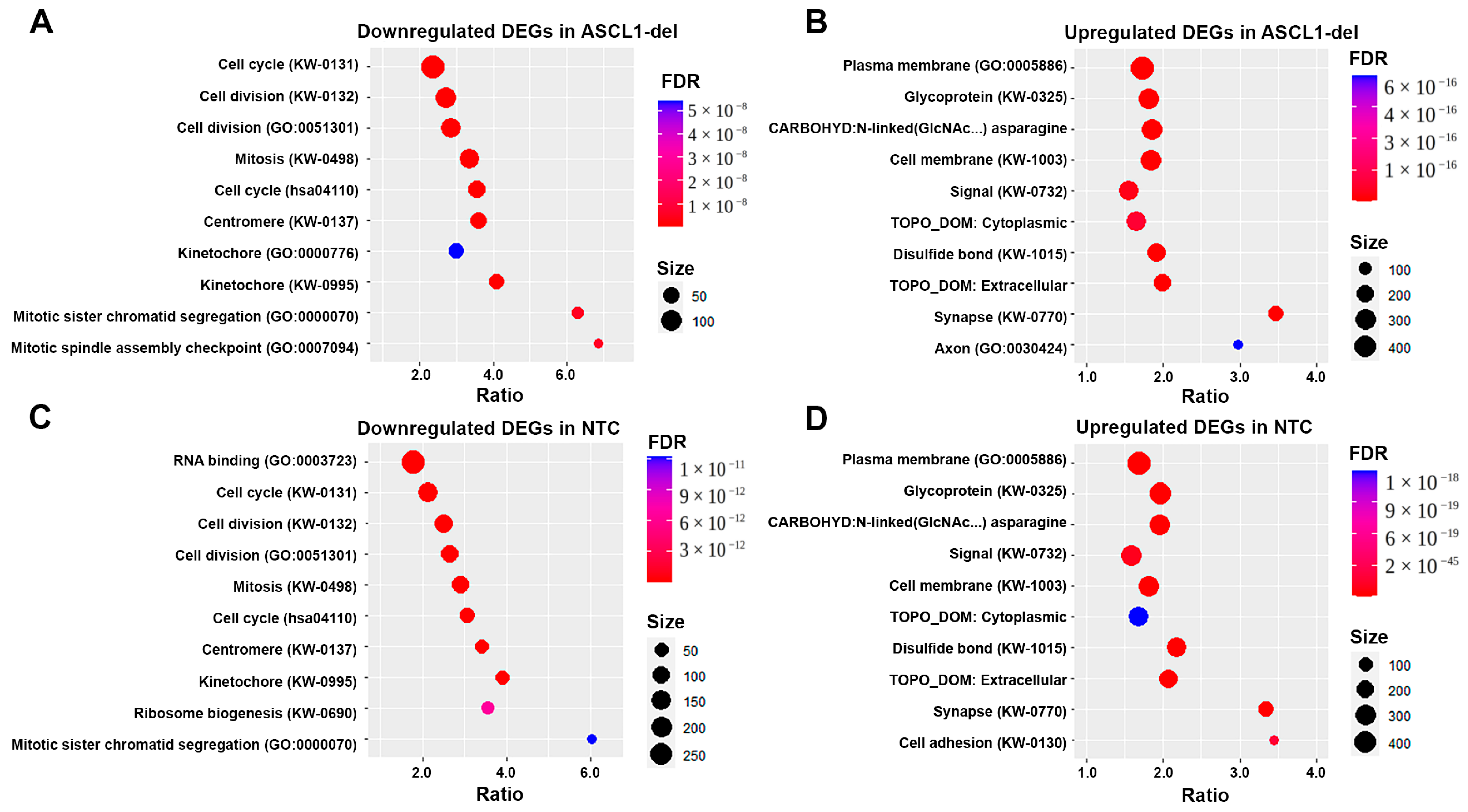
2.4. Differential Changes in the Transcriptome in Undifferentiated SH-SY5Y Cell Lines
2.4.1. Analysis of Transcription Factor Enrichment
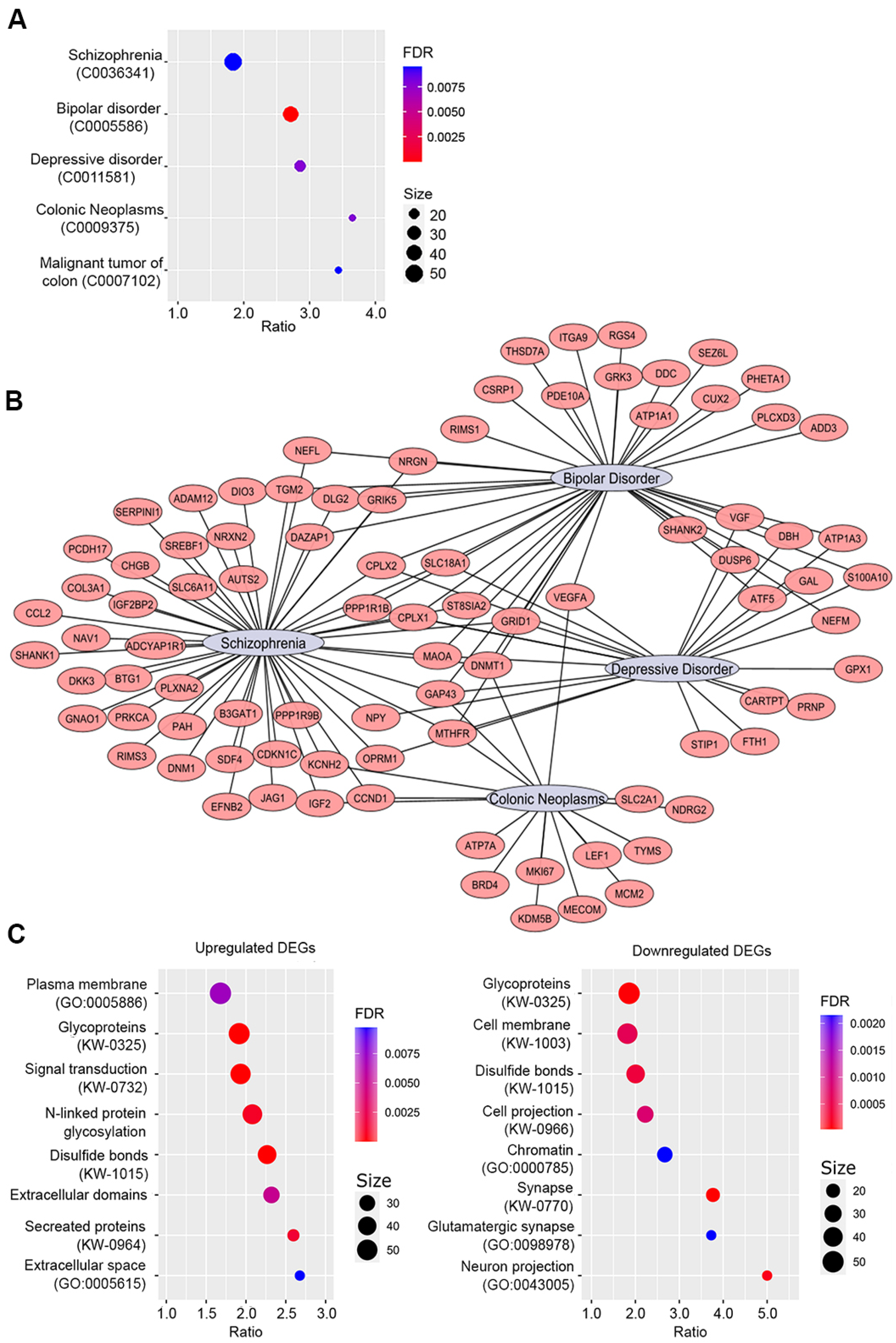
2.4.2. Enrichment Analyses by Disease and Gene Ontology
2.4.3. Gene-Set Enrichment Analysis (GSEA) of DEGs
2.4.4. Gene Interaction Network Analysis
2.5. Changes in the Transcriptome of SH-SY5Y Lines during RA-Induced Differentiation Procedure
3. Discussion
4. Materials and Methods
4.1. Cell Culturing
4.2. Construction of Plasmids
4.3. Transfections, Lentivirus Assembly, and Transductions
4.4. Differentiation of SH-SY5Y by RA Treatment
4.5. Western Blotting
4.6. Fluorescence Microscopy
4.7. RNA Isolation, RT-qPCR, and RNA-seq Library Preparation
4.8. RNAseq Data Analysis
4.9. Capture-C Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C. Genetics in psychiatry: Methods, clinical applications and future perspectives. Psychiatry Clin. Neurosci. Rep. 2022, 1, e6. [Google Scholar] [CrossRef]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Kondratyev, N.V.; Alfimova, M.V.; Golov, A.K.; Golimbet, V.E. Bench research informed by GWAS results. Cells 2021, 10, 3184. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Hill, W.D.; Trampush, J.W.; Yu, J.; Knowles, E.; Davies, G.; Stahl, E.; Huckins, L.; Liewald, D.C.; Djurovic, S.; et al. Pleiotropic meta-analysis of cognition, education, and schizophrenia differentiates roles of early neurodevelopmental and adult synaptic pathways. Am. J. Hum. Genet. 2019, 105, 334–350. [Google Scholar] [CrossRef]
- Aberg, K.A.; Liu, Y.; Bukszár, J.; McClay, J.L.; Khachane, A.N.; Andreassen, O.A.; Blackwood, D.; Corvin, A.; Djurovic, S.; Gurling, H.; et al. A comprehensive family-based replication study of schizophrenia genes. JAMA Psychiatry 2013, 70, 573–581. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Tyler, T.; Dragicevic, K.; Mei, S.; Rydbirk, R.; Petukhov, V.; Deviatiiarov, R.; Sedmak, D.; Frank, E.; Feher, V.; et al. Upper cortical layer-driven network impairment in schizophrenia. Sci. Adv. 2022, 8, eabn8367. [Google Scholar] [CrossRef]
- Woods, L.M.; Ali, F.R.; Gomez, R.; Chernukhin, I.; Marcos, D.; Parkinson, L.M.; Tayoun, A.N.A.; Carroll, J.S.; Philpott, A. Elevated ASCL1 activity creates de novo regulatory elements associated with neuronal differentiation. BMC Genom. 2022, 23, 255. [Google Scholar] [CrossRef]
- Ma, L.; Du, Y.; Hui, Y.; Li, N.; Fan, B.; Zhang, X.; Li, X.; Hong, W.; Wu, Z.; Zhang, S.; et al. Developmental programming and lineage branching of early human telencephalon. EMBO J. 2021, 40, e107277. [Google Scholar] [CrossRef]
- Johnson, J.E.; Birren, S.J.; Anderson, D.J. Two rat homologues of Drosophila achaete-scute specifically expressed in neuronal precursors. Nature 1990, 346, 858–861. [Google Scholar] [CrossRef]
- Lo, L.C.; Johnson, J.E.; Wuenschell, C.W.; Saito, T.; Anderson, D.J. Mammalian achaete-scute homolog 1 is transiently expressed by spatially restricted subsets of early neuroepithelial and neural crest cells. Genes Dev. 1991, 5, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chan, J.A.; Schuurmans, C. Proneural BHLH genes in development and disease. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 110, pp. 75–127. ISBN 978-0-12-405943-6. [Google Scholar]
- Liu, F.; Zhang, Y.; Chen, F.; Yuan, J.; Li, S.; Han, S.; Lu, D.; Geng, J.; Rao, Z.; Sun, L.; et al. Neurog2 directly converts astrocytes into functional neurons in midbrain and spinal cord. Cell Death Dis. 2021, 12, 225. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Miao, Q.; Yuan, J.; Han, S.; Zhang, P.; Li, S.; Rao, Z.; Zhao, W.; Ye, Q.; Geng, J.; et al. Ascl1 converts dorsal midbrain astrocytes into functional neurons in vivo. J. Neurosci. 2015, 35, 9336–9355. [Google Scholar] [CrossRef] [PubMed]
- Parras, C.M.; Schuurmans, C.; Scardigli, R.; Kim, J.; Anderson, D.J.; Guillemot, F. Divergent functions of the proneural genes Mash1 and Ngn2 in the specification of neuronal subtype identity. Genes Dev. 2002, 16, 324–338. [Google Scholar] [CrossRef]
- Păun, O.; Tan, Y.X.; Patel, H.; Strohbuecker, S.; Ghanate, A.; Cobolli-Gigli, C.; Llorian Sopena, M.; Gerontogianni, L.; Goldstone, R.; Ang, S.-L.; et al. Pioneer factor ASCL1 cooperates with the MSWI/SNF complex at distal regulatory elements to regulate human neural differentiation. Genes Dev. 2023, 37, 218–242. [Google Scholar] [CrossRef]
- Vue, T.Y.; Kollipara, R.K.; Borromeo, M.D.; Smith, T.; Mashimo, T.; Burns, D.K.; Bachoo, R.M.; Johnson, J.E. ASCL1 regulates neurodevelopmental transcription factors and cell cycle genes in brain tumors of glioma mouse models. Glia 2020, 68, 2613–2630. [Google Scholar] [CrossRef]
- Golov, A.K.; Abashkin, D.A.; Kondratyev, N.V.; Razin, S.V.; Gavrilov, A.A.; Golimbet, V.E. A modified protocol of capture-C allows affordable and flexible high-resolution promoter interactome analysis. Sci. Rep. 2020, 10, 15491. [Google Scholar] [CrossRef]
- Abashkin, D.A.; Kurishev, A.O.; Karpov, D.S.; Golimbet, V.E. Cellular models in schizophrenia research. Int. J. Mol. Sci. 2021, 22, 8518. [Google Scholar] [CrossRef]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Linkner, T.R.; Ambrus, V.; Kunkli, B.; Szojka, Z.I.; Kalló, G.; Csősz, K.; Kumar, A.; Emri, M.; Tőzsér, J.; Mahdi, M. Cellular proteo-transcriptomic changes in the immediate early-phase of lentiviral transduction. Microorganisms 2021, 9, 2207. [Google Scholar] [CrossRef]
- Hulsen, T.; De Vlieg, J.; Alkema, W. BioVenn—A Web Application for the comparison and visualization of biological lists using area-proportional venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; I Furlong, L. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists. Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Matsumoto, M.; Iijima, K.; Sumiyoshi, T. Specificity and continuity of schizophrenia and bipolar disorder: Relation to biomarkers. Curr. Pharm. Des. 2020, 26, 191–200. [Google Scholar] [CrossRef]
- Wang, L.; Tan, T.K.; Durbin, A.D.; Zimmerman, M.W.; Abraham, B.J.; Tan, S.H.; Ngoc, P.C.T.; Weichert-Leahey, N.; Akahane, K.; Lawton, L.N.; et al. ASCL1 is a MYCN- and LMO1-dependent member of the adrenergic neuroblastoma core regulatory circuitry. Nat. Commun. 2019, 10, 5622. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417. [Google Scholar] [CrossRef]
- Parkinson, L.M.; Gillen, S.L.; Woods, L.M.; Chaytor, L.; Marcos, D.; Ali, F.R.; Carroll, J.S.; Philpott, A. The proneural transcription factor ASCL1 regulates cell proliferation and primes for differentiation in neuroblastoma. Front. Cell Dev. Biol. 2022, 10, 942579. [Google Scholar] [CrossRef]
- Miao, L.; Wang, Y.; Xia, H.; Yao, C.; Cai, H.; Song, Y. SPOCK1 is a novel transforming growth factor-β target gene that regulates lung cancer cell epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2013, 440, 792–797. [Google Scholar] [CrossRef]
- Sun, L.; Li, S.; Guo, Q.; Zhou, W.; Zhang, H. SPOCK1 involvement in epithelial-to-mesenchymal transition: A new target in cancer therapy? Cancer Manag. Res. 2020, 12, 3561–3569. [Google Scholar] [CrossRef]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef]
- Meister, B.; Grünebach, F.; Bautz, F.; Brugger, W.; Fink, F.-M.; Kanz, L.; Möhle, R. Expression of vascular endothelial growth factor (VEGF) and its receptors in human neuroblastoma. Eur. J. Cancer 1999, 35, 445–449. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network analysis and visualization of proteomics data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Huang, Y.; Lin, X.; Lin, S. Neuropeptide Y and metabolism syndrome: An update on perspectives of clinical therapeutic intervention strategies. Front. Cell Dev. Biol. 2021, 9, 695623. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef]
- Ranneva, S.V.; Maksimov, V.F.; Korostyshevskaja, I.M.; Lipina, T.V. Lack of synaptic protein, calsyntenin-2, impairs morphology of synaptic complexes in mice. Synapse 2020, 74, e22132. [Google Scholar] [CrossRef]
- Centanni, T.M.; Sanmann, J.N.; Green, J.R.; Iuzzini-Seigel, J.; Bartlett, C.; Sanger, W.G.; Hogan, T.P. The role of candidate-gene CNTNAP2 in childhood apraxia of speech and specific language impairment. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168, 536–543. [Google Scholar] [CrossRef]
- Ji, W.; Li, T.; Pan, Y.; Tao, H.; Ju, K.; Wen, Z.; Fu, Y.; An, Z.; Zhao, Q.; Wang, T.; et al. CNTNAP2 is significantly associated with schizophrenia and major depression in the Han Chinese population. Psychiatry Res. 2013, 207, 225–228. [Google Scholar] [CrossRef]
- Biedler, J.L.; Helson, L.; Spengler, B.A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973, 33, 2643–2652. [Google Scholar]
- Wang, C.Y.; Shahi, P.; Huang, J.T.; Phan, N.N.; Sun, Z.; Lin, Y.-C.; Lai, M.-D.; Werb, Z. Systematic analysis of the achaete-scute complex-like gene signature in clinical cancer patients. Mol. Clin. Oncol. 2017, 6, 7–18. [Google Scholar] [CrossRef]
- Amirfallah, A.; Calibasi Kocal, G.; Unal, O.U.; Ellidokuz, H.; Oztop, I.; Basbinar, Y. DPYD, TYMS and MTHFR genes polymorphism frequencies in a series of Turkish colorectal cancer patients. J. Pers. Med. 2018, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Petrone, I.; Bernardo, P.S.; dos Santos, E.C.; Abdelhay, E. MTHFR C677T and A1298C polymorphisms in breast cancer, gliomas and gastric cancer: A review. Genes 2021, 12, 587. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chanda, S.; Marro, S.; Ng, Y.-H.; Janas, J.A.; Haag, D.; Ang, C.E.; Tang, Y.; Flores, Q.; Mall, M.; et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 2017, 14, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Chiapponi, C.; Piras, F.; Piras, F.; Caltagirone, C.; Spalletta, G. GABA System in schizophrenia and mood disorders: A mini review on third-Generation imaging studies. Front. Psychiatry 2016, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Benes, F. GABAergic Interneurons implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001, 25, 1–27. [Google Scholar] [CrossRef]
- de Jonge, J.C.; Vinkers, C.H.; Hulshoff Pol, H.E.; Marsman, A. GABAergic mechanisms in schizophrenia: Linking postmortem and in vivo studies. Front. Psychiatry 2017, 8, 118. [Google Scholar] [CrossRef]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal GABAergic inhibitory interneurons. Physiol. Rev. 2017, 97, 1619–1747. [Google Scholar] [CrossRef]
- Purves-Tyson, T.D.; Brown, A.M.; Weissleder, C.; Rothmond, D.A.; Shannon Weickert, C. Reductions in midbrain GABAergic and dopamine neuron markers are linked in schizophrenia. Mol. Brain 2021, 14, 96. [Google Scholar] [CrossRef]
- Benes, F.M.; Kwok, E.W.; Vincent, S.L.; Todtenkopf, M.S. A reduction of nonpyramidal cells in sector CA2 of schizophrenics and manic depressives. Biol. Psychiatry 1998, 44, 88–97. [Google Scholar] [CrossRef]
- Lee, S.; Lee, B.; Lee, J.W.; Lee, S.-K. Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier CBP. Neuron 2009, 62, 641–654. [Google Scholar] [CrossRef]
- Ribes, V.; Stutzmann, F.; Bianchetti, L.; Guillemot, F.; Dolle, P.; Le Roux, I. Combinatorial signalling controls Neurogenin2 expression at the onset of spinal neurogenesis. Dev. Biol. 2008, 321, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Ragland, J.D.; Carter, C.S. Memory and cognition in schizophrenia. Mol. Psychiatry 2019, 24, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, P.F. Mechanism of aromatic amino acid hydroxylation. Biochemistry 2003, 42, 14083–14091. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. Oxf. Engl. 2015, 29, 97–115. [Google Scholar] [CrossRef]
- Bergen, S.E.; Fanous, A.H.; Walsh, D.; O’Neill, F.A.; Kendler, K.S. Polymorphisms in SLC6A4, PAH, GABRB3, and MAOB and modification of psychotic disorder features. Schizophr. Res. 2009, 109, 94–97. [Google Scholar] [CrossRef]
- Gao, Z.; Ure, K.; Ding, P.; Nashaat, M.; Yuan, L.; Ma, J.; Hammer, R.E.; Hsieh, J. The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J. Neurosci. 2011, 31, 9772–9786. [Google Scholar] [CrossRef]
- Tang, B.L. REST regulation of neural development: From outside-in? Cell Adhes. Migr. 2009, 3, 141–142. [Google Scholar] [CrossRef]
- Hwang, J.-Y.; Zukin, R.S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef]
- de Pontual, L.; Nepote, V.; Attie-Bitach, T.; Al Halabiah, H.; Trang, H.; Elghouzzi, V.; Levacher, B.; Benihoud, K.; Auge, J.; Faure, C.; et al. Noradrenergic neuronal development is impaired by mutation of the proneural HASH-1 gene in congenital central hypoventilation syndrome (Ondine’s curse). Hum. Mol. Genet. 2003, 12, 3173–3180. [Google Scholar] [CrossRef]
- Mallolas, J.; Vilaseca, M.A.; Pavia, C.; Lambruschini, N.; Cambra, F.J.; Campistol, J.; Gomez, D.; Carrio, A.; Estivill, X.; Mila, M. Large de novo deletion in chromosome 12 affecting the PAH, IGF1, ASCL1, and TRA1 genes. J. Mol. Med. 2001, 78, 721–724. [Google Scholar] [CrossRef]
- Ashe, K.; Kelso, W.; Farrand, S.; Panetta, J.; Fazio, T.; De Jong, G.; Walterfang, M. Psychiatric and cognitive aspects of phenylketonuria: The limitations of diet and promise of new treatments. Front. Psychiatry 2019, 10, 561. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Kageyama, R. bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron 2014, 82, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ghazale, H.; Park, E.; Vasan, L.; Mester, J.; Saleh, F.; Trevisiol, A.; Zinyk, D.; Chinchalongporn, V.; Liu, M.; Fleming, T.; et al. Ascl1 phospho-site mutations enhance neuronal conversion of adult cortical astrocytes in vivo. Front. Neurosci. 2022, 16, 917071. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Abbate, J.; Lacayo, J.C.; Prichard, M.; Pari, G.; McVoy, M.A. Bifunctional protein conferring enhanced green fluorescence and puromycin resistance. Biotechniques 2001, 31, 336–340. [Google Scholar] [CrossRef]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; E McConkey, M.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef]
- Cheng, A.W.; Jillette, N.; Lee, P.; Plaskon, D.; Fujiwara, Y.; Wang, W.; Taghbalout, A.; Wang, H. Casilio: A versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016, 26, 254–257. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Abe, A.; Miyanohara, A.; Friedmann, T. Polybrene increases the efficiency of gene transfer by lipofection. Gene Ther. 1998, 5, 708–711. [Google Scholar] [CrossRef]
- Berggren, T. General spinfection. StemBook 2014. [Google Scholar] [CrossRef]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y human neuroblastoma cell line. J. Vis. Exp. 2016, 108, e53193. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinforma. Oxf. Engl. 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. Bioinformatics 2016. [Google Scholar] [CrossRef]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef]
- Zou, Z.; Ohta, T.; Miura, F.; Oki, S. ChIP-Atlas 2021 update: A data-mining suite for exploring epigenomic landscapes by fully integrating ChIP-Seq, ATAC-Seq and Bisulfite-Seq Data. Nucleic Acids Res. 2022, 50, W175–W182. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J.; Freire-Pritchett, P.; Wingett, S.W.; Várnai, C.; Dimond, A.; Plagnol, V.; Zerbino, D.; Schoenfelder, S.; Javierre, B.-M.; Osborne, C.; et al. CHiCAGO: Robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 2016, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Set Name, TF (Cell Line) | p-Value | FDR | Odds Ratio |
|---|---|---|---|
| REST (HCT 116) | 1.83 × 10−9 | 1.01 × 10−6 | 3.711 |
| TCF7L2 (HEK293) | 3.08 × 10−5 | 2.12 × 10−3 | 1.734 |
| GATA3 (SK-N-SH) | 8.41 × 10−5 | 4.64 × 10−3 | 1.910 |
| GATA2 (HUVEC) | 2.93 × 10−4 | 1.35 × 10−2 | 1.623 |
| CBX2 (K562) | 4.91 × 10−4 | 2.08 × 10−2 | 1.600 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abashkin, D.A.; Karpov, D.S.; Kurishev, A.O.; Marilovtseva, E.V.; Golimbet, V.E. ASCL1 Is Involved in the Pathogenesis of Schizophrenia by Regulation of Genes Related to Cell Proliferation, Neuronal Signature Formation, and Neuroplasticity. Int. J. Mol. Sci. 2023, 24, 15746. https://doi.org/10.3390/ijms242115746
Abashkin DA, Karpov DS, Kurishev AO, Marilovtseva EV, Golimbet VE. ASCL1 Is Involved in the Pathogenesis of Schizophrenia by Regulation of Genes Related to Cell Proliferation, Neuronal Signature Formation, and Neuroplasticity. International Journal of Molecular Sciences. 2023; 24(21):15746. https://doi.org/10.3390/ijms242115746
Chicago/Turabian StyleAbashkin, Dmitrii A., Dmitry S. Karpov, Artemii O. Kurishev, Ekaterina V. Marilovtseva, and Vera E. Golimbet. 2023. "ASCL1 Is Involved in the Pathogenesis of Schizophrenia by Regulation of Genes Related to Cell Proliferation, Neuronal Signature Formation, and Neuroplasticity" International Journal of Molecular Sciences 24, no. 21: 15746. https://doi.org/10.3390/ijms242115746