
Abstract
Curcuma longa L., is recognized worldwide as a medicinally and economically important plant species due to its curcumin content which is an industrially important compound. In this study, a total of 329 accessions were collected from four states of India and planted in the experimental farm of CSIR-NEIST, Jorhat, India, in augmented design. Among these, 152 high curcumin (> 1.50%) accessions were screened for molecular divergence study using 39 SSR primers. The primers showed the most efficient outcome with 2–8 allele/ loci and a total 163 number of alleles with 100% polymorphism. Cluster analysis revealed the construction of three clusters, out of which one cluster was geographically dependent, and germplasm was particularly from Assam state. Jaccard's pairwise coefficient showed maximum genetic dissimilarity of (0.75) between accession RRLJCL 3 and RRLJCL 126, indicating high variation as it was from two different states viz Arunachal Pradesh and Nagaland respectively and minimum genetic dissimilarity of (0.09) between RRLJCL 58 and RRLJCL 59 indicating significantly less variation as the two accessions were from same state, i.e., Arunachal Pradesh. Analysis of Molecular Variance (AMOVA) revealed high molecular variation within the population (87%) and significantly less variation among the population (13%). Additionally, Neighbour Joining dendrogram, Principal Component Analysis (PCA), and bar plot structure revealed similar clustering of germplasm. This diversity assessment will help in selecting the trait-specific genotypes, crop improvement program, conservation of gene pool, marker-assisted breeding, and quantitative trait loci identification. Moreover, to the best of our knowledge, it is the first molecular diversity report among 152 high curcumin lines of C. longa from North East India using 39 SSR primers.
Similar content being viewed by others

Introduction
In the present era of great scientific studies, acknowledging the importance of medicinal crops and shifting towards therapeutic agents is increasing tremendously. Among all, the genus Curcuma, which belongs to the Zingiberaceae family, is acknowledged medicinally and economically as one of the most predominate plant species1,2.Curcuma longa L. generally known as turmeric, is widely distributed throughout South and South East Asia, with the most diversity concentrated in Thailand and India, followed by Bangladesh, Vietnam, Myanmar, and Indonesia3. It is an interbreed triploid species (3n = 63) which is procreate vegetatively using the below-ground rhizome4. Morphologically, turmeric is a perennial herb that measures up to 1 m in height with a wee stem, having funnel-shaped yellow flowers and oblong, pointed leaves5.
Since time immemorial, turmeric as flavoring spice powder has been used continuously in the preparation of both vegetarian and non-vegetarian food due to its digestive properties6. In addition to its uses in religious practices, the rhizome part of C. longa has numerous masteries in traditional medicines like stimulants, diabetic wounds, jaundice, to treat liver complaints, stomachic, arthritic, muscular disorders, blood purifiers, rheumatism, cough, and sinusitis like diseases7,8. The C. longa rhizomes are also widely practiced in treating sprains and swelling caused due to different injuries9. The primary compound of turmeric is curcuminoid which is the most active component, and it prevails in three polyphenolic forms, namely curcumin, demethoxycurcumin, and bisdemethoxycurcumin10,11,12. The chemical compounds like curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione), which is also better known as diferuloylmethane is one of the key natural polyphenols found in the turmeric rhizome. It exists in two tautomeric forms: enolic in organic solvents and keto in water13. Curcumin is the foremost essential component present in the C. longa rhizomes, which is responsible for the yellow hue. It is responsible for several pharmacological activities like anti-inflammatory, anti-hepatotoxic, anti-microbial, anti-rheumatic, anti-fibrotic, choleretic, anti-venomous, hypercholesteremic, anti-diabetic, insect repellent as well as anti-cancerous properties4,7,14.
Additionally, the essential oil (EO) of C. longa, which is extracted from the leaf, flower, root, and rhizome of the plant, is considered as a highly valuable product in different pharmaceutical and cosmetic industries. The essential oil (EO) is epitomized by the existence of secondary metabolites, which possess a strong odour, volatile terpene, and hydrocarbon15. The chemical composition present in the EO of plants can vary due to various factors like nutrients present in the soil, temperature, humidity, altitude, ultraviolet radiation, luminosity, seasonality, circadian cycle, and portion of the plant16,17,18. The prime essential oil constituents of Curcuma longa consist of monoterpenoids and sesquiterpenoids19. Earlier studies outlined that in C. longa, rhizome essential oil comprised of constituents like ar-turmerone, turmerone, curlone, ar-curcumene, and root consists of ar-turmerone, dihydro curcumene, and ar-curcumene where components like turmerone were absent. Moreover, the major constituents found in leaf oil were p-cymene, terpinolene, α-phellandrene, 1,8- cineole, and flower oil consist of terpinolene, p-cymen-8-ol, 1,8-cineole20. These significant constituents were reported to exhibit numerous health benefits, which help to strengthen the immune system, expedite toxin elimination, and elevate blood circulation21,22,23.
High rhizome yield, disease resistance, and high curcumin content are the desirable characteristics for the turmeric varietal developmental programme, and North East India being a biodiversity hotspot, has ample variability of C. longa L. which needs a meticulous study. For screening of the elite germplasm, the study on genetic diversity as well as their association study is of immense significance. Though the value of this golden crop has been recognized since ages, but the demand for this crop in the domain of the food and pharmaceutical industry is still increasing24,25,26. Therefore, genetic diversity analysis was performed in order to enhance a deeper study in the productivity and conservation of this crop which forms the basis of the crop breeding program27. The application of phenotypic attribute in germplasm conservation is meagred due to the influence of environment and genotype interactions28; therefore, studies have been performed on molecular diversity using various markers like Simple Sequence Repeat (SSR), Randomly Amplified Polymorphic DNA (RAPD), Amplified Fragment Length Polymorphism (AFLP). The application of RAPD provides insightful information about genetic diversity29 but yields a low level of polymorphism as relative to SSR markers30. Molecular diversity was successfully studied in many crops for phylogenetic assessment and genetic diversity using SSR marker31,32,33,34 as SSR markers are locus-specific, co-dominant markers and have the capability to perceive high levels of miscellany in the genome34,35,36.
The present study was designed to estimate the genetic divergence of C. longa germplasm using a large number of accessions (152) gathered from different regions of North East India using SSR markers. A very few reports have been found where Singh et al.37 evaluated 30 genotypes of C. longa using 9 SSR markers, Sahoo et al.38 studied 88 accessions using 50 EST SSR primers, out of which only 11 primers showed polymorphic banding patterns. Till now, to our supreme cognizance, no molecular diversity survey has been performed on such a large number of accessions, and hence this study will help in the progress of worthier germplasm with more yield-related traits and crossing of parents with preferable traits34. This will provide more reliable information in improving traits and will be very effective for the selection and conservation of trait-specific germplasm, which can be further implemented in the study of the food and pharmaceutical industry.
Results
Selection of high curcumin lines
Among the 329 accessions collected from four states of India, 152 accessions were selected based on curcumin content (> 1.50%) for further molecular analysis. The total curcuminoids content of selected lines ranged between 1.86 and 12.97%, which is charted in (Table 1).
SSR primer competency
For screening of efficient primer, a total of 44 SSR Primers were identified based on review of previous literature on the family Zingiberaceae. Primer testing was executed using all 44 SSR primers by maintaining the melting temperatures of the selected primers, of which 39 primers showed efficient results like sound amplification, better reproducible pattern, number of polymorphic fragments per assay, and level of polymorphism detected. The primer sequences, polymorphism percentage, Polymorphism Information Centre (PIC), Resolving Power (Rp), and Marker index (MI) outcomes of the screened primers are depicted in Table 2.
The screened 39 primers showed good amplified bands, which ranged between 2 and 8 alleles per individual in all the loci. A total of 163 alleles were achieved, of which all (163) were polymorphic, and no monomorphic bands were observed. The polymorphism percentage of all the primers showed 100%, which is unique to this study. Furthermore, PIC, Rp, and MI values were evaluated to check the proficiency of the screened primers. In the current survey, the PIC value of all primers ranged between 0.20 and 0.89, of which primer (MAGN27) showed minimum PIC value and primer (CuMiSat 31) showed maximum PIC value. The primer proficiency was further analyzed by calculating the Marker Index (MI) and Resolving Power (Rp). The average MI was estimated to be 2.82, of which primer (MAGN27) showed a minimum of 0.40, and CuMiSat 22 showed a maximum of 6.32. Similarly, the Rp value was also evaluated where Rp is the resolving power which objective is to discriminate between genotypes, and from the calculated study, it was observed that the average Rp value was 1.204, of which primer (RM 171) showed a minimum Rp value (0) and primer (CuMiSat 37) showed maximum Rp value (3.46). The gel image of the SSR primer (CuMiSat-37) profile of all the 152 accessions of C. longa is represented in Fig. S1.
Genetic diversity of inter and intra population
The genotypes of selected lines were splitted into four populations, namely Pop 1 from Assam, Pop 2 from Arunachal Pradesh, Pop 3 from Nagaland, and Pop 4 from Manipur, based on the geographical location. The genetic variability parameters like the number of observed alleles (na), Nei’s gene diversity (h), number of effective alleles (ne), Shannon’s information index (I), genetic diversity within the population (Hs), genetic differentiation degree (Gst), genetic diversity in the population (Ht), gene flow (Nm) are computed and mentioned in Table 3. The parameters (na, ne, h, I) showed highest in Population 1 of Assam (1.93, 1.42, 0.26, 0.40), followed by Population 2 of Arunachal Pradesh (1.89, 1.43. 0.26, 0.39), Population 3 of Nagaland (1.76, 1.41, 0.24, 0.36) and lowest from Population 4 of Manipur (1.50, 1.30, 0.17, 0.26). Moreover, significant genetic diversity was confirmed from the result of total species diversity within the population (Ht) and among the population (Hs): 0.27 ± 0.03 and 0.23 ± 0.02 respectively, with genetic differentiation degree (Gst) of 1.24 and gene flow (Nm) of 3.51 which is significantly higher.
Cluster analysis
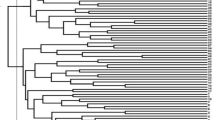
Neighbor joining method based on Jaccard’s pairwise distance matrix was used to construct the dendrogram of 152 accessions of C. longa rich in curcumin content analyzed through SSR marker. A comprehensive study on dendrogram revealed a total of three clusters, out of which cluster I consist of 58 accessions, cluster II consists of 92 accessions, and cluster III consists of 2 accessions which are represented in (Fig. 1). In cluster I, out of 58 accessions, 12 accessions were collected from Assam, 27 from Arunachal Pradesh, and 19 from Manipur. The cluster II is again split up into two sub-clusters: cluster IIa and Cluster IIb where Cluster IIa consists of 82 accessions which are again split up into two minor sub-clusters: cluster IIa(1) consists of 52 accessions gathered from the states of Nagaland (11), Arunachal Pradesh (14), Assam (27) while cluster IIa(2) consist of 30 accessions from the states of Arunachal Pradesh (1), Assam (6), Nagaland (23). Additionally, cluster II(b) consists of 10 accessions comprising from the states of Assam (3) and Arunachal Pradesh (7). Cluster III has a total of 2 accessions (RRLJCL 78 and RRLJCL 76) from Assam.

Dendrogram constructed based on N-J method determining three clusters of C. longa germplasm; numbers depicted in the clusters represent the code number of 152 accessions.
Jaccard’s Pairwise coefficient outlined maximum genetic dissimilarity of (0.75) between accession RRLJCL 3 of cluster IIa(1) and RRLJCL 126 of cluster IIa(2), indicating high variation between these two lines as it was from two different states viz Arunachal Pradesh and Nagaland respectively and minimum genetic dissimilarity of (0.09) between cluster I of line RRLJCL 58 and RRLJCL 59 indicating significantly less variation as the two lines were from same state, i.e., Arunachal Pradesh.
Based on Nei’s genetic study, genetic distance and genetic identity was calculated between four population, where it was revealed that maximal genetic identity (0.9853) was observed between Pop 1 (Assam) and Pop 2 (Arunachal Pradesh) and maximum genetic distance (0.1220) was noticed between Pop 3 (Nagaland) and Pop 4 (Manipur) which explains that genetic similarity was very high between Pop 1 and Pop 2 whereas genetic variation is high between Pop 3 and Pop 4 (depicted in Table 4; Fig. 2).

Dendrogram differentiating the genetic divergence among the population based on Nei's Genetic distance: Method = UPGMA.
Principal Component analysis (PCA) was also performed for 152 accessions of C. longa to check variability and relationship among them. The highest Eigen value calculated from the first three groups was (3.90, 2.52, 1.34) respectively, which provides more information about the divergence among the accessions. The sum cumulative variance perceived was 34.01%, of which 17.53%, 11.34%, and 6.03% were for the first three principal components (charted in Table 5). The Principal Component Analysis plots coincide mostly with the dendrogram clusters except for a few lines (RRLJCL 60, RRLJCL 86, RRLJCL 96, RRLJCL 97) plotted distance apart, as shown in the PCA plot (Fig. 3).

Principal Component Analysis plot of all the accessions of C. longa collected from different regions of North East, India.
Population structure
Based on the structure Harvester software analysis, a total of three subpopulations was constructed, which is shown in (Figs. 4 and 5). Accessions scoring more than 0.80 can be considered as genetically pure accession, and not more than 0.80 can be contemplated as intermixture accession34. In this study, almost all accessions were found to be pure accessions except nine accessions which were intermixture in nature. The admixture accession of population structure 1 (red colour) were RRLJCL-86, RRLJCL-97, RRLJCL-60; and RRLJCL-67, RRLJCL-76, RRLJCL-92, RRLJCL-69, RRLJCL-85, RRLJCL-96 were the admixture accession of population structure 3 (blue colour). All the accessions clustered in population structure 2 (green colour) were pure accessions, as shown in (Fig. 4). The Fst mean value of all three populations (1, 2, 3) were given as 0.507, 0.217, and 0.488, respectively. Also, the allele frequency variance between the populations computed using point estimation of P is presented in Table 6.

Approximation of relevant number of population in 152 accessions of C.longa.

Model-based population structure analysis of C. longa.
Analysis of molecular variance
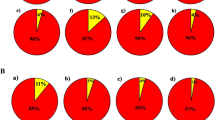
Analysis of Molecular Variance (AMOVA) was conducted for C. longa to assess the difference in population, where it was observed that molecular variation was high within the population (87%) and significantly less variation among the population (13%) mentioned in the (Table 7 and Fig. 6).

Pie chart of AMOVA (Analysis of Molecular Variance) among and within the population of C. longa.
Discussion
C. longa holds industrial significance worldwide and hence needs improvement through breeding program.The collection and assessment of germplasm diversity are imperative to facilitate genetic improvement in the crop. The present study scrutinized high curcumin lines from 329 accessions where 152 accessions showed curcumin content > 1.50%. Among the 152 accessions, 59 accessions showed curcuminoid content > 9%, out of which 29 accessions were from the state of Assam, 18 accessions were from Arunachal Pradesh, 10 accessions were from Nagaland and 2 accessions were from Manipur. The elevated curcumin content was observed mostly in the accessions grown in Assam and Arunachal Pradesh which can be attributed to the favorable environmental conditions in Assam as the state shares a moderate climate condition, characterized by an average temperature of 25.67 °C and an average humidity of 75.15% with an annual rainfall of 2244 mm. This aligns with the findings of Sandeep et al.39 who also noted that the curcumin content in turmeric is influenced by the specific environmental zones in which the plants are cultivated. Thus, it can be stated that soil and environmental factors plays a significant role in the accumulation of curcumin in turmeric plants.
Keeping in view the effect of environmental factors on the genotypes of C. longa which hinders the phenotypic attribute in germplasm variation, a molecular diversity study on 152 high curcumin accessions were conducted using SSR markers. The molecular markers or primers serve as indicators of polymorphism in unveiling genetic distinctions among individual organisms or species which may emerge from its genetic constitution, nucleotide alterations or genome locus mutations40. A total of 39 SSR primers were screened, where all the primers showed good amplified bands with 100% polymorphic percentage. Likewise, Singh et al.37 studied 9 SSR primers on 30 genotypes, out of which only 6 primers pairs showed 100% polymorphism with banding patterns ranging between 1–2 allele/loci. A study on 60 genotypes of turmeric was reported where average polymorphism resulted 91.4% and 95.4%, respectively, for 11 RAPD and 6 ISSR primers41. Again 15 DAMD and 13 ISSR primers were studied by Verma et al.42 where the average polymorphism percentage reported was 84.4% and 79.2%, respectively; Sumi et al.43 reported 64.4% average polymorphism percentage on 8 genotypes by using 12 RAPD primers. Aswasthi et al.44 also worked on 56 primers which include both SSR and ISSR on 18 improved varieties. Similarly, in the present survey, all the screened 39 primers represent a high level of polymorphism percentage (100%) as compared to earlier studies, which outlines an indication towards high level of genetic variation among the individual accessions. In addition to that, PIC, MI, and Rp values were evaluated where the PIC value for all the primers ranged between 0.20 and 0.89, where MAGN 27 showed minimum PIC value, and CuMiSat 31 showed maximum PIC value. The average PIC value obtained from 39 primers is 0.64, out of which 31 primers showed a PIC value higher than 0.50 signifying that SSR primers used in this study are highly informative as PIC value greater than 0.50 (> 0.50) illustrate high efficiency of primers34,45. The primer proficiency was further analyzed through MI and Rp where the average MI was 2.82, with the lowest value showed by primer (MAGN 27) and the highest value showed by primer (CuMiSat 22). Again, average Rp was calculated to be 1.204, where the lowest value showed by primer (RM 171), and the highest value showed by primer (CuMiSat 37). Based on study and observation of data for all the screened primers, eleven primers, namely CuMiSat-19, CuMiSat-21, CuMiSat-22, CuMiSat-31, CuMiSat-35, RM125, RM135, RM153, RM154, CBT-05, and CBT-08 showed most efficient outcome with PIC, Rp and MI values more than the mean values (PIC = 0.64, MI = 2.82 and Rp = 1.20).The findings of the above analysis unveiled efficiency of the primers which will serve as a resource for future conductance of molecular diversity analysis in different species of Zingiberaceae family.
The genetic diversity analysis led to the construction of cluster dendrogram using Neighbor-Joining (N-J) method as it is a rapid tool and reliable in nature46. The N-J method is a distance-based method that relies on assessing shared alleles to calculate distances between taxa, creating distance relationships of different accessions or germplasms in representation of tree47. In the present investigation, the results obtained through cluster dendrogram revealed the presence of geographically independent clusters as majority of the accessions did not exclusively grouped according to geographical provinces, and the reason may be due to the migration of genotypes from one location to another caused by overexploitation of natural habitat34,48. However, region-specific clustering was restricted to cluster III, where 2 lines (RRLJCL78 AND RRLJCL76) were from Assam, and the reason may be due to uninterrupted wild varieties in natural habitat1. Likewise, Verma et al.42 identified two prime clusters from the dendrogram constructed on account of the combination of DAMD and ISSR markers, where it was revealed that genotypes in the clusters grouped independently of their geographical location which align with our study. Again, Sahoo et al.49 worked on 10 Curcuma species using EST SSR primer, revealed the presence of two main clusters, one of which consists of all nine species and the other of only Curcuma longa species. The clustering of accessions in different clusters unveiled more genetic distance, revealing more genetic variation which would be conducive to the future hybridization programme. The genetic diversity analysis of intra and inter-population genotypes were also studied in the four populations which was divided based on its collection site. All the populations showed good polymorphism, with only moderate polymorphism in the Manipur population, which may be due to the low number of accessions collected from Manipur compared to other populations used in the study. All the genetic parameters for diversity revealed highest variation in population 1 (Assam) and lowest in population 4 (Manipur). Here, the reason for highest variation in population 1 may be due to the exchange of accessions from the neighboring states or random mutation. Moreover, significant genetic diversity was confirmed from the results of total species diversity within the population (Ht) (0.27 ± 0.03) and among the population (Hs) (0.23 ± 0.02), with genetic differentiation degree (Gst) of 1.24. The parameter like gene flow (Nm) is the shifting of genetic distinction from one population to another, and when gene flow increases among populations, it indicates less variation and more homogeneity whereas when gene flow increases within population, it indicates high variation and less homogeneity. In this study, gene flow (Nm) was found to be 3.51, which is significantly higher than the threshold value (Nm ≤ 1.0)50, and represents high gene flow, whereas Sahoo et al.49 reported working on genetic variation of Curcuma species using EST SSR primer where Nm value observed was 0.0781 which revealed that whole distribution is highly constricted and genetic distinction is highly noteworthy. Again, Basak et al.51 while working on turmeric cultivars using ISSR and RAPD markers reported the Nm value to be between 0.33 and 0.37 for both markers. Here the present study revealed moderate genetic heterozygosity and genetic differentiation degree, which determine moderate genetic variation among and within the populations, and it can be concluded in context to the previous reports34,52. The results in gene exchange in the present study may be due to human intrusion like genetic swamping, introgression and hybridization1.
Through Nei’s genetic study, genetic identity among the population ranges from 0.9853 to 0.1220 with maximal genetic identity of 0.9853 was found between Pop 1 (Assam) and Pop 2 (Arunachal Pradesh) and the minimal genetic identity of 0.1220 was found between Pop 3 (Nagaland) and Pop 4 (Manipur), and the reason for low variation, i.e., maximal genetic identity between Assam and Arunachal Pradesh population may be due to gene exchange or duplication of germplasm between adjacent states as gene exchange among two population leads to decrease of genetic variation and increase of homogeneity53. Similarly, Nei’s genetic study on Cymbopogon winterianus was studied by Munda et al.,34 where low variation was found among the population and mentioned that the reason for low genetic distance was due to gene exchange among the adjoining population. Moreover may have occurred through human intrusion since gene exchange cannot be facilitated through pollen or seed because the showy flower which blooms in C. longa plant is not a true flower but is actually a bract. Thus, the data procured through this study could be used as baseline data for selection of desired traits in breeding programmes and conservation of highly variable germplasm54.
In addition to that, Principal Component Analysis (PCA) is broadly used to study the structure of data55, where PCA is useful in clustering of similar accession while non-similar accession plot distance apart in the presentation56. In the present survey, the PCA plots closely align with the dendrogram clusters, except for a few outliers (RRLJCL 60, RRLJCL 86, RRLJCL 96, RRLJCL 97) that are plotted at a significant distance from the main cluster. The PCA and cluster analysis provide more reliable information if the first three component scores more than 25% variance57. In the present study, the first three components (17.53%, 11.34%, and 6.03%) come up with 34.01% variability, which is higher than previous reports and indicates more reliable data. Unfortunately, there is currently limited data available regarding the intra and inter-specific relationships and genetic diversity within large number of germplasm of C. longa collected from North East India. Thus, this study will contribute a novel insight to researchers and breeders towards development of superior variety.
Further, the structure analysis employs a Bayesian clustering approach, assigning individuals to grouped as populations based on their genotypes and seeking population structures characterized by linkage equilibrium and Hardy–Weinberg equilibrium47. From the population structure survey, a total of three population structure were constructed where population 1 is represented with red colour, population 2 is represented with green colour and population 3 is represented with blue colour. As accessions scoring more than 0.80 can be considered as pure accessions, here in the present analysis, almost all accessions were found to be pure accession except nine accessions (RRLJCL-86, RRLJCL-97, RRLJCL-60, RRLJCL-67, RRLJCL-76, RRLJCL-92, RRLJCL-69, RRLJCL-85, RRLJCL-96) were found intermixture in nature. The admixture accessions (RRLJCL-86, RRLJCL-97, RRLJCL-60) were found in the population structure 1 (red colour) and the admixture accessions (RRLJCL-67, RRLJCL-76, RRLJCL-92, RRLJCL-69, RRLJCL-85, RRLJCL-96) were found in the population structure 3 (blue colour) and remaining all the accessions were found to be pure accessions.In the population structure 2 (green colour), no intermixture accessions were found. The pure accessions which were retrieved from this study could be used for conservation of gene pool due to distinct genetic makeup for longer survival of the turmeric species, and this could be later applied for cross breeding programme for development of superior genotype. Through this study, it can be concluded that the SSR markers used in this survey for the genetic divergence of C. longa accessions is suitable and satisfactory for population studies. The reason for the genetic divergence or intermixture accessions found within the populations is mainly due to several factors like gene flow, mating process, selection, mutation1. Moreover, accessions grouping from bar plot resembled mostly with dendrogram clusters in the current survey which provide more reliability and satisfactory result towards genetic diversity studies. Thus, the neighbor-joining tree and Structure analysis of the SSR data complement each other, offer distinct methodologies for investigating the relationships among different accessions and together yield a robust analysis.
Furthermore, AMOVA study revealed high molecular variation within the population (87%) and significantly less variation among the population (13%). Similarly, Sigrist et al.26 also performed genetic divergence of turmeric using microsatellites where genetic divergence within the population was 75.29% and among the population was 24.71% which belongs only to Brazilian states. In contrast, when the accessions were collected from different countries like Brazil, India, and Puerto Rico, the genetic variability was observed to be 63.42% among the countries and 27.05% within the countries. Singh et al.41 also conducted the analysis of molecular variance using the RAPD marker, which showed 42% genetic variability among the population, and the remaining 58% was within the population, whereas for the ISSR marker, 48% showed genetic variability among the population and 52% was within the population. Similarly, in present investigation, molecular variation within the population was notably more extensive than among populations, serving as the primary source of overall genetic variation. The lower diversity among populations suggested a heightened level of gene exchange and the higher diversity within the population may be due to random drift, mutation, transgressive segregation58 or collection of all the accessions from adjoining states of India which is in accordance to Sigrist et al.26 study. As per the genetic theory of populations, an increase in diversity enhances a species potential to adapt to evolving environments. Hence it becomes necessary to delve into knowledge of genetics, as the loss of heterogeneity could risk the population's sustainability and ultimately lead to the extinction of the species1.
To devise effective conservation strategies for various species, understanding the genetic makeup within populations is of utmost significance and to prevent the depletion of genetic foundations, selection of genetically diverse genotypes, analysis of plant molecular diversity is an indispensable tool54,59. From an evolutionary perspective and for the long-term survival of species, population genetic diversity of C. longa holds immense importance. Therefore, through this genetic assessment study, eleven most efficient primers were found which showed PIC, Rp and MI values greater than the average PIC, Rp and MI values. A total of three clusters were formed through Neighbour Joining dendrogram, structure bar plot and PCA analysis which resembles with each other. Analysis of variance revealed high molecular variation within the population (87%) and very less variation among the population (13%) and maximal intra-population diversity was distinguished in Pop 1 (Assam) and minimal in Pop 4 (Manipur). Hence it is necessary to conserve the highly variable population through both in-situ and ex-situ conservation, which may otherwise lead to a loss of heterogeneity in a population, causing species extermination. In the present study, population 1 (Assam)which showed highest variation can be invaluable for hybridization efforts in future crop improvement programs and the identification of closely related species which are represented as pure accessions can serve as major sources of genes for conservation of gene pool and future breeding programs. Therefore, result obtained through genetic assessment study of Curcuma longa L. will provide a handful of resources and great potential for enhancing crop improvement programs, facilitating the selection of desired genotypes, aiding in marker-assisted breeding, identifying quantitative trait loci, and preserving the genetic diversity. The growing global demand for curcumin, the most active component found in turmeric, due to its numerous health benefits underscores the importance of conserving and cultivating curcumin-rich superior genotype and hence urgent steps must be taken to effectively conserve and breed these industrially significant plants. Hence, the present research report will play a pivotal role in promoting the sustainable growth of this economically important medicinal species.
Methods
Plantation of collected germplasm of C. longa
A sum of 329 accessions of C. longa was collected from four states of India, specifically from Assam, Nagaland, Arunachal Pradesh, and Manipur during the year 2019 (presented in Fig. 7). The sample specimens were identified by Dr. Mohan Lal, Principal Scientist, CSIR-NEIST, Jorhat. The specimen herbariums with voucher no. RRLJCL-1 to RRLJCL-152 were deposited at the departmental herbarium. The collected rhizome was then planted in augmented design during the month of March 2020 at the Experimental farm of CSIR-NEIST Jorhat. The experimental site received an annual rainfall of 2244 mm, a mean temperature of 25.67 °C, and mean humidity of 75.15%. The soil pH was 4.9, sandy loam in nature, and NPK concentration was nourished at 226, 116, and 144 kg/ha, respectively. The spacing of one plant from another plant was maintained at 45 cm, and row-to-row spacing was also 45 cm. After harvesting, each germplasm rhizome (300 gm) was dried for 7 days, and the curcuminoid content was measured for each. Among them, 152 high curcumin content (> 1.5%) lines were selected and again planted in Randomized Block Design (RBD) with three replications during the month of March 2021, which were used for molecular diversity assessment. All the experimental research and field studies performed on 329 accessions of Curcuma longa L. were carried out in accordance with relevant guidelines.

Experimental trial of 329 accessions of Curcuma longa L. collected from four states of North East India.
High curcumin lines selection from collected germplasm
All the planted accessions were estimated for their total curcuminoids content as per the below-described procedure. Firstly the fresh rhizomes were cut into small pieces and dried in a hot air oven for seven days at 59 ̊ C and then were grind to make fine powder. The fine powder sample was further used for solvent extraction with acetone (100 mL) using the Soxhlet apparatus. The solvent extract was allowed to evaporate through the rotary evaporator to obtain the dry mass. The dry mass was put forwarded for HPLC analysis where a Thermo Scientific Dionex Ultimate 3000 HPLC system was used with Syncronis C-18 column (Dim. 150 × 4.6 (mm), SN: 10,879,123, LOT: 16,092, Particle size (µ) 3), acetonitrile and 0.1% formic acid as mobile phase, the flow rate of 1 mL/min, injection volume of 20 µL, UV: 426 nm, temperature: 25ºC as conditions. For the development of a standard calibration curve, 1 mg of standard total curcuminoids batch number ANE/CL/01/047/21 (purchased from Indica Neutraceuticals, India) was dissolved in 1 mL of HPLC grade acetonitrile from which different concentrations were prepared (20, 40, 60, 80 and 100 ppm) and injected into the system. Samples were prepared by using 1 mg of dried extract in 1 ml of HPLC grade acetonitrile, filtered with 0.2 μm nylon membrane, and injected into the system. Chromatographic data were obtained from which data were analyzed using Thermo Scientific Dionex Chromeleon Chromatography Data System version 7.2.
Extraction of genomic DNA from C. longa accessions
Fresh tender leaves of all the selected high curcumin-rich accessions (152 no.) of Curcuma longa were collected in separate zip lock bags from the experimental field at CSIR NEIST, Jorhat, Assam. Leaf samples were then washed, cleaned properly, and lyophilized at − 40 °C for 48 h. Isolation of plant DNA was carried out using HiMedia Kit (HiMedia Mumbai) by modifying the CTAB method (Cetyltrimethylammonium Bromide) as instructed in the kit. Agarose gel 0.8% was used to check the purity of extracted DNA, and bands were observed in the gel documentation system (Vilber E-Box, France). To check the quantification of stock DNA concentration, 3 µl of DNA sample was assessed in a Nano Bio Spectrophotometer (Eppendorf, Germany) at λ260/λ280 ratio.
PCR analysis of C. longa accessions using SSR Primer
PCR analysis of C. longa accession outlined 39 primer pairs out of a sum of 44 pairs of SSR primers which showed the best amplification for genetic study analysis. The primers were selected based on the earlier literatures on molecular diversity analysis in the Zingiberaceae family, which was then obtained from Bioserve Biotechnologies (India) Private Limited, Hyderabad, India. The primers obtained were in lyophilized form, which was then prepared to stock solution followed by preparation of working solution at a concentration of 10 picomol. The extracted DNA of all the accessions was also converted to a working solution with a concentration of 30 ng. The required mixture for PCR amplification consists of 5μL of DNA sample, 1μL each of forward and reverse primer, 10 μL of master mix (HiChrome), and 3μL of doubled distilled water, making a final volume of 20μL. For amplification, Prima 96 thermocycler (HiMedia, India) was used, which was conditioned at 95 °C for 1 min for initial denaturation, then continued with 35 cycles of 95 °C for 55 s (denaturation), 55 s with primer melting temperature ± 5 °C (annealing), 72 °C for 5 min (extension), and finally by 72 °C for 8 min as a concluding extension. The amplified results were then observed in Agarose gel (2%) using 1X TBE buffer, and electrophoresis was run for 1 h at 90 constant voltage. The amplified bands were then observed in a gel documentation system (Vilber E-Box, France).
Statistical data analysis
In order to obtain genetic variation, the banding patterns were scored after observing in the gel documentation system. The data was obtained by using input as 1 for the presence of band and 0 for the absence of band for each primer in the genotypes. The Jaccard's similarity coefficient was evaluated to obtain all pairwise differences between the accessions to build a genetic dissimilarity matrix. The final cluster dendrogram and principal coordinate analysis (PCA) were plotted based on the dissimilarity matrix and Neighbor Joining (N-J) method using Darwin software version 6.0.
Polymorphic Information Content (PIC) values were calculated by using the formula PIC = 1 − \({\sum pi}^{2}\) on the basis of the polymorphism of the bands where Pi = frequency of ith allele45. Marker Index (MI) was calculated by the formula MI = EMR × PIC where EMR is the Effective Multiplex Ratio and is defined by the multiplication of a number and a fraction of polymorphic loci60,61 while Resolving Power (Rp) is the summation of the band informativeness calculated by the formula [Ib = 1 − [2 ×|0.5 − P|] where p = proportion of the individuals containing the bands61. In addition, the POPGENE (Version-1.31) software was utilized to study the genetic diversity variables such as genetic differentiation degree (Gst), genetic diversity in the population (Ht), gene flow (Nm), genetic diversity within the population (Hs), Shannon’s information index (I), number of observed alleles (na), number of effective alleles (ne) and Nei’s gene diversity (h). Through Analysis of Molecular Variance (AMOVA), inter and intra-population diversities were analyzed by implementing GenAlex software version 6.5. To evaluate the genetic relationship among 152 accessions using 39 SSR primer STRUCTURE software version 2.3.4 has been utilized to construct a model-based population structure where the software was pass multiple times to evaluate the number of populations among all the accession. The most foreseeable genetic population group obtained from the study was computed by using an online tool named Structure Harvester.
Data availability
The datasets used and/or analysed during the current study would be available from the corresponding author on reasonable request.
References
Lal, M., Munda, S., Bhandari, S., Saikia, S., Begum, T. & Pandey, S. K. Molecular genetic diversity analysis using SSR marker amongst high solasodine content lines of Solanum khasianum CB Clarke, an industrially important plant. Ind. Crops Prod. 184, 115073. https://doi.org/10.1016/j.indcrop.2022.115073 (2022).
Paw, M., Munda, S., Borah, A., Pandey, S. K. & Lal, M. Estimation of variability, genetic divergence, correlation studies of Curcuma caesia Roxb. J. Appl. Res. Med. Aromat. Plants. 17, 100251. https://doi.org/10.1016/j.jarmap.2020.100251 (2020).
Jan, H. U., Rabbani, M. A. & Shinwari, Z. K. Assessment of genetic diversity of indigenous turmeric (Curcuma longa L.) germplasm from Pakistan using RAPD markers. J. Med. Plant Res. 5(5), 823–830 (2011).
Sasikumar, B. Genetic resources of Curcuma: diversity, characterization and utilization. Plant Genet. Res. 3(2), 230–251. https://doi.org/10.1079/PGR200574 (2005).
Chanda, S. & Ramachandra, T. V. Phytochemical and pharmacological importance of turmeric (Curcuma longa): A review.RRJoP, 9(1), 16–23 (2019).
Govindarajan, V. S. Turmeric-chemistry, technology and quality. Crit. Rev. Food Sci. Nutr. 12, 199–301. https://doi.org/10.1080/10408398009527278 (1980).
Araujo, C. A. C. & Leon, L. L. Biological activities of Curcuma longa L. Mem. Inst. Oswaldo Cruz. 96, 723–728. https://doi.org/10.1590/S0074-02762001000500026 (2001).
Omosa, L. K., Midiwo, J. O. & Kuete, V. Curcuma longa. In Medicinal spices and vegetables from Africa. Academic press, (pp. 425–435).https://doi.org/10.1016/B978-0-12-809286-6.00019-4 (2017).
Ammon, H. P. & Wahl, M. A. Pharmacology of Curcuma longa. Planta Med. 57, 1–7. https://doi.org/10.1055/s-2006-960004 (1991).
Satyavati, G.V., Raina, M.R. & Sharma, M. Medicinal plants of India. Indian Council of Medical Research, New Delhi, (1976).
Ravindranath, V. & Satyanaraya, M. N. An unsymmetrical diarylheptanoid from Curcuma longa. Phytochem. 19, 2031–2032. https://doi.org/10.1016/0031-9422(80)83033-0 (1980).
Niranjan, A. & Prakash, D. Chemical constituents and biological activities of turmeric (Curcuma longa L.)—a review. J. Food Sci. Technol. 109, (2008).
Rathore, S. et al. Curcumin: A review for health benefits. Int. J. Res. Rev. 7(1), 273–290 (2020).
Ravindran, P. N., Babu, K. N., & Sivaraman, K. (Eds.). Turmeric: the genus Curcuma. CRC press. https://doi.org/10.1201/9781420006322 (2007).
Baptista-Silva, S., Borges, S., Ramos, O. L., Pintado, M. & Sarmento, B. The progress of essential oils as potential therapeutic agents: A review. J. Essent. Oil Res. 32(4), 279–295. https://doi.org/10.1080/10412905.2020.1746698 (2020).
Gobbo-Neto, L. & Lopes, N. P. Medicinal plants: factors of influence on the content of secondary metabolites. Quim. Nova. 30, 374–381. https://doi.org/10.1590/S0100-40422007000200026 (2007).
Sandeep, I. S. et al. Agroclimatic zone based metabolic profiling of turmeric (Curcuma longa L.) for phytochemical yield optimization. Ind. Crops Prod. 85, 229–240. https://doi.org/10.1016/j.indcrop.2016.03.007 (2016).
Gomes, A. F. et al. Seasonal variation in the chemical composition of two chemotypes of Lippia alba. Food Chem.273, 186–193. https://doi.org/10.1016/j.foodchem.2017.11.089 (2019).
Xiang, H. et al. Phytochemical profiles and bioactivities of essential oils extracted from seven Curcuma herbs. Ind Crops Prod. 111, 298–305.https://doi.org/10.1016/j.indcrop.2017.10.035 (2018).
Leela, N. K., Tava, A., Shafi, P. M., Sinu, P. J. & Chempakam, B. Chemical composition of essential oils of turmeric. Acta Pharm. 52, 137–141 (2002).
Sacchetti, G. et al. Comparative evaluation of 11 essential oils of different origin as functional antioxidants, antiradicals and anti-microbials in foods. Food Chem. 91(4), 621–632. https://doi.org/10.1016/j.foodchem.2004.06.031 (2005).
Liju, V. B., Jeena, K. & Kuttan, R. An evaluation of antioxidant, anti-inflammatory, and antinociceptive activities of essential oil from Curcuma longa L. Indian J. Pharmacol. 43(5), 526. https://doi.org/10.4103/2F0253-7613.84961 (2011).
Raut, J. S. & Karuppayil, S. M. A status review on the medicinal properties of essential oils. Ind Crops Prod. 62, 250–264. https://doi.org/10.1016/j.indcrop.2014.05.055 (2014).
Meenakshi, N. & Sulikeri, G.S. Effect of different planting materials on growth, yield and productivity of turmeric (Curcuma longa L.). Inter. J. Trop. Agric.21, 37–44, (2003).
May, A., Cecilio-Filho, A.B., Cavarianni, R.L. & Barbosa, J.C. Turmeric (Curcuma longa L) development and productivity in function at nitrogen and potassium doses. Rev. Bras. Plant Med. 7, 73–78 (2005).
Sigrist, M. S., Pinheiro, J. B., Azevedo Filho, J. A. & Zucchi, M. I. Genetic diversity of turmeric germplasm (Curcuma longa; Zingiberaceae) identified by microsatellite markers. Genet. Mol. Res. 10(1), 419–428. https://doi.org/10.4238/vol10-1gmr1047 (2011).
Nass, L.L. Utilização de Recursos Genéticos Vegetais no Melhoramento. In: Recursos Genéticos e Melhoramento - Plantas (Nass LL, Valois ACC, Melo IS and Valadares-Inglis MC, eds.). Fundação MT, Rondonópolis, 29–56 (2001).
Ferreira, M.E., Moretzsohn, M.C. & Buso, G.S.C. Fundamentos de Caracterização Molecular de Germoplasma Vegetal. In: Recursos Genéticos Vegetais (Nass LL, ed.). Embrapa, Brasília, 858 (2007).
Pinheiro, J. B., Zucchi, M. I., Teles, F. L. & Azara, N. A. Diversida degenética molecular emacessos de açafrãoutilizandomarcadores RAPD. Acta Sci. Agron. 25, 195–199. https://doi.org/10.4025/actasciagron.v25i1.2671 (2003).
Powell, W. et al. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 2, 225–238. https://doi.org/10.1007/BF00564200 (1996).
Naghavi, M. R. et al. Comparison of genetic variation among accessions of Aegilops tauschii using AFLP and SSR markers. Genet. Resour. Crop. Evol. 54, 237–240. https://doi.org/10.1007/s10722-006-9143-z (2007).
Henkrar, F. et al. Genetic diversity reduction in improved durum wheat cultivars of Morocco as revealed by microsatellite markers. Sci. Agric. 73, 134–141. https://doi.org/10.1590/0103-9016-2015-0054 (2016).
Abbasov, M. et al. Genetic relationship of diploid wheat (Triticum spp.) species assessed by SSR markers. Genet. Resour. Crop Evol. 65, 1441–1453. https://doi.org/10.1007/s10722-018-0629-2 (2018).
Munda, S. et al. Evaluation of genetic diversity based on microsatellites and phytochemical markers of core collection of Cymbopogon winterianus Jowitt. germplasm. Plants. 11(4), 528. https://doi.org/10.3390/plants11040528 (2022).
Song, Q. J. et al. Development and mapping of microsatellite (SSR) markers in wheat. Theor. Appl. Genet. 110(3), 550–560. https://doi.org/10.1007/s00122-004-1871-x (2005).
Baruah, J., Pandey, S.K., Sarmah, N. & Lal, M. Assessing molecular diversity among high capsaicin content lines of Capsicum chinense Jacq. using simple sequence repeat marker. Ind. Crops Prod. 141, 111769. https://doi.org/10.1016/j.indcrop.2019.111769 (2019).
Singh, T. J., Patel, R. K., Patel, S. N. & Patel, P. A. Molecular diversity analysis in Turmeric (Curcuma longa L.) using SSR markers. Int. J. Curr. Microbiol. Appl. Sci. 7(11), 552–560. https://doi.org/10.20546/ijcmas.2018.711.066 (2018).
Sahoo, A. et al. Qualitative and quantitative evaluation of rhizome essential oil of eight different cultivars of Curcuma longa L. (Turmeric). J. Essent. Oil-Bear. Plants. 22(1), 239–247. https://doi.org/10.1080/0972060X.2019.1599734 (2019).
Sandeep, I. S. et al. Differential expression of CURS gene during various growth stages, climatic condition and soil nutrients in turmeric (Curcuma longa): Towards site specific cultivation for high curcumin yield. Plant Physiol. Biochem. 118, 348–355. https://doi.org/10.1016/j.plaphy.2017.07.001 (2017).
Idrees, M. & Irshad, M. Molecular markers in plants for analysis of genetic diversity: a review. Eur. J. Acad. Res. 2(1), 1513–1540 (2014).
Singh, S., Panda, M. K. & Nayak, S. Evaluation of genetic diversity in turmeric (Curcuma longa L.) using RAPD and ISSR markers. Ind Crops Prod. 37(1), 284–291. https://doi.org/10.1016/j.indcrop.2011.12.022 (2012).
Verma, S. et al. Assessment of genetic diversity in indigenous turmeric (Curcuma longa) germplasm from India using molecular markers. Physiol. Mol. Biol. Plants. 21(2), 233–242. https://doi.org/10.1007/s12298-015-0286-2 (2015)
Sumi, S. A., Wahab, M. A., Islam, A. & Houqe, M. E. Molecular diversity analysis of different turmeric (Curcuma longa L.) genotypes using RAPD markers. J. Glob. Biosci. 9(9), 7897–7911 (2020).
Aswathi, A. P., Raghav, S. B. & Prasath, D. Assessment of genetic variation in turmeric (Curcuma longa L.) varieties based on morphological and molecular characterization. Genet. Resour. Crop Evol. 1–12. https://doi.org/10.1007/s10722-022-01417-3 (2022).
Botstein, D., White, R. L., Skolnick, M. & Davis, R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331 (1980).
Saitou, N. & Nei, M. The neighbour joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454 (1987).
Barkley, N. A., Roose, M. L., Krueger, R. R. & Federici, C. T. Assessing genetic diversity and population structure in a citrus germplasm collection utilizing simple sequence repeat markers (SSRs). Theor. Appl. Genet. 112, 1519–1531. https://doi.org/10.1007/s00122-006-0255-9 (2006).
Spooner, D. M. et al. Comparison of four molecular markers in measuring relationships among the wild potato relatives Solanum section E tuberosum (subgenus Potatoe). Theor. Appl. Genet. 92, 532–540 (1996).
Sahoo, A. et al. EST-SSR marker-based genetic diversity and population structure analysis of Indian Curcuma species: Significance for conservation. Rev. Bras. Bot. 44, 411–428 (2021).
Slatkin, M. Gene flow and geographic structure of natural populations. Science. 236, 787–792. https://doi.org/10.1126/science.3576198 (1987).
Basak, S. et al. Assessment of genetic variation among nineteen turmeric cultivars of Northeast India: Nuclear DNA content and molecular marker approach. Acta Physiol. Plant. 39(2), 45. https://doi.org/10.1007/s11738-016-2341-1 (2017).
Zheng, W. H. et al. Conservation and population genetic diversity of Curcuma wenyujin (Zingiberaceae), a multifunctional medicinal herb. Genet. Mol. Res. 14, 10422–10432 (2015).
Harsono, T., Pasaribu, N., Fitmawati, F., Sobir, S. & Prasetya, E. Genetic variability and classification of Gandaria (Bouea) in Indonesia based on inter simple sequence repeat (ISSR) markers. SABRAO J. Bred. Genet. 50, 129–144 (2018).
Tamang, R., Munda, S., Darnei, R. L., Begum, T. & Lal, M. Genetic diversity evaluation of core collection gene bank using simple sequence repeat marker of Acorus calamus L.: An important aromatic species. Ind Crops Prod, 204, 117292. https://doi.org/10.1016/j.indcrop.2023.117292 (2023).
Luo, C., Chen, D., Cheng, X., Liu, H., Li, Y. & Huang, C. SSR analysis of genetic relationship and classification in Chrysanthemum germplasm collection. Hortic. Pl. J. 4, 73–82 (2018).
Sangwan, N. S., Yadav, U. & Sangwan, R. S. Molecular analysis of genetic diversity in elite Indian cultivars of essential oil trade types of aromatic grasses (Cymbopogon species). Plant Cell Rep. 20, 437–444. https://doi.org/10.1007/s002990100324 (2001).
Rajwade, A. V. et al. Relatedness of Indian flax genotypes (Linum usitatissimum L.): An inter-simple sequence repeat (ISSR) primer assay. Mol. Biotechnol. 45, 161–170. https://doi.org/10.1007/s12033-010-9256-7 (2010).
Singh, P. & Narayanan, S. S. Biometrical techniques in plant breeding (Kalyani Publishers, 1997).
Wu, F. et al. Genetic diversity and population structure analysis in a large collection of white clover (Trifolium repens L.) germplasm worldwide. PeerJ 9, e11325. https://doi.org/10.7717/peerj.11325 (2021).
Milbourne, D. et al. Comparison of PCR-based marker systems for the analysis of genetic relationships in cultivated potato. Mol. Breed. 3, 127–136. https://doi.org/10.1023/A:1009633005390 (1997).
Prevost, A. & Wilkinson, M. J. A new system of comparing PCR primers applied to ISSR fingerprinting of potato cultivars. Theor. Appl. Genet. 98, 107–112. https://doi.org/10.1007/s001220051046 (1999).
Acknowledgements
The authors are thankful to the Director of CSIR-NEIST, Jorhat for giving us the opportunity to conduct the field and lab experiment as required for the study under CSIR Aroma Mission (HCP007). The authors would like to extend their sincere appreciation to the Researchers Supporting Project number (RSPD2023R686), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
A.G. and M.L. conceptualized the study, A.G. and M.P. conducted the experiment, S.M., M.H.S., A.R.Z.G., M.S.K. performed software analysis, A.G. wrote and prepared original writing, T.B., S.M., M.H.S., A.R.Z.G., M.S.K. reviewed and edited the writing, M.L. supervised the study. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gogoi, A., Munda, S., Paw, M. et al. Molecular genetic divergence analysis amongst high curcumin lines of Golden Crop (Curcuma longa L.) using SSR marker and use in trait-specific breeding. Sci Rep 13, 19690 (2023). https://doi.org/10.1038/s41598-023-46779-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46779-5
This article is cited by
-
Conservation Strategies for Aquilaria sinensis: Insights from DNA Barcoding and ISSR Markers
Plant Foods for Human Nutrition (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
