
Abstract
Congenital hypo-dysfibrinogenemia is a rare autosomal dominant or co-dominant genetic disorder. This study was designed to analyze the clinical phenotype and genetic mutations of a patient with congenital hypo-dysfibrinogenemia. Prothrombin time (PT), activated partial thromboplastin time (APTT), thrombin time (TT), and D-dimer were measured using an automated coagulation analyzer. Fibrinogen (Fg) was assessed by the Clauss and PT-Fg methods. Sanger sequencing was conducted to identify the mutations in FGA, FGB, and FGG genes. The proband and proband’s mother, maternal uncle, and maternal grandfather exhibited a prolonged TT, decreased Clauss Fg and PT-Fg, and normal PT, APTT, and D-dimer without abnormalities in liver and kidney function. The ratio of Clauss Fg to PT-Fg was less than 0.7. A novel c.180 + 1G > A mutation was detected in FGA gene by Sanger sequencing in the proband and proband’s mother, maternal uncle, and maternal grandfather. FGA c.180 + 1G > A variant leads to decreased transcription of FGA. The patient was diagnosed with congenital hypo-dysfibrinogenemia presenting a novel c.180 + 1G > A mutation in FGA, causing a decrease in FGA transcription in proband’s peripheral blood.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital dysfibrinogenemia (MIM: 616,004) is a rare autosomal dominant or co-dominant genetic disorder caused by mutations in FGA, FGB, and FGG that encode fibrinogen proteins [1,2,3]. Hypo-fibrinogenemia is a subcategory of the disease that is mostly autosomal dominant and a few autosomal recessive [4]. The mutation FGA Arg35 (Arg16) in exon 2 and the mutation FGA Arg301 (Arg275) in exon 8 are the two mutation “hot spots” of major significance in screening for abnormal fibrinogenemia, but other mutations are also common in the surrounding residues [5]. In this study, we reported a 4-year-old boy with a prolonged thrombin time (TT) and decreased fibrinogen activity (Fg:Ac) and PT-Fg, without abnormalities in the liver and kidney function. FGA c.180 + 1G > A variant was sequenced in the proband and the proband’s mother, maternal uncle, and maternal grandfather, which caused a decreased mRNA expression of FGA. The proband and the proband’s mother, maternal uncle, and maternal grandfather were then diagnosed with congenital.
Case presentation
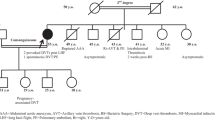
The proband of interest was a 4-year-old boy who presented with recurrent abdominal pain for 6 months. Three generations of the family were investigated, including the proband and the proband’s elder sister, mother, father, maternal uncle, maternal grandfather, and grandmother. The mother of the proband described that she had difficulty in stopping the bleeding during the delivery of her second child (the proband). In addition, the proband, his maternal grandfather, and maternal uncle showed a slightly long duration for hemostasis after injury. Other family members showed no spontaneous bleeding and thrombosis, as well as other abnormalities. The abdominal pain symptoms were due to inflammation of the mesenteric lymph nodes and were not associated with hypo-fibrinogenemia.
Peripheral venous blood specimens (5 mL) were collected from each participant. The partial collection of blood samples was carried out with sodium citrate as anticoagulation and was used for coagulation tests (2 mL), RNA extraction (500 μL), and DNA extraction (500 μL). The other samples (2 mL) were used for liver and kidney function determination. For coagulation tests, blood specimens were centrifuged at 3500 r/min for 15 min, and the supernatant was utilized for determination of prothrombin time (PT), activated partial thromboplastin time (APTT), thrombin time (TT), and D-dimer on an Sysmex CS-5100 analyzer (Sysmex, Kobe, Japan). Fibrinogen (Fg) was measured by the Clauss and PT-Fg methods. Serum liver and kidney functions were evaluated using a Beckman-Coulter AU-5800 auto analyzer (Beckman, Brea, CA, USA). As shown in Table 1, the proband exhibited a prolonged TT (26.8 s; reference interval: 15.0–22.0 s) and decreased Fg (0.5 g/L; Clauss method) and PT-Fg (1.55 g/L; PT-derived method) (reference interval: 1.8–4.0 g/L). The proband showed normal PT (12.2 s; reference interval: 9.0–14.0), APTT (23.8 s; reference interval: 23.0–38.0 s), and D-dimer (0.1 μg/mL; reference interval: < 0.55). The proband’s grandmother, maternal grandmother, father, and sister showed a normal coagulation phenotype. However, the proband’s mother, maternal uncle, and maternal grandfather showed increased TT and decreased Fg:Ac and PT-Fg, which was similar to that of the proband (Table 1). Routine blood examination revealed no abnormalities. No abnormalities were detected in the liver or kidney function of each participant.
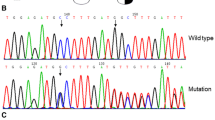
Genomic DNA was extracted from peripheral venous blood samples for DNA sequence analysis. Sanger sequencing revealed FGA c.180 + 1G > A variant in the proband, and FGA c.180 + 1G > A variant caused the splice site mutation (Fig. 1). FGA c.180 + 1G > A variant was identified in the proband’s mother, maternal uncle, and maternal grandfather. However, no variant was identified in the proband’s father, sister, grandmother, and maternal grandmother. These results suggested that the proband inherited FGA c.180 + 1G > A variant from the proband’s mother (Fig. 2). The proband’s mother and maternal uncle inherited the variant from the proband’s maternal grandfather (Fig. 2).

Sanger sequencing map

Pedigree of the family
Then FGA mRNA expression was compared between the proband and his sister. Initially, total RNA was extracted from the peripheral blood samples using the RNA extraction kit (Liferiver, Shanghai, China). The cDNA was reversely transcribed, and then PCR amplification was carried out in a total volume of 25 μL containing 2 μL cDNA, 0.5 μL each primer, 9.5 μL double-distilled water, and 12.5 μL TB green (TAKARA, Japan). Amplification was performed in a 7500 real-time PCR system (ABI, USA). The reaction included 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 34 s, and 95 °C for 15 s. PCR primers were FGA (forward 5′-CTCGCCCTGTCAGAGGTATC-3′ and reverse 5′-CTGCCCCCAAGGAACTTAC-3′) and GAPDH (forward 5′-AGATCCCTCCAAAATCAAGTGG-3′ and reverse 5′-GGCAGAGATGATGACCCTTTT-3′). FGA mRNA expression was calculated using the 2−ΔΔCT method. GAPDH was used as the housekeeping gene. As presented in Fig. 3, the mRNA expression of FGA was significantly decreased in the proband compared to the proband’s sister (P < 0.05).

mRNA expression of FGA in peripheral blood of the proband and healthy individuals
Discussion
Fibrinogen protein consists of two groups of Aα, Bβ, and γ-chains bridged by disulfide bonds and are an extended 45-nm structure [6]. Fibrinogen is synthesized by hepatocytes and secreted into the circulation, playing a crucial role in coagulation, fibrinolysis, cellular and matrix interactions, inflammatory responses, wound healing, and biological activity of tumor [7]. In this study, a novel splicing mutation FGA c.180 + 1G > A occurring in exon 2 and intron 2 boundaries was identified in a 4-year-old boy with congenital hypo-dysfibrinogenemia. There is no record of this mutation in the Fg gene mutation database (http://www.geht.org/databaseang/fibrinogen), so the mutation is considered to be an unreported mutation. The mutation caused a decrease in FGA transcription in the peripheral blood in the proband.
Mutations in the gene encoding fibrinopeptides, such as large deletions, promoter, splice site, frameshift, nonsense, and missense mutations, are reported to result in structural and functional changes in the fibrinogen [8]. Structural and functional abnormalities impede the release of fibrin peptide A/B, polymerization of fibrin monomers, or factor XIIIa-mediated fibrin cross-linking [9]. In this study, Sanger sequencing identified a splice site mutation of FGA c.180 + 1G > A in the proband. The proband’s mother, maternal uncle, and maternal grandfather had the FGA c.180 + 1G > A mutation, which was not detected in the proband’s father, sister, and maternal grandmother. Therefore, it was concluded that the FGA c.180 + 1G > A mutation in the proband was derived from the proband’s mother. Besides, the patient’s mother inherited the FGA c.180 + 1G > A mutation from the proband’s maternal grandfather. FGA c.180 + 1G > A mutation has not been annotated in either ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) or HGMD (http://www.hgmd.cf.ac.uk/ac/index.php).
Similar splicing mutations of FGA have been reported in several other literatures. Site-specific mutations in intron 1 (c.IVS1 + 1G > A and c.IVS1 + 3A > G) and in intron 3 (c.IVS3 + 1_ + 4delGTAA) affect the consensus sequence of the conserved donor splice site [10,11,12,13]. In addition, studies revealed three mutations in the same splice site GT (c.180 + 1G > C, c.180 + 1G > T, and c.180 + 2 T > C) that have been reported to be associated with hypo-fibrinogenemia [14, 15]. Further, a mutation site (FGA c.169_180 + 2del) was reported by Zhou et al. [16]. Unlike the present case this variant was due to deletion disrupting the splice consensus sequence GT at the junction of exon 2 and intron 2, resulting in disruption of normal processing of mRNA transcripts and impaired coagulation activity [16]. However, the expression and secretion of fibrinogen were not hindered [16]. Hypo-abnormal fibrinogenemia can be caused by different molecular mechanisms. Heterozygosity for a single mutation leads to the synthesis of an abnormal fibrinogen chain, which is secreted less efficiently than normal fibrinogen. Compound heterozygous mutation results in fibrinogen deficiency and abnormal molecular function [8]. More studies are required to investigate the pathogenicity and molecular pathogenic mechanisms of FGA c.180 + 1G > A.
Laboratory tests for PT, APTT, TT, and fibrinogen and genetic analysis are essential for the diagnosis of congenital hypo-dysfibrinogenemia. However, patients with congenital hypo-dysfibrinogenemia showed normal PT and APTT. TT is the primary screening indicator for the disease. Abnormal fibrinogen inhibits the release of fibrinopeptides A/B or impedes fibrin monomer polymerization, resulting in prolonged TT in patients [4]. However, a case of shortened TT due to abnormal fibrinogen has been reported previously [17]. The diagnosis of congenital hypo-dysfibrinogenemia can be further verified by the fibrinogen activity/antigen ratio. Clauss method and PT-Fg methods are the main methods to determine fibrinogen. The Clauss method was used to determine the functional activity of fibrinogen, while the PT-Fg method was used to determine the fibrinogen concentration indirectly through the reaction curve of PT. When Clauss Fg/PT-Fg was less than 0.7 or more than 1.43, the sensitivity and specificity for the diagnosis of dysfibrinogenemia was 100% [18, 19]. Clauss fibrinogen levels of the proband and the proband’s mother, maternal uncle, and maternal grandfather were less than 1.8 g/L. The PT-Fg tests yielded higher values than the Clauss results, and the degree of the disparity may be related to the calibrator and the turbidity of the assayed samples. The diagnosis of abnormal blood fibrinogen was then confirmed by excluding abnormal liver function. Confirmation of the diagnosis of congenital hypo-dysfibrinogenemia requires sequencing of the fibrinogen-encoding gene in patients and families. The results of RT-PCR showed that the mRNA expression of FGA in the peripheral blood of the proband was significantly reduced compared with that the proband’s sister, verifying that FGA c.180 + 1G > A mutation affected FGA transcription. This splicing site mutation causes a low level of FGA protein in the proband.
Mutant fibrinogen molecules are present in heterozygous forms in patients as homodimers (i.e., both chains are mutant) and heterodimers (i.e., one chain is mutant), resulting in differences in the ratio of homodimers to heterodimers and leading to clinical heterogeneity of dysfibrinogenemia [20]. Analysis of 102 cases of dysfibrinogenemia by Zhou et al. revealed that 68.6% of the patients displayed no clinical symptoms, 27.5% had bleeding manifestations, and 3.9% developed thrombosis [21]. Bleeding manifestations include nose bleeding, easy bruising, excessive menstruation, joint hematomas, postoperative bleeding, postpartum bleeding, antepartum bleeding, and delayed wound healing. Defective binding of abnormal fibrinogen to thrombin leads to elevated thrombin levels, and abnormal fibrinogen can form a fibrin clot that resists degradation by fibrinolytic enzymes, which may be the molecular mechanism that leads to thrombosis [15]. Therefore, female patients with dysfibrinogenemia are of particular clinical concern because the disease may lead to excessive menstrual bleeding, obstetric bleeding, and postpartum thrombosis.
Conclusions
Congenital dysfibrinogenemia is a clinical asymptomatic and rare disease, which may be likely to be misdiagnosed. In this study, the proband and the proband’s mother, maternal uncle, and maternal grandfather exhibited a prolonged TT and decreased Clauss Fg and PT-Fg, without abnormalities in the liver and kidney function. Sanger sequencing detected a novel mutation FGA c.180 + 1G > A in the proband and the proband’s mother, maternal uncle, and maternal grandfather. FGA c.180 + 1G > A variant caused the decreased mRNA expression of FGA. Clinical should conduct genealogical survey and genetic analysis to diagnose congenital hypo-dysfibrinogenemia.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Flood VH, Al-Mondhiry HA, Farrell DH (2006) The fibrinogen Aalpha R16C mutation results in fibrinolytic resistance. Br J Haematol 134(2):220–226
Koopman J, Haverkate F, Lord ST, Grimbergen J, Mannucci PM (1992) Molecular basis of fibrinogen Naples associated with defective thrombin binding and thrombophilia. Homozygous substitution of B beta 68 Ala––Thr. J Clin Invest. 90(1):238–44
Bantia S, Mane SM, Bell WR, Dang CV (1990) Fibrinogen Baltimore I: polymerization defect associated with a gamma 292Gly––Val (GGC––GTC) mutation. Blood 76(11):2279–2283
de Moerloose P, Casini A, Neerman-Arbez M (2013) Congenital fibrinogen disorders: an update. Semin Thromb Hemost 39(6):585–595
de Moerloose P, Neerman-Arbez M (2009) Congenital fibrinogen disorders. Semin Thromb Hemost 35(4):356–366
Mosesson MW (2005) Fibrinogen and fibrin structure and functions. J Thromb Haemost 3(8):1894–1904
Kaur J, Jain A (2021). Fibrinogen. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
Casini A, Neerman-Arbez M, Ariëns RA, de Moerloose P (2015) Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management. J Thromb Haemost 13(6):909–919
Siebenlist KR, Mosesson MW (1994) Progressive cross-linking of fibrin gamma chains increases resistance to fibrinolysis. J Biol Chem 269(45):28414–28419
Monaldini L, Asselta R, Malcovati M, Tenchini ML, Duga S (2005) The DNA-pooling technique allowed for the identification of three novel mutations responsible for afibrinogenemia. J Thromb Haemost 3(11):2591–2593
Neerman-Arbez M, de Moerloose P, Bridel C, Honsberger A, Schönbörner A, Rossier C et al (2000) Mutations in the fibrinogen aalpha gene account for the majority of cases of congenital afibrinogenemia. Blood 96(1):149–152
Neerman-Arbez M, de Moerloose P, Honsberger A, Parlier G, Arnuti B, Biron C et al (2001) Molecular analysis of the fibrinogen gene cluster in 16 patients with congenital afibrinogenemia: novel truncating mutations in the FGA and FGG genes. Hum Genet 108(3):237–240
Fang Y, Wang XF, Wang HL, Fu QH, Wu WM, Ding QL et al (2003) Genetic analysis of a Chinese family with inherited afibrinogenemia. Zhonghua Yi Xue Za Zhi 83(23):2054–2057
Neerman-Arbez M, de Moerloose P (2007) Mutations in the fibrinogen gene cluster accounting for congenital afibrinogenemia: an update and report of 10 novel mutations. Hum Mutat 28(6):540–553
Hanss M, Biot F (2001) A database for human fibrinogen variants. Ann N Y Acad Sci 936:89–90
周景艺. 遗传性异常纤维蛋白原血症的临床和分子发病机制研究: 上海交通大学; 2016
Cunningham MT, Brandt JT, Laposata M, Olson JD (2002) Laboratory diagnosis of dysfibrinogenemia. Arch Pathol Lab Med 126(4):499–505
Mosesson MW (1991) Index of variant human fibrinogens. Blood Coagulation & Fibrinolysis. 2(5):681–682
Xiang L, Luo M, Yan J, Liao L, Zhou W, Deng X et al (2018) Combined use of Clauss and prothrombin time-derived methods for determining fibrinogen concentrations: screening for congenital dysfibrinogenemia. J Clin Lab Anal 32(4):e22322
Galanakis DK, Henschen A, Peerschke EI, Kehl M (1989) Fibrinogen Stony Brook, a heterozygous A alpha 16Arg––Cys dysfibrinogenemia. Evaluation of diminished platelet aggregation support and of enhanced inhibition of fibrin assembly. J Clin Invest. 84(1):295–304
Zhou J, Ding Q, Chen Y, Ouyang Q, Jiang L, Dai J et al (2015) Clinical features and molecular basis of 102 Chinese patients with congenital dysfibrinogenemia. Blood Cells Mol Dis 55(4):308–315
Author information
Authors and Affiliations
Contributions
Jintu Lou contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Zhe Li, Wei Li, and Lin Chen. The first draft of the manuscript was written by Zhe Li and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of the Children’s Hospital, Zhejiang University School of Medicine.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Z., Lou, J., Li, W. et al. A newly detected c.180 + 1G > A variant causes a decrease of FGA transcription in patients with congenital hypo-dysfibrinogenemia. J Hematopathol 15, 259–263 (2022). https://doi.org/10.1007/s12308-022-00518-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-022-00518-3